
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and in vitro leishmanicidal activity of novel [1,2,3]triazolo[1,5-a]pyridine salts†
Álvaro Martín-Montes‡
 a,
Rafael Ballesteros-Garrido‡bc,
Rubén Martín-Escolanoa,
Clotilde Marína,
Ramón Guitiérrez-Sánchezd,
Belén Abarca*b,
Rafael Ballesteros*b and
Manuel Sanchez-Moreno*a
a,
Rafael Ballesteros-Garrido‡bc,
Rubén Martín-Escolanoa,
Clotilde Marína,
Ramón Guitiérrez-Sánchezd,
Belén Abarca*b,
Rafael Ballesteros*b and
Manuel Sanchez-Moreno*a
aDepartamento de Parasitología, Instituto de Investigación Biosanitaria (ibs.GRANADA), Hospitales Universitarios de Granada/Universidad de Granada, Granada, Spain. E-mail: msanchem@ugr.es
bDepartamento de Química Orgánica, Facultad de Farmacia, Universidad de Valencia, Avda. Vicente Andrés Estellés s/n, Burjassot, 46100 Valencia, Spain. E-mail: belen.abarca@uv.es; rafael.ballesteros@uv.es
cICMol Instituto de Ciencia Molecular, Universidad de Valencia, C. Catedrático José Beltrán 2, Paterna, 46100 Valencia, Spain
dDepartment of Statistics, University of Granada, Severo Ochoa s/n, E-18071 Granada, Spain
First published on 9th March 2017
Abstract
Leishmaniasis remains a significant worldwide problem; it is of great interest to develop new drugs to fight this disease. Recently we described some [1,2,3]triazolo[1,5-a]pyridine compounds with significant leishmanicidal activity. The importance of water solubility in drug action made us realise that we could transform non charged triazolopyridines into charged analogues that could increase the degree of water solubility. With this objective we report here the synthesis of novel [1,2,3]triazolo[1,5-a]pyridinium salts 2–7 from triazolopyridines 1, and the study of their in vitro leishmanicidal activity. The activity was tested on Leishmania infantum, Leishmania braziliensis and Leishmania donovani parasites, using promastigote and intracellular amastigote forms. The cytotoxicity of the tested compounds on J774.2 macrophage cells was also measured. Five of the tested compounds (2b, 4a, 4c, 6, 7d) showed selectivity indexes higher than those of the reference drug Glucantime for the three Leishmania species. Moreover, the data on infection rate and on amastigotes showed that these compounds are the most active against the three Leishmania species. The changes in the excretion product profiles of parasites treated with the compounds were also consistent with substantial cytoplasmic alterations. On the other hand, the most active compounds were potent inhibitors of Fe-SOD in the three parasite species considered whereas their impact on human CuZn-SOD was low.
Introduction
Leishmaniases are a group of parasitic diseases caused by different protozoan species of the genus Leishmania, transmitted by phlebotomine sand flies. In humans, the disease is presented as three main clinical forms, depending on the involved species of Leishmania: cutaneous, mucocutaneous and visceral leishmaniasis, with the latter being the most severe and life-threatening form. Leishmaniases are prevalent in tropical and subtropical areas; they currently affect 98 countries with 12 million cases, and a further 350 million people are presently at risk; the annual incidence is estimated at 1–1.5 million cases of cutaneous disease and 500![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cases of visceral disease.1–4
000 cases of visceral disease.1–4
Treatment of leishmaniases has been based for many years on pentavalent antimonials, which are still the first-line drugs. The second-line drugs include pentamidine, amphotericin B, miltefosine and paromomycin, and more recently, sitamaquine has shown very promising properties.2,3,5,6
As in the case of other neglected tropical diseases, most of the current therapies are inadequate essentially due to several factors such as the low therapeutic indexes leading to high toxicities and unacceptable side-effects, the emergence of resistant parasites, high prices that are unaffordable for the affected countries, etc. These drawbacks of the current therapies, together with the fact that a vaccine is an unachieved goal, make the search for new drugs urgently needed.1,3,5 In the last few years, many compounds showing leishmanicidal properties have been reported in the literature, and furthermore, several potential drug targets have been proposed and validated.3,5,6 Nevertheless, owing to the low income of the affected population, investment in the development of new drugs against leishmaniases has not been financially attractive for pharmaceutical companies, and the interest of academic institutions is rather limited.3,5
1,2,3-Triazole compounds do not exist in natural substances but they have been widely investigated because they are considered privileged scaffolds in medicinal chemistry.7,8 This structure is present in molecules described to have antibiotic,9,10 antineoplastic,11,12 anti-HIV,13 or antifungal activities.14 Furthermore, some molecules containing 1,2,3-triazole structure have been recently described as antileishmanial agents.15–19 In the last years, several compounds containing 1,2,3-triazole scaffold, such as quinolone triazoles,20 triazole naphthalimides,21 or triazoloacridones,22 able to interact with DNA have been described. The anticancer activity of the latter family of compounds is remarkable.23
During last years, the research of some of us has been focused on the functionalization of [1,2,3]triazolo[1,5-a] pyridines 1 and on the study of the potential applications of the derivatives obtained.24 Related of its possible biological activity, recently we have reported the first [1,2,3]triazolo[1,5-a]pyridine compounds with leishmanicidal activity, their interaction with DNA could be a mechanism to explain the activity found.25 We have also studied some triazolopyridopyrimidines, a fluorescent family of compounds, that have demonstrated to present photoinduced DNA cleavage, and antiprotozoal activity against different types of Leishmania spp.26 Moreover, in our group we have synthesized a series of triazoles known as [1,2,3]triazolo[1,5-a]pyridyl pyridyl ketones, that by the presence of the carbonyl group, being able to be reduced and form a free radical that could interact with molecular oxygen generating ROS. As Leishmania species as vulnerable to the effects of ROS we considered interesting to study the leishmania activity of these compounds. The results showed that the series are reduced electrochemically at radical species that were observed inside the parasite by ESR. Furthermore, the compounds are able to affect the metabolic pathway of ergosterol indicating mainly in CYP51 inhibition where enzyme lanosterol increased metabolite was observed. The antiproliferative effect would be also related to the inhibition of this pathway.
Supported by these hopeful results we decided to design a new series of compounds containing the triazolopyridine scaffold to explore its potential activity as anti Leishmania drugs. The importance of water solubility in drug action made us think to develop triazolopyridine analogues that they could increase the degree of water solubility. A general method to improve this property is the formation of salts. It is known that the triazolopyridines can make quaternary salts.27 We report here the synthesis of new triazolopyridine disalts 2–6 and monosalts 7 (Scheme 1), and the study of their Leishmanicidal activity in vitro against L. infantum, L. donovani and L. braziliensis (promastigote and amastigote forms) as representative species causing visceral and cutaneous leishmaniasis, respectively; unspecific cytotoxicity against mammalian cells of all compounds, as well as infectivity assays for compounds, were carried out using macrophages. Furthermore, a 1H NMR study has been conducted in order to observe changes in the nature and percentage of metabolites excretion directed to obtain information about the effect of our compounds on the glycolytic pathway of parasites; finally, we have also studied inhibition of the parasitic Fe-SOD, and human CuZn-SOD enzymes was tested and compared.
 | ||
| Scheme 1 | ||
Results and discussion
Chemistry
The synthesis of triazolopyridines salts 2–7 has been carried out from [1,2,3]triazolo[1,5-a]pyridine 1a and 3-methyl-[1,2,3]triazolo[1,5-a]pyridine 1b, that are easily prepared from commercial products.28 The disalts 2–6 were prepared by a reaction between the corresponding triazolopyridine (1a, 1b) (4 eq) and a dibromo compound (1 eq). Similar procedure was applied for the preparation of compounds 7, in this case an excess of a monobromo derivative was employed. The alkylation of triazolopyridines 1, in all the cases studied have been regioselective in N2,27 as shown in compound 4a by NOEDIF studies (see ESI†) (Scheme 1). Purification involved three consecutives cleanings with dimethylformamide and three more with ethyl acetate. Then the corresponding salts were dried under vacuum. This methodology ensured the complete removal of alkylating agents and triazolopyridines which are soluble, compared to the salts. Purity was confirmed by NMR and elementary analysis.Biological evaluation
We report now on the results obtained concerning the toxic activity of the triazolopyridines salts 2–7 against three species of Leishmania (L. infantum, L. braziliensis and L. donovani).In vitro antileishmanial evaluation
We measured in vitro biological activity of 21 triazolopyridine derivatives on both extra- and intracellular forms of the parasites. Extracellular forms are used more commonly due to ease of working with them, but are less indicative of leishmanicidal activity. Use of intracellular forms is more laborious but allow us to obtain more accurate results, as they are transformed into amastigotes in vertebrate host cells.29 Intracellular assays were performed by infecting macrophage cells with promastigotes, which transformed into amastigotes within 1 day after infection. Table 1 shows the IC50 values obtained after 72 h of exposure to L. infantum, L. braziliensis and L. donovani species.| Compounds | IC50a μM | Toxicity IC50 macrophage (μM) | |||||
|---|---|---|---|---|---|---|---|
| Leishmania infantum | Leishmania braziliensis | Leishmania donovani | |||||
| Promastigote forms | Amastigote forms | Promastigote forms | Amastigote forms | Promastigote forms | Amastigote forms | ||
| a Results are averages of four separate determinations. IC50: is the concentration required to give 50% inhibition, calculated by linear regression analysis from the Kc values at the concentrations employed (1 to 2000 μM).b Results are averages of four separate determinations. Against J774.2 macrophages after 72 h of culture. | |||||||
| Glucantime | 18.0 ± 1.3 | 24.2 ± 2.6 | 25.6 ± 1.7 | 30.4 ± 6.1 | 22.1 ± 2.3 | 18.4 ± 2.8 | 15.2 ± 1.0 |
| 1a | 209.7 ± 8.9 | Nd | 176.2 ± 7.9 | Nd | 263.1 ± 16.8 | Nd | 812.4 ± 70.2 |
| 1b | 56.8 ± 5.6 | Nd | 63.2 ± 2.5 | Nd | 64.7 ± 0.7 | Nd | 916.7 ± 37.5 |
| 2a | 245.0 ± 17.3 | Nd | 122.8 ± 7.3 | Nd | 429.7 ± 17.4 | Nd | 843.6 ± 66.8 |
| 2b | 10.6 ± 0.9 | 15.8 ± 1.7 | 15.7 ± 0.8 | 19.7 ± 0.8 | 19.3 ± 0.9 | 13.2 ± 1.8 | 693.9 ± 54.7 |
| 2c | 230.4 ± 11.5 | Nd | 227.5 ± 4.1 | Nd | 233.7 ± 18.2 | Nd | 804.7 ± 63.8 |
| 2d | 263.8 ± 28.6 | Nd | 89.6 ± 5.5 | Nd | 84.4 ± 1.1 | Nd | 1538.3 ± 100.2 |
| 3a | 315.9 ± 17.2 | Nd | 261.7 ± 12.3 | Nd | 302.5 ± 15.3 | Nd | 774.3 ± 36.9 |
| 3b | 275.1 ± 15.3 | Nd | 115.9 ± 6.9 | Nd | 215.8 ± 7.8 | Nd | 806.3 ± 22.8 |
| 3c | 200.1 ± 8.7 | Nd | 207.5 ± 10.0 | Nd | 159.5 ± 10.0 | Nd | 636.3 ± 40.9 |
| 4a | 15.2 ± 1.2 | 20.7 ± 3.6 | 16.8 ± 0.6 | 14.6 ± 2.6 | 67.2 ± 3.0 | Nd | 942.9 ± 33.7 |
| 4b | 215.3 ± 8.8 | Nd | 253.7 ± 13.5 | Nd | 157.8 ± 4.7 | Nd | 408.5 ± 15.8 |
| 4c | 12.3 ± 3.2 | 7.8 ± 3.8 | 54.8 ± 2.1 | Nd | 67.5 ± 2.4 | Nd | 683.1 ± 37.5 |
| 4d | 109.3 ± 5.6 | Nd | 179.3 ± 6.8 | Nd | 227.9 ± 5.9 | Nd | 1445.6 ± 77.4 |
| 5a | 85.1 ± 7.3 | Nd | 72.0 ± 4.9 | Nd | 155.4 ± 1.9 | Nd | 1236.7 ± 88.5 |
| 5b | 64.6 ± 4.4 | Nd | 58.4 ± 3.6 | Nd | 50.4 ± 1.6 | Nd | 743.6 ± 23.4 |
| 5c | 115.4 ± 7.1 | Nd | 118.7 ± 7.9 | Nd | 162.9 ± 12.4 | Nd | 993.5 ± 47.3 |
| 6 | 12.5 ± 3.1 | 8.7 ± 2.0 | 5.3 ± 0.8 | 6.3 ± 1.1 | 13.4 ± 2.5 | 9.1 ± 0.7 | 755.3 ± 56.9 |
| 7a | 186.5 ± 8.5 | Nd | 180.4 ± 8.4 | Nd | 215.2 ± 35.8 | Nd | 1724.6 ± 43.8 |
| 7b | 66.6 ± 7.6 | Nd | 62.9 ± 1.5 | Nd | 86.8 ± 4.1 | Nd | 1223.0 ± 118.5 |
| 7c | 71.5 ± 2.5 | Nd | 63.9 ± 0.3 | Nd | 57.6 ± 3.6 | Nd | 1023.6 ± 66.4 |
| 7d | 17.3 ± 2.7 | 11.6 ± 1.6 | 6.3 ± 1.7 | 9.5 ± 2.5 | 204.4 ± 12.6 | Nd | 1435.7 ± 81.1 |
Toxicity values against J774.2 macrophage after 72 h of culture were also calculated, and the selectivity indexes (SI = IC50 macrophages toxicity/IC50 activity of extracellular or intracellular forms of the parasite) are shown in the Table 2. Results obtained for the reference drug Glucantime were included for comparison.
| Compounds | SI50a μM | |||||
|---|---|---|---|---|---|---|
| Leishmania infantum | Leishmania braziliensis | Leishmania donovani | ||||
| Promastigote forms | Amastigote forms | Promastigote forms | Amastigote forms | Promastigote forms | Amastigote forms | |
| a Selectivity index = IC50 macrophages toxicity/IC50 activity on extracellular or intracellular forms of the parasite. In brackets: number of times the compound SI exceeded the reference drug SI. | ||||||
| Glucantime | 0.8 | 0.6 | 0.6 | 0.6 | 0.7 | 0.8 |
| 1a | 4 (5) | Nd | 5 (8) | Nd | 3 (4) | Nd |
| 1b | 16 (20) | Nd | 14 (23) | Nd | 14 (20) | Nd |
| 2a | 3 (4) | Nd | 7 (11) | Nd | 3 (5) | Nd |
| 2b | 65 (82) | 44 (73) | 44 (74) | 35 (59) | 36 (51) | 53 (66) |
| 2c | 3 (4) | Nd | 3 (6) | Nd | 3 (5) | Nd |
| 2d | 6 (7) | Nd | 17 (28) | Nd | 18 (26) | Nd |
| 3a | 2 (3) | Nd | 3 (5) | Nd | 2 (4) | Nd |
| 3b | 3 (4) | Nd | 7 (12) | Nd | 4 (5) | Nd |
| 3c | 3 (4) | Nd | 3 (5) | Nd | 4 (6) | Nd |
| 4a | 62 (77) | 45 (76) | 56 (93) | 65 (108) | 14 (20) | Nd |
| 4b | 2 (2) | Nd | 2 (3) | Nd | 3 (4) | Nd |
| 4c | 55 (69) | 88 (146) | 12(21) | Nd | 10 (14) | Nd |
| 4d | 13 (16) | Nd | 8 (13) | Nd | 6 (9) | Nd |
| 5a | 14 (18) | Nd | 17 (29) | Nd | 8 (11) | Nd |
| 5b | 11 (14) | Nd | 13 (21) | Nd | 15 (21) | Nd |
| 5c | 9 (11) | Nd | 8 (14) | Nd | 6 (9) | Nd |
| 6 | 60 (75) | 87 (145) | 143 (237) | 120 (200) | 56 (80) | 83 (104) |
| 7a | 9 (12) | Nd | 9.6 (16) | Nd | 8 (11) | Nd |
| 7b | 18 (23) | Nd | 19 (32) | Nd | 14 (20) | Nd |
| 7c | 14 (18) | Nd | 16 (27) | Nd | 18 (26) | Nd |
| 7d | 83 (104) | 124 (206) | 228 (380) | 42 (71) | 7 (10) | Nd |
It was shown that leishmanicidal activities against both the extra- and intracellular forms of the parasites by the some triazolopyridines salts were higher than those seen with Glucantime.
However, more interesting are the toxicity values in mammalian cells, since all compounds tested were found to be much less toxic for macrophages than the reference drug Glucantime. Their values range from 101-fold for compound 2d to 27-fold for compound 4b, less toxic than the parasites by the some triazolopyridines salts were higher than those seen with Glucantime. Toxicity values substantially influence the more informative selectivity index (SI) Table 2 values, so the best values were again obtained by compounds 2b, 4a, 4c, 6, and 7d, in tests performed on L. infantum species.
SI exceeded that of the reference drug by 82- and 73-fold for the extra- and intracellular forms in the case of 2b, by 77- and 76-fold with 4a, 69-fold and 146-fold with 4c, 75-fold and 145-fold with 6, 104-fold and 206-fold with 7d for L. infantum, whereas for L. braziliensis the respective values obtained were 74- and 59-fold with 2b, 93- and 108-fold with 4a, 237- and 200-fold with 6, 380- 71-fold with 7d for the extra- and intracellular forms, respectively, and in the case of L. donovani similar results were obtained: 51- and 66-fold with 2b, 80- and 104-fold with 6, respectively. It should be noted that a SI value more than 20-fold that of the reference drug is one of the usual basic criteria for considering primarily screened compounds as candidates for more advanced testing in vitro and in vivo animal models.30
The tests described above represent only a first crude approach to the leishmanicidal properties of the compounds assayed. In order to gain better insight into the activity of compounds, their effect on the infectivity and intracellular replication of amastigotes was subsequently determined. In accordance with the usual working procedure, compounds without an IC50 value lesser or circa 10 μM and with a SI smaller than 20-fold of the reference drug were not included in this second stage. In this sense the following compounds were selected for the case of L. infantum 2b, 4a, 4c, 6 and 7d, for L. braziliensis 2b, 4a, 6 and 7d, and in the case of L. donovani 2b and 6.
Infectivity assays
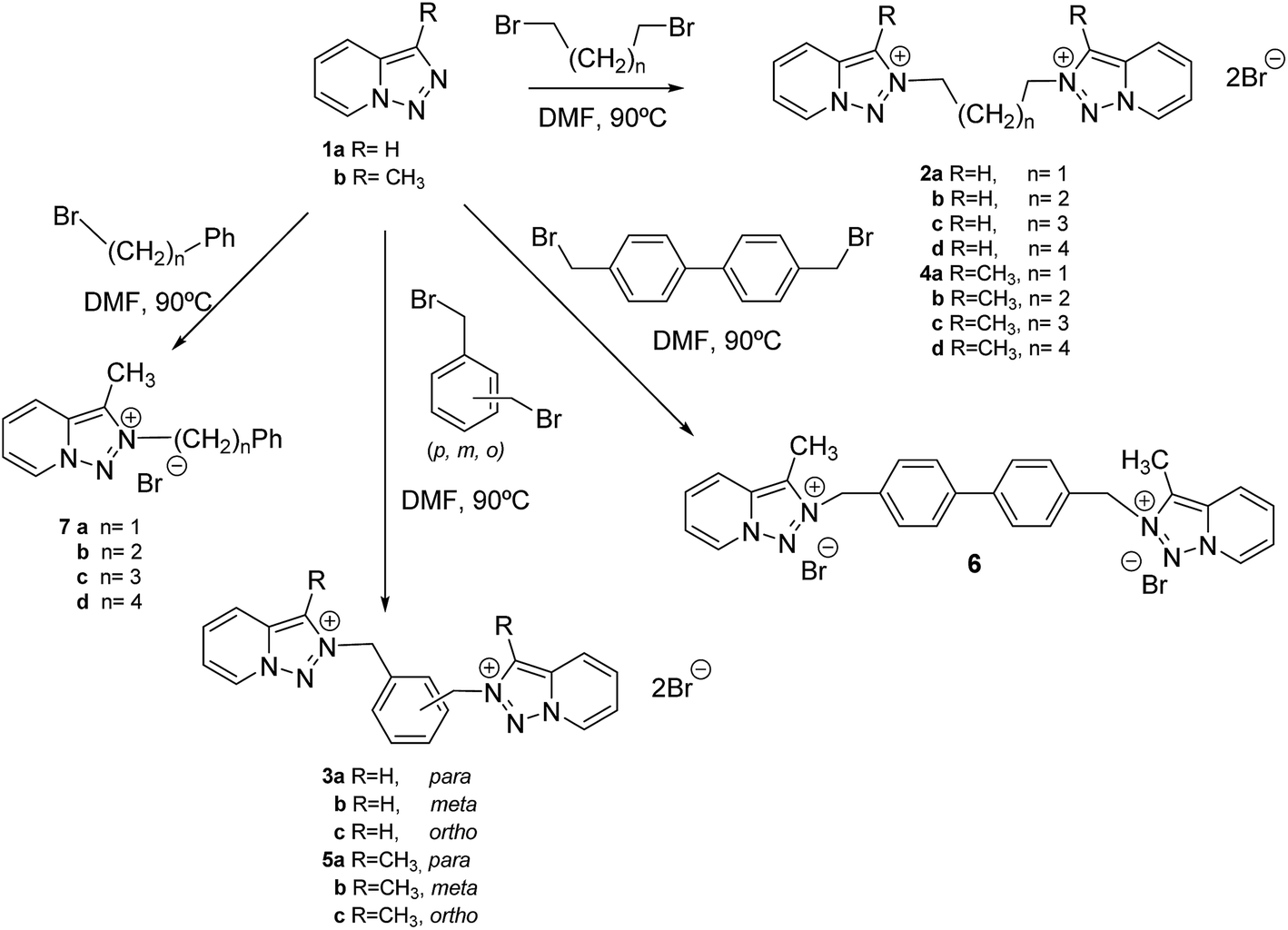
Macrophage cells were grown and infected with promastigotes in the stationary phase. The parasites invaded the cells and underwent morphological conversion to amastigotes within 1 day after infection. On day 10, the rate of host cell infection reached its maximum (control experiment). We used the IC25 of each product as the test dosage.As shown in Fig. 1A, when triazolopyridine salts were added to macrophages infected with L. infantum promastigotes, the infection rate decreased significantly with respect to the control and, furthermore, the six compounds were also more effective in decreasing infectivity than Glucantime (74%, 73%, 72%, 59% and 51% for 2b, 6, 4c, 4a and 7d respectively, opposite to 29% for the reference drug). A measure of the average number of amastigotes per infected macrophage (Fig. 1B) led to similar conclusions: all compounds were more effective than Glucantime and their order of effectiveness followed the same pattern as that seen in the infectivity measures, although the differences between them were less pronounced (48%, 46%, 52%, 39% and 36% for 2b, 6, 4c, 4a and 7d respectively).
 | ||
| Fig. 1 Reduction of the infection of L. infantum in macrophage cells treated with Glucantime and triazolopyridine salts. (A) Rate of infection, (B) means number of amastigotes per infected macrophage cells. Measured at IC25. Values are the means of the three separate experiments. | ||
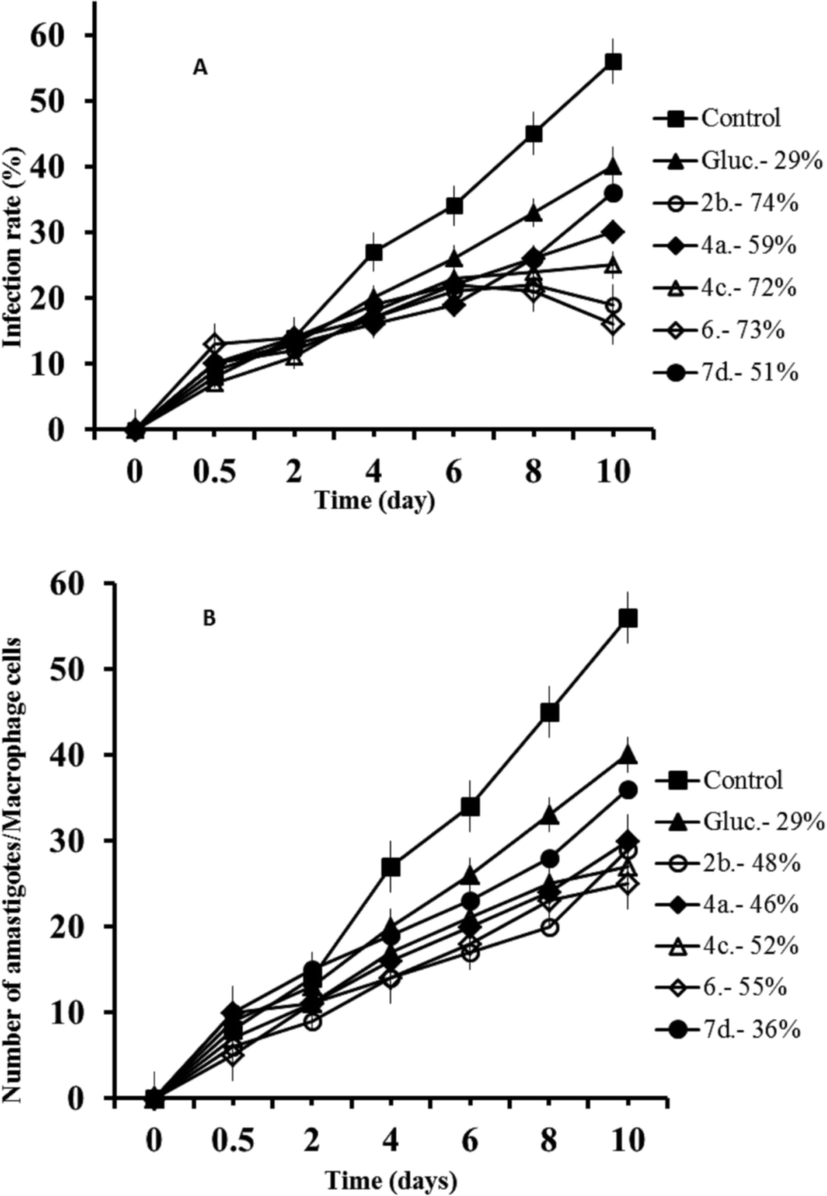
The same experiment was performed with L. braziliensis, and the results obtained concerning infection rates (a) and amastigote numbers (b) can be observed in Fig. 2. In both cases, the compounds were also more effective than Glucantime, and also in both cases the order of effectiveness was now 6 > 2b > 4a > 7d, and since the infectivity rates calculated from Fig. 2A were: 77%, 67%, 63% and 54% respectively, and 37% for Glucantime; and the decrease in amastigote numbers was: 69%, 52%, 39%, 21% and 11% for 6, 7d, 2b, 4a, and Glucantime, respectively (Fig. 2B).
 | ||
| Fig. 2 Reduction of the infection of L. braziliensis in macrophage cells treated with Glucantime and triazolopyridine salts. (A) Rate of infection, (B) means number of amastigotes per infected macrophage cells. Measured at IC25. Values are the means of the three separate experiments. | ||
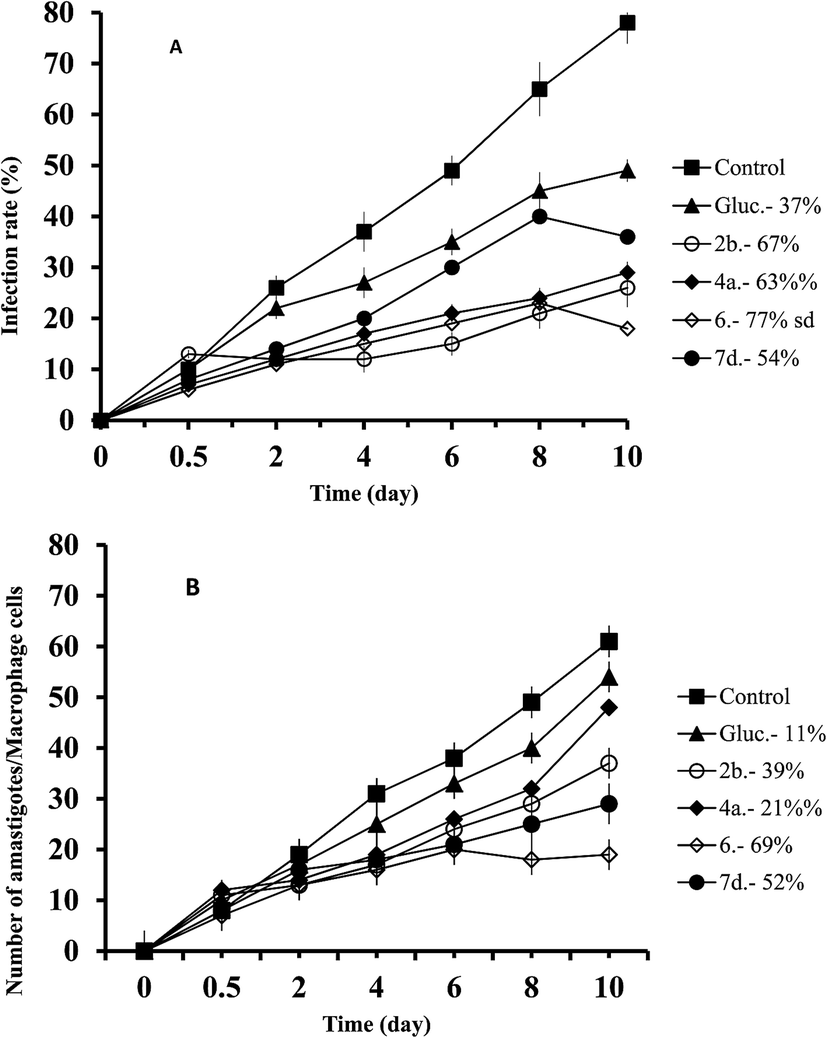
In the case of L. donovani, the order of effectiveness obtained were (Fig. 3): 6 > 2b > Glucantime (Fig. 3A) with the infectivity values of: 76%, 55 and 28% respectively, and the decrease in amastigote (Fig. 3B) numbers was: 66%, 61%, and 38% for 6, 2b and Glucantime, respectively.
 | ||
| Fig. 3 Reduction of the infection of L. donovani in macrophage cells treated with Glucantime and triazolopyridine salts. (A) Rate of infection, (B) means number of amastigotes per infected macrophage cells. Measured at IC25. Values are the means of the three separate experiments. | ||
Metabolite excretion
Since trypanosomatids are unable to completely degrade glucose to CO2, they excrete a considerable portion of its hexose skeleton as partially oxidised fragments in the form of fermented metabolites, whose nature and percentage depend on the pathway used for glucose metabolism.31,32 The catabolism products in Leishmania are usually CO2, succinate, acetate, pyruvate, D-lactate, L-alanine, and, to a minor grade, ethanol.33 Detection of large amounts of succinate as a major end product is an usual feature, because it rules on glycosomal redox balance, enabling reoxidation of NADH produced in the glycolytic pathway. Succinic fermentation requires only half of the phosphoenolpyruvate produced to maintain the NAD+/NADH balance, and the remaining pyruvate is converted inside the mitochondrion and the cytosol into acetate, D-lactate, L-alanine, or ethanol according to the degradation pathway followed by each species.34In order to obtain some information concerning the effect of the tested compounds on glucose metabolism in the parasites, we registered the 1H NMR spectra of promastigotes from L. infantum, L. braziliensis and L. donovani after treatment with the selected compounds triazolopyridine salts, and the final excretion products were identified qualitatively and quantitatively. The results were compared with those found with promastigotes maintained in a medium (control) for three days after inoculation with the parasite. The characteristic presence of acetate, D-lactate, succinate and L-alanine was confirmed in the control experiments performed on the three species. As expected, succinate and acetate were the most abundant end products identified. However, after treatment of the parasites with compounds, the excretion of catabolites was substantially altered at the dosages employed (IC25). Fig. 4–6 displays these modifications with respect to the control observed at the height of the spectral peaks corresponding to the most representative final excretion products.
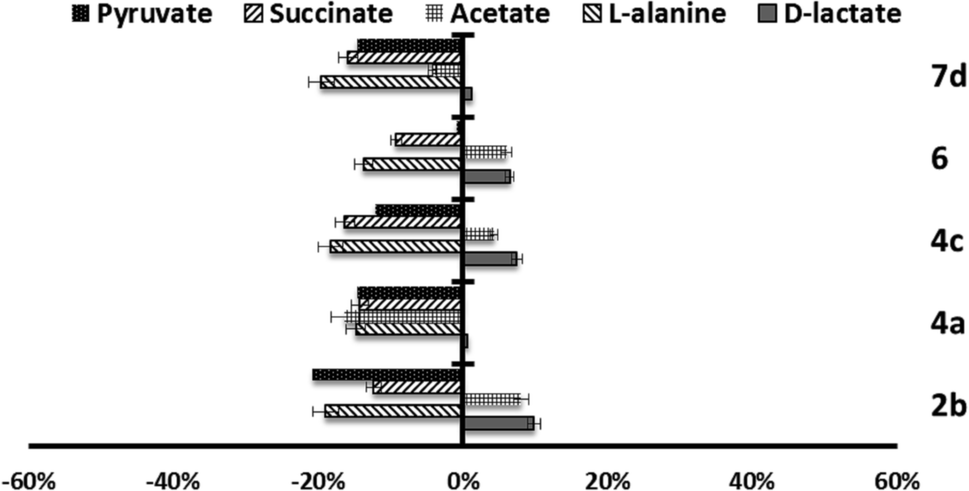 | ||
| Fig. 4 Variation percentages in the area of the peaks corresponding to excreted catabolites by L. infantum promastigotes in the presence of compounds 2b, 4a, 4c, 6 and 7d at their IC25 compared to a control sample after 96 h of incubation. | ||
Marked differences in the catabolic pathway appeared, and that seemed to be connected with the leishmanicidal activity commented above. Mainly, each compound has a certain effect in catabolite production. In the case of L. infantum (Fig. 4), compounds 2b, 4c and 6, trigger an increase in the production of acetate and D-lactate. It is well known that acetate, D-lactate, L-alanine and ethanol originate from the transformation of PEP in pyruvate in the presence of pyruvate kinase or pyruvate phosphate dikinase.32 Therefore, it seems possible that these compounds were interacting with the pyruvate kinase enzymes and modifying the glucose metabolism of the parasite at the pyruvate stage.32 Compounds 4a and 7d do not trigger the increase of those catabolites, instead, there is a remarkable reduction in their production.
On the other hand, in the case of L. braziliensis (Fig. 5), the action of the compounds is different. All excreted metabolites: pyruvate, acetate, D-alanine and D-lactate, yield an enhanced production as mentioned above for compounds 2b, 4c and 6 in L. infantum. This effect also occurs in compound 2b for L. donovani (Fig. 6) what makes us think that those compounds are acting over pyruvate kinase in L. braziliensis and L. donovani. In the case of L. donovani catabolite production is strongly inhibited by compounds 6.
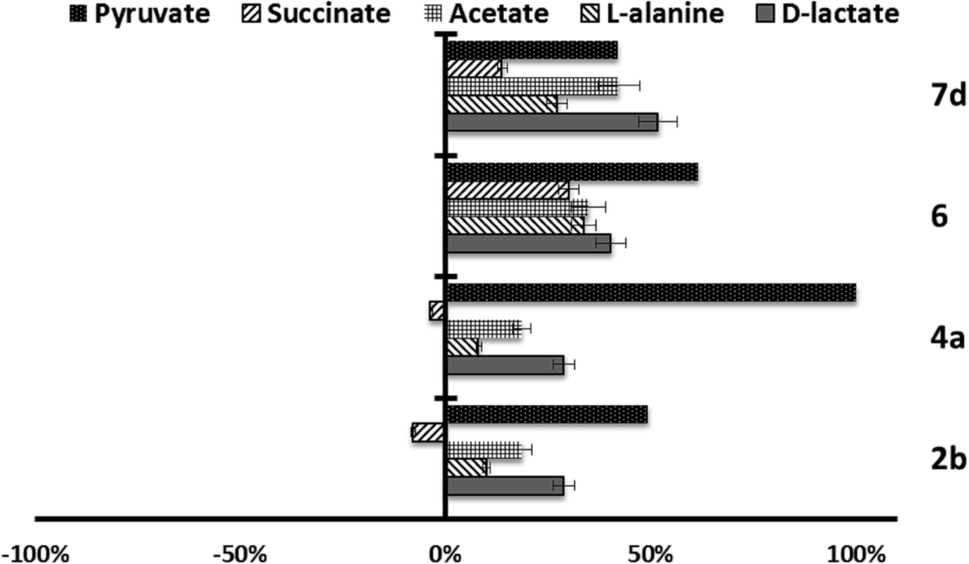 | ||
| Fig. 5 Variation percentages in the area of the peaks corresponding to excreted catabolites by L. brazilinesis promastigotes in the presence of compounds 2b, 4a, 6 and 7d at their IC25 compared to a control sample after 96 h of incubation. | ||
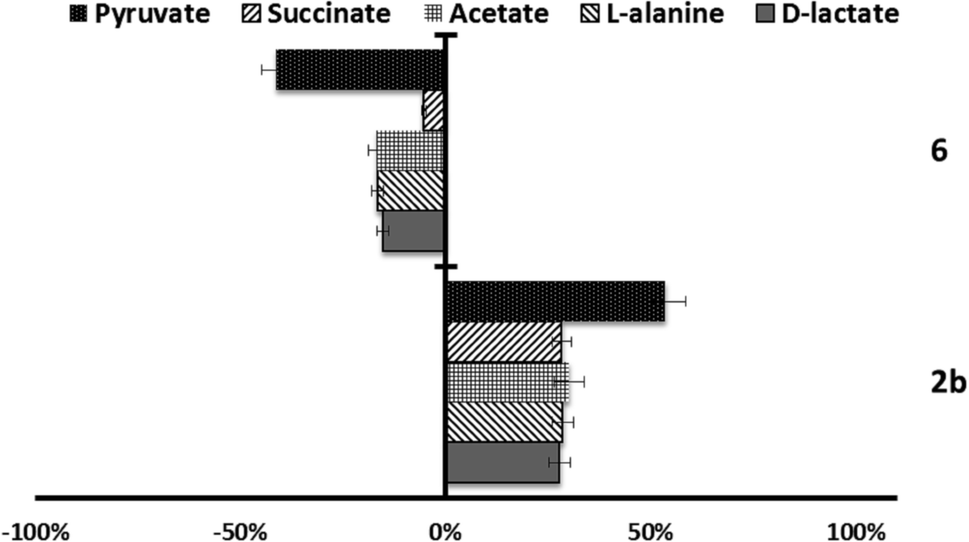 | ||
| Fig. 6 Variation percentages in the area of the peaks corresponding to excreted catabolites by L. donovani promastigotes in the presence of compounds 2b and 6 at their IC25 compared to a control sample after 96 h of incubation. | ||
SOD enzymatic inhibition in the parasites and in human erythrocytes
We tested the effect of compounds on Fe-SOD isolated from the three species of Leishmania over a range of concentrations from 1 to 100 μM. We used promastigote forms of the three species, which excreted Fe-SOD when cultured in a medium lacking inactive FBS.35 The inhibition values data obtained are displayed in Fig. 7–10, and the corresponding IC50 values are included in order to make interpretation the results easier. For comparison Fig. 7 shows the effect of the same compounds on CuZn-SOD obtained from human erythrocytes.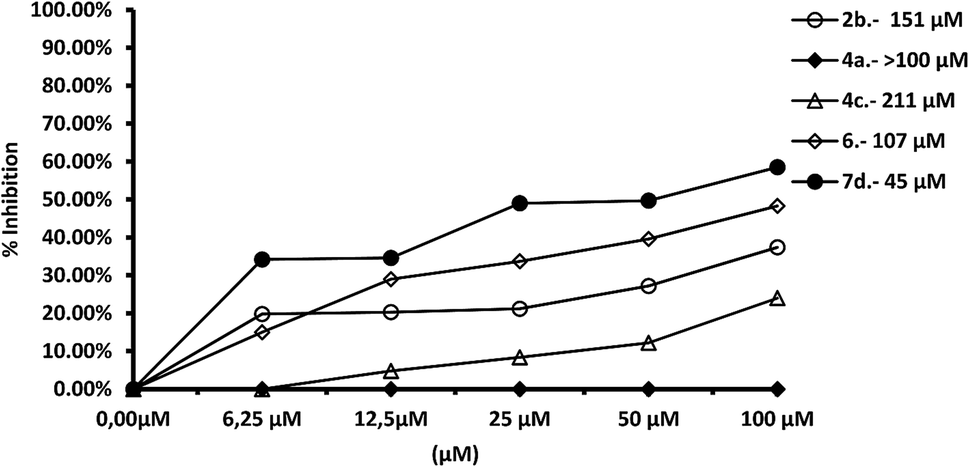 | ||
| Fig. 7 In vitro inhibition (%) of CuZn-SOD from human erythrocytes for compounds 2b, 4a, 4c, 6, 7d. Differences between the activities of the control homogenate and those ones incubated with compounds were obtained according to the Newman–Keuls test. Values are the average of three separate rate determinations. | ||
The most remarkable result (Fig. 8) was the significant inhibition of Fe-SOD activity found by the compounds 2b, 4a, 4c, 6 and 7d, whereas their inhibition of human CuZn-SOD was clearly lower. If we consider the IC50 calculated for L. infantum, inhibition of Fe-SOD by compounds 2b, 4a, 4c, 6 and 7d was respectively 15.1-, >19.6-, 13.2-, 5.3- and 45-fold higher than inhibition of CuZn-SOD, and respective values of 37.8-, 19.6-, 586.1-, 42800-, 12857.1-fold higher were obtained in the case of L. braziliensis (Fig. 9).
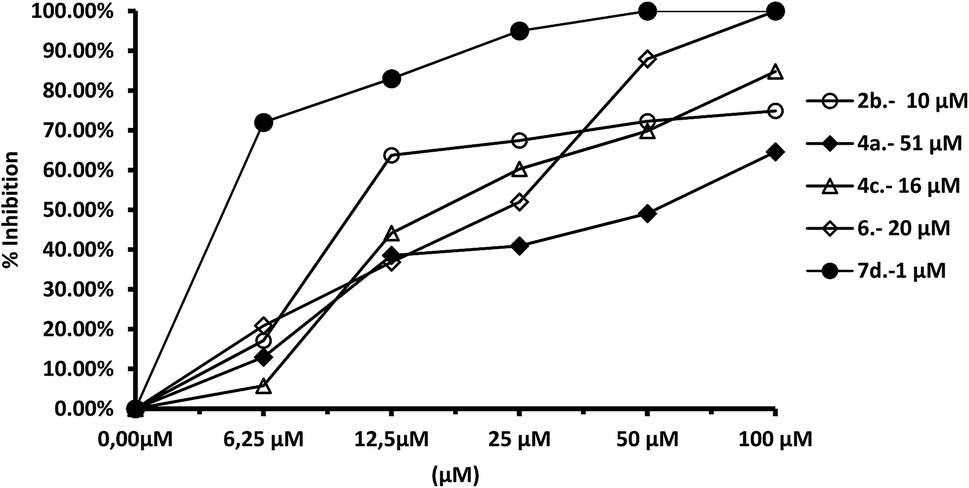 | ||
| Fig. 8 In vitro inhibition (%) of Fe-SOD from L. infantum promastigotes for the same compounds. Differences between the activities of the control homogenate and those ones incubated with compounds were obtained according to the Newman–Keuls test. Values are the average of three separate rate determinations. | ||
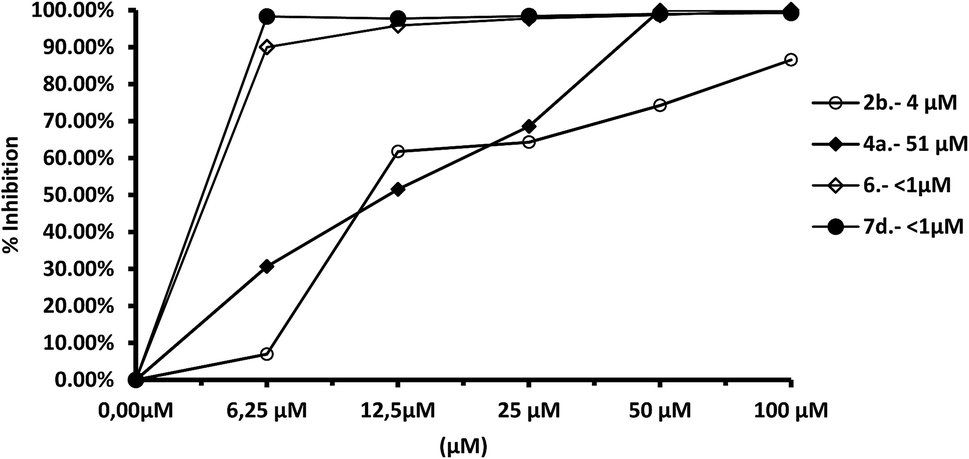 | ||
| Fig. 9 In vitro inhibition (%) of Fe-SOD from L. braziliensis promastigotes for compounds 2b, 4a, 6, 7. Differences between the activities of the control homogenate and those ones incubated with compounds were obtained according to the Newman–Keuls test. Values are the average of three separate rate determinations. | ||
In the case of L. donovani, (Fig. 10) results for compounds 2b and 6 were 3.5- and 6.7-fold higher than those in the case of Cu/Zn-SOD. We can also conclude that compound 6 is a more efficient Fe-SOD inhibitor in L. braziliensis than in L. infantum and L donovani (IC50 0.0025 μM, 20 μM and 16 μM, respectively). The same occurs with compound 7d, that is a major inhibitor of Fe-SOD of L. braziliensis than in L. infantum.
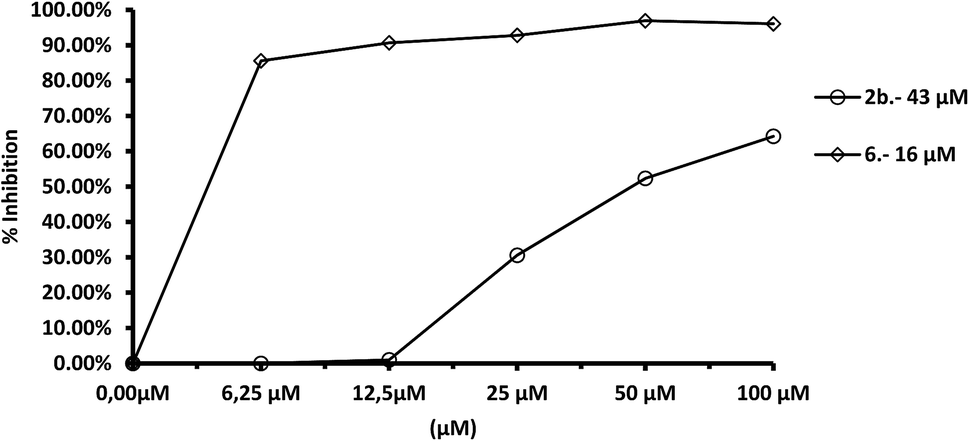 | ||
| Fig. 10 In vitro inhibition (%) of Fe-SOD from L. donovani promastigotes for compounds 2b, 6. Differences between the activities of the control homogenate and those ones incubated with compounds were obtained according to the Newman–Keuls test. Values are the average of three separate rate determinations. | ||
Conclusion
In summary, we described a novel family of triazolopyridinium salts with interesting antileishmanial activity. The experiments allowed us to select five compounds (2b, 4a, 4c, 6, 7d) that showed improved efficiency and lower toxicity (selectivity indexes higher) than those of the reference drug Glucantime for the three Leishmania species. In addition, these compounds are potent inhibitors of Fe-SOD in the three parasite species considered, whereas their impact on human CuZn-SOD was low. The high activity, low toxicity, stability and solubility, low cost of the starting materials and straightforward synthesis make these compounds appropriate molecules for the development of affordable antileishmanicidal agents. Furthermore, these compounds show a significative action on the glucose metabolism of the parasites, in addition to the Fe-SOD inhibitory action.Experimental
Materials and measurements
Starting materials, if commercially available, were purchased and used as such, provided that suitable checks (melting ranges, refractive indices and gas chromatography) had confirmed the claimed purity. When known compounds had to be prepared by literature procedures, pertinent references are given. Melting points or ranges (m.p.) given were determined on a Büchi B-545 heated stage and found to be reproducible after recrystallization, unless stated otherwise (“decomp”). 1H and (1H decoupled), 13C nuclear magnetic resonance (NMR) spectra were recorded at 500 or 300 and 101 or 75 MHz, respectively. Chemical shifts are reported in δ units, parts per million (ppm), and were measured relative to the signals for residual water/methanol. Coupling constants (J) are given in Hz. COSY experiments were performed for all compounds. IR spectra were recorded using a Thermoscientific Nicolet FT IR iS10 ATR. HRMS TRIPLETOFT5600 (ABSciex): LC/MS. [m/2z]for disalts or [m/z] for monosalts. Elemental analyses (C, H, N) were run on a CE Instrument EA 1110 CHNS analyzer. The solvents used were of spectroscopic or equivalent grade. Water was twice distilled and passed through a Millipore apparatus. The compounds were isolated as amorphous solids with variable amounts of hydratation.General procedure
At 25 °C [1,2,3]triazolo[1,5-a]pyridine 1a or 3-methyl[1,2,3]triazolo[1,5-a]pyridine 1b (2 mmol, 4 eq) and the corresponding alkylating agent (0.5 mmol, 1 eq) were diluted in DMF (1 mL) and heated at 90° without stirring for 9 h in a sealed tube (see Table 3). Then the reaction mixture was allowed to reach 25 °C and a solid was precipitated. The remaining liquid phase was removed by decantation, the solid was washed 3 times with DMF and then 3 more times with ethyl acetate, ultrasonication was employed each time for the complete removal of remaining reagents. The solid material was then dried under reduced pressure for 24 h. In Table 3 salts yields are given. See ESI† for all figures of NMR spectra.| Trizolopyridine | Alkylating agent | Product (mg) | Yield | ||
|---|---|---|---|---|---|
| 1a | 240 mg | 1,3-Dibromopropane | 100 mg | 2a 140 | 69% |
| 1a | 241 mg | 1,4-Dibromobutane | 106 mg | 2b 180 | 79% |
| 1a | 242 mg | 1,5-Dibromopentane | 112 mg | 2c 150 | 63% |
| 1a | 243 mg | 1,6-Dibromohexane | 124 mg | 2d 120 | 50% |
| 1a | 244 mg | 1,4-Bis(bromomethyl)benzene | 130 mg | 3a 171 | 68% |
| 1a | 245 mg | 1,3-Bis(bromomethyl)benzene | 130 mg | 3b 235 | 93% |
| 1a | 246 mg | 1,2-Bis(bromomethyl)benzene | 130 mg | 3c 175 | 70% |
| 1b | 260 mg | 1,3-Dibromopropane | 100 mg | 4a 140 | 60% |
| 1b | 260 mg | 1,4-Dibromobutane | 106 mg | 4b 143 | 59% |
| 1b | 260 mg | 1,5-Dibromopentane | 112 mg | 4c 134 | 52% |
| 1b | 260 mg | 1,6-Dibromohexane | 124 mg | 4d 150 | 58% |
| 1b | 260 mg | 1,4-Bis(bromomethyl)benzene | 130 mg | 5a 240 | 90% |
| 1b | 260 mg | 1,3-Bis(bromomethyl)benzene | 130 mg | 5b 242 | 90% |
| 1b | 260 mg | 1,2-Bis(bromomethyl)benzene | 130 mg | 5c 171 | 64% |
| 1b | 260 mg | 4,4′-Bis(bromomethyl)-1,1′-biphenyl | 170 mg | 6 190 | 62% |
| 1b | 130 mg | Benzylbromide | 240 mg | 7a 185 | 60% |
| 1b | 130 mg | (2-Bromoethyl)benzene | 255 mg | 7b 190 | 61% |
| 1b | 130 mg | (3-Bromopropyl)benzene | 267 mg | 7c 170 | 51% |
| 1b | 130 mg | (4-Bromobutyl)benzene | 270 mg | 7d 170 | 49% |
Biological evaluation
The cytotoxicity testing on macrophages was performed by flow cytometric analysis according to a method previously described.29 The percentage of viable cells was calculated with respect to the control culture. The IC50 was calculated using linear regression analysis from the Kc values of the concentrations employed.
:1 and maintained for 24 h at 37 °C in air containing 5% CO2. Non-phagocytosed parasites were removed by washing, and the infected cultures were incubated with the testing compounds (concentrations ranging from 1 to 100 μM) and then cultured for 72 h in MEM plus glutamine (2 mM) and 20% inactive FCS. Compound activity was determined from the percentage reductions in amastigote number in treated versus untreated cultures in methanol-fixed and Giemsa-stained preparations. Values are the means of three separate determinations.37:1. The compounds to be tested (IC25 concentrations) were added immediately after infection, and incubated for 12 h at 37 °C in 5% CO2. Nonphagocytosed parasites and compounds were removed by washing, and then the infected cultures were grown for 10 days in fresh medium. Cultures were washed every 48 h and fresh culture medium was added. Compound activity was determined on the basis of both the percentage of infected cells and the number of amastigotes per infected cell in treated and untreated cultures in methanol-fixed and Giemsa-stained preparations. The percentage of infected cells and the mean number of amastigotes per infected cell were determined by analysing more than 500 host cells distributed in randomly chosen microscopic fields. Values are the means of three separate determinations.After 24 h, the promastigote culture was centrifuged (1500 × g for 10 min) and the supernatant was filtered (Minisart®,Φ 20 μm). The filtered supernatant was subjected to ice-cold ammonium sulphate precipitation at 35% salt concentration. Following centrifugation, the resultant supernatant was then treated with 85% ice-cold ammonium sulphate and the second precipitate collected. The resulting precipitate was finally dissolved in 2.5 mL of distilled water and desalted by chromatography in a Sephadex G-25 column (GE Healthcare Life Sciences®, PD 10 column), previously equilibrated with 25 mL of distilled water, bringing it to a final volume of 3.5 mL (Fraction P85e).
The protein content was quantified using the Sigma Bradford test, which uses bovine serum albumin (BSA) as a standard.39 Iron and copper–zinc superoxide dismutases activities were determined using a previously described method,40 that measures the reduction in nitroblue tetrazolium (NBT) by superoxide ions. According to the protocol, 845 μL of stock solution [3 mL of L-methionine (300 mg, 10 mL−l), 2 mL of NBT (1.41 mg, 10 mL−1) and 1.5 mL of Triton X-100 1% (v/v)] were added into each well, along with 30 μL of the parasite homogenate fraction, 10 μL of riboflavin (0.44 mg, 10 mL−l), and an equivalent volume of the different concentrations of the compounds being tested. Seven different concentrations were used for each agent, from 0.1 to 100 μM. In the control experiment the volume was made up to 1000 μL with 50 mM potassium phosphate buffer (pH 7.8, 3 mL), and 30 μL of the parasite homogenate fraction were added to the mixtures containing the compounds. Then, the absorbance (A0) was measured at 560 nm in a UV spectrophotometer. Afterward, each well was illuminated with UV light for 10 min under constant stirring and the absorbance (A1) was measured again. The human CuZn-SOD and substrates used in these assays were obtained from Sigma Chemical Co. The resulting data were analysed using the Newman–Keuls test.
Acknowledgements
We are grateful to the Ministerio de Ciencia e Innovación (Spain) (Project CONSOLIDER-INGENIO SUPRAMED CSD 2010-00065), Generalitat Valenciana (PROMETEO 2015/002) and to Universidad de Valencia (Valencia, Spain) (Project UV-INV-AE15-332846) for its financial support. R.B-G. acknowledges to Ministerio de Economía y Competitividad (Spain) for a Contrato de Formación Posdoctoral 2013.Notes and references
- S. L. Croft, S. Sundar and A. H. Fairlamb, Clin. Microbiol. Rev., 2006, 19, 111–126 CrossRef CAS PubMed.
- D. R. L. dos Santos, C. L. Vitral, V. S. de Paula, R. S. Marchevsky, J. F. Lopes, A. M. C. Gaspar, T. M. Saddi, N. C. d. M. Júnior, F. d. R. Guimarães, J. G. C. Júnior, L. L. L. Ximenes, F. J. D. Souto and M. A. Pinto, Vet. J., 2009, 182, 474–480 CrossRef PubMed.
- A. Cavalli and M. L. Bolognesi, J. Med. Chem., 2009, 52, 7339–7359 CrossRef CAS PubMed.
- Control of the Leishmaniases: Report of a Meeting of the WHO Expert Committee on the Control of Leishmaniases, Geneva, 22–26 March 2010, ed. WHO Expert Committee on the Control of the Leishmaniases and World Health Organization, WHO technical report series, World Health Organization, Geneva, 2010 Search PubMed.
- S. Espuelas, D. Plano, P. Nguewa, M. Font, J. A. Palop, J. M. Irache and C. Sanmartín, Curr. Med. Chem., 2012, 19, 4259–4288 CrossRef CAS PubMed.
- N. Singh, M. Kumar and R. K. Singh, Asian Pac. J. Trop. Med., 2012, 5, 485–497 CrossRef CAS PubMed.
- G. C. Tron, T. Pirali, R. A. Billington, P. L. Canonico, G. Sorba and A. A. Genazzani, Med. Res. Rev., 2008, 28, 278–308 CrossRef CAS PubMed.
- S. G. Agalave, S. R. Maujan and V. S. Pore, Chem.–Asian J., 2011, 6, 2696–2718 CrossRef CAS PubMed.
- G. L. Almajan, S.-F. Barbuceanu, E.-R. Almajan, C. Draghici and G. Saramet, Eur. J. Med. Chem., 2009, 44, 3083–3089 CrossRef CAS PubMed.
- O. A. Phillips, E. E. Udo, M. E. Abdel-Hamid and R. Varghese, Eur. J. Med. Chem., 2009, 44, 3217–3227 CrossRef CAS PubMed.
- K. S. Bauer, K. J. Cude, S. C. Dixon, E. A. Kruger and W. D. Figg, J. Pharmacol. Exp. Ther., 2000, 292, 31–37 CAS.
- A. Kumar, I. Ahmad, B. S. Chhikara, R. Tiwari, D. Mandal and K. Parang, Bioorg. Med. Chem. Lett., 2011, 21, 1342–1346 CrossRef CAS PubMed.
- M. J. Giffin, H. Heaslet, A. Brik, Y.-C. Lin, G. Cauvi, C.-H. Wong, D. E. McRee, J. H. Elder, C. D. Stout and B. E. Torbett, J. Med. Chem., 2008, 51, 6263–6270 CrossRef CAS PubMed.
- N. G. Aher, V. S. Pore, N. N. Mishra, A. Kumar, P. K. Shukla, A. Sharma and M. K. Bhat, Bioorg. Med. Chem. Lett., 2009, 19, 759–763 CrossRef CAS PubMed.
- V. Patil, W. Guerrant, P. C. Chen, B. Gryder, D. B. Benicewicz, S. I. Khan, B. L. Tekwani and A. K. Oyelere, Bioorg. Med. Chem., 2010, 18, 415–425 CrossRef CAS PubMed.
- A. Tahghighi, S. Razmi, M. Mahdavi, P. Foroumadi, S. K. Ardestani, S. Emami, F. Kobarfard, S. Dastmalchi, A. Shafiee and A. Foroumadi, Eur. J. Med. Chem., 2012, 50, 124–128 CrossRef CAS PubMed.
- T. T. Guimarães, M. d. C. F. R. Pinto, J. S. Lanza, M. N. Melo, R. L. do Monte-Neto, I. M. M. de Melo, E. B. T. Diogo, V. F. Ferreira, C. A. Camara, W. O. Valença, R. N. de Oliveira, F. Frézard and E. N. da Silva, Eur. J. Med. Chem., 2013, 63, 523–530 CrossRef PubMed.
- S. N. Suryawanshi, A. Tiwari, S. Kumar, R. Shivahare, M. Mittal, P. Kant and S. Gupta, Bioorg. Med. Chem. Lett., 2013, 23, 2925–2928 CrossRef CAS PubMed.
- (a) E. N. da Silva, R. F. S. Menna-Barreto, M. d. C. F. R. Pinto, R. S. F. Silva, D. V. Teixeira, M. C. B. V. de Souza, C. A. De Simone, S. L. De Castro, V. F. Ferreira and A. V. Pinto, Eur. J. Med. Chem., 2008, 43, 1774–1780 CrossRef CAS PubMed; (b) V. Patil, W. Guerrant, P. C. Chen, B. Gryder, D. B. Benicewicz, S. I. Khan, B. L. Tekwani and A. K. Oyelere, Bioorg. Med. Chem., 2010, 18, 415–425 CrossRef CAS PubMed.
- (a) T. Aravinda, H. S. B. Naik and H. R. P. Naik, Int. J. Pept. Res. Ther., 2009, 15, 273 CrossRef CAS; (b) S.-F. Cui, Y. Ren, S.-L. Zhang, X.-M. Peng, G. L. V. Damu, R.-X. Geng and C.-H. Zhou, Bioorg. Med. Chem. Lett., 2013, 23, 3267–3272 CrossRef CAS PubMed.
- X. Li, Y. Lin, Q. Wang, Y. Yuan, H. Zhang and X. Qian, Eur. J. Med. Chem., 2011, 46, 1274–1279 CrossRef CAS PubMed.
- (a) K. Lemke, M. Wojciechowski, W. Laine, C. Bailly, P. Colson, M. Baginski, A. K. Larsen and A. Skladanowski, Nucleic Acids Res., 2005, 33, 6034–6047 CrossRef CAS PubMed; (b) E. Zabost, A. M. Nowicka, Z. Mazerska and Z. Stojek, Phys. Chem. Chem. Phys., 2012, 14, 3408–3413 RSC.
- W. M. Cholody, S. Martelli and J. Konopa, J. Med. Chem., 1990, 33, 2852–2856 CrossRef CAS PubMed.
- B. Abarca and R. Ballesteros-Garrido, in Chemistry of 1,2,3-triazoles, ed. W. Dehaen and V. A. Bakulev, Springer International Publishing, 2014, pp. 325–378 Search PubMed.
- R. Adam, P. Bilbao-Ramos, S. López-Molina, B. Abarca, R. Ballesteros, M. E. González-Rosende, M. A. Dea-Ayuela and G. Alzuet-Piña, Bioorg. Med. Chem., 2014, 22, 4018–4027 CrossRef CAS PubMed.
- R. Adam, P. Bilbao-Ramos, B. Abarca, R. Ballesteros, M. E. González-Rosende, M. A. Dea-Ayuela, F. Estevan and G. Alzuet-Piña, Org. Biomol. Chem., 2015, 13, 4903–4910 CAS.
- (a) B. Abarca, A. Asensio, R. Ballesteros, J. Bosch, G. Jones, F. Mojarrad, M. R. Metni and C. M. Richardson, J. Chem. Res., Miniprint, 1990, 347–354 (J. Chem. Res., Synop., 1990, 9) Search PubMed; (b) G. Jones, C. M. Richardson, P. C. Yates, G. Hajós and G. Timari, Tetrahedron, 1993, 49, 4307–4314 CrossRef CAS.
- J. D. Bower and G. R. Ramage, J. Chem. Soc., 1957, 4506–4510 RSC.
- M. Sánchez-Moreno, C. Marín, P. Navarro, L. Lamarque, E. García-España, C. Miranda, O. Huertas, F. Olmo, F. Gómez-Contreras, J. Pitarch and F. Arrebola, J. Med. Chem., 2012, 55, 4231–4243 CrossRef PubMed.
- S. Nwaka, B. Ramirez, R. Brun, L. Maes, F. Douglas and R. Ridley, PLoS Neglected Trop. Dis., 2009, 3, e440 Search PubMed.
- M. L. Ginger, Protist, 2005, 156, 377–392 CrossRef CAS PubMed.
- F. Bringaud, L. Rivière and V. Coustou, Mol. Biochem. Parasitol., 2006, 149, 1–9 CrossRef CAS PubMed.
- J. J. Cazzulo, FASEB J., 1992, 6, 3153–3161 CAS.
- P. A. M. Michels, F. Bringaud, M. Herman and V. Hannaert, Biochim. Biophys. Acta, 2006, 1763, 1463–1477 CrossRef CAS PubMed.
- S. S. Longoni, M. Sánchez-Moreno, J. E. R. López and C. Marín, Comp. Immunol. Microbiol. Infect. Dis., 2013, 36, 499–506 CrossRef PubMed.
- P. González, C. Marín, I. Rodríguez-González, A. B. Hitos, M. J. Rosales, M. Reina, J. G. Díaz, A. González-Coloma and M. Sánchez-Moreno, Int. J. Antimicrob. Agents, 2005, 25, 136–141 CrossRef PubMed.
- M. Sánchez-Moreno, A. M. Sanz, F. Gómez-Contreras, P. Navarro, C. Marín, I. Ramírez-Macias, M. J. Rosales, F. Olmo, I. Garcia-Aranda, L. Campayo, C. Cano, F. Arrebola and M. J. R. Yunta, J. Med. Chem., 2011, 54, 970–979 CrossRef PubMed.
- M. Sánchez-Moreno, F. Gómez-Contreras, P. Navarro, C. Marín, F. Olmo, M. J. R. Yunta, A. M. Sanz, M. J. Rosales, C. Cano and L. Campayo, J. Med. Chem., 2012, 55, 9900–9913 CrossRef PubMed.
- M. M. Bradford, Anal. Biochem., 1976, 72, 248–254 CrossRef CAS PubMed.
- W. F. Beyer and I. Fridovich, Anal. Biochem., 1987, 161, 559–566 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra01070b |
| ‡ A. M-M. and R. B-G. contributed equally. |
| This journal is © The Royal Society of Chemistry 2017 |