
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A comparative study of the photovoltaic performances of terpolymers and ternary systems†
Zewdneh Genene ac,
Junyi Wangb,
Xiaofeng Xuc,
Renqiang Yang*b,
Wendimagegn Mammo*a and
Ergang Wang*c
ac,
Junyi Wangb,
Xiaofeng Xuc,
Renqiang Yang*b,
Wendimagegn Mammo*a and
Ergang Wang*c
aDepartment of Chemistry, Addis Ababa University, P.O. Box 33658, Addis Ababa, Ethiopia. E-mail: wendimagegn.mammo@aau.edu.et
bCAS Key Laboratory of Bio-based Materials, Qingdao Institute of Bioenergy and Bioprocess Technology, Chinese Academy of Sciences, Qingdao 266101, China. E-mail: yangrq@qibebt.ac.cn
cDepartment of Chemistry and Chemical Engineering, Chalmers University of Technology, SE-412 96 Göteborg, Sweden. E-mail: ergang@chalmers.se
First published on 24th March 2017
Abstract
Random terpolymers were synthesized from the electron-rich unit thiophene as the donor and two electron-deficient units with complementary absorption as the acceptor. Polymer solar cells (PSCs) fabricated from these terpolymers were compared with those fabricated from the ternary blends of two alternating polymers to explore the best strategy for extending the light absorption range. The two approaches showed similar open-circuit voltages (Voc) but different short-circuit current densities (Jsc). The terpolymer strategy broadened the light absorption range and provided a high power conversion efficiency (PCE) of 5.8%. This is due to a high Jsc and high hole mobility. The device fabricated from the ternary blend exhibited a lower PCE (3.5%) compared to those fabricated from the terpolymers and alternating polymer blends due to the morphological incompatibility of the donor polymers. Our results illustrate the potential of the terpolymer systems as a promising strategy to effectively increase the light absorption and thereby performance of PSCs by combining two morphologically incompatible polymers.
Introduction
Bulk-heterojunction (BHJ) polymer solar cells (PSCs) comprising π-conjugated polymers as donors and fullerene derivatives as acceptors are promising for the realization of low-cost solar energy conversion due to their attractive properties of light weight, easy fabrication, and the ability to be fabricated into flexible large-area devices.1–3 In the past few years, remarkable progress has been made in improving the performances of PSCs, with PCEs reaching over 11%.4 Designing conjugated polymers that exhibit broad and strong light absorption, high charge carrier mobility, and suitable energy level matching with fullerene derivatives has been an efficient strategy to obtain high performance PSCs.5,6A broader and stronger absorption of the photoactive layer is valuable to harvest more photons from solar light, which is a crucial prerequisite for attaining high-performance PSCs. Great efforts have been devoted to extending the absorption range into the visible to near-infrared region and enhancing the performances of BHJ solar cells. One strategy to broaden the absorption ranges of the active layers involves the use of ternary systems containing two donors and one acceptor, or one donor and two acceptors, through complementary absorption of the two donors or two acceptors.7–10 This provides a potentially effective route to achieve high Jsc and thus high PCE.11–15 However, enhancing Jsc, and thereby PCE, is quite challenging because the ternary system has a complicated phase separation behavior. Phase separation has been attributed to the unfavorable interaction between two donors, resulting in severe molecular disorder and large domain size, which acts as the charge recombination trap site.8
Random terpolymers have been prepared by the copolymerization of two different electron-rich units and one electron-deficient unit16–18 or one electron-rich unit and two different electron-deficient units.19–22 This has been used as an alternative strategy towards broadening the absorption of the solar spectrum and tuning the highest occupied molecular orbital (HOMO)/lowest unoccupied molecular orbital (LUMO) energy levels by tweaking the ratio between three different components.22,23 Fan et al.24 reported the syntheses and photovoltaic applications of terpolymers, which provided PCEs over 6% with broad light absorption. Kim et al.25 also reported a random terpolymer that exhibited an extended absorption range, yielding a PCE of 7.2%. Furthermore, a terpolymer, which provided a Jsc of over 15 mA cm−2 and PCE exceeding 8%, was reported.19,20,26 Terpolymers have also been used to tune the molecular packing by introducing components that favor π–π stacking.27,28 Moreover, random terpolymers have some other advantages such as fine-tuning the crystallinity and enhancing the solubility from a range of solvents (including non-halogenated solvents), which provides a wide processing window for reproducible solution-based roll-to-roll production processes.29–31 Although a number of terpolymers have been developed in the past few years, there is a lack of an accurate understanding of compositional effects owing to the random arrangement of the monomer units, which could result in poor reproducibility of the polymer backbone and the resulting performance.
The two abovementioned strategies have gained extensive attention because several polymer combinations can be employed in the active layers of the ternary blend solar cells and a large variety of terpolymers can be synthesized.32 However, a direct comparison of the strategies, in which same molecular constituents were incorporated into the solar cells fabricated from ternary and terpolymer blends, has not been systematically explored. Moreover, these studies are very limited, and the results are mixed.33–36 A comparative study of binary blend systems (chemical blends) based on terpolymers and ternary blend systems (physical blends) was conducted by Lee and co-workers.36 It was found that binary systems exhibited higher PCEs than ternary systems. In our pervious study, we have reported that a random terpolymer containing isoindigo and quinoxaline in the repeat unit exhibited a higher device efficiency (4.37%) than the corresponding ternary blend (2.40%).33 Khlyabich et al.34 reported similar efficiencies for both the ternary blend and the random terpolymers containing thienopyrroledione and diketopyrrolopyrrole acceptors. Recently, Zhang et al.35 obtained a PCE of 4.1% for ternary blend PSCs that is higher than that for random terpolymer blend PSCs. Thus, these comparative studies have to be extensively conducted to further explore the applications and limitations of these two methods (i.e., ternary blend and terpolymer blend). In addition, less extensive comparisons of ternary systems containing similar ratios of the donor polymers with those of random terpolymers have been conducted.36 These studies are vital for understanding the compositional effect on the device performance in terms of charge generation, transport, and structural properties.
In this study, we designed and compared new random terpolymer-based BHJ solar cells and alternating polymer-based ternary solar cells. The random terpolymers were synthesized from fluorinated quinoxaline and isoindigo. We preferred these two electron-deficient acceptor units because they are easy to synthesize and are also ideal for producing polymers with broad light absorption properties due to the difference between their electron-withdrawing strengths. In addition, both units have been successfully used in donor (D)–acceptor (A) and D–A1–D–A2 copolymers to produce high-performance PSCs.37–41 Compared to alternating polymers, terpolymers showed broad absorption. PSCs based on terpolymers showed a higher PCE (5.8%) than the parent binary (PTI-1:PC61BM and TQF2-1:PC61BM) and ternary blends. This is due to the lower Jsc of the ternary blend-based devices than that of the terpolymer-based devices. The lower Jsc of ternary blend-based devices can be ascribed to the poor morphology. Our results indicated that the random terpolymer blend is an effective approach to extend the absorption and improve the device efficiency.
Results and discussion
Synthesis and characterization
To investigate the effect of copolymer composition on the properties of the random terpolymers, three random terpolymers with different molar ratios of fluorinated quinoxaline 1 to isoindigo 2 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 = 3:1, 1:1, and 1:3) were synthesized via the palladium-catalyzed Stille coupling reaction, as shown in Scheme 1. Two D–A alternating polymers (TQF2-1 and PTI-1) were also synthesized for comparison via the procedures reported in our previous work.35,36
:2 = 3:1, 1:1, and 1:3) were synthesized via the palladium-catalyzed Stille coupling reaction, as shown in Scheme 1. Two D–A alternating polymers (TQF2-1 and PTI-1) were also synthesized for comparison via the procedures reported in our previous work.35,36
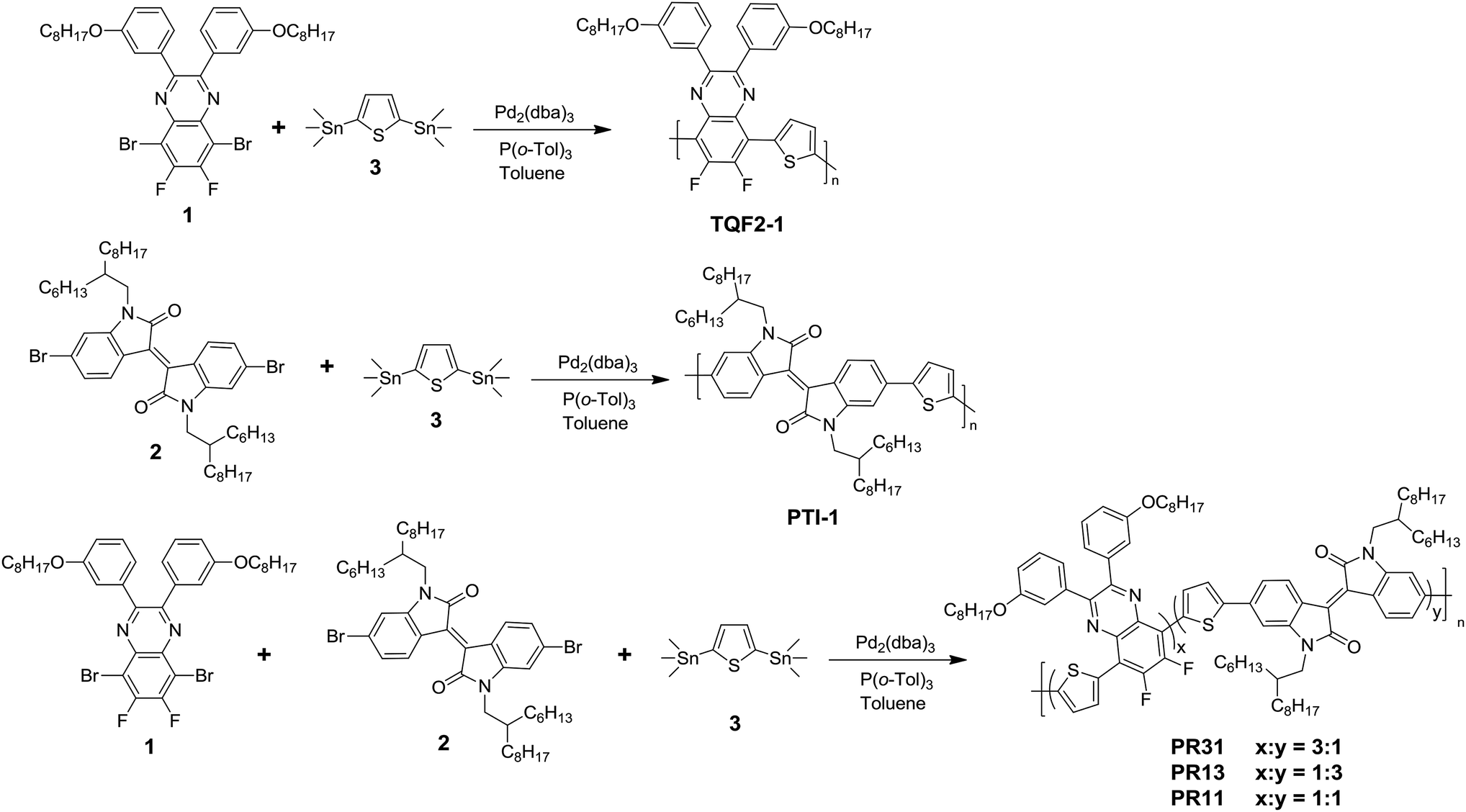 | ||
| Scheme 1 Syntheses of polymers TQF2-1, PTI-1, PR31, PR13, and PR11. | ||
All the polymers showed high solubility in common organic solvents such as chloroform, chlorobenzene (CB), and o-dichlorobenzene (o-DCB). The molecular weights and polydispersity indices (PDI) of the polymers, determined by gel permeation chromatography (GPC) at 150 °C with 1,2,4-trichlorobenzene as the eluent, are summarized in Table 1. TQF2-1 exhibited a low molecular weight, mainly due to the low reactivity of the quinoxaline unit when strongly electron-withdrawing fluorine atoms were introduced. The random terpolymers, especially PR13 with more isoindigo chromophore, exhibited high molecular weights, which can be explained by the more disordered backbones and high entropy as disclosed in our previous work.30,42
| Polymer | Mn (kDa) | Mw (kDa) | PDI | λmax (film) (nm) | Eopg (eV) | Td (°C) |
|---|---|---|---|---|---|---|
| a From ref. 39. | ||||||
| TQF2-1 | 26.9 | 64.4 | 2.4 | 585, 615 | 1.80 | 439 |
| PTI-1 | 44.0 | 127.0 | 2.9 | 633, 685 | 1.60 | 408 |
| PR31 | 44.3 | 123.6 | 2.8 | 626 | 1.60 | 419 |
| PR13 | 64.9 | 199.5 | 3.1 | 636, 688 | 1.58 | 411 |
| PR11 | 34.9 | 90.7 | 2.6 | 638, 685 | 1.58 | 406 |
| PTQTI-Fa | 37.0 | 134.6 | 3.6 | 628, 674 | 1.68 | — |
Thermal properties
The thermal properties of the polymers were investigated by thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC). All polymers possessed excellent thermal stability with the decomposition onset temperatures (Td) above 400 °C (Fig. S1 and Table 1, ESI†). The alternating polymer TQF2-1 showed the highest thermal stability compared to PTI-1 and terpolymers. DSC revealed that TQF2-1 exhibited a noticeable melting transition upon heating and a crystallization transition upon cooling in the temperature range from 50 to 350 °C (Fig. S2†), indicating that this polymer has semi-crystalline properties. This is different from the amorphous property of TQ1, a polymer lacking fluorine atoms, as investigated in our previous work.42 The semi-crystalline property of TQF2-1 may be caused by the interaction between fluorine and hydrogen (F⋯H) on the adjacent thiophene, which promotes planarity of the backbone and π–π stacking.43,44 The polymer PTI-1 also presented a clear melting transition upon heating, which agrees with the previous studies that most isoindigo polymers are semi-crystalline due to the strong interaction of isoindigo units.45 However, the terpolymers PR13 and PR11 had no obvious thermal transition in the temperature range from 50 to 350 °C, which indicates that the semi-crystallinity individually exhibited by the parent polymers (TQF2-1 and PTI-1) was mutually suppressed in the terpolymer. PR31 showed clear melting and a crystallization transition due to the higher mole fraction of fluorinated quinoxaline, which make the thermal property of PR31 to be similar to that of TQF2-1. The DSCs of TQF2-1:PTI-1 blends with 1:3 and 1:1 ratios showed no obvious thermal transition, indicating that the crystallinity of the two polymers was suppressed in these blends, whereas the blend with 3:1 ratio showed a clear melting transition upon heating due to the higher mole fraction of fluorinated quinoxaline (Fig. S2, ESI†).
Optical and electrochemical properties
The UV-vis absorption spectra of the terpolymers and alternating polymers in dilute chloroform solution and as thin films are shown in Fig. 1, and the related parameters are summarized in Table 1. All the polymers showed distinct absorption bands. The UV-vis spectra of the thin films (Fig. 1b) of all the polymers exhibited broader absorptions with red-shifts as compared to their solution spectra. Varying the copolymer composition resulted in changes in the absorption behaviors of both the thin films and solutions. The alternating polymer PTI-1 exhibited broad absorption in the range of 550–750 nm with the absorption onset at 775 nm in the solid state, corresponding to an optical bandgap (Eopg) of 1.60 eV. On the other hand, TQF2-1 showed strong absorption in the range of 450–675 nm with the onset of absorption at the shorter wavelength (688 nm) in the solid state, corresponding to an Eopg of 1.80 eV. This indicates that the absorptions of PTI-1 and TQF2-1 are considerably complementary. The random terpolymer PR31 exhibited a slightly broader absorption with a red-shift of the λmax compared to the alternating polymer TQF2-1. PR13, with the isoindigo-rich composition, exhibited a broader absorption range (450–785 nm) with a red-shift of the absorption compared to PR31. Similarly, PR11, with equal composition of fluorinated quinoxaline and isoindigo, also showed a red-shift of the λmax compared to PR31. For the regular D–A1–D–A2 terpolymer PTQTI-F, a sharp onset of the electronic transitions and a narrow band were observed in our previous studies.39 In contrast, the random terpolymers PR13 and PR11 showed much broader spectra with the onsets of absorption at lower energies. It can be conjectured that longer segments, enriched in fluorinated quinoxaline or isoindigo units, led to local differences in the HOMO and LUMO energy levels along the conjugated backbones of the random terpolymers and thus broadened the absorptions.46,47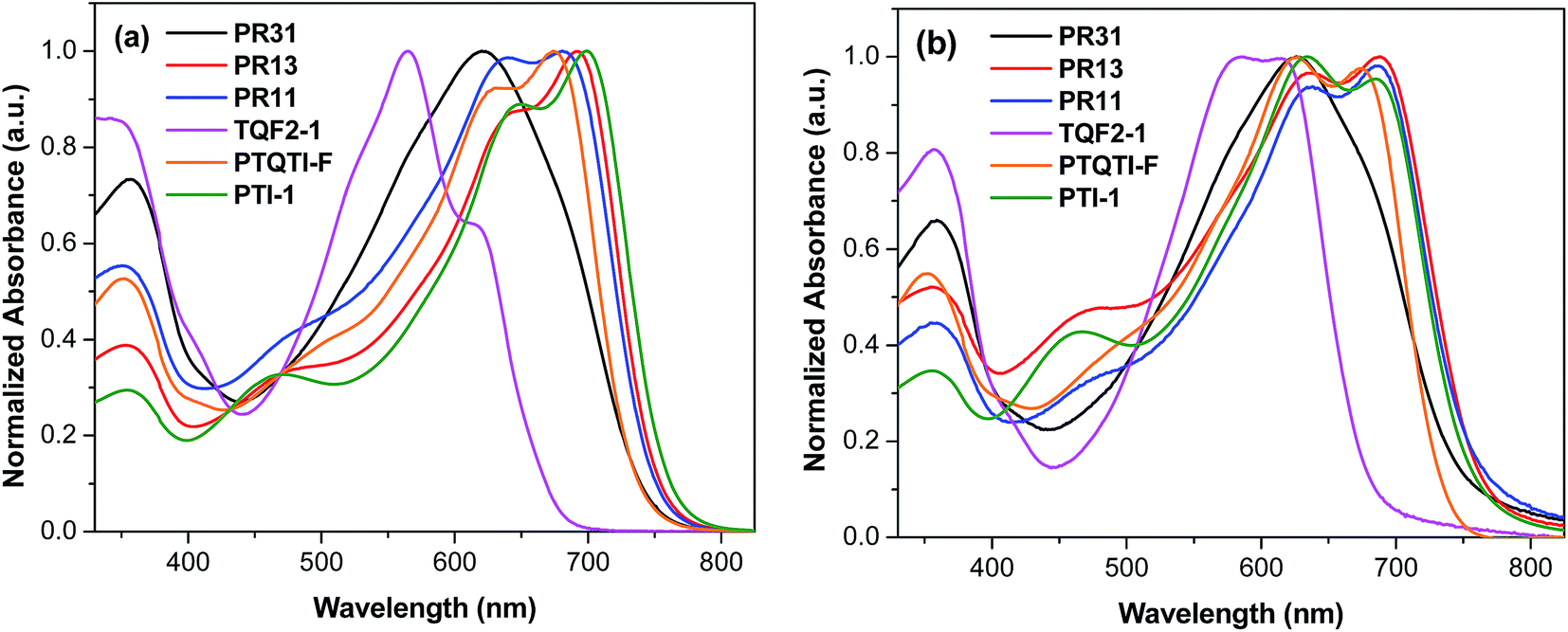 | ||
| Fig. 1 UV-vis absorption spectra of PR31, PR13, PR11, TQF2-1, PTI-1, and PTQTI-F in (a) chloroform solutions and (b) thin films. | ||
The HOMO and LUMO energy levels of all polymers were measured by cyclic voltammetry (Fig. 2). The HOMO levels of TQF2-1 and PTI-1 were −6.18 and −5.78 eV, respectively, which are consistent with the literature values,48,49 whereas PR31, PR13, and PR11 have HOMO levels of −5.92, −5.89, and −5.88 eV, respectively. The three random terpolymers showed similar HOMO levels, which stem from the dominant contribution of the electron-donating thiophene units and two-electron withdrawing units. The low-lying HOMO energy levels of all the polymers are desirable to maximize the open circuit voltage (Voc) values of the corresponding BHJ solar cells. The LUMO levels of PR31, PR13, and PR11 are −3.66, −3.83, and −3.98 eV, respectively.
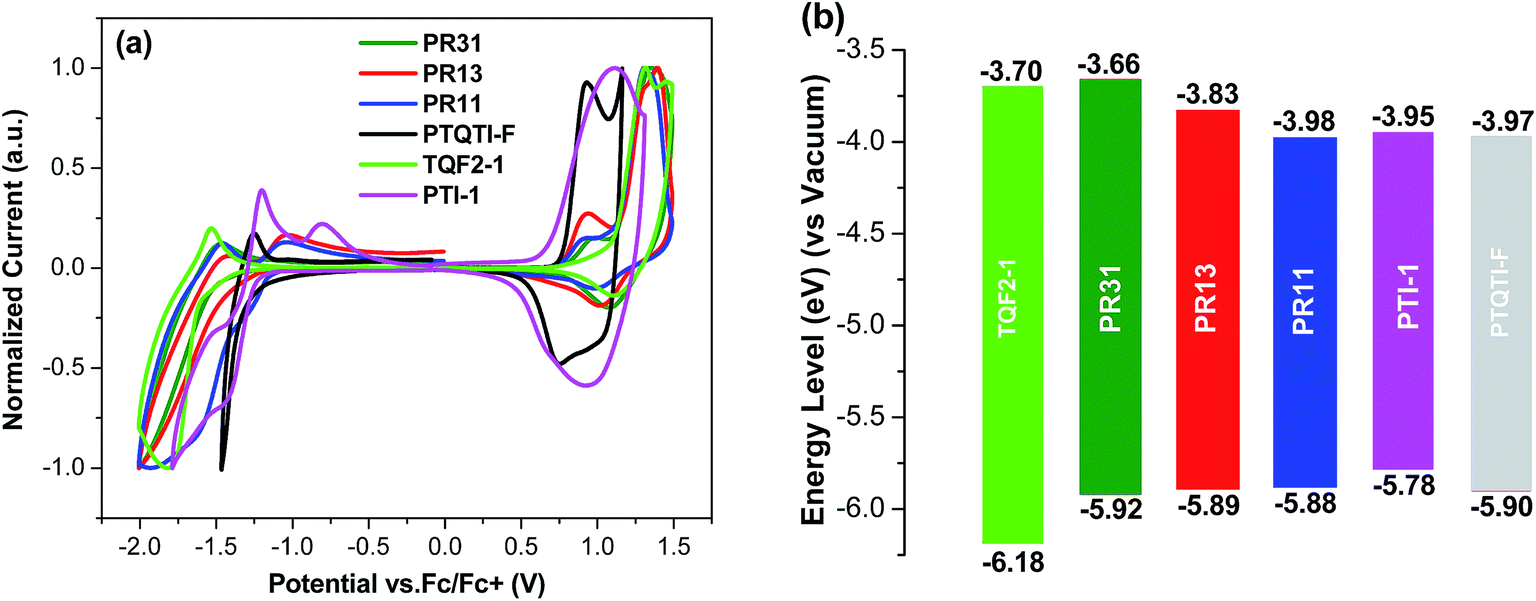 | ||
| Fig. 2 (a) Cyclic voltammograms of the polymers and (b) energy level diagram of the polymers. | ||
Photovoltaic performances
The photovoltaic properties of all five polymers were measured by fabricating BHJ solar cells with the conventional structure ITO/PEDOT:PSS/active layer/Ca/Al, where the active layer was composed of polymer:PC61BM blends. To establish a meaningful comparison of the photovoltaic properties of all the polymers, we applied identical fabrication conditions including the processing solvent (o-DCB), ratio of polymer:PC61BM (1:1), and thicknesses of the active layers (100 nm).
Fig. 3a shows the current density–voltage (J–V) characteristics of optimized PSCs, and the relevant photovoltaic properties are listed in Table 2. The best PCEs of PR31, PR13, and PR11 were 5.5%, 5.8%, and 4.0%, respectively. For comparison, PSCs were also fabricated from the two alternating polymers TQF2-1 and PTI-1. The optimum amount of 1,8-diiodooctane (DIO) used for TQF2-1:PC61BM and PTI-1:PC61BM blends was 2.5%. Devices based on TQF2-1:PC61BM (1:1), optimized by adding 2.5% DIO, provided a Voc of 0.93 V, a Jsc of 8.5 mA cm−2, and a fill factor (FF) of 0.58, resulting in a PCE of 4.6%. The device fabricated without the addition of DIO yielded a higher Voc of 1.00 V and a PCE of 1.9% (Table S1, ESI†), which are comparable to the values reported in the literature.49 Although the Voc value of the device with DIO as an additive slightly decreased, the improved PCE can be attributed to the increase in both Jsc and FF, as promoted by the optimized morphology. On the other hand, the device fabricated from PTI-1:PC61BM (1:1) showed a PCE of 3.7%, which is lower than the values obtained for terpolymers PR31, PR13, and PR11, as shown in Table 2. TQF2-1 and terpolymers had high Voc values due to their deeper HOMO levels, which was also observed in other fluorinated polymer systems.39,41,50,51 PR13 showed the highest Jsc compared to PR31 and PR11, which indicates that the 1:3 ratio of fluorinated quinoxaline to isoindigo is the optimum composition of the acceptor units. These high Voc values (>0.9 V) obtained herein are among the highest reported for the terpolymer-based BHJ solar cells.
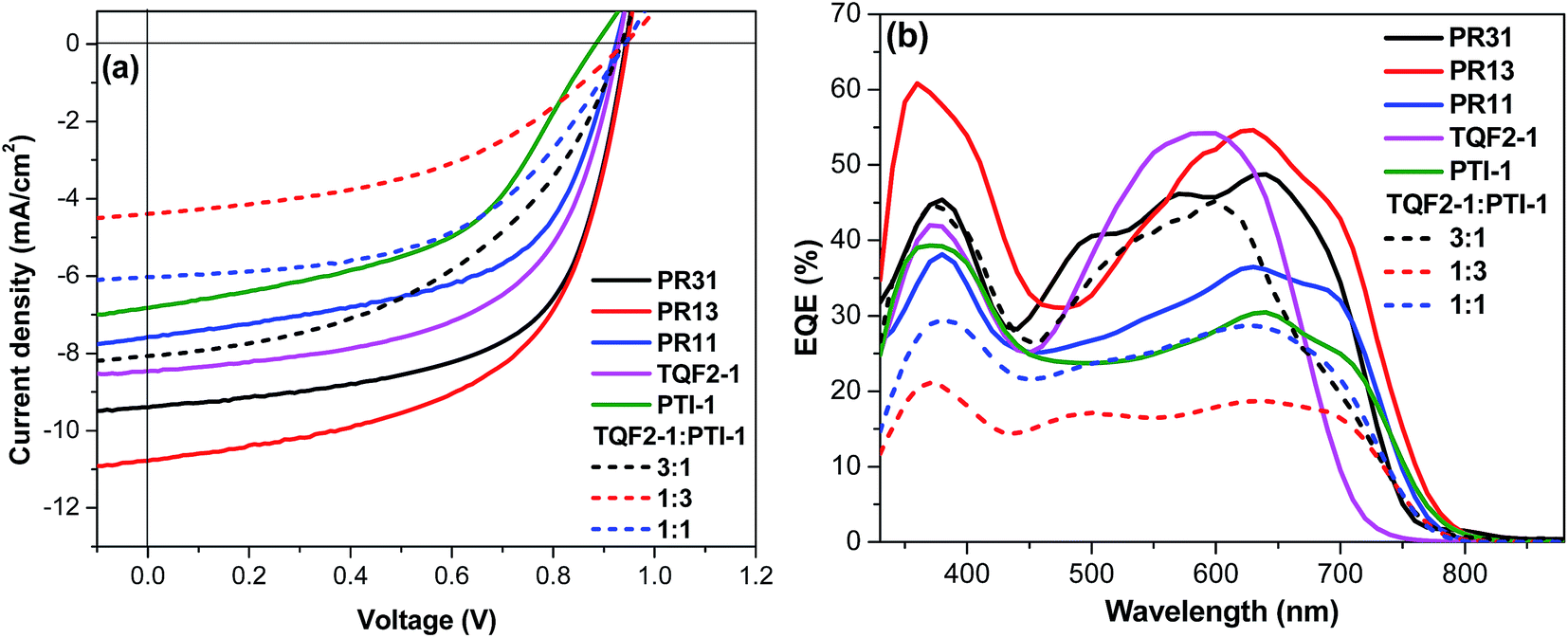 | ||
| Fig. 3 (a) J–V curves and (b) external quantum efficiency spectra of the binary and ternary blend solar cells. | ||
:1) and ternary blends of TQF2-1:PTI-1:PC61BM (1:1)
| Polymer | DIO | Voc (V) | Jsc (mA cm−2) | FF | PCE (%) | SCLC μh (cm2 V−1 s−1) |
|---|---|---|---|---|---|---|
| a From ref. 39.b Integrated Jsc from EQE spectra. | ||||||
| TQF2-1 | 2.5% | 0.93 | 8.5 (8.1)b | 0.58 | 4.6 | 2.7 × 10−5 |
| PTI-1 | 2.5% | 0.88 | 6.8 (6.4)b | 0.62 | 3.7 | 3.1 × 10−4 |
| PR31 | 2% | 0.94 | 9.4 (9.2)b | 0.62 | 5.5 | 4.6 × 10−4 |
| PR13 | 2% | 0.95 | 10.8 (10.3)b | 0.57 | 5.8 | 8.7 × 10−4 |
| PR11 | 2% | 0.92 | 7.6 (7.2)b | 0.57 | 4.0 | 3.7 × 10−4 |
| PTQTI-Fa | 2.5% | 0.93 | 12.6 | 0.54 | 6.3 | — |
| TQF2-1:PTI-1 (3:1) |
2.5% | 0.93 | 8.1 (7.7)b | 0.47 | 3.5 | — |
| TQF2-1:PTI-1 (1:3) |
2.5% | 0.94 | 4.4 (4.0)b | 0.45 | 1.9 | — |
| TQF2-1:PTI-1 (1:1) |
2.5% | 0.94 | 6.0 (5.7)b | 0.52 | 2.9 | — |
Among all the polymers investigated in this study, PR13 exhibited the highest PCE value of 5.8%, which can be attributed to the increased Voc value of 0.95 V and the high Jsc value of 10.8 mA cm−2, probably due to its broad absorption. This PCE (5.8%) is among the highest efficiencies reported for random terpolymer-based PSCs containing one electron-rich and two different electron-deficient moieties. The random terpolymer PR31 also showed better efficiency than the corresponding D–A alternating polymers. Furthermore, PR31 showed higher Jsc and thereby higher efficiency than PR11 although PR31 exhibited a narrower absorption with a blue-shift compared to PR11. We inferred that the semi-crystalline property of PR31 might have facilitated hole transport and thus afforded higher Jsc.52 However, the PCEs of the terpolymers decreased with the increasing volume ratio of DIO. The performances of all the PSCs based on the terpolymers reported herein are lower than that of the regular terpolymer PTQTI-F reported in our previous study.39 Binary blend solar cells with all five polymers as donors and PC61BM as the acceptor showed optimal device performance at the polymer:PC61BM ratio of 1:1 under the same processing conditions (Table S1, ESI†).
For comparison, photovoltaic devices containing ternary blends, with TQF2-1 and PTI-1 as donors and PC61BM as the acceptor, in the same device configuration were also fabricated. In the ternary blend solar cells, the overall polymer:fullerene ratio was kept constant at 1:1, and the ratio between TQF2-1 and PTI-1 was maintained at 3:1, 1:1, and 1:3 for the three ternary devices, similar to the feed ratio of the corresponding components in the random terpolymers. Film thicknesses were kept constant at 100 nm for direct comparison with the five binary BHJ solar cells. As can be seen from Table 2, Voc of the ternary blend solar cells was similar to those of the binary BHJ solar cells based on terpolymers, which is consistent with our previous observation.33 On the other hand, Jsc was found to decrease with the increasing PTI-1 concentration in the three-component blends (Fig. 3a). The ternary blend solar cell with high TQF2-1 concentration exhibited a PCE of 3.5%. It was noticed that the PCEs of all the ternary blend solar cells were lower than those of the terpolymer- and alternating polymer-based binary BHJ solar cells. This is due to the noticeably lower Jsc and FF values of the ternary blend devices compared to those of the terpolymer- and alternating polymer-based devices.
The external quantum efficiency (EQE) spectra of the optimized binary solar cells and ternary blend solar cells were obtained to further confirm the accuracy of the J–V measurements. As shown in Fig. 3b, the EQEs of the terpolymer-based devices were in the range of 38–60% over the broad wavelength region from 400 to 800 nm. The ternary blend-based devices showed much lower EQE; however, the contributions from both donor components can be clearly observed. The calculated Jsc from EQE profiles, integrated with the solar spectrum, are included in Table 2, which are in a good agreement with the corresponding Jsc values obtained from the J–V measurements of all the devices. To assess the charge transport properties of the blends, hole mobilities were evaluated by space-charge-limited current (SCLC) measurements. The hole mobility of PR13 was higher as compared to those of PR31, PR11, and the alternating polymers (Table 2). Therefore, the broad light absorption and high hole mobility of PR13 are probably the main reasons for its highest Jsc value.
Morphology study
The film morphology of the BHJ layer can significantly influence the photovoltaic performance of a PSC.6,53 Thus, to further understand the differences between the performances of ternary blend-based solar cells and the parent binary-based solar cells, atomic force microscopy (AFM) was utilized to probe the morphologies of these polymer:fullerene blends. Fig. 4 displays the AFM height images of the binary and ternary blend BHJ solar cells. Distinct film morphologies were observed for these blend films. Smaller nanoscale domains and a more continuous interpenetrating network were observed for the PR31:PC61BM and PR13:PC61BM blend films, with root mean square (RMS) roughness of 1.58 and 2.72 nm, respectively. In contrast, some aggregations with fibrous nanostructures across the entire film of the PR11:PC61BM blend could be clearly observed (Fig. 4). In addition, the PR11:PC61BM blend film exhibited much rougher film morphology with an RMS value of 9.25 nm. This poor morphology of the PR11:PC61BM blend could explain its lower Jsc and photovoltaic performance compared to those of the PR31- and PR13-based devices.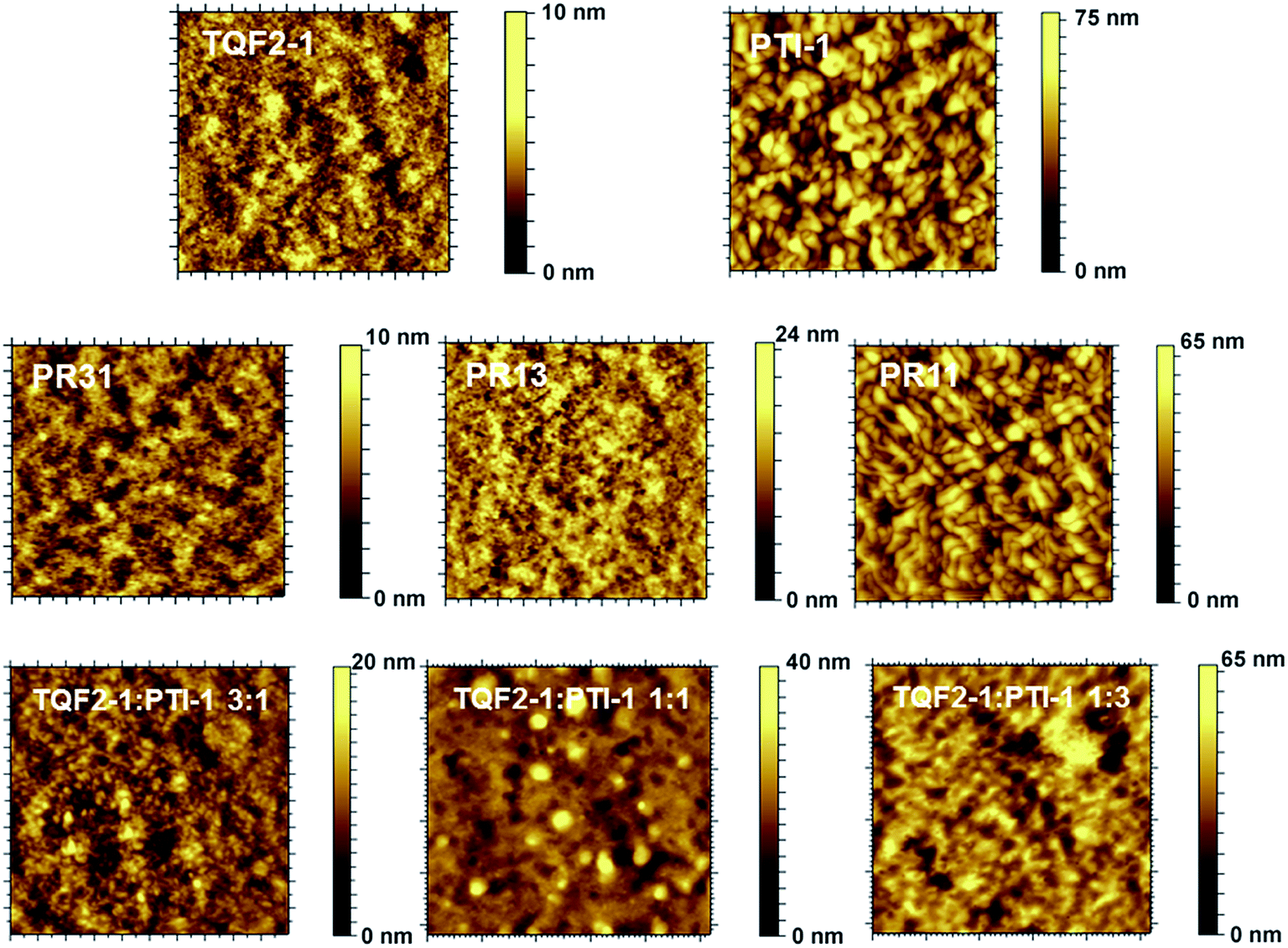 | ||
| Fig. 4 AFM height images of the binary and ternary blend films (5 × 5 μm2). | ||
On the other hand, AFM images of the ternary blend films showed increased domain sizes as the concentration of PTI-1 increased, probably indicating serious phase separation. The corresponding RMS values of 2.21, 4.15, and 8.15 nm were obtained for the ternary blend films of TQF2-1:PTI-1(3:1):PC61BM, TQF2-1:PTI-1(1:1):PC61BM, and TQF2-1:PTI-1(1:3):PC61BM, respectively. This is consistent to the much higher roughness (RMS = 11.4) of PTI-1 than that of TQF2-1 (RMS = 1.01) when mixed with PC61BM. The ternary blend systems featured larger scale phase-separated polymer–PC61BM domains as compared to the binary blend systems. Considering the large domain sizes, particularly in the TQF2-1:PTI-1(1:3):PC61BM and TQF2-1:PTI-1(1:1):PC61BM blend films, and the limited diffusion length of the excitons (10 nm),54 most photogenerated excitons in the ternary blend could not reach the interfaces of the donor and acceptor, which reduced charge carrier generation and led to much lower Jsc and FF. In the case of the binary blend films of the terpolymers, smooth and uniform morphologies indicate much better miscibility between the polymers and PC61BM, which led to improved Jsc and FF.
For ternary BHJs, relatively similar phase separation behaviors of both donor polymers with the fullerene derivative are a reasonable requirement for compatible morphology and good performance.55 The two donor polymers TQF2-1 and PTI-1 that have different frameworks of the PC61BM phase in their binary films showed large phase separation in their ternary films, presumably because of morphological incompatibility. As a result, the ternary blends exhibited lower performance. Our results suggest that the terpolymer system consisting of two incompatible polymers could be a good strategy to improve the light absorption and to further improve the efficiency of the PSCs.
Conclusion
In summary, we synthesized alternating copolymers and random terpolymers from one donor unit and two different acceptor units to achieve PSCs with panchromatic light absorption. The optical, electrochemical, and photovoltaic properties of the random terpolymers were optimized by varying the ratios of the two acceptor units in the copolymers. PSCs based on these terpolymers were compared with the PSCs based on the alternating polymers as well as on the ternary blends of the parent polymers. The terpolymers showed extended absorption compared to the parent polymers. BHJ solar cells based on the terpolymers and ternary blends showed comparably high Voc values. However, the Jsc behaviors were significantly different. The ternary systems showed lower Jsc values due to large scale phase separation observed in the morphologies of the ternary blends. As a result, PSCs fabricated from the terpolymers performed better than those fabricated from the ternary blends. Under optimized conditions, PR13-based PSC exhibited high PCE of 5.8% with a Voc of 0.95 V, a Jsc of 10.8 mA cm−2, and a FF of 0.57, which is superior to the values obtained from the devices fabricated from the alternating polymers and ternary blends. A study of the morphologies of the alternating polymers showed that the two binary blends have relatively different phase separation, which makes them incompatible polymers for ternary systems. Our study demonstrated that terpolymer system could be a good strategy to combine two incompatible polymers.Experimental
Synthesis
All reagents and starting materials were purchased from commercial sources and used without further purification unless otherwise noted. 2,5-Bis-(trimethylstannyl)thiophene (3) was purchased from Solarmer Materials Inc. 5,8-Bis(5-bromothiophen-2-yl)-6,7-difluoro-2,3-bis(3-octyloxyphenyl)quinoxaline (1)39 and (E)-6,6′-dibromo-1,1′-bis(2-hexyldecyl)-[3,3′-biindolinylidene]-2,2′-dione (2)48 were synthesized by following the literature procedures.Synthesis of polymer PR31
In a dry 25 mL flask, monomer 1 (220 mg, 0.3 mmol), monomer 2 (87 mg, 0.1 mmol), monomer 3 (164 mg, 0.4 mmol), tris(dibenzylideneacetone) dipalladium(0) (Pd2(dba)3) (4.58 mg), and tri(o-tolyl)phosphine (P(o-Tol)3) (6.08 mg) were dissolved in anhydrous toluene (8 mL) under nitrogen atmosphere. The reaction mixture was vigorously stirred at 100 °C for 1 h. 2-Bromothiophene and tributyl(thiophen-2-yl)stannane were added to the solution as end-cappers. After cooling down to room temperature, the polymer was precipitated by pouring the solution into methanol, filtered through a Soxhlet thimble, and then subjected to Soxhlet extraction with acetone, diethyl ether, and chloroform. The chloroform fraction was purified by passing it though a short silica gel column followed by precipitation from acetone. Finally, the polymer PR31 was obtained by filtering the precipitate through a 0.45 μm Teflon filter and drying overnight under vacuum at 40 °C. Yield: 251 mg (90.6%).Synthesis of polymer PR13
Polymer PR13 was prepared from monomer 1 (73.3 mg, 0.1 mmol), monomer 2 (260.7 mg, 0.3 mmol), and monomer 3 (164 mg, 0.4 mmol) by following a similar procedure as described above for the synthesis of PR31, but the reaction time was 0.5 h. The yield was 95.4%.Synthesis of polymer PR11
Polymer PR11 was prepared from monomer 1 (146.5 mg, 0.2 mmol), monomer 2 (173.8 mg, 0.2 mmol), and monomer 3 (164 mg, 0.4 mmol) by following a similar procedure as described above for the synthesis of PR31, but the reaction time was 6 h. The yield was 95.2%.Synthesis of polymer TQF2-1
In a dry 25 mL flask, monomer 1 (234.4 mg, 0.32 mmol), monomer 3 (131.1 mg, 0.32 mmol), Pd2(dba)3 (9 mg), and P(o-Tol)3 (15 mg) were dissolved in anhydrous toluene (8 mL) under nitrogen atmosphere. The reaction mixture was vigorously stirred at 110 °C for 48 h. 2-Bromothiophene and tributyl(thiophen-2-yl)stannane were added to the solution as end-cappers. After cooling down to room temperature, the polymer was precipitated by pouring the solution into methanol, filtered through a Soxhlet thimble, and then subjected to Soxhlet extraction with acetone, diethyl ether, and chloroform. The chloroform fraction was purified by passing it through a short silica gel column followed by precipitation from acetone. Finally, the polymer TQF2-1 was obtained by filtering the precipitate through a 0.45 μm Teflon filter and drying overnight under vacuum at 40 °C. Yield: 180 mg (90.6%).Characterization
1H NMR (400 MHz) and 13C NMR (100 MHz) spectra were acquired using a Varian Inova 400 MHz NMR spectrometer. Tetramethylsilane was used as an internal reference with deuterated chloroform as the solvent. Size exclusion chromatography (SEC) was performed using a Waters Alliance GPCV2000 with a refractive index detector. Columns used are as follows: Waters Styragel HT GE ×1 and Waters Styragel HMW GE ×2. The eluent was 1,2,4-trichlorobenzene. The working temperature was 150 °C and the resolution time was 2 h. The concentration of the samples was 0.5 mg mL−1, which were filtered (filter: 0.45 μm) prior to the analysis. The molecular weights were calculated according to the relative calibration with polystyrene standards. UV-vis absorption spectra were obtained using a Perkin Elmer Lambda 900 UV-vis-NIR absorption spectrometer. AFM images were acquired via an Agilent-5400 scanning probe microscope with a Nanodrive controller in the tapping mode using MikroMasch NSC-15 AFM tips with resonant frequencies ∼300 kHz. Cyclic voltammetry (CV) measurements were carried out via a CH-Instruments 650A Electrochemical Workstation. A three-electrode setup was used with platinum wires both as the working electrode and the counter electrode, and Ag/Ag+ was used as the reference electrode calibrated with the ferrocene/ferrocenyl couple (Fc/Fc+). A 0.1 M solution of tetrabutylammonium hexafluorophosphate (Bu4NPF6) in anhydrous acetonitrile was used as the supporting electrolyte. The polymer was deposited onto the working electrode from the chloroform solution. To remove oxygen from the electrolyte, the system was bubbled with nitrogen prior to each experiment. The nitrogen inlet was then moved to above the liquid surface and left there during the scans. HOMO and LUMO levels were estimated from the onset potentials of the third scan by setting the oxidative onset potential of Fc/Fc+ vs. the normal hydrogen electrode (NHE) to 0.63 V,56 and the NHE vs. the vacuum level to 4.5 V.57 The HOMO and LUMO levels were calculated according to the formula HOMO = −(Eox + 5.13) eV and LUMO = −(Ered + 5.13) eV, where Eox and Ered were determined from the oxidation and reduction onsets, respectively.58 Hole mobility was measured using the space charge limited current (SCLC) model, with the device configuration of ITO/PEDOT:PSS/active layer/MoO3/Al by taking current–voltage in the range of 0–5 V and fitting the results to a space charge limited form for hole-only device. In the presence of carrier traps in the active layer, a trap-filled-limit (TFL) region existed between the ohmic and trap-free SCLC regions. The SCLC behavior in the trap-free region can be characterized using the Mott–Gurney square law.59Device fabrication and characterization
The structure of the solar cells was glass/ITO/PEDOT:PSS/active layer/Ca/Al. As a buffer layer, the conductive polymer PEDOT:PSS (Baytron P VP Al 4083) was spin-coated onto ITO-coated glass substrates, followed by annealing at 160 °C for 30 minutes to remove water. The thickness of the PEDOT:PSS layer was about 40 nm, as determined by a Dektak 150 surface profilometer. The active layer (100 nm) consisting of polymers and PC61BM was spin-coated from o-DCB solution onto the PEDOT:PSS layer. Spin-coating was carried out in a glove box and the material was directly transferred to a vapor deposition system mounted inside the glove box. Ca (10 nm) and Al (100 nm) were used as top electrodes and were deposited via a mask in vacuum onto the active layer. The accurate area of every device (10 mm2), defined by the overlap of the ITO and metal electrode, was carefully measured by microscope imaging. PCE was calculated from the J–V characteristics obtained by a Keithley 2420 source meter under illumination of an AM 1.5G solar simulator with an intensity of 100 mW cm−2 (Newport Oriel 1000 W). The light intensity was determined by a standard silicon photodiode. EQEs of solar cells were measured using a Newport 2931-C coupled with a 300 W xenon lamp.Acknowledgements
We acknowledge the financial support received from the Swedish Research Council, the Swedish Research Council Formas and Chalmers Area of Advance Energy. W. M. and Z. G. acknowledge the financial support received from the International Science Programme, Uppsala University, Sweden. R. Y. thanks the National Natural Science Foundation of China (51573205). The research leading to these results has received funding from the People Programme (Marie Curie Actions) of the European Union's Seventh Framework Programme (FP7/2007–2013) under REA grant agreement no. 608743. This publication reflects only the view of the authors and the European Union is not liable for any use that may be made of the information contained herein.Notes and references
- G. Li, R. Zhu and Y. Yang, Nat. Photonics, 2012, 6, 153 CrossRef CAS.
- E. Wang, W. Mammo and M. R. Andersson, Adv. Mater., 2014, 26, 1801 CrossRef CAS PubMed.
- L. Lu, T. Zheng, Q. Wu, A. M. Schneider, D. Zhao and L. Yu, Chem. Rev., 2015, 115, 12666 CrossRef CAS PubMed.
- J. Zhao, Y. Li, G. Yang, K. Jiang, H. Lin, H. Ade, W. Ma and H. Yan, Nat. Energy, 2016, 1, 15027 CrossRef CAS.
- Z. Genene, J. Wang, X. Meng, W. Ma, X. Xu, R. Yang, W. Mammo and E. Wang, Adv. Electron. Mater., 2016, 2, 1600084 CrossRef.
- Y. Huang, E. J. Kramer, A. J. Heeger and G. C. Bazan, Chem. Rev., 2014, 114, 7006 CrossRef CAS PubMed.
- N. Felekidis, E. Wang and M. Kemerink, Energy Environ. Sci., 2016, 9, 257 CAS.
- Q. An, F. Zhang, J. Zhang, W. Tang, Z. Deng and B. Hu, Energy Environ. Sci., 2016, 9, 281 Search PubMed.
- L. Lu, M. A. Kelly, W. You and L. Yu, Nat. Photonics, 2015, 9, 491 CrossRef CAS.
- L. Yang, L. Yan and W. You, J. Phys. Chem. Lett., 2013, 4, 1802 CrossRef CAS PubMed.
- P. Cheng, Y. Li and X. Zhan, Energy Environ. Sci., 2014, 7, 2005 CAS.
- L. Lu, T. Xu, W. Chen, E. S. Landry and L. Yu, Nat. Photonics, 2014, 8, 716 CrossRef CAS.
- Y. Wang, H. Ohkita, H. Benten and S. Ito, Phys. Chem. Chem. Phys., 2015, 17, 27217 RSC.
- M. Zhang, F. Zhang, Q. An, Q. Sun, J. Wang, L. Li, W. Wang and J. Zhang, Sol. Energy Mater. Sol. Cells, 2015, 141, 154 CrossRef CAS.
- T. Liu, L. Huo, X. Sun, B. Fan, Y. Cai, T. Kim, J. Y. Kim, H. Choi and Y. Sun, Adv. Energy Mater., 2016, 6, 1502109 CrossRef.
- Z. Zhu, D. Waller, R. Gaudiana, M. Morana, D. Mühlbacher, M. Scharber and C. Brabec, Macromolecules, 2007, 40, 1981 CrossRef CAS.
- M.-C. Yuan, M.-Y. Chiu, C.-M. Chiang and K.-H. Wei, Macromolecules, 2010, 43, 6270 CrossRef CAS.
- J. Li, K.-H. Ong, P. Sonar, S.-L. Lim, G.-M. Ng, H.-K. Wong, H.-S. Tan and Z.-K. Chen, Polym. Chem., 2013, 4, 804 RSC.
- B. Burkhart, P. P. Khlyabich and B. C. Thompson, ACS Macro Lett., 2012, 1, 660 CrossRef CAS.
- J.-M. Jiang, H.-C. Chen, H.-K. Lin, C.-M. Yu, S.-C. Lan, C.-M. Liu and K.-H. Wei, Polym. Chem., 2013, 4, 5321 RSC.
- T. E. Kang, H.-H. Cho, H. j. Kim, W. Lee, H. Kang and B. J. Kim, Macromolecules, 2013, 46, 6806 CrossRef CAS.
- J. Zhou, S. Xie, E. F. Amond and M. L. Becker, Macromolecules, 2013, 46, 3391 CrossRef CAS.
- T. E. Kang, K.-H. Kim and B. J. Kim, J. Mater. Chem. A, 2014, 2, 15252 CAS.
- Q. Fan, Y. Liu, M. Xiao, W. Su, H. Gao, J. Chen, H. Tan, Y. Wang, R. Yang and W. Zhu, J. Mater. Chem. C, 2015, 3, 6240 RSC.
- K.-H. Kim, S. Park, H. Yu, H. Kang, I. Song, J. H. Oh and B. J. Kim, Chem. Mater., 2014, 26, 6963 CrossRef CAS.
- T. Jiang, J. Yang, Y. Tao, C. Fan, L. Xue, Z. Zhang, H. Li, Y. Li and W. Huang, Polym. Chem., 2016, 7, 926 RSC.
- L. Fang, Y. Zhou, Y.-X. Yao, Y. Diao, W.-Y. Lee, A. L. Appleton, R. Allen, J. Reinspach, S. C. B. Mannsfeld and Z. Bao, Chem. Mater., 2013, 25, 4874 CrossRef CAS.
- W.-H. Chang, J. Gao, L. Dou, C.-C. Chen, Y. Liu and Y. Yang, Adv. Energy Mater., 2014, 4, 1300864 CrossRef.
- S. Berny, N. Blouin, A. Distler, H.-J. Egelhaaf, M. Krompiec, A. Lohr, O. R. Lozman, G. E. Morse, L. Nanson, A. Pron, T. Sauermann, N. Seidler, S. Tierney, P. Tiwana, M. Wagner and H. Wilson, Adv. Sci., 2016, 3, 1500342 CrossRef PubMed.
- Z. Li, X. Xu, W. Zhang, X. Meng, W. Ma, A. Yartsev, O. Inganäs, M. R. Andersson, R. A. J. Janssen and E. Wang, J. Am. Chem. Soc., 2016, 138, 10935 CrossRef CAS PubMed.
- Y.-J. Hwang, T. Earmme, S. Subramaniyan and S. A. Jenekhe, Chem. Commun., 2014, 50, 10801 RSC.
- P. P. Khlyabich, A. E. Rudenko, R. A. Street and B. C. Thompson, ACS Appl. Mater. Interfaces, 2014, 6, 9913 CAS.
- W. Sun, Z. Ma, D. Dang, W. Zhu, M. R. Andersson, F. Zhang and E. Wang, J. Mater. Chem. A, 2013, 1, 11141 CAS.
- P. P. Khlyabich, A. E. Rudenko, B. Burkhart and B. C. Thompson, ACS Appl. Mater. Interfaces, 2015, 7, 2322 CAS.
- Q. Zhang, M. A. Kelly, A. Hunt, H. Ade and W. You, Macromolecules, 2016, 49, 2533 CrossRef CAS.
- S. J. Park, J. M. Cho, W.-B. Byun, J.-C. Lee, W. S. Shin, I.-N. Kang, S.-J. Moon and S. K. Lee, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 4416 CrossRef CAS.
- Q. Tao, Y. Xia, X. Xu, S. Hedström, O. Bäcke, D. I. James, P. Persson, E. Olsson, O. Inganäs, L. Hou, W. Zhu and E. Wang, Macromolecules, 2015, 48, 1009 CrossRef CAS.
- D. Dang, W. Chen, S. Himmelberger, Q. Tao, A. Lundin, R. Yang, W. Zhu, A. Salleo, C. Müller and E. Wang, Adv. Energy Mater., 2014, 4, 1801 Search PubMed.
- D. Dang, W. Chen, R. Yang, W. Zhu, W. Mammo and E. Wang, Chem. Commun., 2013, 49, 9335 RSC.
- D. Dang, P. Zhou, L. Duan, X. Bao, R. Yang and W. Zhu, J. Mater. Chem. A, 2016, 4, 8291 CAS.
- D. Liu, W. Zhao, S. Zhang, L. Ye, Z. Zheng, Y. Cui, Y. Chen and J. Hou, Macromolecules, 2015, 48, 5172 CrossRef CAS.
- E. Wang, J. Bergqvist, K. Vandewal, Z. Ma, L. Hou, A. Lundin, S. Himmelberger, A. Salleo, C. Müller, O. Inganäs, F. Zhang and M. R. Andersson, Adv. Energy Mater., 2013, 3, 806 CrossRef CAS.
- T. L. Nguyen, H. Choi, S. J. Ko, M. A. Uddin, B. Walker, S. Yum, J. E. Jeong, M. H. Yun, T. J. Shin, S. Hwang, J. Y. Kim and H. Y. Woo, Energy Environ. Sci., 2014, 7, 3040 CAS.
- Y.-S. Lee, J. Y. Lee, S.-M. Bang, B. Lim, J. Lee and S.-I. Na, J. Mater. Chem. A, 2016, 4, 11439 CAS.
- Z. Ma, W. Sun, S. Himmelberger, K. Vandewal, Z. Tang, J. Bergqvist, A. Salleo, J. W. Andreasen, O. Inganäs, M. R. Andersson, C. Müller, F. Zhang and E. Wang, Energy Environ. Sci., 2014, 7, 361 CAS.
- H. Heo, H. Kim, D. Lee, S. Jang, L. Ban, B. Lim, J. Lee and Y. Lee, Macromolecules, 2016, 49, 3328 CrossRef CAS.
- K. H. Hendriks, G. H. L. Heintges, M. M. Wienk and R. A. J. Janssen, J. Mater. Chem. A, 2014, 2, 17899 CAS.
- E. Wang, Z. Ma, Z. Zhang, P. Henriksson, O. Inganäs, F. Zhang and M. R. Andersson, Chem. Commun., 2011, 47, 4908 RSC.
- G. K. Dutta, T. Kim, H. Choi, J. Lee, D. S. Kim, J. Y. Kim and C. Yang, Polym. Chem., 2014, 5, 2540 RSC.
- Q. Peng, X. Liu, D. Su, G. Fu, J. Xu and L. Dai, Adv. Mater., 2011, 23, 4554 CrossRef CAS PubMed.
- S. Albrecht, S. Janietz, W. Schindler, J. Frisch, J. Kurpiers, J. Kniepert, S. Inal, P. Pingel, K. Fostiropoulos, N. Koch and D. Neher, J. Am. Chem. Soc., 2012, 134, 14932 CrossRef CAS PubMed.
- J. W. Jung, F. Liu, T. P. Russell and W. H. Jo, Energy Environ. Sci., 2013, 6, 3301 CAS.
- J. S. Moon, J. Jo and A. J. Heeger, Adv. Energy Mater., 2012, 2, 304 CrossRef CAS.
- O. V. Mikhnenko, H. Azimi, M. Scharber, M. Morana, P. W. M. Blom and M. A. Loi, Energy Environ. Sci., 2012, 5, 6960 CAS.
- J. Mai, T.-K. Lau, J. Li, S.-H. Peng, C.-S. Hsu, U. S. Jeng, J. Zeng, N. Zhao, X. Xiao and X. Lu, Chem. Mater., 2016, 28, 6186 CrossRef CAS.
- V. V. Pavlishchuk and A. W. Addison, Inorg. Chim. Acta, 2000, 298, 97 CrossRef CAS.
- A. J. Bard and L. R. Faulkner, Electrochemical methods: fundamentals and applications, Wiley, New York, 2nd edn, 2001 Search PubMed.
- S. Hellstrom, F. Zhang, O. Inganäs and M. R. Andersson, Dalton Trans., 2009, 10032 RSC.
- G. G. Malliaras, J. R. Salem, P. J. Brock and C. Scott, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, R13411 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra01418j |
| This journal is © The Royal Society of Chemistry 2017 |