
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Highly dispersed CoOx in layered double oxides for oxidative dehydrogenation of propane: guest–host interactions†
Ming-Xiang Huang ,
Xin Wu,
Xiao-Dong Yi,
Guo-Bin Han,
Wen-Sheng Xia* and
Hui-Lin Wan*
,
Xin Wu,
Xiao-Dong Yi,
Guo-Bin Han,
Wen-Sheng Xia* and
Hui-Lin Wan*
State Key Laboratory of Physical Chemistry of Solid State Surfaces, National Engineering Laboratory for Green Chemical Productions of Alcohols-Ethers-Esters, Fujian Province Key Laboratory of Theoretical and Computational Chemistry, College of Chemistry and Chemical Engineering, Xiamen University, Xiamen, Fujian 361005, P. R. China. E-mail: wsxia@xmu.edu.cn; hlwan@xmu.edu.cn; Fax: +86-592-2183047; Tel: +86-592-2181937
First published on 7th March 2017
Abstract
Co–Al layered-double-oxides (LDOs) derived from inorganic anion (nitrate, carbonate, sulfate and phosphate) pillared Co–Al layered-double-hydroxides (LDHs) were synthesized. These LDO samples exhibited impressive catalytic performances in oxidative dehydrogenation of propane (ODHP) at low temperatures. Characterization techniques such as XRD, TEM, H2-TPR and O2-TPD were used to measure the physicochemical properties of the samples. The results showed that the LDO samples consisted of homogeneously mixed cobalt spinel oxides (Co2AlOx) containing both Co(III) and Co(II). Co(III) instead of Co(II) was linked with the active sites for ODHP at low temperatures. The sulfate and phosphate anions were preserved in the corresponding LDO samples, unlike the nitrate and carbonate pillars. Sulfate (phosphate) around the cobalt decreased the size of the spinel oxide particles more relative to nitrate and carbonate, and so the dispersion of CoOx in the LDO and the selectivity to propylene in ODHP over these catalysts were improved. Both surface adsorbed oxygen species and surface lattice oxygen species were significantly found over the phosphate-pillared LDO, but only lattice oxygen species were available over the sulfate-pillared LDO. Different mechanisms of propane activation were exhibited over these LDO samples for ODHP.
1. Introduction
Oxidative dehydrogenation of propane (ODHP) into propylene is an important reaction as an alternative route to the most largely used pyrolysis processes. Pyrolysis suffers from some drawbacks, such as thermodynamic limitations, rapid coking and high reaction temperature.1 However, the target product, propylene, in ODHP is more active than the reactant, and so is prone to being further oxidized into products such as CO2 in the presence of oxygen, resulting in low selectivity toward propylene. Thus the development of catalysts for ODHP is a great challenge.Among all the studied catalysts, vanadium and molybdenum-based catalysts show relatively good performances for ODHP because of their unique redox properties,2 but high operating temperatures (above 500 °C) are necessary, resulting in a significant probability of extensive transformation of the target product, such as through C–C bond cleavage. Comparatively, cobalt oxides have been used due to their ability to catalyze reactions at low temperatures, such as CO oxidation3 and partial oxidation of light hydrocarbons.4 Performing the reactions under mild conditions can reduce the risk of extensive transformation of the product, along with the amount of energy wasted, and thus increase the product selectivity.
Furthermore, researchers have used active-site isolation to improve the product selectivity for the selective oxidation of light alkanes. For example, high yields of propylene in ODHP were obtained over vanadium oxides supported on meso-cellulose silica foams, due to the increased concentration of dispersed V species.5 Isolated monomeric VOx species were also introduced through the incorporation of Sb atoms, leading to enhanced activity and selectivity.6 A similar strategy of isolating the active sites was also achieved in cerium phosphate catalysts.7
On the other hand, we noted that hydrotalcite-like layered-double-hydroxides (LDHs) contain positively charged brucite-like layers and exchangeable anions in the interlayer, which are conducive to the preparation of highly dispersed catalysts. The general molecular formula of an LDH is [M2+1−xM3+x(OH)2](An−)x/n·mH2O, where M2+ and M3+ are di- and trivalent metal cations, An− is a charge compensating inorganic or organic anion, and x is normally between 0.2 and 0.4. The M2+ and M3+ cations are uniformly dispersed in an orderly manner within the layers in the LDH lattice, and the derived metal oxides (layered-double-oxides, LDOs) with improved stability and dispersion can be obtained through thermal treatment of the LDH precursors. LDOs have been widely used as supports or precursors in catalysis.8 Therefore, we here introduced cobalt into an LDO to combine a highly active component (Co) with a layer-structured material to synthesize a catalyst exhibiting a good dispersion of active sites, i.e., Co–Al LDO. To the best of our knowledge, this is the first report on the use of Co–Al LDO catalysts in ODHP, especially those being pillared by different anions. In this work, the inorganic anion-pillared Co–Al LDO samples derived from LDHs featuring well dispersed lamellar structures were investigated in ODHP at low temperatures. The textural, redox and structural properties of the catalysts were studied to account for their catalytic performances.
2. Experimental section
2.1. Synthesis of the catalysts
The Co–Al LDH pillared with nitrate was prepared using a modified co-precipitation method followed by hydrothermal treatment.9 Typically, 0.08 mmol of Co(NO3)2·6H2O and 0.04 mmol of Al(NO3)3·9H2O were dissolved into 120 mL of decarbonated water, and the pH was adjusted to 10 by adding 2 M NaOH aqueous solution. Care was taken to avoid CO2 contamination during the synthesis. The suspension was magnetically stirred at room temperature for 0.5 h, and afterwards was poured into a Teflon-lined autoclave, which was heated at 100 °C in a convection oven for 24 h. The precipitate was centrifuged, washed several times with decarbonated water and then dried at 80 °C for 12 h in a vacuum oven. The obtained sample was referred to as Co–AlNO3− LDH and denoted as CA-N LDH. A co-precipitated sample (named CA-co) was also prepared without the hydrothermal process and used as a reference sample.For the preparation of the other anion-pillared Co–Al LDHs, CA-N LDH in the form of a wet suspension was used, and anion nitrate exchange was performed with the pertinent anions in solution. A solution (100 mL) of the ammonium salt of the relevant anion was added to the Co–Al LDH suspension, and the solution was stirred for 24 h at room temperature. The precipitate was centrifuged, washed several times with decarbonated water and then dried at 80 °C for 12 h in a vacuum oven. The carbonate-, sulfate- and phosphate-pillared Co–Al LDH samples were named as CA-C, CA-S and CA-P LDH, respectively.
LDO samples were obtained by calcination of the LDH samples in air at 400 °C for 4 h, and used as the catalysts for ODHP. A pure Co3O4 catalyst was also prepared using a similar procedure to that used for CA-co LDO but without the addition of Al(NO3)3.
2.2. Characterization of the catalysts
X-ray diffraction (XRD) patterns were recorded on a Panalytical X'pert PRO powder diffractometer. The samples were ground into a fine powder, loaded as a flat layer onto a glass sheet with a square groove and scanned in the 2θ range from 5 to 80°. Cu Kα radiation (λ = 0.15406 nm) generated at 40 kV and 30 mA was used as the X-ray source.Scanning electron microscopy (SEM) characterization of the samples was performed on a Hitachi S-4800 FEGSEM microscope equipped with an energy dispersive X-ray (EDX) microprobe at an acceleration voltage of 15 kV. The samples were dispersed in ethanol and loaded onto a silicon wafer before the test.
The content of Co, Al, S and P was determined by EDX spectroscopy. The carbon and nitrogen content was determined on an Elementar Vario EL III element analyzer.
N2 adsorption–desorption isotherms were recorded at −196 °C on automated Micromeritics Tri-Star 3020 apparatus. Surface areas were calculated using the multipoint BET equation. Pore size was calculated using the BJH method, using the desorption branch of the isotherm. The samples were outgassed for 3 h under vacuum at 300 °C before the measurements were carried out.
Transmission electron microscopy (TEM) characterization of the samples was performed on an FEI Tecnai-F30 FEG instrument at an acceleration voltage of 300 kV.
Thermal gravimetric and differential thermal gravimetric (TG-DTG) experiments were performed in air on NETZSCH TG 209 F1 apparatus. The LDH samples were heated from 30 to 800 °C in a flowing air stream at a heating rate of 10 °C min−1. The flow rate of air was 20 mL min−1.
Laser Raman spectroscopy was performed using a Renishaw UV-vis Raman 1000 system equipped with a CCD detector and a Leica DMLM microscope. The 532 nm line of an Ar+ laser was used for excitation.
The Fourier transform-infrared (FT-IR) spectra of the samples in the form of KBr pellets were recorded at room temperature on an FT-IR Nicolet Avatar 330 spectrometer.
H2 temperature-programmed reaction (H2-TPR) analysis of the samples was carried out using flow apparatus with a 5% H2/Ar mixture flowing at 20 mL min−1. The heating rate was 10 °C min−1. Hydrogen consumption was monitored with a thermal conductivity detector (TCD) equipped to a Haixin GC-950 gas chromatograph after removing the formed water. All the samples were pretreated in 10% O2/He at 400 °C for 0.5 h before the measurements.
X-ray photoelectron spectroscopy (XPS) experiments were performed using an Omicron Sphera II hemispherical electron energy analyzer at a base pressure of 5.0 × 10−9 mbar. The instrument was equipped with a monochromatic Al Kα radiation source operated at a power of 300 W. The spectra were corrected using the C 1s signal located at 284.5 eV.
The electron paramagnetic resonance (EPR) spectra of the samples (100 mg) were measured at −179 °C on a Bruker EMX X-band 10/12 spectrometer with a resonance frequency of 9.43 GHz and a modulation frequency of 100 kHz.
NH3 temperature-programmed desorption (NH3-TPD) and O2 temperature-programmed desorption (O2-TPD) analyses of the samples were performed with an AutoChem II 2920 equipped with a Balzers Omnistar mass analyzer. In the O2-TPD experiment, the LDO sample (0.3 g) was calcined at 400 °C in 20% O2/He atmosphere for 1 h, and then cooled to 40 °C. After purging with He at 40 °C for 1 h to make the baseline flat, the TPD plot was obtained from 50 to 800 °C (heating rate: 10 °C min−1). In the NH3-TPD experiment, the LDO sample (0.2 g) was treated at 300 °C in a He atmosphere for 1 h, cooled to 100 °C, and exposed to pure NH3 for 1 h at 100 °C. After purging with He for 1 h at 100 °C to make the baseline flat, the TPD plot was obtained from 100 to 800 °C (heating rate: 10 °C min−1).
CO2-TPD analyses of the samples were performed in a gas flow system equipped with a HIDEN QIC-20 mass analyzer. The LDO sample (0.15 g) was calcined at 400 °C in a He atmosphere for 0.5 h, cooled to 50 °C, and exposed to pure CO2 for 1 h at 50 °C. After purging with He for 1 h at 100 °C to make the baseline flat, the TPD plot was obtained from 50 to 850 °C (heating rate: 10 °C min−1).
C3H6-TPD and C3H8-TPD analyses of the samples were performed in a gas flow system equipped with a HIDEN QIC-20 mass analyzer. The LDO sample (0.2 g) was degassed at 400 °C in an Ar atmosphere for 0.5 h, cooled to 30 °C, and exposed to pure C3H6 or C3H8 for 0.5 h at 30 °C. After purging with Ar for 1 h at 30 °C to make the baseline flat, the TPD plot was obtained from 30 to 800 °C (heating rate: 10 °C min−1).
Surface acidity properties were determined by monitoring the adsorption of pyridine in a Thermo Nicolet Nexus 470 FT-IR spectrometer. The samples were degassed in situ in a cell with CaF2 windows at 400 °C for 0.5 h under vacuum. The pyridine vapor was adsorbed at 100 °C and the spectrum recorded after degassing.
2.3. Catalytic reaction
The catalytic performances of the samples were investigated with a mixture of propane, oxygen and helium (C3H8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :O2:He = 1:1:4, the molar ratio) as a reaction feed. The reaction was carried out in a fixed-bed quartz tubular reactor at atmospheric pressure and at a total flow rate of 50 mL min−1. The reactor was packed with 100 mg of LDO sample diluted with 100 mg of quartz sand (40–60 mesh size) between quartz wool plugs. The temperature was measured by a thin quartz wall thermocouple well fixed in the middle of the catalytic bed. The performances of the catalysts were evaluated over the temperature range of 200–550 °C with a gas hourly space velocity (GHSV) of 30000 mL g−1 h−1. The products were analyzed by online gas chromatographs (Shimadzu GC-14C; Shimadzu GC 2010), equipped with an alumina column and a carbon sieve column (for the detection of CH4, O2/He, CO and CO2) and an RT-PLOTQ column (for oxygenates). The conversion of propane and oxygen (XC3H8 and XO2), the selectivity for different products (Si) and the yield of propylene (YC3H6) were defined as:
:O2:He = 1:1:4, the molar ratio) as a reaction feed. The reaction was carried out in a fixed-bed quartz tubular reactor at atmospheric pressure and at a total flow rate of 50 mL min−1. The reactor was packed with 100 mg of LDO sample diluted with 100 mg of quartz sand (40–60 mesh size) between quartz wool plugs. The temperature was measured by a thin quartz wall thermocouple well fixed in the middle of the catalytic bed. The performances of the catalysts were evaluated over the temperature range of 200–550 °C with a gas hourly space velocity (GHSV) of 30000 mL g−1 h−1. The products were analyzed by online gas chromatographs (Shimadzu GC-14C; Shimadzu GC 2010), equipped with an alumina column and a carbon sieve column (for the detection of CH4, O2/He, CO and CO2) and an RT-PLOTQ column (for oxygenates). The conversion of propane and oxygen (XC3H8 and XO2), the selectivity for different products (Si) and the yield of propylene (YC3H6) were defined as: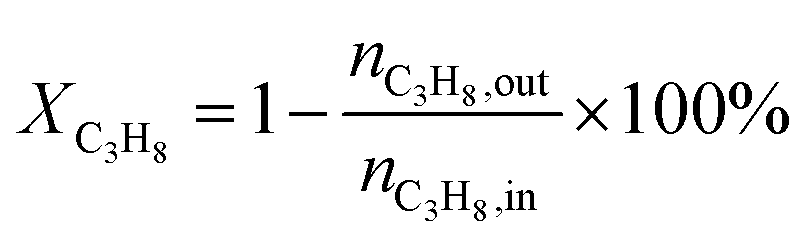
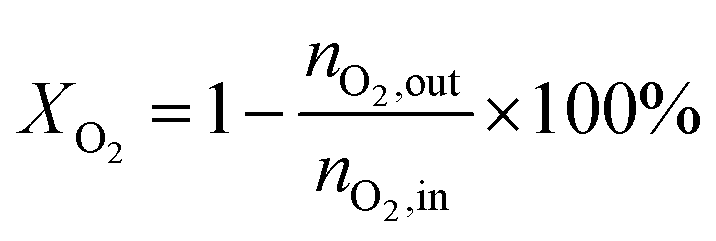
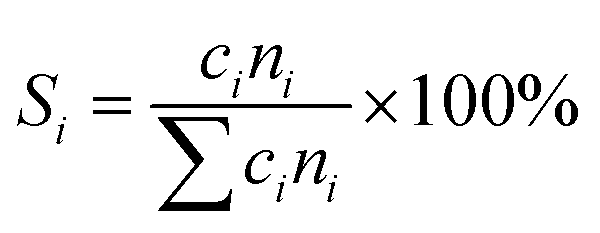
| YC3H6 = XC3H8 × SC3H6 |
The carbon balance was satisfactory in all runs with an error margin of ±5%.
3. Results and discussion
3.1. Textural properties and chemical composition of LDH
The successful preparation of the Co–Al LDH samples pillared with different anions was confirmed by their XRD patterns as shown in Fig. 1. A series of sharp and symmetric characteristic reflections are seen for all of the hydrothermally treated samples (CA-N, CA-C, CA-S and CA-P LDH), indicating that hexagonal Co–Al hydrotalcite-like structures are the only phase.9 The peaks for CA-co LDH are relatively widened, indicating that the structure of CA-co LDH is not as well defined as for the other counterparts. From the S(T)EM images in Fig. S1 of the ESI,† the CA-N, CA-C, CA-S and CA-P LDH samples are further found to be hexagonal plate-like sheets with a diameter of ∼150 nm, a thickness of ∼15 nm, and well-defined grain boundaries, but the CA-co LDH sample contains stacked amorphous particles with sizes in the range of 15–25 nm.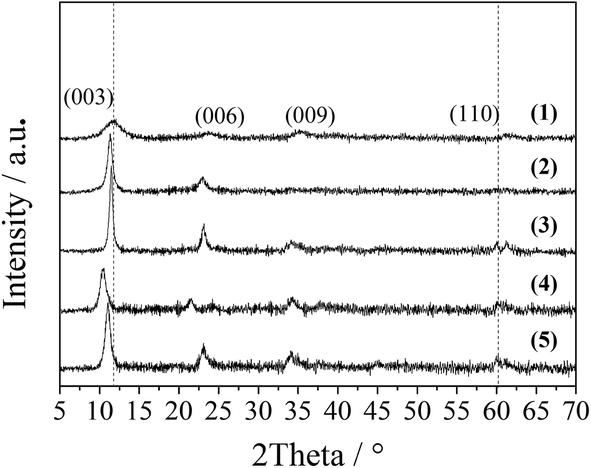 | ||
| Fig. 1 XRD patterns of the LDH samples: CA-co (1), CA-N (2), CA-C (3), CA-S (4), and CA-P (5). | ||
The lattice parameters (a and c) of the samples are presented in Table 1. The a value is almost the same for all of these LDH samples, while the c value is 2.33, 2.32, 2.43 and 2.36 nm for the nitrate, carbonate, sulfate and phosphate intercalated LDH, respectively. Furthermore, the interlayer spacing decreased in the order of CA-S > CA-P > CA-N > CA-C, indicating that the isolation of metal ions by the anion interlayer changed depending on the pillared anion. These interlayer anions were identified by FT-IR (Fig. S2, ESI†).10,11
| Sample | Formulaa | LP-LDHb/nm | LP-LDOb/nm | Co/Al molar ratioc | Al/anion molar ratio | |||
|---|---|---|---|---|---|---|---|---|
| a | c | a | LDH | LDO | LDH | LDO | ||
| a The amount of H2O was determined by TG-DTG experiments.b Determined by XRD, a = 2d(110), c = 3dspace, dspace = (d(003) + 2d(006))/2.c Determined by EDX or elemental analyses. | ||||||||
| CA-co | [Co0.66Al0.34(OH)2](NO3)0.24(CO3)0.05·0.64H2O | 0.304 | 2.270 | 0.795 | 1.91 | 1.93 | 1.39 | — |
| CA-N | [Co0.67Al0.33(OH)2](NO3)0.28(CO3)0.025·0.35H2O | 0.307 | 2.330 | 0.797 | 2.02 | 2.00 | 1.16 | — |
| CA-C | [Co0.67Al0.33(OH)2](CO3)0.165·0.78H2O | 0.307 | 2.320 | 0.799 | 1.97 | 2.01 | 2.04 | — |
| CA-S | [Co0.66Al0.34(OH)2](SO4)0.15(CO3)0.02·0.51H2O | 0.306 | 2.430 | 0.797 | 1.93 | 2.05 | 2.17 | 2.25 |
| CA-P | [Co0.66Al0.34(OH)2](PO4)0.113·0.55H2O | 0.306 | 2.360 | 0.798 | 1.94 | 2.03 | 2.90 | 2.96 |
The chemical composition of the LDH samples was measured by elemental chemical analysis. The value of the Co/Al ratio for CA-co, CA-N, CA-C, CA-S and CA-P LDH is 1.91, 2.02, 1.97, 1.93 and 1.94, respectively, close to the expected value (Co/Al ratio = 2). The value of the Al/anion ratio for CA-N, CA-C, CA-S and CA-P LDH is 1.16, 2.04, 2.17 and 2.90, respectively. Thus, the chemical formula for the LDH samples could be derived (see Table 1).
3.2. Physicochemical properties of the LDO catalysts
| Sample | Specific surface area/m2 g−1 | Average pore diameter/nm | Pore volume/cm3 g−1 | Size of spinel oxide particles for LDO/nm | ||||
|---|---|---|---|---|---|---|---|---|
| LDH | LDO | LDH | LDO | LDH | LDO | Dav,XRDa | Dav,TEM | |
| a Calculated from the position of the reflection of the (311) facet in Fig. 3 using the Scherrer formula.b The Scherrer formula is not applicable. | ||||||||
| CA-co | 172.0 | 100.1 | 5.2 | 11.6 | 0.32 | 0.40 | 14.7 | 14.2 ± 3.3 |
| CA-N | 56.6 | 137.1 | 15.4 | 11.6 | 0.28 | 0.49 | 10.1 | 7.6 ± 2.8 |
| CA-C | 58.6 | 158.4 | 18.5 | 9.8 | 0.29 | 0.49 | 6.3 | 5.8 ± 1.0 |
| CA-S | 30.6 | 115.7 | 20.1 | 9.4 | 0.17 | 0.24 | 5.0 | 4.3 ± 0.7 |
| CA-P | 55.8 | 110.6 | 17.0 | 12.8 | 0.27 | 0.28 | N.D.b | 3.9 ± 0.6 |
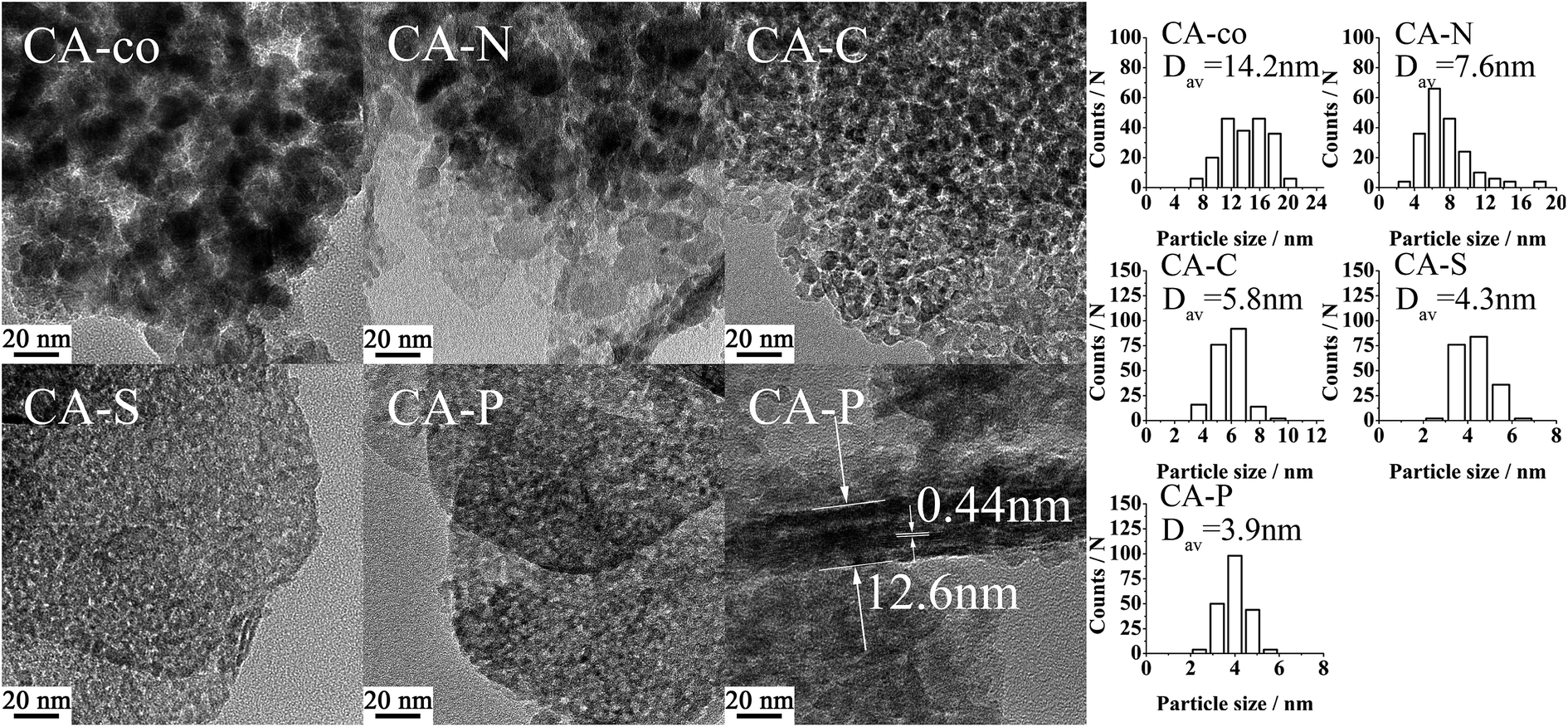 | ||
| Fig. 2 TEM images of the LDO samples. | ||
The XRD patterns of the LDO samples are shown in Fig. 3, where the characteristic peaks are assigned as a cubic cobalt spinel phase. It seems that this phase can be assumed to be a very homogeneous and stable spinel-like mixed oxide, Co(Co,Al)2O4, in which Al isomorphically substitutes Co(III) of the Co3O4 phase,12 as it is difficult to identify if the phase is Co3O4, CoAl2O4, or Co2AlO4 because these phases exhibit similar XRD reflections. The Raman spectra of the LDO samples in Fig. 4 show the characteristic peaks of F12g, Eg, F22g, F32g and A1g vibration modes of Co3O4-like crystals (194, 480, 521, 617 and 684 cm−1, respectively), with a blue shift compared to the spectrum of Co3O4. This is significantly different from the peaks of CoAl2O4 (198, 412, 480, 519, 619, 690, and 753 cm−1).13 Thus, in combination with the chemical formula of the precursor LDH and the fact that no peak related to amorphous Al2O3 was observed in the Raman spectra,14 we think that the bulk phase of the LDO samples consists of a homogeneous mixed cobalt spinel, Co2AlOx, containing both Co(II) and Co(III) species.
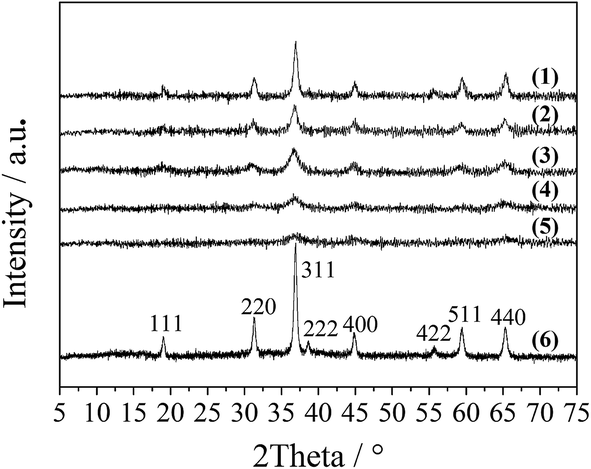 | ||
| Fig. 3 XRD patterns of the LDO samples: CA-co (1), CA-N (2), CA-C (3), CA-S (4), CA-P (5) and Co3O4 (6). | ||
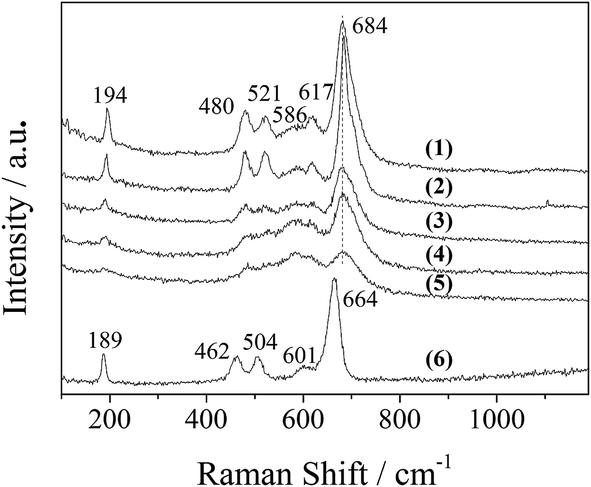 | ||
| Fig. 4 Raman spectra of the LDO samples: (1) CA-co, (2) CA-N, (3) CA-C, (4) CA-S, (5) CA-P and (6) Co3O4. | ||
Furthermore, the Co 2p XPS spectrum of the LDO samples is shown in Fig. 5. The spin–orbital splitting peaks at 780.3 and 795.4 eV were assigned to Co 2p3/2 and Co 2p1/2, respectively, along with an energy separation (Co 2p3/2–Co 2p1/2) of 15.1 eV. These features are similar to the reported data for Co3O4,15 and related to the Co(Co,Al)2O4 spinel phase.
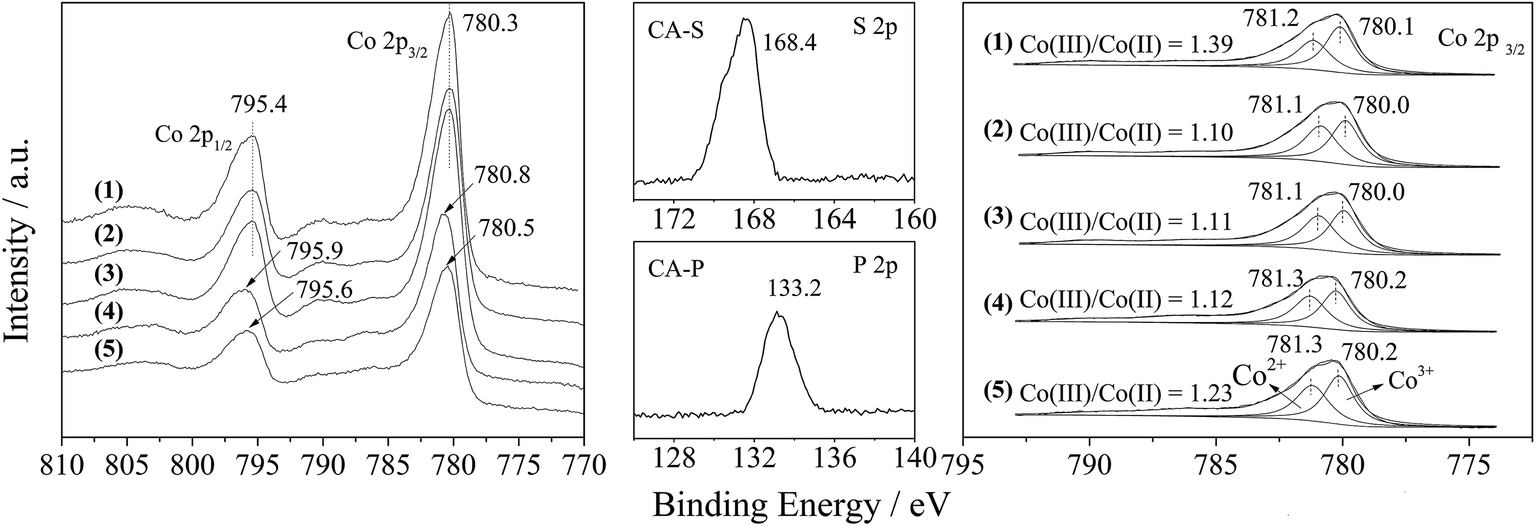 | ||
| Fig. 5 XPS spectra of the LDO samples: CA-co (1), CA-N (2), CA-C (3), CA-S (4) and CA-P (5). | ||
The presence of sulfate and phosphate in the CA-S and CA-P LDO samples was substantiated by the appearance of S 2p and P 2p peaks at the binding energies of 168.4 and 133.2 eV (middle part of Fig. 5), which was assigned to chemisorbed SO42− and PO43−, respectively.16,17 These anions caused the Co 2p3/2 and Co 2p1/2 peaks for the CA-S and CA-P LDO samples to be shifted to a higher binding energy relative to the CA-N and CA-C LDO samples (left part of Fig. 5).
By deconvoluting the Co 2p3/2 peaks of the LDO samples according to reported data,18 we found that the ratio of Co(III)/Co(II) on the surface of CA-co, CA-N, CA-C, CA-S and CA-P LDO was 1.39, 1.10, 1.11, 1.12 and 1.23, respectively (right part of Fig. 5). The highest amount of Co(III) was found on the surface of CA-co LDO, because less Co(III) is isomorphically substituted by Al in this sample. In the hydrothermally treated samples, CA-N, CA-C, CA-S and CA-P LDO, the amount of Co(III) increased gradually in the sequence of CA-N < CA-C < CA-S < CA-P. Thus, host–guest interactions led to a relatively higher amount of Co(III), especially for CA-P LDO.
Additionally, FT-IR spectra could provide some information on the bonding state and the coordination around the metal ion in the LDO samples. As shown in Fig. 6, the bands at 669 and 566 cm−1 are typical of a normal II–III spinel compound.19 The former (669 cm−1) was attributed to the M–O stretching vibration mode for the tetrahedrally coordinated divalent metal ions, and the latter (566 cm−1) was assigned to the octahedrally coordinated trivalent metal ion. These two bands for CA-co LDO appeared at 673 and 574 cm−1, with a blue shift relative to the other LDO samples. This was related to the surroundings of the metal ions in CA-co LDO, which has a relatively lower Co/Al ratio. Interestingly, the presence of sulfate and phosphate for CA-S and CA-P LDO was supported by the broad bands at 1124 and 1069 cm−1 assigned to ν3 mode S–O and P–O stretching vibration of tetrahedral SO42− and PO43−, respectively.20,21 The M–O stretching vibration bands were weakened, but the peak positions did not change. This can be explained by the lowered dipole of M–O caused by the oxygen of the anions coordinated to the metal.22 Thus, the divalent Co(II) and trivalent Co(III) or Al3+ were located at the tetrahedral and octahedral sites, respectively, and metal ions were connected with the oxygen of the pillared anions (sulfate or phosphate) for CA-S and CA-P LDO.
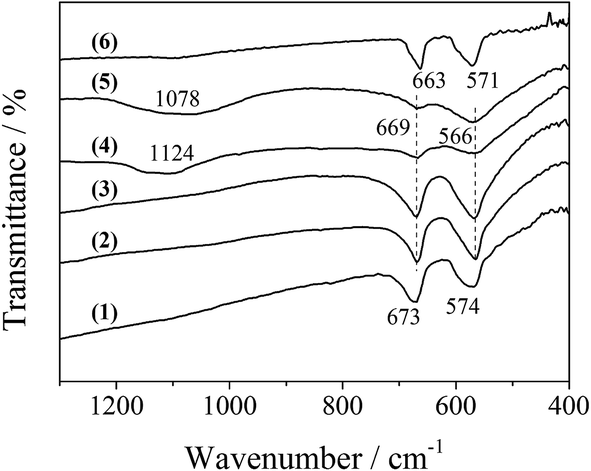 | ||
| Fig. 6 FT-IR spectra of the LDO samples: CA-co (1), CA-N (2), CA-C (3), CA-S (4), CA-P (5) and Co3O4 (6). | ||
The metal was coordinated by the pillared anions, meaning that the cobalt spinel oxides of the samples were isolated. The size of the isolated cobalt spinel oxides should be relatively small. As shown in Table 2, the size of the spinel oxide particles for these LDO samples decreased from CA-co, to CA-N, CA-C, CA-S and CA-P LDO (Dav,XRD: 14.7, 10.1, 6.3, 5.0, N.D. nm; Dav,TEM: 14.2, 7.6, 5.8, 4.3, 3.9 nm, respectively), and the spinel oxide for CA-P LDO displayed the smallest crystallite size. Both the XRD and TEM measured values are consistent. Moreover, the TEM images showed that the uniformity and dispersion of the spinel oxides increased from CA-co, to CA-N, CA-C, CA-S and CA-P LDO. The uniformity and size of the spinel oxides for CA-C LDO were not much different from those of CA-S and CA-P LDO, indicating that the carbonate anion also plays a significant role in improving the dispersion of the spinel oxide, though the carbonate was not found in CA-C LDO, which was prepared by calcination of CA-C LDH. However, Javier Perez-Ramirez12 found that the collapse of the layered structure occurred before complete decarbonation during calcination, meaning that the carbonate anion would physically obstruct the dispersion of the spinel oxides. Thus, the dispersion of the spinel oxides for these LDO samples is closely related to the pillared anions.
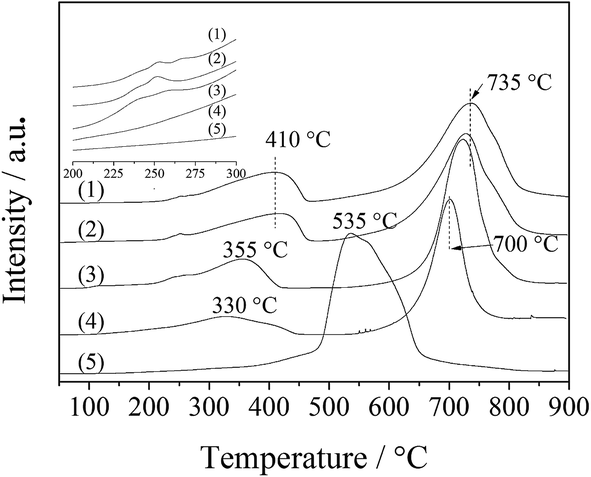 | ||
| Fig. 7 H2-TPR profiles of the LDO samples: CA-co (1), CA-N (2), CA-C (3), CA-S (4) and CA-P (5). | ||
For CA-S, a different H2-TPR process occurred. It seemed that Co(III) was reduced along with Co(II). Only a broad peak at ∼535 °C was situated between the Co(III) and Co(II) reduction peaks, suggesting that Co(III) was more difficult to reduce in CA-S LDO than in the other LDO samples. This could be associated with its stronger host–guest interaction (spinel oxide–anion interlayer) relative to the other LDO samples, as suggested by the above XPS results. A similar case was found for the reduction of sulfate-modified Co/Al2O3 catalysts.25
The interaction of the spinel oxides with oxygen was measured with O2-TPD. As shown in Fig. 8, the oxygen desorption curves for the LDO samples were divided into two regions, 200–400 °C and above 400 °C, which corresponded to desorption of surface adsorbed oxygen species and surface lattice oxygen, respectively.26 For the CA-co, CA-N, CA-C and CA-P LDO samples, a small broad peak appeared at 200–400 °C, assigned to the surface adsorbed oxygen species; and a wide broad peak was found at above 400 °C, assigned to the surface lattice oxygen. However, for the CA-S LDO sample, the two significant peaks appeared at 530 °C and 820 °C, ascribed to the surface lattice oxygen and the decomposition of sulfate, respectively, meaning that surface adsorbed oxygen species were observed for the CA-S LDO sample. These results are in agreement with a recent report that sulfate can hinder the formation of oxygen vacancies and reduce the mobility of surface oxygen species on Pt/MnOx–CeO2 catalysts.27 As the peak area for the surface lattice oxygen was far greater than that for the surface adsorbed oxygen species, surface lattice oxygen was predominant in these samples.
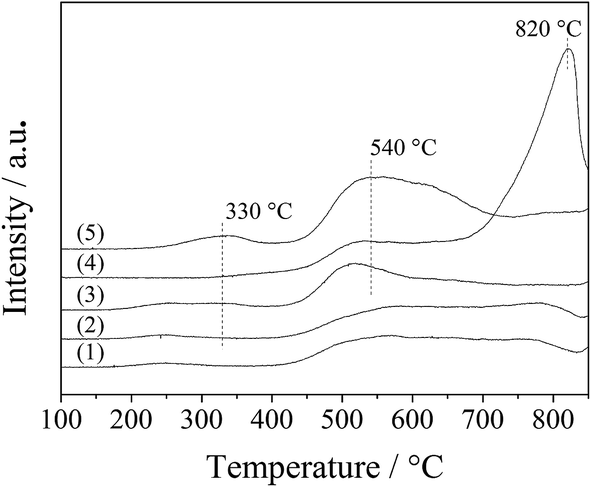 | ||
| Fig. 8 O2-TPD profiles of the LDO samples: CA-co (1), CA-N (2), CA-C (3), CA-S (4) and CA-P (5). | ||
Moreover, both the surface lattice oxygen and the surface adsorbed oxygen species were more prominent over the CA-P LDO sample than the other LDO samples, revealing that there were more oxygen vacancies over the CA-P LDO sample than the other LDO samples. This was confirmed further by EPR results (Fig. S4, ESI†), where the signal with a g-factor of about 2.0 was attributed to oxygen vacancies.28,29
3.3. Catalytic performances
The catalytic performances of the LDO samples in ODHP were tested. The optimized ratio of propane to oxygen in the feed was also investigated at T = 400 °C and GHSV = 30000 mL gcat−1 h−1. It was found that propylene selectivity increased and propane conversion decreased with the ratio (Table S1, ESI†). The highest yield of propylene was reached at C3H8:O2 = 1:1 for CA-N and CA-C LDO in ODHP, and at an even lower ratio of propane to oxygen for CA-S and CA-P LDO. As a result, we chose a 1:1 ratio of propane to oxygen in the reactant feed for the following comparison among the LDO samples in ODHP.
As shown in Table 3, the ODHP reaction over the LDO samples occurred at low temperatures (240–400 °C) with a significant conversion of propane and selectivity to propylene. The conversion of propane at 400 °C is 41.1, 28.4, 24.2, 25.8, 24.5 and 27.2% over Co3O4, CA-co, CA-N, CA-C, CA-S and CA-P, respectively. Propylene was not found at the selected temperatures (240–400 °C) over Co3O4, but the propylene selectivity at 400 °C was 18.8, 21.5, 22.1, 31.4, and 36.0% over CA-co, CA-N, CA-C, CA-S and CA-P, respectively. That is to say, the LDO samples with the segregation of Al on the surface displayed improved propylene selectivity in ODHP. These results also reveal the good efficiency of the reaction at low temperatures.
| Sample | T/°C | X/% | S/% | YC3H6/% | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| C3H8 | O2 | C3H6 | CH4 | C2H4 | CO | CO2 | Othersb | |||
| a The data were obtained after 30 min on stream (reaction conditions: C3H8:O2:He = 1:1:4; GHSV = 30000 mL gcat−1 h−1).b Others = C3H4O + CH3CH2CHO + CH3COCH3. |
||||||||||
| Co3O4 | 240 | 0.4 | 2.1 | 0.0 | 0.1 | 0.1 | 0.0 | 89.7 | 10.1 | 0.0 |
| 250 | 20.9 | 100 | 0.0 | 0.4 | 0.1 | 0.0 | 99.4 | 0.1 | 0.0 | |
| 300 | 24.3 | 100 | 0.0 | 2.8 | 0.0 | 3.1 | 93.9 | 0.1 | 0.0 | |
| 350 | 27.8 | 100 | 0.0 | 11.2 | 0.0 | 5.6 | 83.1 | 0.1 | 0.0 | |
| 400 | 41.1 | 100 | 0.0 | 18.2 | 0.1 | 8.0 | 73.4 | 0.0 | 0.0 | |
| CA-co | 250 | 0.4 | 1.9 | 0.5 | 0.0 | 1.7 | 0.0 | 84.8 | 13 | 0.0 |
| 260 | 17.8 | 100 | 11.3 | 0.1 | 1.0 | 0.0 | 87.4 | 0.2 | 2.0 | |
| 300 | 18.5 | 100 | 11.9 | 0.1 | 0.9 | 0.0 | 86.8 | 0.2 | 2.2 | |
| 350 | 19.4 | 100 | 15.6 | 0.2 | 2.1 | 0.0 | 81.9 | 0.1 | 3.0 | |
| 400 | 20.3 | 100 | 18.8 | 0.3 | 3.9 | 0.0 | 76.8 | 0.2 | 3.8 | |
| 425 | 21.6 | 100 | 6.7 | 0.5 | 9.2 | 16.7 | 66.7 | 0.3 | 1.4 | |
| 450 | 28.7 | 100 | 0.0 | 1.6 | 0.0 | 37.2 | 61.0 | 0.1 | 0.0 | |
| 500 | 30.8 | 100 | 0.0 | 1.2 | 0.0 | 46.9 | 51.9 | 0.1 | 0.0 | |
| 550 | 58.6 | 100 | 0.0 | 26.5 | 0.0 | 45.1 | 28.4 | 0.0 | 0.0 | |
| CA-N | 250 | 0.3 | 1.6 | 1.2 | 0.0 | 2.1 | 0.0 | 80.7 | 15.7 | 0.0 |
| 260 | 19.0 | 100 | 11.2 | 0.1 | 1.2 | 0.0 | 87.4 | 0.1 | 2.8 | |
| 300 | 19.2 | 100 | 12.3 | 0.1 | 1.3 | 0.0 | 86.1 | 0.1 | 2.4 | |
| 350 | 19.6 | 100 | 16.1 | 0.2 | 2.6 | 0.0 | 80.8 | 0.2 | 3.2 | |
| 400 | 20.4 | 100 | 21.5 | 0.4 | 3.7 | 0.0 | 74.3 | 0.1 | 4.4 | |
| 425 | 21.3 | 100 | 10.5 | 0.3 | 1.1 | 18.8 | 69.2 | 0.2 | 2.2 | |
| 450 | 31.5 | 100 | 0.0 | 0.2 | 2.0 | 33.2 | 64.6 | 0.1 | 0.0 | |
| 500 | 34.3 | 100 | 0.0 | 5.3 | 0.0 | 54.1 | 40.6 | 0.0 | 0.0 | |
| 550 | 43.2 | 100 | 0.0 | 10.9 | 0.0 | 52.6 | 36.4 | 0.0 | 0.0 | |
| CA-C | 240 | 0.4 | 1.7 | 1.3 | 0.1 | 2.2 | 0.0 | 83.3 | 13.1 | 0.0 |
| 250 | 19.0 | 100 | 16.9 | 0.3 | 3.2 | 0.0 | 79.5 | 0.1 | 3.2 | |
| 300 | 20.8 | 100 | 17.0 | 0.2 | 3.4 | 0.0 | 79.3 | 0.1 | 3.5 | |
| 350 | 20.8 | 100 | 19.1 | 0.3 | 5.7 | 0.0 | 74.8 | 0.1 | 4.0 | |
| 400 | 23.0 | 100 | 22.1 | 0.4 | 8.1 | 1.1 | 68.2 | 0.1 | 5.1 | |
| 450 | 23.8 | 100 | 21.5 | 0.6 | 10.6 | 1.7 | 65.3 | 0.2 | 5.1 | |
| 500 | 25.6 | 100 | 23.5 | 0.8 | 11.3 | 3.9 | 60.4 | 0.1 | 6.0 | |
| 550 | 25.4 | 100 | 25.9 | 1.0 | 11.9 | 6.4 | 54.7 | 0.1 | 6.6 | |
| CA-S | 350 | 2.3 | 13.5 | 51.6 | 0.0 | 0.8 | 0.0 | 45.9 | 1.7 | 1.2 |
| 360 | 23.0 | 88.8 | 34.1 | 0.2 | 4.4 | 1.6 | 59.3 | 0.3 | 7.8 | |
| 375 | 24.3 | 97.0 | 32.3 | 0.3 | 4.5 | 1.9 | 60.9 | 0.2 | 7.8 | |
| 400 | 24.6 | 100 | 31.4 | 0.5 | 4.2 | 2.1 | 61.6 | 0.2 | 7.7 | |
| 450 | 25.0 | 100 | 31.6 | 0.6 | 4.6 | 2.3 | 60.8 | 0.2 | 7.9 | |
| 500 | 25.3 | 100 | 30.3 | 0.7 | 5.8 | 3.5 | 59.6 | 0.2 | 7.7 | |
| 550 | 29.2 | 100 | 28.4 | 1.2 | 8.3 | 5.9 | 56.0 | 0.2 | 8.3 | |
| CA-P | 265 | 1.3 | 6.7 | 77.4 | 0.0 | 0.6 | 0.0 | 19.3 | 2.6 | 1.2 |
| 275 | 23.2 | 100 | 28.4 | 0.0 | 0.9 | 0.0 | 70.6 | 0.1 | 6.6 | |
| 300 | 24.2 | 100 | 30.0 | 0.0 | 1.1 | 0.0 | 68.8 | 0.1 | 7.2 | |
| 350 | 26.4 | 100 | 33.9 | 0.0 | 1.6 | 1.6 | 62.8 | 0.1 | 9.0 | |
| 400 | 27.9 | 100 | 36.0 | 0.1 | 2.3 | 4.4 | 57.1 | 0.1 | 10.0 | |
| 450 | 28.0 | 100 | 34.8 | 0.1 | 3.0 | 7.6 | 54.3 | 0.1 | 9.7 | |
| 500 | 28.3 | 100 | 32.4 | 0.1 | 4.4 | 12.0 | 51.0 | 0.1 | 9.2 | |
| 550 | 28.5 | 100 | 31.2 | 0.2 | 6.5 | 15.4 | 46.5 | 0.1 | 8.9 | |
The catalytic behavior of the LDO samples in ODHP at low temperatures (400 °C below) is actually derived from the reduction of Co(III) related species. As shown by H2-TPR measurements (Fig. 7), the reduction peaks of both Co(III) to Co(II) and Co(II) to Co, for CA-co, CA-N, CA-C and CA-P LDO, appeared at below 400 °C and above 700 °C, respectively, and the reduction peak of Co(III) and Co(II) for CA-S LDO emerged at around 500 °C. Li et al.30 found that Co(III) species with spinel-like structures were responsible for the ODHP activity. In our work, the ratio of Co(III)/Co(II) was tuned by the pillared anions in the LDOs, and extra Co(III) on the LDO (especially for CA-P LDO) could facilitate the redox cycling of Co(III)/Co(II) and oxygen activation.
The ignition point of ODHP was 250, 260, 260, 250, 360, and 275 °C over Co3O4, CA-co, CA-N, CA-C, CA-S and CA-P LDO, respectively. This is related to the highly active CoOx over CA-co, CA-N and CA-C LDO, which corresponded to the weakened peaks at 225–275 °C shown in the inset of Fig. 7. Over CA-P LDO, the species that activate propane at low temperatures could be surface adsorbed oxygen species, which were linked with the peak located at 330 °C in the O2-TPD measurements (Fig. 8). For CA-S LDO, the reduction peak of Co(III) shifted up to a higher temperature (∼500 °C), and there were only lattice oxygen species available. The oxidation activity of CA-S LDO is the lowest among the investigated LDO samples, and so the ignition point of ODHP over CA-S LDO is relatively high.
Comparing the catalytic performances over the CA-co, CA-N, CA-C, CA-S and CA-P LDO samples, we found that the highest yield of propylene at the investigated temperatures was 3.8, 4.4, 6.6, 8.3 and 10.0%, respectively, and the selectivity to propylene increased in the same sequence. The structures of these samples, in turn, became well defined, and the size of the cobalt spinel oxide particles gradually decreased, and so the dispersion of CoOx was increased. The propylene selectivity in ODHP seems to be correlated with the particle size and the dispersion of cobalt oxides.
However, the size of the spinel oxides for CA-C LDO (5.8 nm) is close to that for CA-S and CA-P LDO (4.3 and 3.9 nm), and propane conversion is not much different over these LDO samples, but the selectivity to propylene over CA-C is significantly less than that over CA-S and CA-P (22.1, 31.4 and 36.0% at 400 °C, respectively). Therefore, the particle size and the dispersion of oxides may not account completely for the enhanced catalytic performances of these samples in ODHP, implying that the pillared anions play a significant role.
As the pillared anions (sulfate and phosphate) are repulsive to the π electrons of propylene, the residual time for propylene is shorter on the surfaces of CA-S and CA-P LDO than CA-C and CA-N, LDO, and so oxidation of the produced propylene in ODHP over CA-S and CA-P LDO is avoided more efficiently. This is confirmed by the C3H6-TPD result that the desorption of propylene over CA-P occurred at a lower temperature than over CA-C (580 vs. 625 °C)31 (Fig. 9A). The large peak at 107 °C was attributed to weakly bound propylene on acid sites,32 similar to the case of C3H8-TPD (Fig. 9B), and the peak temperature for CA-P was slightly higher than that for CA-C. These peaks were localized at ∼110 °C, far lower than the ignition point of ODHP over the LDO samples, and the effect of the species that these peaks represent on the reactivity of the samples in ODHP can be ignored. On the other hand, the interaction of cobalt with the anions, especially sulfate, reduced the oxygen mobility of the LDOs where the presence of lattice oxygen species was predominant, and the oxygen reactivity was lowered. For instance, oxygen conversion in the initial activation of propane over the CA-S LDO sample occurred at higher temperatures relative to the other samples (Table 3). The decreased reactivity of oxygen would reduce the probability of the extensive transformation of the targeted products. Thus, the presence of the anions on the catalyst surfaces is beneficial to an improvement of the selectivity to propylene in ODHP.
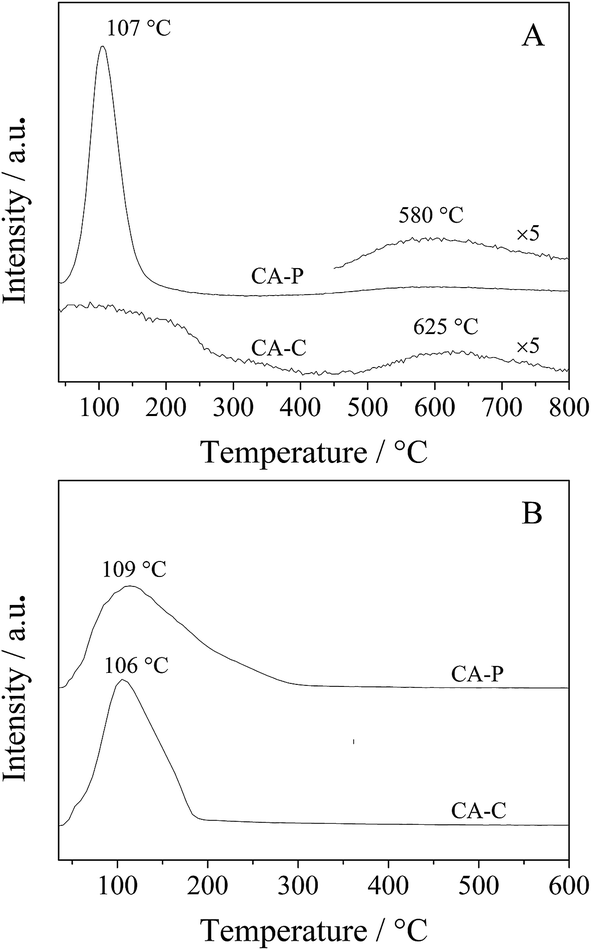 | ||
| Fig. 9 (A) C3H6- and (B) C3H8-TPD profiles of the CA-C and CA-P LDOs. | ||
Although the oxygen mobility in CA-S LDO was lowered by the sulfate anion, propane conversion over the CA-S LDO sample (24.6%) was greater than that over the other CA-co, CA-N, and CA-C LDO samples (20.3, 20.4, and 23.0%) at around 400 °C. The increased selectivity to propylene induced by the anions around cobalt was accompanied by a decrease in selectivity to CO2 (the deep oxidation product), and the oxygen saved due to the decreased CO2 selectivity would transform more propane, provided that the amount of oxygen in the reactant is limited. Thus, the conversion of propane was increased over CA-S and CA-P, though the oxygen mobility in CA-S was lower than in the CA-co, CA-N and CA-C samples.
The dependence of propane conversion on the selectivity to propylene was further investigated over the LDO samples at 400 °C by varying GHSV in the range of 30000–300000 mL gcat−1 h−1. As shown in Fig. 10, different selectivities to propylene were observed over the LDO samples. CA-S and CA-P LDO exhibited much higher selectivity to propylene than the other LDO samples, illustrating that the pillared anions in the LDO samples were beneficial to improving the selectivity to propylene in ODHP. The propylene selectivity over CA-S LDO decreased with propane conversion, whereas the opposite trend was observed over the other LDO samples (Fig. 10), implying that the mechanisms of propane activation were different over these LDO samples. Propylene would be the primary product over CA-S LDO if the conversion of propane were extrapolated to zero, suggesting that the formation of deep oxidation products (CO and CO2) was derived from the extensive oxidation of propylene. This is consistent with the above argument that the reduced (lattice) oxygen mobility (oxygen reactivity) results in increased selectivity to propylene. So the ODHP reaction over CA-S was performed according to the Mars-Van Krevelen mechanism.33 For CA-co, CA-N and CA-C LDO, the selectivity to propylene would be near zero if the propane conversion in ODHP were extrapolated to zero, indicating that the CO and CO2 formed over these LDO samples resulted from the direct oxidation of propane. For CA-P LDO, similar extrapolation showed that the primary products included propylene, CO and CO2. As there were surface adsorbed/lattice oxygen species over CA-co, CA-N, CA-C and CA-P LDO, and only lattice oxygen species over CA-S LDO, the surface adsorbed oxygen species (electrophilic oxygen species) were associated with the deep oxidation products (COx), and the lattice oxygen species (nucleophilic oxygen species) were linked with the formation of propylene. This meant that the activation of propane over CA-co, CA-N, CA-C and CA-P LDO was related to both the lattice oxygen species and the surface adsorbed species.
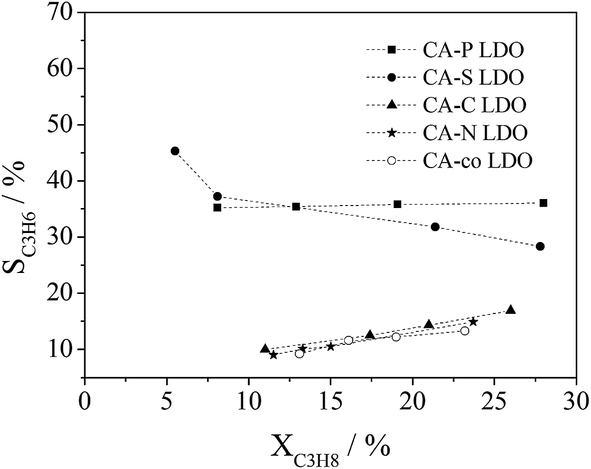 | ||
| Fig. 10 Correlation of propane conversion with propylene selectivity in ODHP over LDOs (reaction conditions: C3H8:O2:He = 1:1:4 (molar ratio); GHSV = 30000–300000 mL gcat−1 h−1; T = 400 °C). | ||
The acidity and basicity of surfaces are often used in tuning the activity and selectivity of catalysts for selective oxidation reactions.34 In this work, we noted that there were mainly weakly acidic and basic sites over the surfaces of the LDO samples (Fig. S5, ESI†). Desorption peaks in NH3- and CO2-TPD experiments appeared at 150–180 °C and 115 °C, respectively. These weakly acidic and basic sites would not have any influence on the desorption of the formed propylene from the surfaces, as the ignition point of the reaction appeared at above 250 °C, and so they do not correspond to the improvement in propylene selectivity over the catalysts for ODHP at low temperatures. There were small amounts of moderate basic sites found for the CA-C and CA-P LDO samples (CO2 desorption at 575 °C, Fig. S5†), which could facilitate propylene desorption and avoid its extensive transformation, such as C–C bond cleavage.
The C–C bond cleavage in propane occurred at relatively high temperatures. As shown in Table 3, a significant amount of methane in ODHP over the Co3O4, CA-co, CA-N, CA-C and CA-S samples is seen at 300, 450, 500, 550 and above 550 °C, respectively, and almost no methane is found over CA-P at 270–550 °C. In other words, C–C bond cleavage occurred at adequately high temperatures, and the temperature of C–C cleavage in ODHP was increased over the anion pillared LDO samples relative to the other counterparts.
Therefore, the pillared anions changed the properties of the LDO samples such as the particle size, the reducibility and the properties of surface oxygen species, and thus led to an improvement in their ODHP performance. The phosphate-pillared LDO sample displayed good catalytic performance for ODHP: the conversion of propane and the selectivity to propylene was 27.9% and 36.0% at 400 °C, respectively (the propylene yield reached to 10.0%). In comparison, the highest propylene yield of 10% was reached at a higher temperature (600 °C) for CoMgAlO mixed oxide catalysts,35 in which well-dispersed CoOx with tetrahedrally coordinated structures acted as the active sites; montmorillonitic clay pillared with Al3+ and/or Cr3+ also showed a high activity at low temperatures (propylene yield at 450 °C: 10.3%).36
3.4. Stability of the catalysts in ODHP
Fig. 11 shows the catalytic performances over the CA-P LDO catalyst as the time-on-stream increased under conditions of T = 400 °C, C3H8:O2:He = 1:1:4, and GHSV = 30000 mL gcat−1 h−1. Both the propane conversion (∼26%) and propylene selectivity (∼39%) were observed to be almost unchanged over the initial 12 hours of time-on-stream (propylene yield was 10%). However, they were changed to ∼6% and ∼77%, respectively, after 50 hours of time-on-stream (propylene yield was ∼5%) and the oxygen conversion decreased gradually from 100% to ∼25%. These results suggest that the properties of the active sites of CA-P LDO varied during a long period of ODHP reaction.
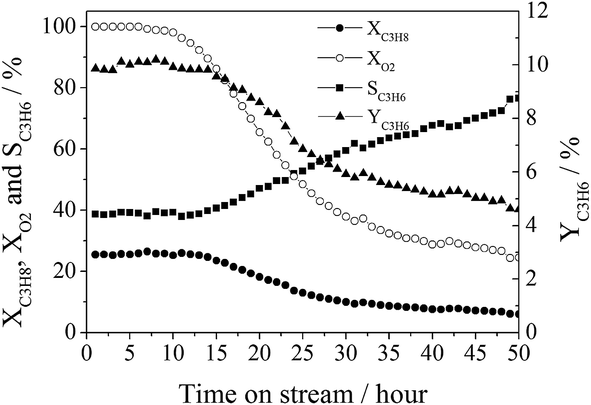 | ||
| Fig. 11 The lifetime test of the CA-P LDO sample for ODHP (reaction conditions: C3H8:O2:He = 1:1:4 (molar ratio); GHSV = 30000 mL gcat−1 h−1; T = 400 °C). | ||
The XRD pattern of the CA-P LDO catalyst after the reaction is shown in Fig. S6 (ESI†). The mixed spinel oxides were still the main phase of CA-P LDO, but the characteristic peaks of the oxides (Co2AlOx) became sharper after 50 hours of reaction. The peaks for CoO were significant. A decreased amount of Co(III) was also observed on the spent catalysts (Fig. S7, ESI†). This indicated that the spinel oxide particles grew, and the Co species was partially reduced after a long period of reaction, which reduced the ability of the catalyst to activate oxygen and propane. Therefore, to improve the stability of the catalysts, we could increase the thermal conductivity of the catalysts and the concentration of the pillared anions or the interaction between CoOx and the anions around it, which will help to prevent the catalyst from being sintered and reduced.
4. Conclusions
Co–Al LDH samples pillared with inorganic anions (nitrate, carbonate, sulfate, and phosphate) were prepared via a hydrothermal method followed by anion exchange. The catalytic performances of the resultant Co–Al LDO samples were tested for ODHP, showing good catalytic behavior relative to Co3O4. Homogeneous mixed cobalt spinel oxides were identified in the LDO samples, and Co(III) at octahedral sites instead of Co(II) at tetrahedral sites was associated with the active sites for ODHP at low temperatures. The size of the spinel oxide particles in the LDOs decreased in the order of CA-co > CA-N > CA-C > CA-S > CA-P, and was linked with the catalytic properties of the LDO samples in ODHP.The pillared anions and the enrichment of aluminum on the surfaces made the active sites isolated, leading to an improvement in the selectivity to propylene in ODHP.
The pillared anions not only increased the dispersion of CoOx, but also modified other properties of the LDO samples, such as reducibility, especially for the preserved anions (sulfate and phosphate) in CA-S and CA-P LDO. Surface adsorbed/lattice oxygen species were found over CA-P LDO, but only lattice oxygen species were available over CA-S LDO. The sulfate around cobalt made the reduction of Co(III) to Co(II) relatively more difficult, and so the mobility of oxygen (lattice) in the LDO was reduced; the phosphate around cobalt provided a significant amount of oxygen vacancies, and enhanced the mobility of oxygen (surface adsorbed oxygen species). As both CA-S and CA-P exhibited better catalytic performances in ODHP than the other LDO samples, the mechanisms of propane activation over CA-S and CA-P must be different. The mechanism over CA-S depended on the mobility of lattice oxygen, and that over CA-P was related to the participation of the surface adsorbed/lattice oxygen in ODHP.
Acknowledgements
Support for this research from the National Natural Science Foundation of China (21373169, 21373168), NFFTBS (No. J1310024), and Program for Changjiang Scholars and Innovative Research Team in the University (No. IRT1036) is gratefully acknowledged.References
- R. B. Watson and U. S. Ozkan, J. Catal., 2000, 191, 12–29 CrossRef CAS.
- Q. Liu, J. Li, Z. Zhao, M. Gao, L. Kong, J. Liu and Y. Wei, Catal. Sci. Technol., 2016, 6, 5927–5941 CAS.
- L. F. Liotta, G. Di Carlo, A. Longo, G. Pantaleo and A. M. Venezia, Catal. Today, 2008, 139, 174–179 CrossRef CAS.
- T. E. Davies, T. Garcia, B. Solsona and S. H. Taylor, Chem. Commun., 2006, 3417–3419, 10.1039/b606973h.
- Y. Liu, W. Feng, T. Li, H. He, W. Dai, W. Huang, Y. Cao and K. Fan, J. Catal., 2006, 239, 125–136 CrossRef CAS.
- H. Zhang, S. Cao, Y. Zou, Y.-M. Wang, X. Zhou, Y. Shen and X. Zheng, Catal. Commun., 2014, 45, 158–161 CrossRef CAS.
- I.-T. Trotuş, C. M. Teodorescu, V. I. Pârvulescu and I.-C. Marcu, ChemCatChem, 2013, 5, 757–765 CrossRef.
- G. Fan, F. Li, D. G. Evans and X. Duan, Chem. Soc. Rev., 2014, 43, 7040–7066 RSC.
- S. Kannan and C. S. Swamy, Preparation and characterisation of cobalt containing layered double hydroxides, Elsevier Science, 1995 Search PubMed.
- A. Halajnia, S. Oustan, N. Najafi, A. R. Khataee and A. Lakzian, Appl. Clay Sci., 2013, 80–81, 305–312 CrossRef CAS.
- J. Qu, L. Zhong, Z. Li, M. Chen, Q. Zhang and X. Liu, Appl. Clay Sci., 2016, 124–125, 267–270 CrossRef CAS.
- J. Perez-Ramirez, G. Mul, F. Kapteijn and J. A. Moulijn, J. Mater. Chem., 2001, 11, 821–830 RSC.
- B. Jongsomjit, J. Panpranot and J. G. Goodwin, J. Catal., 2001, 204, 98–109 CrossRef CAS.
- A. Mortensen, D. H. Christensen and O. F. Nielseo, J. Raman Spectrosc., 1991, 22, 47–49 CrossRef CAS.
- M. Nishibori, W. Shin, N. Izu, T. Itoh and I. Matsubara, Catal. Today, 2013, 201, 85–91 CrossRef CAS.
- S. P. Chenakin, G. Melaet, R. Szukiewicz and N. Kruse, J. Catal., 2014, 312, 1–11 CrossRef CAS.
- N. Sleiman, V. Deluchat, M. Wazne, M. Mallet, A. Courtin-Nomade, V. Kazpard and M. Baudu, Water Res., 2016, 99, 56–65 CrossRef CAS PubMed.
- M. C. Biesinger, B. P. Payne, A. P. Grosvenor, L. W. M. Lau, A. R. Gerson and R. S. C. Smart, Appl. Surf. Sci., 2011, 257, 2717–2730 CrossRef CAS.
- M. Herrero, P. Benito, F. M. Labajos and V. Rives, Catal. Today, 2007, 128, 129–137 CrossRef CAS.
- M. Khaldi, A. D. Roy, M. Chaouch and J. P. Besse, J. Solid State Chem., 1997, 130, 66–73 CrossRef CAS.
- M. Badreddine, A. Legrouri, A. Barroug, A. D. Roy and J. P. Besse, Mater. Lett., 1999, 38, 391–395 CrossRef CAS.
- K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds: Part A: Theory and Applications in Inorganic Chemistry, John Wiley & Sons. Inc., 2009 Search PubMed.
- M. Gabrovska, R. Edreva-Kardjieva, K. Tenchev, P. Tzvetkov, A. Spojakina and L. Petrov, Appl. Catal., A, 2011, 399, 242–251 CrossRef CAS.
- Q. Wang, Y. Peng, J. Fu, G. Z. Kyzas, S. M. R. Billah and S. An, Appl. Catal., B, 2015, 168–169, 42–50 CrossRef CAS.
- Y. Sun, Y. Gao, Y. Wu, H. Shan, G. Wang and C. Li, Catal. Commun., 2015, 60, 42–45 CrossRef CAS.
- F. Teng, M. Chen, N. Li, X. Hua, J. Wang, Q. Zhang, Y. Wang, D. D. Meng and G. Li, RSC Adv., 2013, 3, 743–751 RSC.
- H. Zhang, Z. Hou, Y. Zhu, J. Wang and Y. Chen, Appl. Surf. Sci., 2017, 396, 560–565 CrossRef CAS.
- C. Perdomo, A. Pérez, R. Molina and S. Moreno, Appl. Surf. Sci., 2016, 383, 42–48 CrossRef CAS.
- I. P. Vorona, V. V. Nosenko, N. P. Baran, S. S. Ishchenko, S. V. Lemishko, I. V. Zatovsky and N. Y. Strutynska, Radiat. Meas., 2016, 87, 49–55 CrossRef CAS.
- Z. Li, A. W. Peters, V. Bernales, M. A. Ortuno, N. M. Schweitzer, M. R. DeStefano, L. C. Gallington, A. E. Platero-Prats, K. W. Chapman, C. J. Cramer, L. Gagliardi, J. T. Hupp and O. K. Farha, ACS Cent. Sci., 2017, 3, 31–38 CrossRef CAS PubMed.
- X. Zhang, H.-l. Wan, W.-z. Weng and X.-d. Yi, J. Mol. Catal. A: Chem., 2003, 200, 291–300 CrossRef CAS.
- R. Spinicci and A. Tofanari, J. Therm. Anal., 1982, 23, 45–50 CrossRef CAS.
- C. A. Carrero, R. Schloegl, I. E. Wachs and R. Schomaecker, ACS Catal., 2014, 4, 3357–3380 CrossRef CAS.
- Y.-H. Hou, Y.-L. Lin, Q. Li, W.-Z. Weng, W.-S. Xia and H.-L. Wan, ChemCatChem, 2013, 5, 3725–3735 CrossRef CAS.
- G. Mitran, T. Cacciaguerra, S. Loridant, D. Tichit and I.-C. Marcu, Appl. Catal., A, 2012, 417–418, 153–162 CrossRef CAS.
- M. A. De León, C. De Los Santos, L. Latrónica, A. M. Cesio, C. Volzone, J. Castiglioni and M. Sergio, Chem. Eng. J., 2014, 241, 336–343 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra01190c |
| This journal is © The Royal Society of Chemistry 2017 |