
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of methylated quercetin analogues for enhancement of radical-scavenging activity†
Kohei Imai *ab,
Ikuo Nakanishi*b,
Kei Ohkubo*bc,
Yusuke Ohbaa,
Takuya Araia,
Mirei Mizunoa,
Shunichi Fukuzumide,
Ken-ichiro Matsumotob and
Kiyoshi Fukuharaa
*ab,
Ikuo Nakanishi*b,
Kei Ohkubo*bc,
Yusuke Ohbaa,
Takuya Araia,
Mirei Mizunoa,
Shunichi Fukuzumide,
Ken-ichiro Matsumotob and
Kiyoshi Fukuharaa
aSchool of Pharmacy, Showa University, Hatanodai, Shinagawa-ku, Tokyo 142-8555, Japan. E-mail: imai@pharm.showa-u.ac.jp
bQuantitative RedOx Sensing Team (QRST), Department of Basic Medical Sciences for Radiation Damages, National Institute of Radiological Sciences (NIRS), National Institutes for Quantum and Radiological Science and Technology (QST), Inage-ku, Chiba 263-8555, Japan
cDivision of Innovative Research for Drug Design, Institute of Academic Initiatives, Osaka University, Suita, Osaka 565-0871, Japan
dDepartment of Chemistry and Nano Science, Ewha Womans University, Seoul 120-750, Korea
eFaculty of Science and Technology, Meijo University, SENTAN, Japan Science and Technology Agency (JST), Nagoya, Aichi 468-8502, Japan
First published on 24th March 2017
Abstract
Three quercetin derivatives with enhanced radical-scavenging activity were designed and synthesised. Because the radical-scavenging reaction of quercetin is known to proceed via an electron transfer from quercetin to radicals, producing the corresponding quercetin radical cation intermediate, the introduction of electron-donating groups into the quercetin molecule is expected to enhance its radical-scavenging activity. Thus, methyl groups were introduced into the catechol moiety in the quercetin molecule at either the 2′- or 5′-position, or both. All three quercetin analogues were found to exhibit higher radical-scavenging activity than the parent quercetin. The activity of 5′-methylquercetin is the highest among the three analogues. The optimised structure of 5′-methylquercetin calculated by density functional theory demonstrated a coplanar structure between the 4H-curomen (AC rings) and catechol (B ring) moieties, while dimethylquercetin and 2′-methylquercetin have a twisted structure between the AC and B rings. These results demonstrate that the highest radical-scavenging activity of 5′-methylquercetin is due to the stabilisation of the radical cation intermediate by the electron-donating effect of the methyl group as well as by the planar structure of the molecule.
Introduction
Flavonoids are natural compounds that are commonly found in fruits and vegetables and exhibit numerous beneficial physiological activities.1–4 The antioxidative and radical-scavenging activities of flavonoids have been widely studied, and flavonoid derivatives have been developed to enhance the radical-scavenging activities.5–8 Flavonoids are a natural part of the human diet, but can also be taken as supplements. The intake of flavonoids is believed to help prevention from certain illnesses associated with high levels of reactive radicals, such as Parkinson's disease and Alzheimer's disease.9–11 Reactive radicals are known to be scavenged by antioxidants, such as flavonoids, via hydrogen-transfer reactions from the phenolic OH group to radicals. There are two possibilities for the mechanism of hydrogen-transfer reactions of phenolic antioxidants, namely a one-step hydrogen-atom transfer (route A), or electron transfer followed by proton transfer (route B), as shown in Fig. 1.12–14 Resveratrol15 and flavonoids, such as catechin16,17 and quercetin,18 are known to scavenge radicals via the electron transfer mechanism. In this case, solvents significantly affect the radical-scavenging mechanism.19 In fact, the radical-scavenging reaction of a vitamin E analogue proceeds via the one-step hydrogen-atom transfer in aprotic media, such as acetonitrile,20 while the electron-transfer process is involved in protic solvents, such as methanol.21 In this study, we focused on quercetin (2-(3,4-dihydroxyphenyl)-3,5,7-trihydroxy-4H-chromen-4-one), as a potential therapeutic agent to prevent illnesses caused by reactive oxygen radicals. Quercetin, which is the most common antioxidant found in citrus fruit and onions, exhibits not only the ability to scavenge radicals, but also anti-inflammatory and anticancer effects.22 Wai Mun Loke et al. also reported that quercetin may attenuate atherosclerosis in vivo.23 Furthermore, quercetin has been shown to inhibit the proliferation of breast cancer, human lung cancer and nasopharyngeal carcinoma cells.1,14,24–26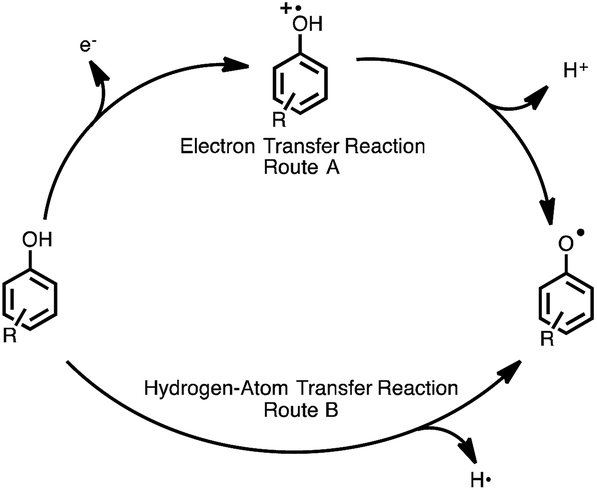 | ||
| Fig. 1 Radical-scavenging mechanism of phenolic antioxidants. | ||
Increased importance of considering antioxidative capabilities in drug development has led to an interest in improving the radical-scavenging activity of quercetin. Furthermore, the chemical modification of natural antioxidants provides valuable fundamental information about the structure–radical scavenging activity relationship of compounds.
Our efforts in derivatising quercetin to enhance its radical-scavenging activity were inspired by the methoxy group in the ferulic acid (caffeic acid structure), due to its proven antioxidative effects.27 The electron-donating group such as methoxy group at the ortho position relative to the hydroxy group stabilises radical cation. In the electron-transfer mechanism, the ionisation potential (IP) of the compound is important for its radical-scavenging efficacy. Introduction of an electron-donating group, such as methyl group, at positions ortho and/or para to the phenolic hydroxy group would decrease the IP of the phenolic compound, and thereby enhance the radical-scavenging activity.
Recently, we reported that resveratrol derivatives, in which methyl groups were introduced at the position ortho to the hydroxy group, showed enhanced radical-scavenging activities. The radical-scavenging activities of resveratrol derivatives 4 and 5 were shown to be about 14- and 36-fold higher than that of the parent resveratrol (Fig. 2).28 Similarly, introduction of methyl groups into the catechol moiety of catechin resulted in an enhanced radical-scavenging activity relative to the underivatised catechin. Specifically, the radical-scavenging activity of the dimethylcatechin derivative 6 (Fig. 2) was shown to be about 28-fold higher than that of (+)-catechin.29 In addition to a methyl group, an isopropyl fragment was also introduced as an electron-donating group to enhance the radical-scavenging activity. A planar-catechin analogue 7 (Fig. 2), in which an isopropyl fragment was introduced into (+)-catechin, exhibited 5-fold more potent radical-scavenging activity compared to (+)-catechin.30 The conformationally constrained epigallocatechin analogue containing an isopropyl fragment 8 (Fig. 2) showed 16-fold more potent radical-scavenging activity than the parent epigallocatechin.31 We report herein the design and synthesis of 2′,5′-dimethylquercetin (1), 2′-methylquercetin (2), and 5′-methylquercetin (3) for enhancement of the radical-scavenging activity relative to the underivatised quercetin. The radical-scavenging activity of the quercetin analogues is expected to be enhanced upon introduction of a methyl group at positions ortho to the catechol hydroxy group, similar to the dimethylcatechin analogue 6. It should be noted that while it is very difficult to control the stereochemistry during the synthesis of the dimethylcatechin analogue 6, the synthesis of quercetin analogues was expected to be easier, because there is no need to control the stereochemistry. Herein, we also demonstrate their enhanced radical-scavenging ability against galvinoxyl radical (GO˙) as a reactivity model of reactive oxygen species.
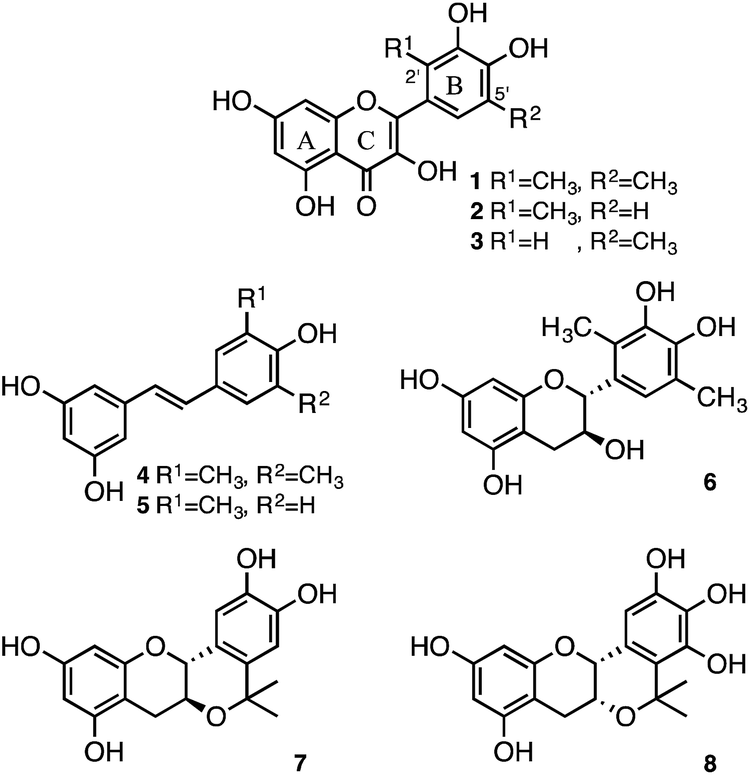 | ||
| Fig. 2 Chemical structures of synthetic antioxidants. | ||
Results and discussion
Design and synthesis of quercetin analogues
Quercetin analogues (1, 2, and 3) were synthesised in which methyl groups were introduced at the positions ortho to the hydroxy groups of the catechol moiety to enhance the radical-scavenging activity. These analogues were designed with reference to the dimethylcatechin analogue 6. Similar to catechin, quercetin scavenges free radicals via an electron-transfer reaction.18 Radical cations formed after the electron-transfer reaction are delocalised and stabilised by the introduction of an electron-donating group. Moreover, the radical cation intermediate of quercetin was expected to be even further stabilised by the effect of both electron donation and hyperconjugation of the methyl group.The retrosynthesis of quercetin analogues 1, 2 and 3 is shown in Scheme 1. The flavone scaffold can be synthesised via the Algar–Flynn–Oyamada (AFO) reaction from the chalcone structure, which is a useful way to form the flavone scaffold.32–34 The chalcone structure can be synthesised by Aldol condensation using an aldehyde and an acetophenone. The acetophenone forms the A ring of the quercetin molecule, while the aldehyde forms the B ring. The target molecules (1, 2 and 3) contain methyl groups in the B ring, which are introduced into the starting aldehyde. Previously, the aldehyde derivative 9a, in which methyl groups were introduced at both positions ortho to the catechol hydroxy groups, had been synthesised to obtain the dimethyl catechin analogue 6. The intermediate (11) of 9a had been obtained by catalytic reduction after introduction of morpholine structure to catechol with Mannich reaction.29 Because the catalytic reduction is a dangerous reaction that is carried out at 70 °C and 5 atm, another route was employed for the synthesis of 9a.
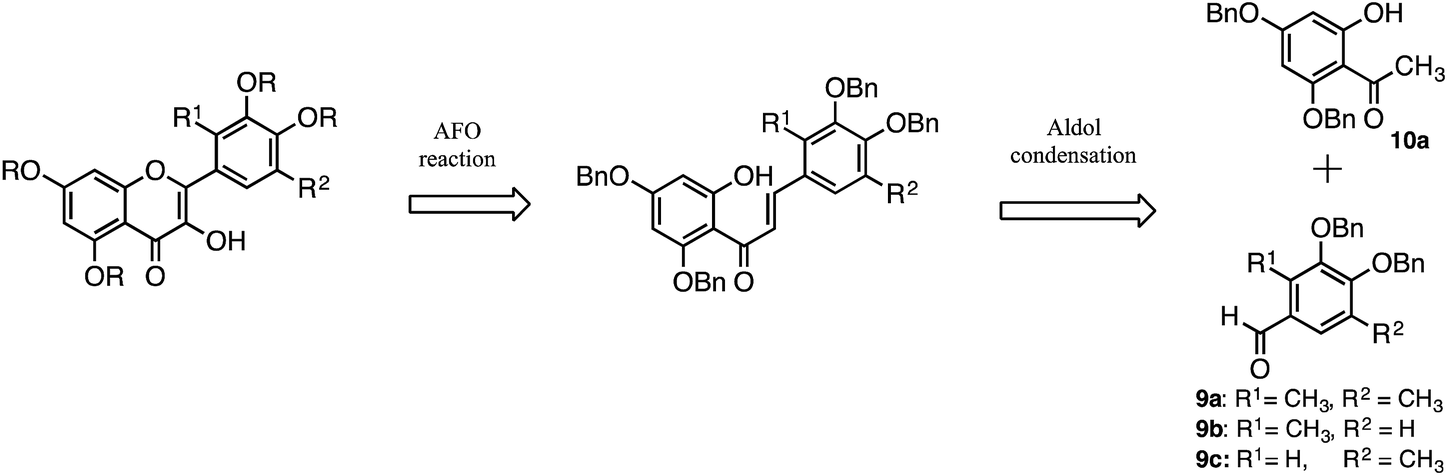 | ||
| Scheme 1 Retrosynthesis of methyl quercetin analogues. | ||
Synthesis of the aldehyde derivative 9a used in this study is shown in Scheme 2. Dimethyl catechol 11 was obtained by the Dakin reaction after formylation of 2,5-dimethylphenol.35 Following formylation of 11 at the 4-position, protection of the hydroxy groups of 13 with benzyl bromide was accomplished to yield 9a. The AFO reaction, after Aldol condensation of 9a and 10a, is shown in Scheme 3. Acetophenone derivative (10a) was obtained by benzylation of the hydroxy groups at the 2- and 4-positions of 2,4,6-trihydroxyacetophenone. The chalcone 14 was obtained by the aldol condensation of 9a with 10a. We initially attempted formation of flavone structure by the AFO reaction; however, the flavone structure was not formed. Instead, the aurone structure (16) was formed, followed by hydrogenation to yield 17.
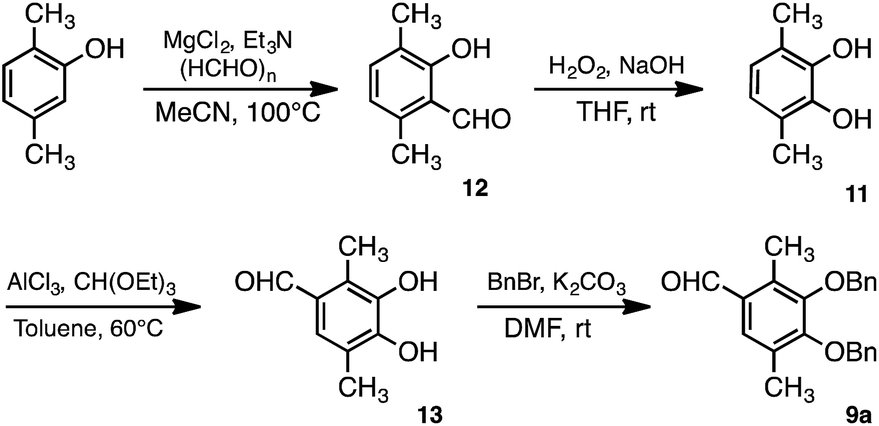 | ||
| Scheme 2 Synthesis of dimethyl aldehyde derivative 9a. | ||
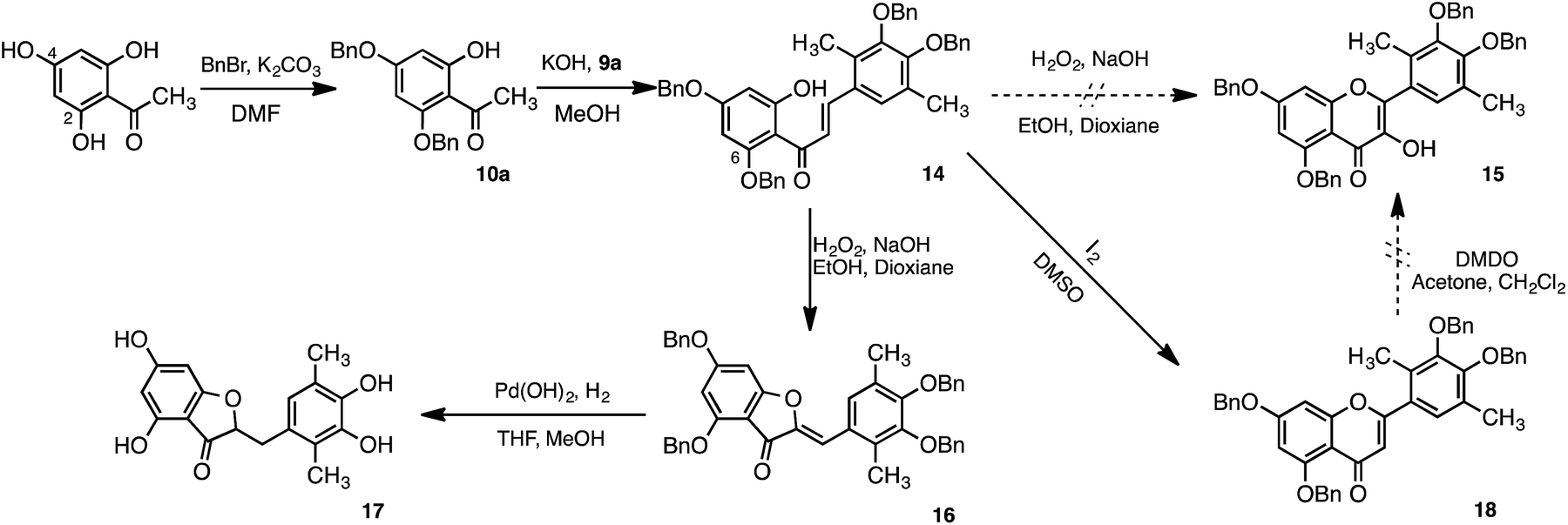 | ||
| Scheme 3 Formation of dimethyl aurone structure 17. | ||
We had deluded ourselves into taking that 15 was obtained by the AFO reaction because the 1H-NMR spectra of 15 and 16 were similar. Therefore, the catalytic reduction to give 1 was carried out, however, the compound 17 instead of 1 was obtained.
When the catalytic reduction was performed, the resulting yellow reaction solution turned colorless. This reduction reaction was performed not only for deprotection, but also for reduction of the unsaturated aurone bond. If, instead, the quercetin structure (15) had been synthesised by the AFO reaction, only the deprotection reaction of the benzyl group would have occurred following catalytic hydrogen reduction, and the color of reaction solution should have remained yellow. However, the reaction solution became colorless upon catalytic hydrogen reduction. Thus, the AFO reaction of 14 yielded the aurone structure (16) without the quercetin structure (15).
Epoxide may be predicted to be formed as an intermediate when the chalcone structure contains a substituent at the 6-position, as in 14. The AFO reaction of chalcone with a substituent at the 6-position has previously been reported by various authors. Formation of the epoxide intermediate takes place by attack of the 2-O group at the α- rather than at the β-position.36,37 Therefore, the AFO reaction of 14 yielded the aurone structure (16) (Scheme 4).
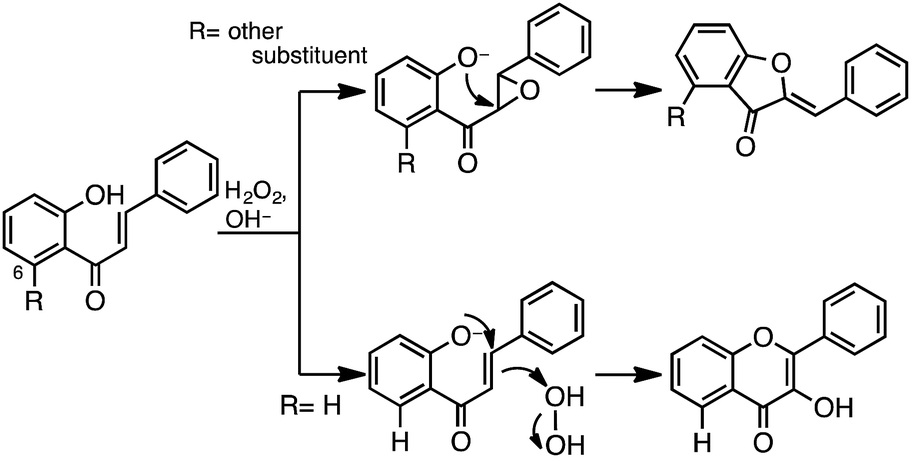 | ||
| Scheme 4 AFO reaction. | ||
We attempted to introduce a hydroxy group to 18 at the 3-position with dimethyldioxirane (DMDO) to yield 15. The chalcone structure (14) was reacted with I2 in DMSO to yield 18. While the cyclisation reaction was successful, hydroxylation of 18 with DMDO failed to yield 15.
Following these initial attempts, quercetin analogues were subsequently synthesised with other scheme which is via the taxifolin framework (22a) intermediate, as shown in Scheme 5. The synthetic scheme was employed with reference to the synthesis of isorhamnetin, tamarixetin, and kaempferide. The protective group of acetophenone derivative (19) was changed from a benzyl group to methoxymethyl (MOM). The protection of all the hydroxy groups of acetophenone allows for termination of the reaction of epoxide conversion to aurone. The MOM was employed to be easily deprotected after formation of epoxide. The taxifolin framework (22a) should be obtained by the deprotection of MOM and cyclisation in a 1-pot reaction with 21a. The 2,4,6-tri-MOM phloracetophenone (19) was first synthesised, in which three hydroxy groups were protected by MOM groups (Scheme 5).38–40 The chalcone derivative 20a was subsequently obtained by condensation of 19 with the aldehyde derivative 9a. Next, epoxidation of 20a with hydrogen peroxide in the presence of sodium hydroxide as a base yielded 21a.41 Cyclisation of 21a was achieved at the same time as deprotection of MOM with hydrochloric acid. Catalytic hydrogenation of 22a with 20% Pd(OH)2 yielded 23a. Finally, oxidation of 23a with potassium metabisulfite solution was performed to yield dimethyl quercetin (1) (Scheme 6).42
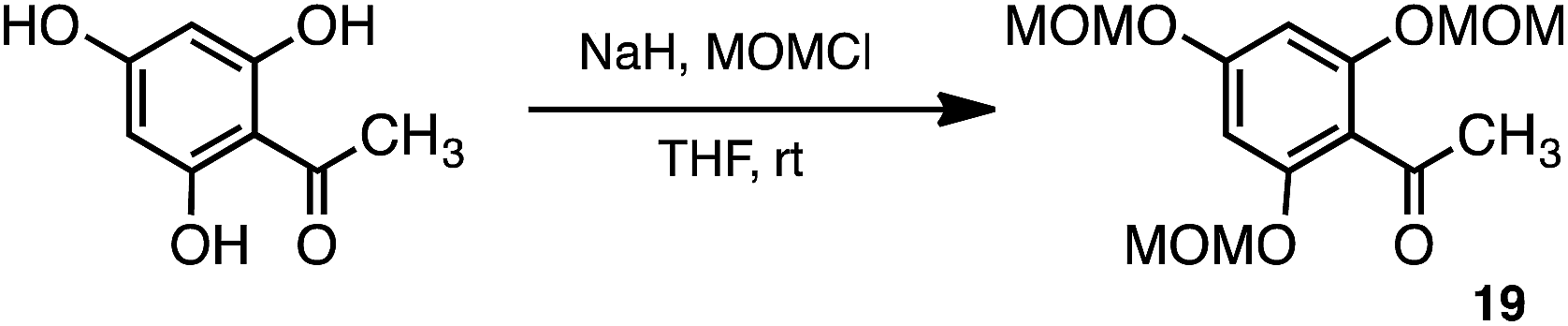 | ||
| Scheme 5 Synthesis of 19. | ||
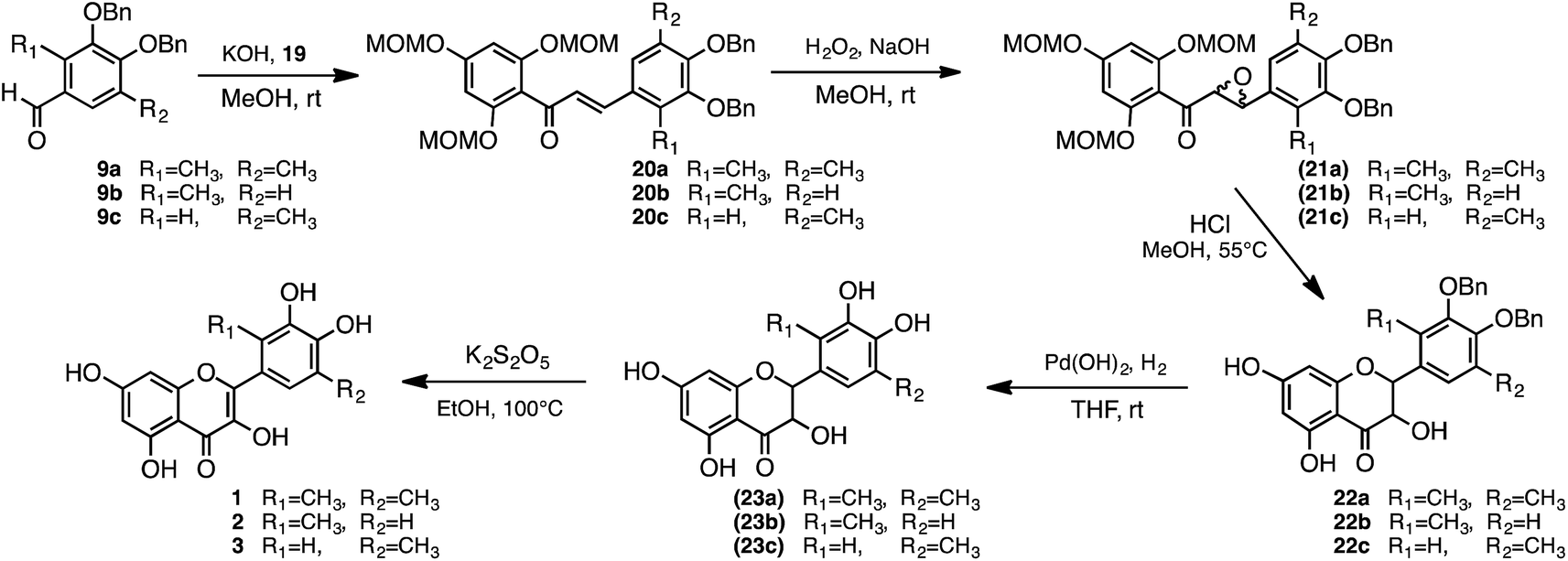 | ||
| Scheme 6 Formation of quercetin analogues 1, 2 and 3. | ||
The aldehyde derivatives 9b and 9c were synthesised to obtain the quercetin analogues 2 and 3, respectively (Scheme 7). Formylation of 24, which was benzylated using 3-methylcatechol, at the 4-position was carried out by the Vilsmeier–Haack reaction to yield 9b. The aldehyde derivative 9c was also synthesised from 3-methylcatechol. Specifically, 3-methylcatechol was first benzylated at the 1-position of the hydroxy group to yield 25. Formylation of 25 at the 5-position was then achieved via the Duff reaction using hexamethylenetetramine (HMTA).43,44 The aldehyde derivative 9c was then obtained by benzylation at the 2-position of the hydroxy group. Quercetin analogues 2 and 3 were synthesised with the resulting 9b and 9c, respectively, by the same scheme used to obtain dimethylquercetin (1) (Scheme 6).
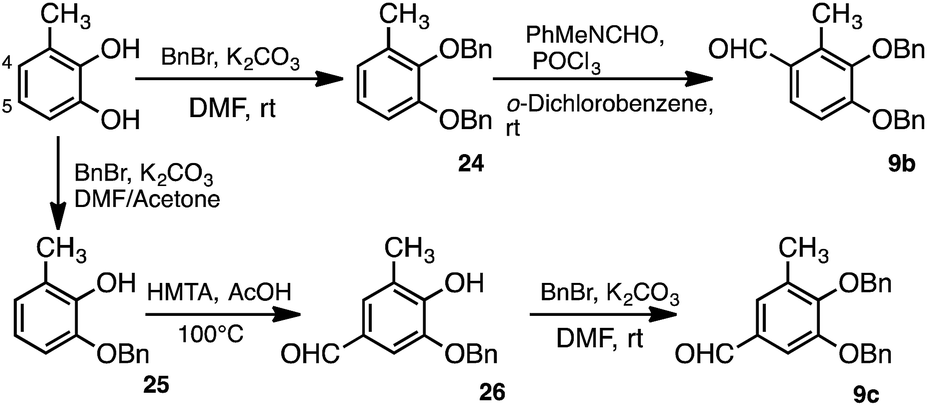 | ||
| Scheme 7 Synthesis of monomethyl aldehyde derivative 9b and 9c. | ||
Radical-scavenging activity of quercetin analogues monitored by the stopped flow technique
The radical-scavenging activity of quercetin analogues was evaluated in a non-aqueous system using the galvinoxyl radical (GO˙) as a reactivity model of reactive oxygen species. GO˙ exhibits a strong absorption band at 428 nm, and a solution of GO˙ appears yellow in color.21,45 Upon mixing of GO˙ with an antioxidant, the visible absorption band immediately vanishes, and the resulting decolorization is stoichiometric with respect to the number of electrons taken up. The rates of GO˙-scavenging by the quercetin analogues were measured based on the color change of GO˙ in the presence of antioxidants. Because this spectral change occurs very fast, the rates of the radical-scavenging reaction of quercetin analogues were measured by monitoring the decrease in absorbance at 428 nm using a stopped flow technique in deaerated acetonitrile at 298 K. The decay of the absorbance at 428 nm obeyed pseudo-first-order kinetics when the concentration of 2 ([2]) was maintained at more than 10-fold excess to the GO˙ concentration. The decay of the absorbance at 428 nm of GO˙ is shown in Fig. 3. The pseudo-first-order rate constant (kobs) increased linearly with an increase in the concentration of quercetin analogues. An extended figure for 1, 2 and quercetin is included as an insert in Fig. 4 because the k value of 3 is too large; the insert shows a magnified axis for kobs from 0 to 2 s−1. From the slope of a linear plot of kobs vs. [2], the second-order rate constant (k) for the radical-scavenging reaction was determined to be 6.1 × 10 M−1 s−1. The k values for dimethylquercetin (1), 5′-methylquercetin (3), and the parent quercetin were determined in the same manner to be 2.1 × 103 M−1 s−1, 1.6 × 105 M−1 s−1, and 1.1 × 10 M−1 s−1, respectively (Fig. 4). These results demonstrate that the k values for 1, 2 and 3 were about 191-, 5.5- and 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500-fold greater than that of the underivatised quercetin, respectively. Thus, structurally modified quercetin analogues (1, 2 and 3) could afford significantly larger k values than the parent quercetin. The k value for 3 is the largest among the quercetin analogues examined in this study. The GO˙-scavenging activity of 3 is about 2600-fold larger than that of 2. The large difference in k values between 2 and 3 demonstrates that the radical-scavenging activity of the quercetin derivatives largely depends on the position of the methyl group. The k values of aurone derivative 17 and dimethylcatechin 6 were 7.2 M−1 s−1 and 1.1 × 103 M−1 s−1, respectively, indicating that delocalisation of the radical cation might operate a high radical-scavenging activity. The delocalisation of radical cation of 6 and 17 may remain limited to the catechol moiety, and these radical cations may not be delocalised throughout the entire molecule.
500-fold greater than that of the underivatised quercetin, respectively. Thus, structurally modified quercetin analogues (1, 2 and 3) could afford significantly larger k values than the parent quercetin. The k value for 3 is the largest among the quercetin analogues examined in this study. The GO˙-scavenging activity of 3 is about 2600-fold larger than that of 2. The large difference in k values between 2 and 3 demonstrates that the radical-scavenging activity of the quercetin derivatives largely depends on the position of the methyl group. The k values of aurone derivative 17 and dimethylcatechin 6 were 7.2 M−1 s−1 and 1.1 × 103 M−1 s−1, respectively, indicating that delocalisation of the radical cation might operate a high radical-scavenging activity. The delocalisation of radical cation of 6 and 17 may remain limited to the catechol moiety, and these radical cations may not be delocalised throughout the entire molecule.
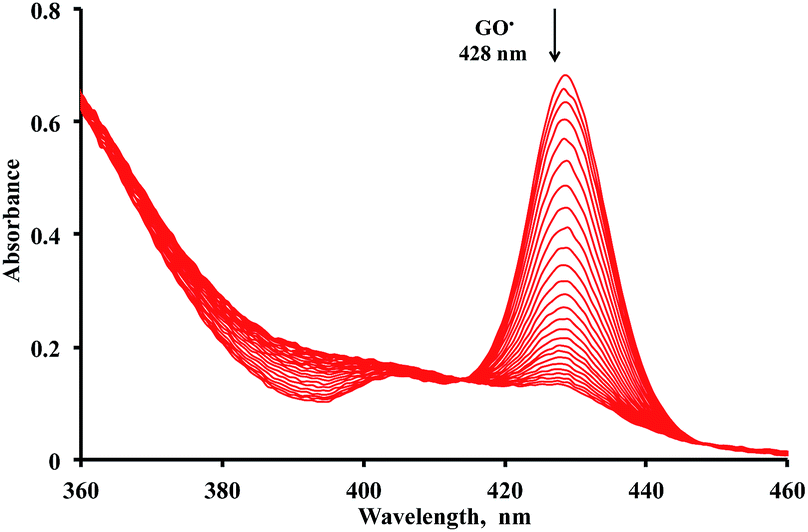 | ||
| Fig. 3 Spectral change (interval: 4 s) observed during the reaction between 2 (4.7 × 10−5 M) and GO˙ (4.7 × 10−6 M) in deaerated MeCN at 298 K. | ||
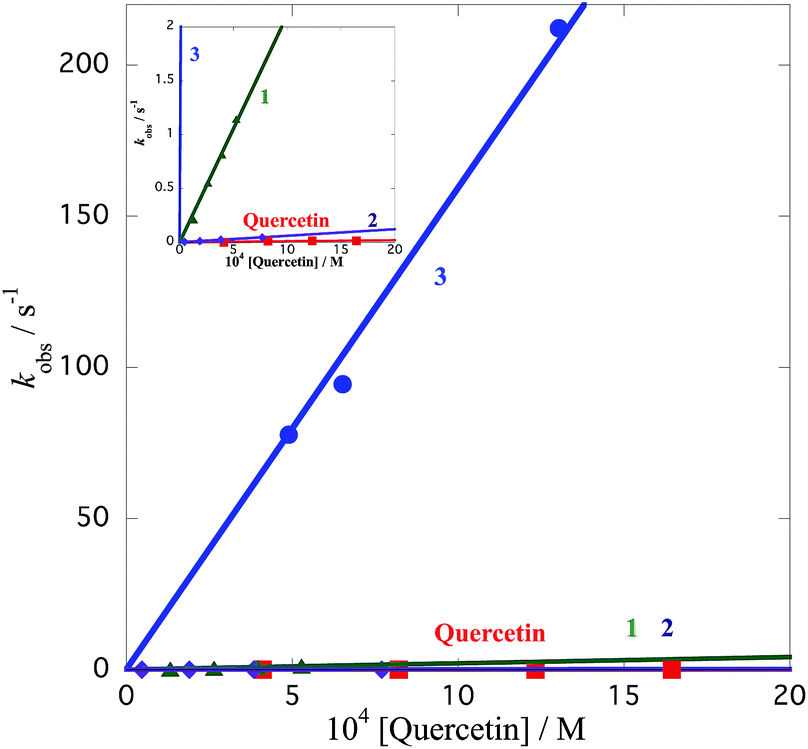 | ||
| Fig. 4 Plots of the pseudo-first-order rate constant (kobs) vs. the concentrations of 1 (green triangles), 2 (purple diamonds), 3 (blue circles) and quercetin (red squares) for radical-scavenging reaction against GO˙. Inset: enlargement of the same plots from kobs values of 0 to 2. | ||
IPs and energy difference values (DHT, HT: hydrogen transfer) between quercetins and the corresponding phenoxyl radicals, which equal the O–H bond dissociation energies (BDE) of their phenolic hydroxy groups, were calculated to elucidate the effect of the methyl groups in the quercetin analogues on their radical-scavenging activity by the density functional theory (DFT). As shown in Fig. 1, phenolic compounds scavenge radicals via two mechanisms. Thus, the IP and DHT values are reliable parameters to distinguish these two different radical-scavenging mechanisms. If the phenolic compound scavenges GO˙ via a one-step hydrogen-atom transfer reaction, i.e., route B, the compound with lower DHT value exhibit the higher radical-scavenging activity. In contrast, the reactivity of the electron-transfer reaction of phenolic compounds, i.e., route A, is related to the IP values. Because, the lower of the IP, the easier is the electron abstraction. The DHT and IP values of quercetin analogues were determined by DFT calculations at the B3LYP/6-31G(d) level and the results are shown in Table 1. The IP value of 3, which exhibits a markedly enhanced radical-scavenging activity, was the lowest among all the quercetin analogues. Therefore, the mechanism of radical-scavenging of 3 may occur via an electron transfer followed by proton transfer. If the radical-scavenging mechanism of 3 is a one-step hydrogen atom-transfer reaction, the DHT value at the 4′-OH position should be the lowest among all the quercetin analogues. However, the DHT value of 3 is higher than that of 1. Thus, enhancement of the radical-scavenging activity of 3 may be the result of electron donation by the introduced methyl group.
|
k (M−1 s−1) | DHT (kcal mol−1) | IP (eV) | |
|---|---|---|---|---|
| 3′-OH | 4′-OH | |||
| 1 | 2.1 × 103 | 390.02 | 388.58 | 6.84 |
| 2 | 6.1 × 10 | 390.19 | 390.46 | 6.92 |
| 3 | 1.5 × 105 | 392.91 | 389.10 | 6.80 |
| Quercetin | 1.1 × 10 | 397.17 | 390.44 | 6.88 |
We also evaluated the most stable structures of quercetin and quercetin analogues by DFT calculations, as shown in Fig. 5. Quercetin exhibits a coplanar structure between the 4H-curomen (AC rings) and catechol (B ring) moieties (Fig. 5a). In contrast to the structure of quercetin, analogues 1 and 2, which contain methyl groups at the 2′-position, exhibit a twisted structure between the AC and B rings due to steric hindrance of the hydroxyl group of the C ring. The torsional angles between the AC and B rings of 1 and 2 were 49.5° and 49.8°, respectively (Fig. 5b and c). On the other hand, 3, which exhibited the highest radical-scavenging activity, formed a coplanar structure similar to the parent quercetin (Fig. 5d). The planar structure of quercetin was maintained even after introduction of a methyl group at the 5′-position. When 3 gives an electron to GO˙ to produce the corresponding radical cation of 3, the planar structure allows for delocalisation of the radical cation throughout the entire molecule, resulting in enhanced radical-scavenging activity. The hydrogen transfer from 3 to GO˙ proceeded via electron transfer from 3 to GO˙ followed by proton transfer from the resultant radical cation intermediate 3˙+ to the one-electron reduced species of GO˙, i.e. GO−. Thus, enhancement of the radical-scavenging activity of 3 is thought to be due to the introduction of an electron-donating methyl group into the catechol moiety. In addition, the planar structure of 3 promoted the enhancement of radical-scavenging activity by stabilisation of 3˙+. The structure of synthetic analogues 1, 2 exhibited a twisted structure by introduction of methyl group at 2′-position. As a result, the radical cation, which was generated by radical-scavenging reaction, was not able to be delocalised throughout the entire molecule. On the other hands, introduction of methyl group at 5′-position did not affect the planar structure. Thus, the quercetin analogue with methyl group at 5′-position exhibited the highest radical-scavenging activity because delocalised radical cation might be stabilised efficiently by the electron-donating methyl group.
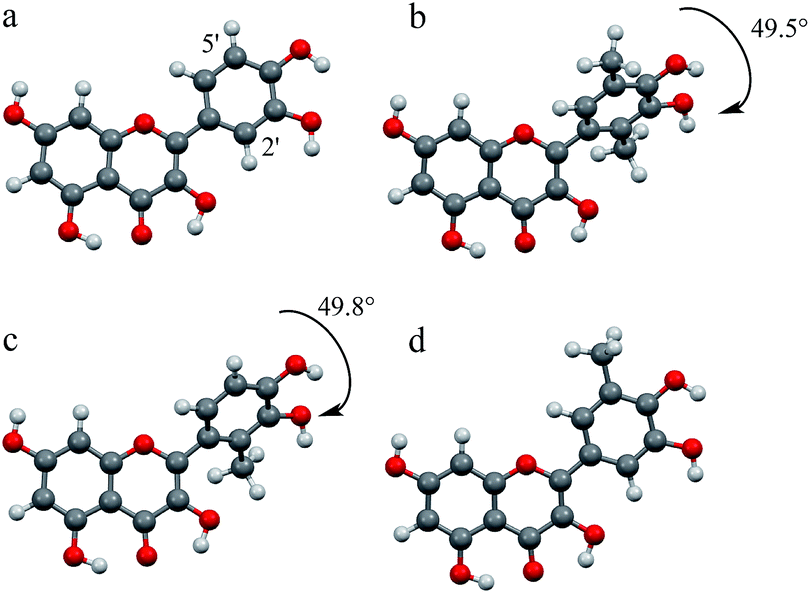 | ||
| Fig. 5 DFT optimised structures of (a) quercetin, (b) 1, (c) 2, and (d) 3 calculated using DFT (B3LYP/6-31G(d) level). | ||
Conclusion
In this study, we designed and synthesised quercetin analogues to enhance radical-scavenging activity of quercetin. The radical-scavenging activity of quercetin, which scavenges free radicals via the electron-transfer process, was enhanced by the introduction of an electron-donating methyl group into the catechol moiety in the quercetin molecule. Although a single methyl group was introduced into both 2 and 3, the radical-scavenging activities of both quercetin derivatives varied greatly as a result of the different positions of the introduced methyl group. The radical-scavenging activity of 3 was enhanced by the electron donating effect. In addition, the radical cation of 3 was found to be stabilised by the planar structure between the AC and B rings. The significant enhancement in radical-scavenging activity observed for 3 was due to both the electron-donating effect and the planar quercetin structure. Results reported herein suggest that future design and synthesis of novel antioxidant agents with enhanced radical-scavenging activities should consider not only the introduction of electron-donating groups but also stabilisation of radical cations by to flatten throughout the entire molecule. The 3 with high radical-scavenging activity would also be studied not only radical-scavenging activity but also physiological effects of quercetin such as anti-inflammatory and anticancer.Experimental section
General methods
Unless otherwise noted, all commercially available compounds were used as provided without further purification. Solvents for chromatography were technical grade, and acetonitrile for antioxidant activity measurements was analytical grade. Proton (1H) NMR spectra and (13C) NMR spectra were recorded on a JEOL JNMAL-400 (400 MHz) instrument and JNM-ECX-500 (500 MHz). Chemical shifts (δ) are given in parts per million (ppm) and are referenced to residual protonated solvent (CHCl3: δ H 7.26 ppm, δ C 77.16 ppm; CH3OH: δ H 3.31 ppm, δ C 49.00 ppm; (CH3)2CO: δ H 2.05 ppm, δ C 29.84 ppm; (CH3)2SO: δ H 2.50 ppm, δ C 39.52 ppm). J values are given in Hz. Mass spectra were obtained FAB mode with m-nitrobenzyl alcohol as a matrix. The progress of all reactions was monitored by thin-layer chromatography (TLC) on silica gel 60 F254 (0.25 nm, Merck). Column chromatography was performed on silica gel 60 (0.063–0.200 mm, Merck).:1 n-hexane–ethylacetate) to give 1.61 g (total yield from 2,5-xylenol: 33%) of compound 9a as yellow liquid; 1H-NMR (CDCl3)29 δ 2.23 (s, 3H), 2.53 (s, 3H), 4.98 (s, 2H), 5.10 (s, 2H), 7.33–7.44 (m, 11H), 10.15 (s, 1H); 13C-NMR (CDCl3) δ 11.2, 16.0, 74.6, 74.9, 128.1, 128.3, 128.3, 128.4, 128.4, 130.0, 130.4, 130.4, 136.8, 150.4, 155.3, 191.7; HR-MS (ESI) calcd for C23H23O3 [M + H]+: 347.1687, found 347.1647.13: 1H-NMR (acetone-d6) δ 2.24 (s, 3H), 2.53 (s, 3H), 5.00 (s, 2H), 5.11 (s, 2H), 7.34–7.44 (m, 11H), 10.15 (s, 1H); [M − H]− 165.
:2:0 to 4:3:1 n-hexane–CH2Cl2–ethyl acetate) to give 118 mg (97%) of 24 as white solid; mp 49–50 °C; 1H-NMR (CDCl3) δ 2.24 (s, 3H), 5.00 (s, 2H), 5.11 (s, 2H), 6.79 (d, J = 8.0 Hz, 1H), 6.84 (d, J = 8.0 Hz, 1H), 6.93 (t, J = 8.0 Hz, 1H) 7.27 (m, 10H); 13C-NMR (CDCl3) δ 16.2, 70.8, 74.4, 112.0, 123.3, 123.7, 127.4, 127.8, 127.9, 128.1, 128.3, 128.3, 128.5, 132.6, 137.2, 137.9, 146.7, 151.9; HR-MS (ESI) calcd for C21H21O2 [M + H]+: 305.1542, found 305.1520.:1 n-hexane–ethyl acetate) to give 2.43 g (44%) of 9b as white solid; mp 92–94 °C; 1H-NMR (CDCl3) δ 2.55 (s, 3H), 4.96 (s, 2H), 5.21 (s, 2H), 6.98 (d, J = 8.5 Hz, 1H), 7.31–7.46 (m, 10H), 7.58 (d, J = 8.5 Hz, 1H), 10.10 (s, 1H); 13C-NMR (CDCl3) δ 11.5, 70.6, 74.7, 110.5, 127.5, 128.1, 128.3, 128.4, 128.5, 128.7, 129.8, 135.5, 136.0, 137.2, 146.4, 156.5, 191.5; HR-MS (ESI) calcd for C22H21O3 [M + H]+: 333.14907, found 333.14835.:1 n-hexane–ethyl acetate) to give 338 mg (18%) of 25 as white solid; mp 51–52 °C; 1H-NMR (CDCl3) δ 2.30 (s, 3H), 4.85 (s, 2H), 5.55 (s, 1H), 6.69 (dd, J = 1.2, 8.0 Hz, 1H), 6.76 (dd, J = 1.2, 8.0 Hz, 1H), 6.89 (t, J = 8.0 Hz, 1H), 7.30–7.41 (m, 5H), 13C-NMR (CDCl3) δ 15.4, 71.1, 109.7, 119.1, 123.5, 124.2, 127.7, 128.3, 128.7, 136.5, 144.0, 145.4; HR-MS (ESI) calcd for C14H14O2 [M]+: 214.0994, found 214.0978.:1 n-hexane–ethyl acetate) to give 676.4 mg (56%) of 26 as white solid; mp 113–114 °C; 1H-NMR (acetone-d6) δ 2.28 (s, 3H), 5.25 (s, 2H), 7.36–7.51 (m, 7H), 8.69 (brs, 1H), 9.77 (s, 1H) 13C-NMR (CDCl3) δ 15.4, 71.1, 109.9, 125.2, 127.9, 128.6, 128.6, 129.0, 129.3, 137.2, 147.0, 151.2, 190.8; HR-MS (ESI) calcd for C15H15O3 [M + H]+: 243.1021, found 243.1071.:1 n-hexane–ethyl acetate) to give 841 mg (94%) of 9c as white solid; mp 62–64 °C; 1H-NMR (CD3OD)46,47 δ 2.24 (s, 3H), 5.14 (s, 2H), 5.27 (s, 2H), 7.29–7.44 (m, 9H), 7.51 (d, J = 2.0 Hz, 1H), 7.57–7.58 (m, 2H), 9.87 (s, 1H); 13C-NMR (CD3OD) δ 16.4, 71.3, 74.9, 111.6, 127.1, 128.7, 128.8, 128.8, 129.1, 129.3, 129.3, 133.4, 133.7, 137.8, 138.5, 152.4, 153.2, 191.7; HR-MS (FAB+) calcd for C22H21O3 [M + H]+: 333.1491, found 333.1507.:3 ethyl acetate–n-hexane) to give 2.81 g (43%) of 19 as white solid; mp 37–38 °C; 1H-NMR (CDCl3) δ 2.50 (s, 3H), 3.46 (s, 9H), 5.14 (s, 6H), 6.51 (s, 2H); 13C-NMR (CDCl3) δ 32.5, 56.2, 56.3, 94.4, 94.7, 96.9, 116.8, 155.1, 159.4, 201.4; HR-MS (ESI) calcd for C14H21O7 [M + H]+: 301.1287, found 301.1267.:1 to 2:1 n-hexane–ethyl acetate) to give 1.26 g (85%) of chalcone structure 20a as yellow liquid; 1H-NMR (CDCl3) δ 2.21 (s, 3H), 2.21 (s, 3H), 3.41 (s, 6H), 3.46 (s, 3H), 4.96 (s, 2H), 5.03 (s, 2H), 5.12 (s, 4H), 5.18 (s, 2H), 6.57 (s, 2H), 6.83 (d, J = 16.0 Hz, 1H), 7.25 (s, 1H), 7.32–7.41 (m, 10H), 7.63 (d, J = 16.0 Hz, 1H); 13C-NMR (CDCl3) δ 12.1, 16.2, 56.2, 56.3, 56.3, 74.8, 74.9, 94.5, 94.7, 94.7, 97.0, 97.2, 115.1, 124.5, 128.1, 128.4, 128.4, 128.4, 128.5, 129.2, 130.0, 130.2, 130.8, 137.2, 137.3, 142.3, 150.4, 152.2, 155.8, 159.4, 159.6, 194.0; HR-MS (ESI) calcd for C37H41O9 [M + H]+: 629.2751, found 629.2786.:1 to 1:1 n-hexane–ethyl acetate) to give 1.67 g (91%) of chalcone structure 20b as yellow liquid; 1H-NMR (CDCl3) δ 2.21 (s, 3H), 3.39 (s, 6H), 3.49 (s, 3H), 4.95 (s, 2H), 5.11 (s, 4H), 5.16 (s, 2H), 5.17 (s, 2H), 6.56 (s, 2H), 6.82 (d, J = 16.0 Hz, 1H), 6.88 (d, J = 9.0 Hz, 1H), 7.30–7.46 (m, 11H), 7.63 (d, J = 16.0 Hz, 1H); 13C-NMR (CDCl3) δ 12.2, 56.3, 70.6, 74.6, 94.5, 94.6, 97.1, 111.4, 115.1, 123.0, 127.4, 127.5, 128.0, 128.1, 128.3, 128.4, 128.6, 133.0, 136.5, 137.4, 142.5, 146.4, 153.4, 155.8, 159.5, 194.1; HR-MS (FAB+) calcd for C36H39O9 [M + H]+: 615.2581, found 615.2594.:1 to 2:1 n-hexane–ethyl acetate) to give 838 mg (86%) of chalcone structure 20c as yellow liquid; 1H-NMR (CDCl3) δ 2.20 (s, 3H), 3.38 (s, 6H), 3.51 (s, 3H), 5.04 (s, 2H), 5.11 (s, 4H), 5.13 (s, 2H), 5.18 (s, 2H), 6.57 (s, 2H), 6.85 (d, J = 16.0 Hz, 1H), 6.96 (d, J = 1.0 Hz, 1H), 7.04 (d, J = 2.0 Hz, 1H), 7.25 (d, J = 17.0 Hz, 1H), 7.30–7.46 (m, 10H); 13C-NMR (CDCl3) δ 16.3, 56.3, 70.8, 76.7, 94.5, 97.1, 111.0, 114.8, 127.4, 128.0, 128.0, 128.0, 128.2, 128.3, 128.4, 128.6, 130.3, 132.9, 136.6, 137.4, 145.3, 148.8, 152.0, 155.7, 159.5, 194.3; HR-MS (FAB+) calcd for C36H39O9 [M + H]+: 615.2638, found 615.2594.:1 n-hexane–ethyl acetate) to give 105 mg (total yield from 20a: 40%) of 22a as white solid; mp 207–210 °C; 1H-NMR (acetone-d6) δ 2.22 (s, 3H), 2.27 (s, 3H), 4.36 (m, 1H), 4.89 (d, J = 3.2 Hz, 1H), 4.96 (s, 2H), 5.00 (s, 2H), 5.43 (m, 1H), 5.99 (s, 2H), 7.16 (s, 1H), 7.32–7.49 (m, 10H); 13C-NMR (acetone-d6) δ 11.7, 16.1, 74.8, 75.0, 78.4, 81.6, 95.8, 103.4, 126.9, 128.3, 128.7, 128.8, 129.2, 129.4, 133.2, 128.6, 138.7, 150.3, 150.3, 164.6, 203.1; HR-MS (FAB+) calcd for C31H29O7 [M + H]+: 513.1893, found 513.1913.21a: 1H-NMR (acetone-d6) δ 2.17 (s, 3H), 2.26 (S, 3H), 3.40 (s, 6H), 3.43 (s, 3H), 3.72 (d, J = 1.5 Hz, 1H), 4.05 (d, J = 1.5 Hz, 1H), 4.99 (s, 2H), 4.99 (s, 2H), 5.16 (s, 2H), 5.20 (s, 2H), 6.57 (s, 2H), 6.81 (s, 1H), 7.32–7.46 (m, 10H); 13C-NMR (acetone-d6) δ 11.6, 16.2, 56.3, 56.5, 57.4, 63.9, 75.2, 75.4, 95.1, 95.6, 97.5, 113.5, 122.3, 128.7, 128.8, 129.1, 129.2, 129.5, 130.8, 131.4, 138.5, 138.7, 150.9, 151.2, 157.3, 161.3, 197.4; HR-MS (ESI) calcd for C37H41O10 [M + H]+: 645.2700, found 645.2699.
:1 n-hexane–ethyl acetate) to give 230 mg (total yield from 20b: 32%) of 22b as white solid; mp 164–166 °C; 1H-NMR (acetone-d6) δ 2.31 (s, 3H), 4.68 (d, J = 12.0 Hz, 1H), 4.97 (s, 2H), 5.21 (s, 2H), 5.40 (d, J = 12.0 Hz, 1H), 5.89 (d, 2.0 Hz, 1H), 5.94 (d, J = 2.5 Hz, 1H), 7.08 (d, J = 9.0 Hz, 1H), 7.26–7.53 (m, 11H); 13C-NMR (acetone-d6) δ 12.2, 71.1, 73.2, 75.1, 80.7, 95.9, 97.0, 101.5, 112.1, 124.1, 128.5, 128.6, 128.7, 129.1, 129.2, 129.3, 129.9, 133.2, 138.3, 139.0, 147.0, 153.0, 164.3, 164.8, 167.9, 198.3; HR-MS (FAB+) calcd for C30H27O7 [M + H]+: 499.1752, found 499.1757.21b: 1H-NMR (CDCl3) δ 2.13 (s, 3H), 3.41 (s, 6H), 3.48 (s, 3H), 5.08–5.17 (m, 11H), 5.45 (d, J = 1.7 Hz, 1H), 6.56 (s, 2H), 6.89 (d, J = 9.2 Hz, 1H), 7.30–7.44 (m, 10H), 7.59 (d, J = 9.2 Hz, 1H); 13C-NMR (CDCl3) δ 11.3, 56.3, 56.5, 59.4, 70.6, 74.6, 79.3, 94.3, 94.9, 96.9, 110.6, 111.4, 124.7, 127.4, 127.9, 128.3, 128.5, 129.7, 130.6, 136.8, 137.6, 145.8, 151.7, 156.9, 161.1, 199.7; HR-MS (FAB+) calcd for C36H38O10 [M + H]+: 630.2472, found 630.2465.
:1 to 2:1 n-hexane–ethyl acetate) to give 33 mg (total yield from 20c: 25%) of 22c as white solid; mp 120–121 °C; 1H-NMR (acetone-d6) δ 2.24 (s, 3H), 4.70 (d, 12.0 Hz, 1H), 4.77 (brs, 1H), 5.04 (s, 2H), 5.09 (d, J = 11.0 Hz, 1H), 5.20 (s, 2H), 5.98 (d, J = 1.5 Hz, 1H), 6.01 (d, J = 2.5 Hz, 1H), 7.03 (d, J = 1.5 Hz, 1H), 7.28–7.56 (m, 11H), 9.71 (brs, 1H), 11.71 (s, 1H); 13C-NMR (acetone-d6) δ 17.3, 72.1, 73.8, 75.5, 85.2, 96.8, 97.9, 102.2, 113.3, 124.5, 129.3, 129.4, 129.8, 129.9, 130.0, 133.3, 134.4, 138.9, 139.8, 148.5, 153.4, 164.8, 165.7, 168.5, 196.7; HR-MS (FAB+) calcd for C30H27O7 [M + H]+: 499.1757, found 499.1756.21c: 1H-NMR (CDCl3) δ 2.20 (s, 3H), 3.36 (s, 6H), 3.46 (s, 3H), 3.90 (s, 1H), 3.90 (s, 1H), 4.99 (s, 2H), 5.07–5.11 (m, 4H), 5.14 (s, 2H), 5.17 (s, 2H), 6.53 (s, 2H), 6.78 (s, 1H), 6.84 (d, J = 1.8 Hz, 1H), 7.29–7.47 (m, 10H); 13C-NMR (CDCl3) δ 15.6, 55.6, 55.6, 58.9, 63.7, 70.1, 73.8, 93.7, 94.3, 96.3, 108.2, 112.0, 120.2, 126.9, 127.4, 127.7, 127.9, 128.0, 131.0, 132.1, 136.3, 137.1, 146.3, 151.5, 156.5, 160.3, 195.4; HR-MS (FAB+) calcd for C36H38O10 [M + H]+: 630.2476, found 630.2465.
:1 to 10:1 n-hexane–ethyl acetate) to give 4.516 g (33%) of 10a as white solid; mp 110–111 °C; 1H-NMR (CDCl3) δ 2.54 (s, 3H), 5.04 (s, 2H), 5.05 (s, 2H), 6.09 (d, J = 2.4 Hz, 1H), 6.15 (d, J = 2.4 Hz, 1H), 7.31–7.40 (m, 10H), 14.02 (s 1H); 13C-NMR (CD3Cl) δ 32.9, 69.9, 70.7, 92.0, 94.4, 106.0, 127.3, 127.6, 128.0, 128.1, 128.3, 128.4, 135.2, 135.5, 161.6, 164.7, 167.2, 202.8; HR-MS (ESI) calcd for C22H21O4 [M + H]+: 349.1440, found 349.1440.:1 to 2:1 n-hexane–CH2Cl2) to give 2.70 g (84%) of 14 as yellow liquid; 1H-NMR (CDCl3) δ 1.98 (s, 3H), 2.31 (s, 3H), 4.96 (s, 2H), 5.04 (s, 2H), 5.09 (s, 2H), 5.10 (s, 2H), 6.17 (d, J = 2.5 Hz, 1H), 6.23 (d, J = 2.5 Hz, 1H), 6.79 (s, 1H), 7.26–7.43 (m, 20H), 7.77 (d, J = 15.5 Hz, 1H), 8.03 (d, J = 15.5 Hz, 1H); 13C-NMR (CD3Cl) δ 12.1, 16.2, 70.3, 71.3, 74.8, 74.9, 92.8, 95.0, 106.7, 124.4, 127.5, 127.6, 127.7, 127.7, 128.0, 128.1, 128.4, 128.5, 128.5, 128.6, 128.7, 129.9, 130.4, 131.2, 135.6, 135.9, 137.3, 137.5, 140.2, 150.2, 152.0, 161.6, 165.1, 168.4, 192.8; HR-MS (ESI) calcd for C45H40O6 [M + Na]+: 699.2717, found 699.2686.:4:1 n-hexane–CH2Cl2–MeOH) to give 1.02 g (53.2%) of 16 as a yellow solid; mp 152–155 °C; 1H-NMR (CDCl3) δ 2.29 (s, 3H), 2.36 (s, 3H), 4.98 (s, 2H), 5.06 (s, 2H), 5.11 (s, 2H), 5.28 (s, 2H), 6.25 (d, J = 2.0 Hz, 1H), 6.47 (d, J = 2.0 Hz, 1H), 6.94 (s, 1H), 7.33–7.49 (m, 20H), 7.79 (s, 1H); 13C-NMR (CD3Cl) δ 12.5, 16.4, 70.6, 70.8, 74.8, 75.0, 90.6, 96.4, 106.0, 107.9, 126.7, 127.4, 127.6, 127.9, 128.1, 128.1, 128.4, 128.5, 128.5, 128.6, 128.8, 130.1, 131.8, 135.5, 136.0, 137.3, 137.5, 147.6, 150.4, 151.6, 158.4, 167.6, 168.9, 180.5; HR-MS (ESI) calcd for C45H38O6 [M + Na]+: 697.2561, found 697.2526.:1 to 2:1 n-hexane–ethyl acetate) to give 131.2 mg (82%) of 18 as brown solid; mp 55–57 °C; 1H-NMR (CDCl3) δ 2.28 (s, 3H), 2.31 (s, 3H), 5.04 (s, 2H), 5.07 (s, 2H), 5.09 (s, 2H), 5.24 (s, 2H), 6.28 (s, 1H), 6.52 (d, J = 2.5 Hz, 1H), 6.57 (d, J = 2.5 Hz, 1H), 7.13 (s, 1H), 7.37–7.64 (m, 20H); 13C-NMR (CD3Cl) δ 13.6, 16.1, 70.5, 70.7, 74.9, 74.9, 94.2, 98.3, 109.7, 113.5, 126.6, 126.8, 127.6, 128.2, 128.4, 128.4, 128.4, 128.5, 128.6, 128.8, 128.8, 129.6, 130.6, 135.6, 136.4, 137.2, 137.3, 150.8, 152.3, 159.8, 160.0, 162.7, 162.9, 177.2; HR-MS (ESI) calcd for C45H38O6 [M + H]+: 675.2741, found 675.2710.Antioxidant activity measurements
Since phenoxyl radical generated in the reaction of quercetin analogues with oxyl radicals readily reacts with molecular oxygen, the reactions were carried out under strictly deaerated conditions. A continuous flow of argon gas was bubbled through an acetonitrile solution (3.0 mL) containing galvinoxyl radical (GO˙, 2.0 × 10−6 M) in a square quartz cuvette with a glass tube neck for 7 min. Air was prevented from leaking into the neck of the cuvette with a rubber septum. Typically, an aliquot of quercetin analogues (quercetins were maintained at more than 10-fold excess to the GO˙ concentration), which were dissolved in deaerated acetonitrile, were added to the cuvette with a microsyringe to initiate the reaction of quercetin analogues with GO˙. UV-vis spectral changes associated with the reaction were monitored using an Agilent 8453 photodiode array spectrometer. The rate of the GO˙ scavenging reaction of quercetin analogues was determined by monitoring the absorbance change at 428 nm due to GO˙ (ε = 1.32 × 105 M−1 cm−1) using a stopped flow technique on a UNISOKU RSP-1000-02NM spectrophotometer. The pseudo first-order rate constants (kobs) were determined by least-squares curve fit using an Apple MacBook Pro personal computer. The first-order plots of ln(A − A∞) vs. time (A and A∞ denote the absorbance at the reaction time and the final absorbance, respectively) were linear until three or more half-lives with a correlation coefficient ρ > 0.999.Theoretical calculations
Density functional theory (DFT) calculations were carried out on an 8CPU workstation (PQS, Quantum Cube QS8-2400C-064). The geometry optimisations were performed using the B3LYP/6-31G(d) basis set for the radical with the unrestricted Hartree–Fock (UHF) formalism as implemented in the Gaussian 09 program Revision A. 02. The BDE values were determined by the single point energy calculations at the B3LYP/6-31G(d) basis set with the restricted open-shell Hartree–Fock (ROHF) formalism.Acknowledgements
This work was partially supported by Grant-in-Aid (15K18901 to K. I. and 26460056 to I. N.) from the Ministry of Education, Culture, Sports, Science and Technology, Japan, and the Sasakawa Scientific Research Grant from the Japan Science Society (K. I.). We thank Ms Y. Odanaka and Ms S. Matsubayashi at the Center for Instrumental Analysis of Showa University for assistance with NMR and MS measurements.Notes and references
- Y. Du, G. Wei and R. J. Linhardt, J. Org. Chem., 2004, 69, 2206–2209 CrossRef CAS PubMed.
- S. Renaud and L. M. de, Lancet, 1992, 339, 1523–1526 CrossRef CAS.
- A. Scalbert, C. Manach, C. Morand, C. Rémésy and L. Jiménez, Crit. Rev. Food Sci. Nutr., 2005, 45, 287–306 CrossRef CAS PubMed.
- H. Tanaka, M. M. Stohlmeyer, T. J. Wandless and L. P. Taylor, Tetrahedron Lett., 2000, 41, 9735–9739 CrossRef CAS.
- B. B. Aggarwal, C. Sundaram, S. Prasad and R. Kannappan, Biochem. Pharmacol., 2010, 80, 1613–1631 CrossRef CAS PubMed.
- S. L. Albarracin, B. Stab, Z. Casas, J. J. Sutachan, I. Samudio, J. Gonzalez, L. Gonzalo, F. Capani, L. Morales and G. E. Barreto, Nutr. Neurosci., 2012, 15, 1–9 CrossRef CAS PubMed.
- S. Arranz, G. Chiva-Blanch, P. Valderas-Martinez, A. Medina-Remon, R. M. Lamuela-Raventos and R. Estruch, Nutrients, 2012, 4, 759–781 CrossRef CAS PubMed.
- M. Gonzalez-Vallinas, M. Gonzalez-Castejon, A. Rodriguez-Casado and d. M. A. Ramirez, Nutr. Rev., 2013, 71, 585–599 CrossRef PubMed.
- G. Aliev, M. E. Obrenovich, V. P. Reddy, J. C. Shenk, P. I. Moreira, A. Nunomura, X. Zhu, M. A. Smith and G. Perry, Mini-Rev. Med. Chem., 2008, 8, 1395–1406 CrossRef CAS PubMed.
- W. A. Pryor, Free Radical Biol. Med., 2000, 28, 141–164 CrossRef CAS PubMed.
- H. E. Seifried, D. E. Anderson, E. I. Fisher and J. A. Milner, J. Nutr. Biochem., 2007, 18, 567–579 CrossRef CAS PubMed.
- E. Anouar, C. A. Calliste, P. Kosinova, F. Di Meo, J. L. Duroux, Y. Champavier, K. Marakchi and P. Trouillas, J. Phys. Chem. A, 2009, 113, 13881–13891 CrossRef CAS PubMed.
- K. Inami, Y. Iizuka, M. Furukawa, I. Nakanishi, K. Ohkubo, K. Fukuhara, S. Fukuzumi and M. Mochizuki, Bioorg. Med. Chem., 2012, 20, 4049–4055 CrossRef CAS PubMed.
- S. Quideau, D. Deffieux, C. Douat-Casassus and L. Pouysegu, Angew. Chem., Int. Ed., 2011, 50, 586–621 CrossRef CAS PubMed.
- I. Nakanishi, T. Shimada, K. Ohkubo, S. Manda, T. Shimizu, S. Urano, H. Okuda, N. Miyata, T. Ozawa, K. Anzai, S. Fukuzumi, N. Ikota and K. Fukuhara, Chem. Lett., 2007, 36, 1276–1277 CrossRef CAS.
- S. Mitani, A. Ouchi, E. Watanabe, Y. Kanesaki, S.-I. Nagaoka and K. Mukai, J. Agric. Food Chem., 2008, 56, 4406–4417 CrossRef CAS PubMed.
- I. Nakanishi, K. Miyazaki, T. Shimada, K. Ohkubo, S. Urano, N. Ikota, T. Ozawa, S. Fukuzumi and K. Fukuhara, J. Phys. Chem. A, 2002, 106, 11123–11126 CrossRef CAS.
- O. Dangles, G. Fargeix and C. Dufour, J. Chem. Soc., Perkin Trans. 2, 1999, 1387–1395 RSC.
- M. Leopoldini, T. Marino, N. Russo and M. Toscano, J. Phys. Chem. A, 2004, 108, 4916–4922 CrossRef CAS.
- I. Nakanishi, K. Fukuhara, T. Shimada, K. Ohkubo, Y. Iizuka, K. Inami, M. Mochizuki, S. Urano, S. Itoh, N. Miyata and S. Fukuzumi, J. Chem. Soc., Perkin Trans. 2, 2002, 1520–1524 RSC.
- I. Nakanishi, T. Kawashima, K. Ohkubo, H. Kanazawa, K. Inami, M. Mochizuki, K. Fukuhara, H. Okuda, T. Ozawa, S. Itoh, S. Fukuzumi and N. Ikota, Org. Biomol. Chem., 2005, 3, 626–629 CAS.
- N. S. Alrawaiq and A. Abdullah, Int. J. PharmTech Res., 2014, 6, 933–941 CAS.
- W. M. Loke, J. M. Proudfoot, J. M. Hodgson, A. J. McKinley, N. Hime, M. Magat, R. Stocker and K. D. Croft, Arterioscler., Thromb., Vasc. Biol., 2010, 30, 749–757 CrossRef CAS PubMed.
- A. B. Bentz, Journal of Young Investigators, 2009, 19, 1–14 Search PubMed.
- N. A. Kelsey, H. M. Wilkins and D. A. Linseman, Molecules, 2010, 15, 7792–7814 CrossRef CAS PubMed.
- A. Mattarei, N. Sassi, C. Durante, L. Biasutto, G. Sandonà, E. Marotta, S. Garbisa, A. Gennaro, C. Paradisi and M. Zoratti, Eur. J. Org. Chem., 2011, 2011, 5577–5586 CrossRef CAS.
- M. Salas-Reyes, J. Hernández, Z. Domínguez, F. J. González, P. D. Astudillo, R. E. Navarro, E. Martínez-Benavidez, C. Velázquez-Contreras and S. Cruz-Sánchez, J. Braz. Chem. Soc., 2011, 22, 693–701 CrossRef CAS.
- K. Fukuhara, I. Nakanishi, A. Matsuoka, T. Matsumura, S. Honda, M. Hayashi, T. Ozawa, N. Miyata, S. Saito, N. Ikota and H. Okuda, Chem. Res. Toxicol., 2008, 21, 282–287 CrossRef CAS PubMed.
- K. Imai, I. Nakanishi, A. Ohno, M. Kurihara, N. Miyata, K. Matsumoto, A. Nakamura and K. Fukuhara, Bioorg. Med. Chem. Lett., 2014, 24, 2582–2584 CrossRef CAS PubMed.
- K. Fukuhara, I. Nakanishi, H. Kansui, E. Sugiyama, M. Kimura, T. Shimada, S. Urano, K. Yamaguchi and N. Miyata, J. Am. Chem. Soc., 2002, 124, 5952–5953 CrossRef CAS PubMed.
- K. Imai, I. Nakanishi, K. Anzai, T. Ozawa, N. Miyata, S. Urano, H. Okuda, A. Nakamura and K. Fukuhara, Chem. Lett., 2011, 40, 1417–1419 CrossRef CAS.
- T. Oyamada, Bull. Chem. Soc. Jpn., 1935, 10, 182–186 CrossRef CAS.
- T. Oyamada, J. Chem. Soc. Jpn., 1934, 55, 1256–1261 CrossRef CAS.
- J. Algar and J. P. Flynn, Proc. R. Ir. Acad., Sect. B, 1934, 42, 1–8 Search PubMed.
- M. Iwatsu, D. Urabe and M. Inoue, Heterocycles, 2010, 82, 491–504 CrossRef CAS.
- M. Bennett, A. J. Burke and W. I. O'Sullivan, Tetrahedron, 1996, 52, 7163–7178 CrossRef CAS.
- Z. Wu, S. Cai, W. Fan and Q. Wang, Chin. J. Org. Chem., 2012, 32, 1296 CrossRef CAS.
- W.-J. Jiang, K. I. Ishiuchi, M. Furukawa, T. Takamiya, S. Kitanaka and H. Iijima, Bioorg. Med. Chem., 2015, 23, 6922–6929 CrossRef CAS PubMed.
- N. Pandurangan, C. Bose and A. Banerji, Bioorg. Med. Chem. Lett., 2011, 21, 5328–5330 CrossRef CAS PubMed.
- L. X. Yang, K. X. Huang, H. B. Li, J. X. Gong, F. Wang, Y. B. Feng, Q. F. Tao, Y. H. Wu, X. K. Li, X. M. Wu, S. Zeng, S. Spencer, Y. Zhao and J. Qu, J. Med. Chem., 2009, 52, 7732–7752 CrossRef CAS PubMed.
- M. Sato, K. Murakami, M. Uno, Y. Nakagawa, S. Katayama, K.-i. Akagi, Y. Masuda, K. Takegoshi and K. Irie, J. Biol. Chem., 2013, 288, 23212–23224 CrossRef CAS PubMed.
- A. Masa, M. Vilanova and F. Pomar, J. Chromatogr. A, 2007, 1164, 291–297 CrossRef CAS PubMed.
- C. R. Razafindrabe, S. Aubry, B. Bourdon, M. Andriantsiferana, S. Pellet-Rostaing and M. Lemaire, Tetrahedron, 2010, 66, 9061–9066 CrossRef CAS.
- R. Tanifuji, H. Oguri, K. Koketsu, Y. Yoshinaga, A. Minami and H. Oikawa, Tetrahedron Lett., 2016, 57, 623–626 CrossRef CAS.
- B. A. Cevallos-Casals and L. Cisneros-Zevallos, J. Agric. Food Chem., 2003, 51, 3313–3319 CrossRef CAS PubMed.
- A. K. Sinhababu, A. K. Ghosh and R. T. Borchardt, J. Med. Chem., 1985, 28, 1273–1279 CrossRef CAS PubMed.
- M. Ghosh, G. K. Kar, J. K. Ray and B. G. Chatterjee, Synth. Commun., 1983, 13, 667–675 CrossRef CAS.
- K. Yamasaki, R. Hishiki, E. Kato and J. Kawabata, ACS Med. Chem. Lett., 2011, 2, 17–21 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra02329d |
| This journal is © The Royal Society of Chemistry 2017 |