
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Analysis of single fly ash particles using laser ablation ICP-MS – an approach achieving lateral elemental distribution information via imaging
G. Bauer ,
B. Achleitner,
M. Bonta,
G. Friedbacher and
A. Limbeck*
,
B. Achleitner,
M. Bonta,
G. Friedbacher and
A. Limbeck*
Institute of Chemical Technologies and Analytics, Division of Instrumental Analytical Chemistry, TU Wien, Getreidemarkt 9/164, 1060 Vienna, Austria. E-mail: andreas.limbeck@tuwien.ac.at
First published on 10th April 2017
Abstract
In environmental questions, particles at the micrometre scale are usually analyzed regarding their particle size and morphology using microscopic techniques. Thereby, their composition and trace element contents are neglected. However, when focusing on recycling and re-use of minor and trace elements in environmental particles like fly ash, originating from municipal waste incineration, information on elemental distribution within single particles may be of grave importance. In this work a procedure for the preparation of fly ash particles for LA-ICP-MS analysis is presented that also allows access to the spatial elemental distribution of each single particle. Herein the particles are dispersed and retained on a sticky substrate, followed by fixation. Roughly one half of the particle is ablated to access the particle cross-section. This cross-section is ablated with consecutive line scans to generate sufficient data to create an image. More than 60 particles with diameters between 25 μm and 100 μm have been analyzed using the presented procedure. General differences in the particle composition could be observed and the elemental distribution of minor constituents like Al, Cu, Pb, Si, Ti and Zn could be visualized for single particles with lateral resolutions down to 2 μm per pixel. Cu and Pb were found to be enriched at the surface, easily accessible for recycling techniques, whereas Ti was found predominantly in the particles' insides. Al, Si and Zn were mostly found to be evenly distributed within particles.
Introduction
When discussing single particle analysis with ICP-MS, usually particles at the nanometre scale are taken into consideration.1,2 In this kind of analysis, typically a suspension of the nanoparticles is measured, and the resulting signals are used to determine the nanoparticle's size and number distribution.3–5 However, with this traditional approach for single particle analysis it is challenging to gain information about the particle composition and entirely impossible to determine element distributions within the particles. When considering larger particles at the micrometre scale, such information may become of special interest to gain deeper knowledge about particle formation. Particles with such sizes are generated in various complex industrial but also environmental processes, leading to a much more inhomogeneous nature and composition than engineered nanoparticles.An area of interest, in which large amounts of such particles are produced, is municipal waste incineration (MWI). Residues from MWI are of major importance for various reasons. So far, scientific concern has been focused mostly on processes regarding air pollution, release of toxic elements,6–12 waste disposal and re-use for soil-improvements.13,14 But the scrap also still contains a relevant fraction of elements like rare earths or other metals valuable for industrial re-use15–17 and therefore is a potential source for recycling. To determine recycling potential and efficiency of such remains, they have to be investigated thoroughly with complementary analytical techniques. Up to now, mainly bulk concentrations were determined with ICP-MS techniques.9,18 However, for sufficient characterization, the analytes of interest need to be investigated with a special focus on their prevailing chemical form (e.g., metallic, oxide, sulphate, silicate) and also their relative distribution within the ash particles. Surface-enriched elements can be easily accessed and recovered with leaching, extraction or other processes involving heat, chemicals and/or mechanical procedures.19,20 In recent years also biotechnological methods have been applied.21,22
Already in the 1980s, leaching experiments have shown to deliver shell-selective chemical information on the particle composition.23 Even though very useful, also knowledge on the core material or even more detailed laterally resolved analysis would be of interest.
Two reasons can be identified for this lack of analytical data. Firstly, it is very delicate to prepare particulate samples for single particle analysis without compromising their composition and integrity. Fixation and isolation is mostly done by dispersing powders manually on a sticky medium (e.g., double sided tape).24 A typical problem of this approach is the complete transfer of a representative sample aliquot to the surface, without losing a relevant size or mass fraction; furthermore, reproducibility often cannot be guaranteed. Secondly, analytical techniques applied for particle characterization provide either sufficient spatial resolution or adequate sensitivity, but seldom both. For example, scanning electron microscopy with energy-dispersive X-ray spectroscopy (SEM-EDX) is often utilized25,26 because of its very good lateral resolution. The sensitivity, however, is not sufficient for adequately detecting minor and trace components. In addition, SEM-EDX is a surface sensitive technique, thus only a layer of some μm thickness can be investigated, making analysis of a particle's core impossible when the particles are some tens of micrometres in diameter. One powerful technique often applied for achieving lateral distribution information is secondary ion mass spectrometry (SIMS).27,28 Although SIMS offers excellent sensitivity and depth resolution, the suitability for analysis of fly ash particles is also limited. Besides long analysis times, crater effects and surface roughening are considered to be the major problems.
An analytical technique which offers also the possibility of depth profiling and laterally resolved analysis is Laser-Ablation inductively-coupled Plasma-Mass Spectrometry (LA-ICP-MS).29,30 The use of LA-ICP-MS for quantitative bulk-analysis of trace metals and minor constituents in a variety of environmental samples is frequently described in literature.31,32 The sensitivity of LA-ICP-MS ranges from sub ng g−1 to μg g−1 depending on applied instrumentation, experimental conditions and the elements of interest. For imaging purposes, sensitivity may be reduced due to reduction of ablated material to improve lateral resolution.31,33 In this work, an approach for the preparation of powdered fly-ash samples for spatially resolved analysis using LA-ICP-MS, a technique very well established for inorganic analysis, is presented.34–37 The procedure proposed in this work allows dispersion and fixation of particles without exposing them to harsh sample preparation conditions. After exhibition of cross sections using laser ablation, 2D-imaging of particles with diameters in the range of 25–100 μm could be achieved, indicating distinct differences in the composition of individual fly ash particles but also in the distribution of minor elements, such as Al, Ti and Zn within the particles.
Experimental
Instrumentation
A commercially available Nd:YAG nanosecond laser ablation system (NWR213, ESI, USA) equipped with a standard, fast-washout-cell provided by the manufacturer and suitable for imaging applications31,38–40 was used to ablate the samples. For analysis, the generated sample aerosol was transferred to a quadrupole ICP-MS (iCAP Q, Thermo Fisher Scientific, Bremen, Germany) using PTFE tubing. He (5.0) was used as ablation gas and mixed with a stream of Ar (5.0) as make-up gas prior to introduction into the ICP-MS.Optical microscopy (DM2500M, Leica, Germany) was used to verify sample preparation techniques and scanning electron microscopy (SEM, Quanta 200, FEI, USA) equipped with an energy dispersive X-ray spectrometer (EDX – Octane Pro Silicon Drift Detector, Ametek) was used to validate elemental distributions from laser ablation experiments for the bulk elements Si and Al.
Sample preparation
Fly ash samples were provided by Wien Energie, collected at Spittelau, Vienna's municipal waste incinerator. Samples were characterized regarding particle size distribution using a laser diffraction particle sizer (Mastersizer 2000, Malvern instruments). Most of the fly ash particles were smaller than 100 μm in diameter (d0.9 96.5 μm) with a median at 26.2 μm d0.5. Samples were stored at room temperature and dried (90 °C, 12 h) prior to preparation.Standard petri dishes were used as sample carriers. The petri dishes were coated with a layer of anti-static spray (Antistatik 100, Kontakt Chemie®) to retain the dispersed particles. 10 mg of dried sample was weighed into an in-house constructed glass spray chamber setup. A petri dish, prepared as previously described was placed at the exit orifice of the setup. Subsequently, a constant argon stream was used to disperse the particles inside the spray chamber and to deposit them onto the pre-treated petri dishes. After dispersion of the particles, a layer of hairspray (Drei Wetter Taft, Schwarzkopf) was added for fixation of the particles. The solvent was allowed to evaporate at room temperature. After another fixation step by hairspray and subsequent drying, the so prepared samples were directly analyzed with optical microscopy and LA-ICP-MS.
LA-ICP-MS analysis
Prior to analysis, a daily performance check was conducted by tuning for maximum 115In signal intensity while ablating NIST SRM 612 (trace elements in glass, National Institute of Standards and Technology, Gaithersburg, MD). After sample insertion, the ablation chamber was evacuated and consecutively flushed with He for 15 min. For analysis, the He flow rate was set to 750 mL min−1. Settings were optimized in preliminary experiments. The laser output energy was adjusted to yield optimal ablation behaviour (i.e., limited elemental fractionation as well as reducing surface damage and material re-deposition on adjacent sample areas). Before firing on the sample, the laser was allowed to warm up for 10 s to reach stable output intensity. Detailed instrumental parameters employed for sample analysis are shown in Table 1. Signals were recorded in transient signal mode with Qtegra software (version 1.5.1189.31) provided by Thermo Fisher Scientific.| Laser ablation system | NWR213, New Wave Research | ICP-MS | iCAP Q, Thermo Scientific |
|---|---|---|---|
| Type of laser | Nd:YAG 213 nm | RF power [W] | 1550 |
| Ablation mode | Raster (lines) | Carrier gas flow rate [L min−1] | 0.4 |
| Beam diameter [μm] | 5–20, 3/5 (imaging) | Plasma gas flow rate [L min−1] | 12 |
| Fluence [J cm−2] | 4.5–17 | Auxiliary gas flow rate [L min−1] | 0.8 |
| Frequency [Hz] | 4–20 | Acquisition time [ms] | 10 |
| Scan speed [μm s−1] | 2–5 | Cones | Ni |
| He gas flow [L min−1] | 0.75 | Measured isotopes | 27Al, 29Si, 48Ti, 63Cu, 66Zn, 115In, 208Pb |
LA-ICP-MS analysis of particles with diameters ranging from 25–100 μm was performed using two different procedures. For both approaches access to a cross section of the fly-ash particle is required. Thus, in a first step particles were ablated evenly to expose their cross section with a laser beam diameter exceeding the particle diameter. Usually ten to 25 laser shots with a fluence of 2.5 to 3 J cm−2 were sufficient.
The generated cross section of the particle was then analyzed using line scans with laser beam diameters ranging from 3 μm to 20 μm depending on the size of the investigated particle. Line scan parameters varied between 2 μm s−1 and 5 μm s−1 for scan speed and 4 Hz to 20 Hz for ablation frequency. Alternatively, for imaging purposes the prepared particle cross sections were analyzed by covering the whole surface area with adjacent line scans; laser beam diameters of 3 and 5 μm were used. The intensity data recorded for the measured line scan patterns were exported as csv files and processed with ImageLab (v.1.02, Epina GmbH, Pressbaum, Austria) as suggested by Bonta et al.38 All 2D maps had squared pixels with pixel sizes of 3 × 3 or 5 × 5 μm2, respectively.
Results & discussion
Sample preparation
To retrieve spatial information of particles via LA-ICP-MS or electron microscopy, the particulate sample needs to be transferred to a surface retaining the particles well enough to prevent them from breaking loose during laser irradiation or transfer into a vacuum (as necessary for SEM investigations). Moreover, the particles need to be isolated well enough for individual characterization. If particles are overlapping or agglomerating on the prepared surface, the achieved analytical information cannot be correlated with a specific particle. The development of sample preparation was done in two steps, focusing on particle dispersion and fixation.Prepared samples were investigated using optical microscopy. The quality of sample collection was determined by visual comparison focusing on two objectives: particles on the substrate needed to be isolated well enough for individual analysis and all particle sizes according to particle size distribution should be present. Conditions for dispersion and fixation were modified to achieve well retained, isolated particle sizes in all relevant fractions. Argon with flows between 0.5 and 3 bar were applied as carrier gas and a defined sample amount (5 to 100 mg) was scattered on the prepared mounting base. Best results were achieved when using 10 mg of sample and an Ar stream of 0.5 bar (see Fig. 1). Image analysis (using ImageJ 1.50i, National Institute of Health, USA) of samples after particle deposition was carried out to ensure particle size distribution being comparable with the initial sample. For the optimized conditions, a median of 23.4 μm was found (assuming circular particles), which is not significantly different from the median of 26.2 μm measured for the loose fly-ash particles. However, the larger particles do not seem to adhere quantitatively to the substrate, as the 0.9-percentile (d0.9) is shifted to 44.5 μm compared to 96.5 μm for the initial particles. Still it is possible to sample the particles of interest in a representative manner, which could be shown by quantitative image analysis.
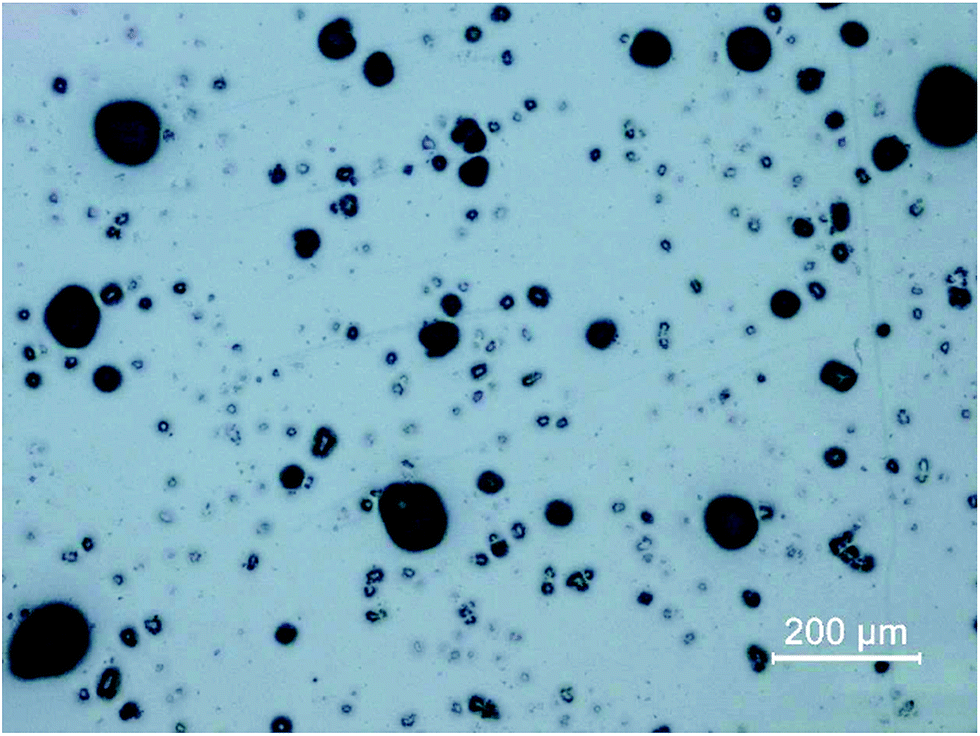 | ||
| Fig. 1 Optical microscopy image of a finalized sample medium, showing the fixation of isolated particles prior to LA-ICP-MS analysis with anti-static spray below the hairspray layer; dispersal of 10 mg of sample, 0.5 bar Ar stream; a representative sample area is shown. | ||
LA-ICP-MS analysis
One of the simplest approaches to achieve spatial information from solid samples is applying a drilling-mode type of analysis. By constantly ablating the surface, the laser beam is penetrating layer by layer, resulting in depth-resolved information when displaying the transient signal. In the present case, however, environmental particles with maximum diameters of 100 μm are the target instead of hard bulk materials like ceramics or metals. Drilling was applied to the particles as initial approach. Even though particles were completely embedded in the hairspray-matrix, they were destroyed in most cases when using a laser beam diameter smaller than the particle itself. This effect can be mainly explained by the usually very rigid nature of the particles, leading to uneven crater formation and breakdown of the outer particle structure when exposed to the laser beam. In addition, thermal effects, such as partial sample melting, could be observed, leading to blurred depth-profiles – a problem which is often encountered when performing direct drilling using ns-LA.41 Even if the number of destroyed particles could be reduced by using low-fluence ablation conditions, this procedure showed to reduce reproducibility of the drilling-mode measurements. Thus, drilling-mode experiments seemed not to offer favorable characteristics for the depth-resolved analysis of single fly-ash particles.Preparation of cross-sections
To overcome the limitations of drilling, a different approach was required. When analyzing materials or ceramics in metallographic analysis, the samples, including the embedding material, are usually cut to achieve a surface which represents the bulk distribution and can be analyzed directly. These newly exposed surfaces are then ground, polished and/or etched and investigated with microscopy or other techniques. In this work, a similar approach has been attempted to be established. However, the environmental particles, embedded in hairspray could not be cut mechanically or treated the same way as hard and stable metallographic samples. The size and mechanical stability of the fly ash particles needed to be considered. Additionally, analyte smearing and cross-contamination might become a problem when grinding these samples. Instead of using mechanical processes, established laser etching techniques like FIB (focused ion beam)42 could be used for particle cutting. Even though FIB is suitable to access cross-sections, when trying to cut larger particles, preparation of a representative number of particles would be extremely time consuming. To reduce time and cost, another method was established in this work. Virgin surfaces were exposed by ablating roughly half of the particle prior to the actual LA-ICP-MS analysis with the LA-laser system itself. Literature suggested good suitability of the 213 nm laser for this purpose.43 These freshly created surfaces could then be analyzed using a line scan pattern without requiring depth-resolved investigations, thus eliminating the problems with depth profiling described in the previous chapter. In Fig. 2 the access of a particle cross section is sketched out.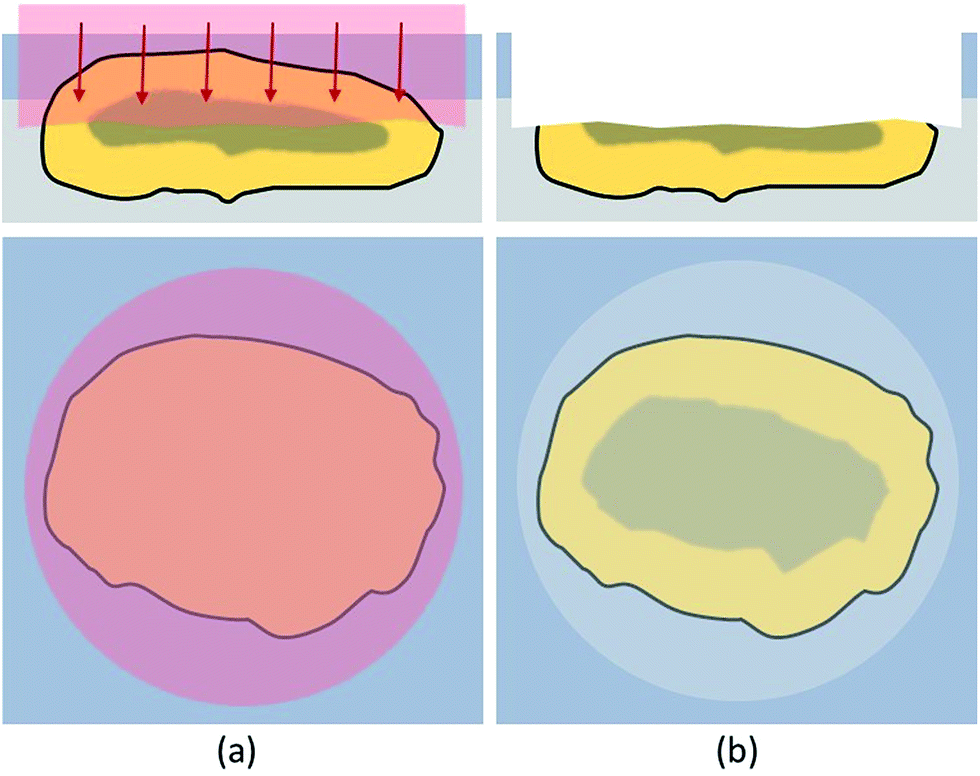 | ||
| Fig. 2 Schematic description of the cross-section mode; the embedded particle is ablated with a laser beam diameter, covering the complete particle diameter and using very mild ablation conditions (a); the newly created cross section surface is subsequently accessible (b). | ||
In this cross-section mode the top of the target particle was ablated with a spot size of 10 μm to 15 μm larger than the actual particle diameter, respectively. This procedure was found to be beneficial to evenly ablate the particle and create a horizontally oriented cross-section. Very sensitive conditions were used to prevent effects like melting or particle break-up which would compromise the bulk distribution information of the cross-sections. By applying 10 to 25 laser shots with a fluence of 2.5–3 J cm−2, depending on particle size, one third to a half of the particle top was removed, resulting in suitable cross-sections available for further analysis. The ablation conditions for accessing the cross-section were optimised by investigating SEM-pictures of ablated cross-sections.
It was initially expected that particles originating from a MWI are very inhomogeneous, which was confirmed by visual inspection of SEM images. When examining a SEM image, it can be observed, that some particles are conglomerates rather than a homogeneous bulk. An exemplary SEM image is shown in Fig. 3. Some areas of the particle appear as structures with a distinctly confined geometry (such as the areas marked by the black circle and the square in Fig. 3) while the major portion is constituted of a crumb-like material. These large inhomogeneities even within single particles already indicate the high complexity of the components of fly-ash. To investigate the origin of this observation, further chemical analysis of the particle's sub-structures is required.
 | ||
| Fig. 3 SEM image of fly ash sample cross-section. | ||
Sample analysis using a single line scan
Line scans were performed across the complete diameter of the particle, starting and ending in embedding medium. A schematic sketch is shown in Fig. 4. This approach allowed a simple and fast analysis of the exposed cross sections. Scan speed (5–40 μm s−1), ablation rate (1–10 Hz), and laser output energy were set to ensure best lateral resolution, depending on the dimensions of each individual particle. The fluence varied between 5 and 18 J cm−2 resulting in much harsher ablation conditions than used for accessing the cross-section.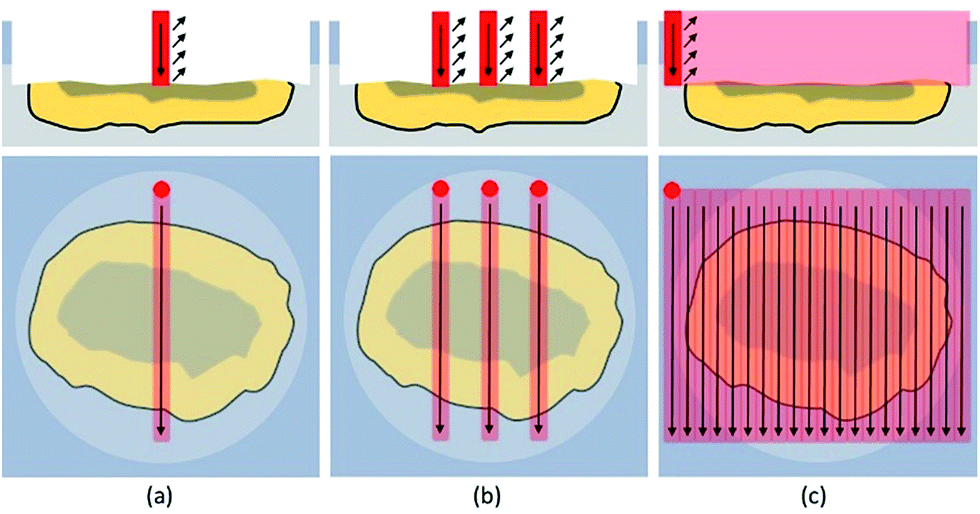 | ||
| Fig. 4 Schematic description of the analysis of the cross section by one single (a), three parallel line scans (b) and by covering the whole particle surface with a pattern of adjacent line scans (c). | ||
When analyzing particles with these parameters, a transient signal with the following course was expected: at first, background levels for target analytes, during ablation of the embedding medium. When reaching the exposed particle cross-section, analyte signals should increase based on the analytes present in the particle. Relative amounts as well as distributions could be derived from these scans. Finally, intensities should drop to background level when again ablating just the embedding material. Exemplary analysis results for one particle are shown in Fig. 5.
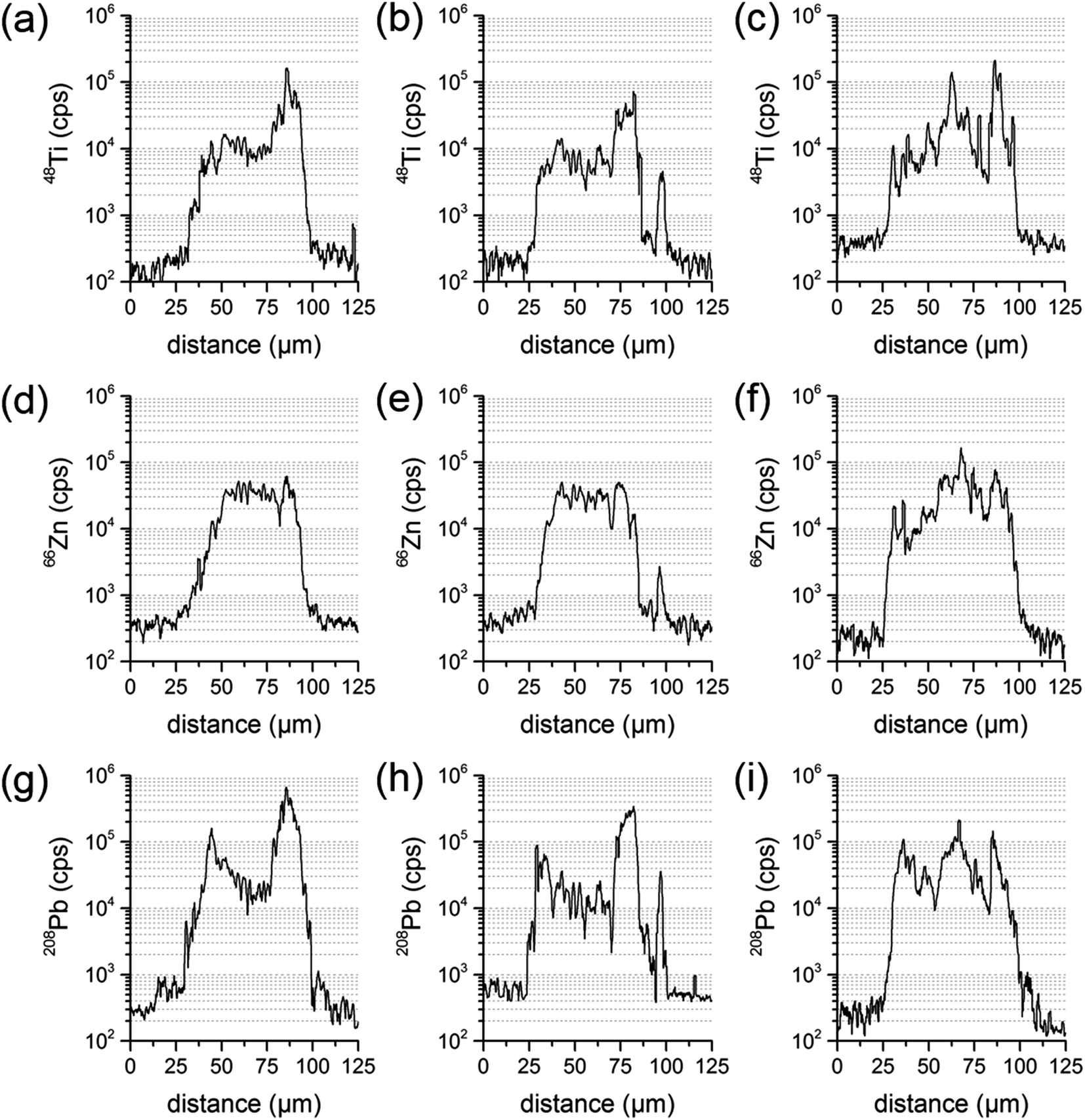 | ||
| Fig. 5 Transient signal courses of three parallel line scans of the same sample particle for the elements Ti (a–c), Zn (d–f) and Pb (g–i). | ||
68 particles were investigated with this cross-section approach. Three types of expected signal courses were found in the analyzed particles: homogeneous analyte distribution, completely inhomogeneous analyte distribution, as well as a core–shell-type signal pattern. Al was found to be evenly distributed in all measurements with a rather constant transient signal over the whole cross-section diameter. In some cases, Ti, Zn, Cu and Pb were significantly enriched in the shell and showed higher intensities in the transient signal (by a factor of 15 to 30) at the edges than at the centre. However, for these elements also signal courses without a specific pattern were achieved. Some particles were found with higher intensities at the edges and some even had maximum intensity values in the centre. Again others were evenly distributed for the named analytes.
Since LA-ICP-MS results are depending on the location analyzed at the sample particle cross-section, one line scan could be insufficient to derive the elemental composition of the whole particle. To investigate the influence of the actual line scan location, three parallel line scans were performed at one and the same particle cross-section. Distance between the central axes of the line scans was set to 15 μm. A particle where an inhomogeneous distribution has already been identified was chosen for that purpose. Transient signal courses differed distinctly for the three scans. The first measurement showed varying concentrations for Pb and Ti, with a higher Ti signal intensity at one edge of the particle, and an increase in Pb at both edges. The second measurement displayed similar signal courses for all analytes and a third line scan showed varying concentrations for Ti, Zn and Pb and signal courses not similar to the ones already measured. All line three scans are presented in Fig. 5.
These investigations were repeated with several other particles, always yielding comparable results. The conclusion is that the enrichment of some elements, especially at the shell, is a highly variable process. Real sample particles are not perfect spheres. Moreover, surface deposits during their formation may not be homogeneous. Most likely, due to condensation processes during the MWI ash generation, some elements coagulate at the particle surface in unevenly distributed areas of the particle. Assuming that analyte enrichment at the surface is not even in the whole shell, these differences are most likely not identified when using only one single line scan per target particle. Thus, single line scans are not suited for representative analysis of the particle composition.
Sample analysis using a line scan pattern – imaging
A different approach is required to investigate the whole cross-section in a comprehensive manner. One way is to completely ablate the particle cross-section with consecutive, adjacent line scans. The data can be further processed to generate a 2D image, showing elemental distributions on the analyzed particle cross section in a two-dimensional manner. A schematic sketch is shown in Fig. 4.For imaging purposes, the ablation parameters were modified to achieve optimum conditions. Scan speed and laser repetition rate were carefully optimized to achieve sufficient lateral resolution in one direction. The spot diameters were decreased to 5 μm (5–6 J cm−2) at first and later on to 3 μm (17–18 J cm−2) with consecutively increasing laser output to maintain a fluence of 10 J cm−2 for guaranteeing optimum resolution (i.e., minimal sample carry-over) between the consecutive line scans. Under optimized conditions, several particles with diameters ranging from 25 μm to 100 μm were analyzed. Results indicate that the analyzed particle pool is highly heterogeneous, consisting of numerous different particle types. Still, some trends could be observed. Al and Si were found to be evenly distributed not only along one lateral line but over most of the cross-section, depending on the nature of the particle. Sometimes higher concentrations (up to two orders of magnitude) were observed in areas with distinct geometrical shapes with regards to a corresponding SEM image. However, in some cases neither Al nor Si were found in the particle bulk. These particles were believed to be carbon-based, a theory which unfortunately cannot be tested with LA-ICP-MS due to sensitivity issues, but was later on confirmed by EDX analysis.
Cu, Pb and Zn were found in different distributions. These elements could be identified in locally enriched sites as well as spread over the whole cross-section. When spread, Cu, Pb and Zn were absent in the particle centre and distributed throughout the outer cross-section ring. Ti was only found in locally enriched, mostly central positioned sites when comparing the elemental distribution image to the SEM image (see Fig. 3 & 6). These 4 elements were found only in the crumb-like cross-section parts and were absent in all more confined and dense-appearing structures.
 | ||
| Fig. 6 SEM images of a cross-section of a fly ash particle; imaging provided by ImageLab, calculated from transient signal ICP-MS data. | ||
These findings show that particles with a difference in shell/core composition can be identified using the presented approach. To validate the found elemental distribution images, matching results with an established comparative analysis technique are required.
Method validation
Since SEM analysis has already been employed, the SEM-EDX elemental mapping feature was applied. With a sub-μm lateral resolution, this technique would be a perfect choice for high-resolution comparative measurements. A representative choice of particles was visually selected. The particles cross-sections were exposed using Laser Ablation, as previously described. Since EDX-analysis is a non-destructive analysis technique, SEM pictures and an EDX-mapping of the particles were done prior to LA-ICP-MS imaging. SEM-EDX analysis was conducted under high vacuum conditions with an operating distance of approximately 10 mm. The accelerating voltage was set to 10 kV. After completing the SEM-EDX measurements, the samples were transferred to LA-ICP-MS and ablated with imaging conditions. SEM-EDX as well as LA-ICP-MS signal data were both exported to the ImageLab software which was used to compare the obtained laterally resolved element information (see Fig. 7).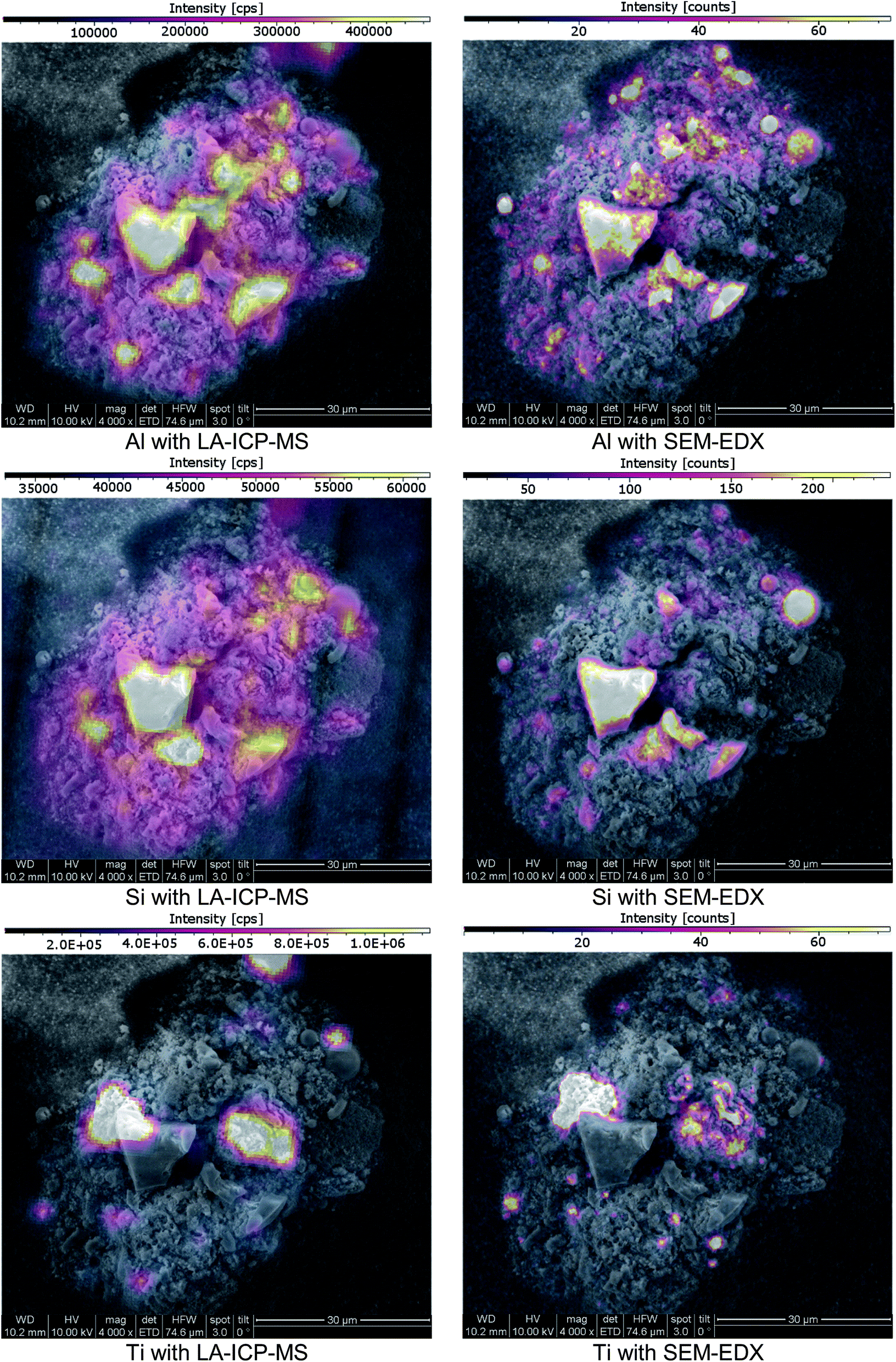 | ||
| Fig. 7 SEM images of a cross-section of a fly ash particle; imaging provided by ImageLab, calculated from transient signal ICP-MS data or EDX-mapping output data. | ||
The analyte choice for method validation was limited to elements with a sufficient concentration for EDX-mapping (sensitivities in the low %-range) and was restricted to those elements accessible with LA-ICP-MS; Al, Si, Ti and Zn met these requirements. When comparing LA-ICP-MS imaging and EDX-mapping of these elements, a very good agreement was found, as indicated in (Fig. 7). Some smaller discrepancies can be explained by the method differences between LA-ICP-MS and SEM-EDX. On the one hand LA-ICP-MS is a very sensitive technique but compared to SEM-EDX, lateral resolution is weaker, making the distribution images appear a bit blurrier. For example, when comparing Al the wider dynamic range of LA-ICP-MS measurements is obvious: even though the colour scale is proportional, EDX shows only areas with elevated concentrations whereas with LA-ICP-MS also minor amounts could be detected. On the other hand, SEM-EDX is able to resolve the lateral information to a much finer extent than LA-ICP-MS with a beam diameter of 3 μm. Therefore, average information from fine structures is sometimes compiled in single pixels of the LA images whereas local enrichments are rather resolved in the EDX images. Concerning carbon, valuable information was provided by EDX analysis which could not be covered by LA-ICP-MS. Furthermore, SEM-EDX also allows the analysis of oxygen, providing additional information about the prevailing general nature of the existing elements – metallic form, oxides or carbonates.
Conclusions
In this paper a method for achieving spatial information from particulate environmental samples by LA-ICP-MS imaging is presented. Sample preparation to finally exhibit cross-sections turned out to be a viable approach. In a first step, particles were cross-sectioned using mild laser ablation conditions followed by the ablation of adjacent consecutive line scans to generate 2D images of target analytes. Thereby, information about the lateral distribution of major and minor metallic constituents of fly ash particles could be obtained. The preparation process is adaptable and can be adjusted to the means necessary for complementary analysis techniques (e.g., SEM-EDX). With this method, particle sizes from 25 μm to 100 μm were investigated regarding their differences in elemental composition. The method was validated by comparison with SEM-EDX analysis. Al, Si, Ti and Zn were found in good agreement between both techniques considering analytical differences and limitations especially in sensitivity.A fly ash sample originating from MWI, was used for method development and validation. Even though more than 60 comparable particles were analyzed, no definitive trends regarding the particle composition and structure were found. Some particles revealed a rather homogeneous analyte distribution, for some particles a core–shell-type signal pattern was observed, and some particles showed a completely inhomogeneous analyte distribution. The investigated particles consisted either of a silicate or a carbon core. Elements like Cu and Pb were often enriched at the particle surfaces whereas Al, Si, and Zn were found to be evenly distributed as well as locally enriched. Ti was mostly found in confined sites inside the particle. As initially stated these results can be used to develop recycling approaches for the element(s) of interest. Derived from these results Cu and Pb can be easily accessed with surface recycling techniques (e.g. leaching). To recover Al, Si, and Zn, recycling needs to process the whole particle. For Ti no surface techniques are applicable because it is only found at the particles interior.
The method may also be suitable to assess toxicity and health risks originating from environmental particles. For that purpose, knowledge about surface composition is crucial. In this case the presence of Pb at the surface could be of interest. For further work in this field the choice of analytes needs to be amended regarding specific interests and a suitable approach for signal quantification has to be developed.
For future development, smaller particles may be accessible with further optimisation of the instrument configuration, considering wash-out cell volume and transfer line arrangement. The preparation approach could also be easily adapted to all kinds of substrates, depending on requirements of the particular analysis technique like transparency or conductivity.
Acknowledgements
The authors acknowledge the TU Wien University Library for financial support through its Open Access Funding Programme.Notes and references
- M. S. Jiménez, M. T. Gómez, E. Bolea, F. Laborda and J. Castillo, Int. J. Mass Spectrom., 2011, 307, 99–104 CrossRef.
- B. Meermann and F. Laborda, J. Anal. At. Spectrom., 2015, 30, 1226–1228 RSC.
- F. Laborda, E. Bolea and J. Jiménez-Lamana, Anal. Chem., 2014, 86, 2270–2278 CrossRef CAS PubMed.
- M. D. Montaño, J. W. Olesik, A. G. Barber, K. Challis and J. F. Ranville, Anal. Bioanal. Chem., 2016, 408, 5053–5074 CrossRef PubMed.
- F. Laborda, J. Jimenez-Lamana, E. Bolea and J. R. Castillo, J. Anal. At. Spectrom., 2011, 26, 1362–1371 RSC.
- M. H. Fulekar and J. M. Dave, Int. J. Environ. Stud., 1986, 26, 191–215 CrossRef CAS.
- D. K. Rigg, M. N. Wacksman, J. Iannuzzi, T. F. Baker and M. Adams, Integr. Environ. Assess. Manage., 2014, 11, 88–101 CrossRef PubMed.
- N. E. Carriker, D. S. Jones, S. J. Walls and A. R. Stojak, Integr. Environ. Assess. Manage., 2014, 11, 80–87 CrossRef PubMed.
- F. J. Kelly and J. C. Fussell, Atmos. Environ., 2012, 60, 504–526 CrossRef CAS.
- Y. E. Yudovich and M. P. Ketris, Int. J. Coal Geol., 2005, 62, 133–165 Search PubMed.
- P. J. A. Borm, Ann. Occup. Hyg., 1997, 41, 659–676 CrossRef CAS PubMed.
- Y. Zhang and E. Kasai, ISIJ Int., 2004, 44, 1457–1468 CrossRef CAS.
- M. S. Shaheen, P. S. Hooda and C. D. Tsadilas, J. Environ. Manage., 2014, 145, 249–467 CrossRef PubMed.
- R. P. Singh, A. K. Gupta, M. H. Ibrahim and A. K. Mittal, Rev. Environ. Sci. Bio/Technol., 2010, 9, 345–358 CrossRef CAS.
- C. Block and R. Dams, Environ. Sci. Technol., 1976, 10, 1011–1017 CrossRef CAS.
- P. Norris, C.-W. Chen and W.-P. Pan, Anal. Chim. Acta, 2010, 663, 39–42 CrossRef CAS PubMed.
- N. Dawle, S. K. Khadse, P. V. Patil and D. Panhekar, Int. J. Adv. Res., 2014, 2, 847–853 Search PubMed.
- P. Eze Chuks, O. Fatoba, G. Madzivire, M. Ostrovnaya Tatyna, F. Petrik Leslie, V. Frontasyeva Marina and N. Nechaev Alexander, Chem.-Didact.-Ecol.-Metrol., 2013, 18, 19 Search PubMed.
- A. S. Meawad, D. Y. Bojinova and Y. G. Pelovski, Waste Manag., 2010, 30, 2548–2559 CrossRef CAS PubMed.
- A. Akcil, F. Vegliò, F. Ferella, M. D. Okudan and A. Tuncuk, Waste Manag., 2015, 45, 420–433 CrossRef CAS PubMed.
- D. Mishra and Y.-H. Rhee, Current Research, Technology and Education Topics in Applied Microbiology and Microbial Biotechnology, 2010, 2, 1289–1292 Search PubMed.
- C. Brombacher, R. Bachofen and H. Brandl, Appl. Microbiol. Biotechnol., 1997, 48, 577–587 CrossRef CAS.
- L. D. Hansen and G. L. Fisher, Environ. Sci. Technol., 1980, 14, 1111–1117 CrossRef CAS.
- T. C. Miller, H. Langley DeWitt and G. J. Havrilla, Spectrochim. Acta, Part B, 2005, 60, 1458–1467 CrossRef.
- O. C. Lind, B. Salbu, K. Janssens, K. Proost, M. García-León and R. García-Tenorio, Sci. Total Environ., 2007, 376, 294–305 CrossRef CAS PubMed.
- J. Ribeiro, K. DaBoit, D. Flores, M. A. Kronbauer and L. F. O. Silva, Sci. Total Environ., 2013, 452–453, 98–107 CrossRef CAS PubMed.
- X. B. Cox, S. R. Bryan, R. W. Linton and D. P. Griffis, Anal. Chem., 1987, 59, 2018–2023 CrossRef CAS.
- J. Pisonero, B. Fernández and D. Günther, J. Anal. At. Spectrom., 2009, 24, 1145–1160 RSC.
- J. S. Becker, M. Zoriy, A. Matusch, B. Wu, D. Salber, C. Palm and J. S. Becker, Mass Spectrom. Rev., 2010, 29, 156–175 CAS.
- P. R. Mason and A. J. Mank, J. Anal. At. Spectrom., 2001, 16, 1381–1388 RSC.
- A. Limbeck, P. Galler, M. Bonta, G. Bauer, W. Nischkauer and F. Vanhaecke, Anal. Bioanal. Chem., 2015, 407, 6593–6617 CrossRef CAS PubMed.
- G. Bauer and A. Limbeck, Spectrochim. Acta, Part B, 2015, 113, 63–69 CrossRef CAS.
- S. F. Durrant and N. I. Ward, J. Anal. At. Spectrom., 2005, 20, 821–829 RSC.
- R. E. Russo, X. Mao, H. Liu, J. Gonzalez and S. S. Mao, Talanta, 2002, 57, 425–451 CrossRef CAS PubMed.
- N. S. Mokgalaka and J. L. Gardea-Torresdey, Appl. Spectrosc. Rev., 2006, 41, 131–150 CrossRef CAS.
- M. Resano, E. Garcia-Ruiz and F. Vanhaecke, Mass Spectrom. Rev., 2010, 29, 55–78 CAS.
- F. A. Orellana, C. G. Gálvez, M. T. Roldán and C. García-Ruiz, TrAC, Trends Anal. Chem., 2013, 42, 1–34 CrossRef CAS.
- M. Bonta, H. Lohninger, M. Marchetti-Deschmann and L. A. Limbeck, Analyst, 2014, 139, 1521–1531 RSC.
- J. Lear, D. Hare, P. Adlard, D. Finkelstein and P. Doble, J. Anal. At. Spectrom., 2012, 27, 159–164 RSC.
- J. S. Becker, U. Kumtabtim, B. Wu, P. Steinacker, M. Otto and A. Matusch, Metallomics, 2012, 4, 284–288 RSC.
- J. Pisonero and D. Günther, Mass Spectrom. Rev., 2008, 27, 609–623 CrossRef CAS PubMed.
- R. Steve and P. Robert, J. Micromech. Microeng., 2001, 11, 287 CrossRef.
- M. Guillong, I. Horn and D. Günther, J. Anal. At. Spectrom., 2003, 18, 1224–1230 RSC.
| This journal is © The Royal Society of Chemistry 2017 |