
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Size- and composition-controlled Pt–Sn bimetallic nanoparticles prepared by atomic layer deposition†
Ranjith K. Ramachandran a,
Matthias Filez‡
b,
Jolien Dendoovena,
Vladimir V. Galvitab,
Hilde Poelmanc,
Eduardo Solanoa,
Emiliano Fondac,
Guy B. Marinb and
Christophe Detavernier*a
a,
Matthias Filez‡
b,
Jolien Dendoovena,
Vladimir V. Galvitab,
Hilde Poelmanc,
Eduardo Solanoa,
Emiliano Fondac,
Guy B. Marinb and
Christophe Detavernier*a
aDepartment of Solid State Sciences, COCOON, Ghent University, Krijgslaan 281/S1, B-9000 Ghent, Belgium. E-mail: Christophe.Detavernier@UGent.be
bLaboratory for Chemical Technology, Ghent University, Technologiepark 914, B-9052 Ghent, Belgium
cSynchrotron SOLEIL, SAMBA Beamline, L'Orme des Merisiers, Saint-Aubin, BP48, 91192 Gif-sur-Yvette, France
First published on 6th April 2017
Abstract
Pt–Sn bimetallic nanoparticles (BMNPs) are used in a variety of catalytic reactions and are widely accepted as a model system for Pt-based bimetallics in fundamental catalysis research. Here, Pt–Sn BMNPs were prepared via a two-step synthesis procedure combining atomic layer deposition (ALD) and temperature programmed reduction (TPR). In situ X-ray diffraction measurements during TPR and ex situ X-ray absorption spectroscopy at the Pt LIII-edge revealed the formation of Pt–Sn bimetallic alloys with a phase determined by the Pt/(Pt + Sn) atomic ratio of the as-deposited bilayer. The size of the BMNPs could be tuned by changing the total thickness of the bilayers, while keeping the Pt/(Pt + Sn) atomic ratio constant. Due to the exceptional control over BMNP size and crystalline phase, the proposed method will enable highly systematic studies of the relation between the structure and the performance of Pt–Sn bimetallic catalysts.
Introduction
Pt–Sn bimetallic catalysts are highly performant in a variety of catalytic reactions, including (de)hydrogenation and oxidation of hydrocarbons.1 In industry, for example, they are used worldwide in the Oleflex (UOP) process for the on-purpose production of propylene.2 Besides their wide applicability, Pt–Sn catalysts have become quintessential model systems in catalysis science as a result of several decades of intense research. In this research history, academia as well as industry have tried to understand the role of the promoting element (Sn) on the overall performance of Pt-based bimetallic catalysts in particular, and metal catalysts in general.3,4 Despite their merits, full elucidation of structure–performance relationships has only been obtained in rare cases, due to the non-uniform nature of the alloyed nanoparticle phase.5,6 More particularly, conventional methods for bimetallic catalyst synthesis – wet impregnation and precipitation – introduce compositional and particle size heterogeneities, hindering unambiguous disentanglement of structure–performance relationships. This missing link between nano-alloy structure, reaction mechanism and the catalyst performance impedes the redesign of current bimetallic catalysts. The fabrication of fundamentally improved bimetallic catalysts therefore relies on the synthesis of bimetallic catalysts which allows for control over the bimetallic nanoparticle (BMNP) composition and particle size. Recently, there have been reports on the controlled synthesis of Pt–Sn nanoparticles employing solution phase methods. While Wang et al.7 reported on controlling the shape of colloidal Pt–Sn nanoparticles, DeSario et al.8 focussed on the low temperature crystallization and ordering in alloys with different compositions.Here, we demonstrate the simultaneous control on both size and composition of the Pt–Sn nanoparticles via a vapor phase method called atomic layer deposition (ALD).9,10 A crucial advantage of ALD is that the amount of deposited material can be controlled at the monolayer level by changing the number of ALD cycles. This is a consequence of the self-limiting nature of the surface reactions determining the ALD process. Moreover, this unique surface chemistry enables conformal depositions on 3D substrates, including mesoporous supports with large surface areas.11
Until recently, the ALD-based synthesis of BMNPs was limited to combinations of two noble metals.12–15 Several groups exploited two characteristics typical of noble metal ALD processes, in casu the island growth mode16,17 and their selective deposition onto other noble metal over oxide sites,14,18,19 to grow Pt–Pd, Pt–Ru and Pd–Ru BMNPs. To broaden the range of possible alloying elements for Pt-based BMNPs, we recently reported an ALD-based synthesis strategy that allows for the size- and composition-controlled preparation of BMNPs containing a non-noble metal next to a noble metal, namely Pt–In and Pt–Ga systems.20 In this work, the generality of the method is further demonstrated by providing proof for its applicability to Pt–Sn BMNPs. Future fundamental catalysis studies, generally employing the Pt–Sn model system, will highly benefit from the atomically precise control over composition and size offered by the ALD method.
Fig. 1 schematically describes the steps involved in the fabrication process of the Pt–Sn BMNPs. First, thin films of SnO2 and Pt are sequentially deposited on a silica substrate by means of ALD, using alternating exposures of tetrakis-(dimethylamino)tin and H2O for SnO2 ALD21 and trimethyl-(methylcyclopentadienyl)platinum and O3 for Pt ALD,16,22 yielding a Pt/SnO2 bilayer structure. These bilayers are then subjected to a temperature programmed reduction (TPR) to 700 °C in 10% H2/N2 at a heating rate of 0.2 °C s−1, which results in the formation of alloyed Pt–Sn particles. The composition and size of the BMNPs can be controlled by profiting from the atomic level thickness control of the ALD technique.
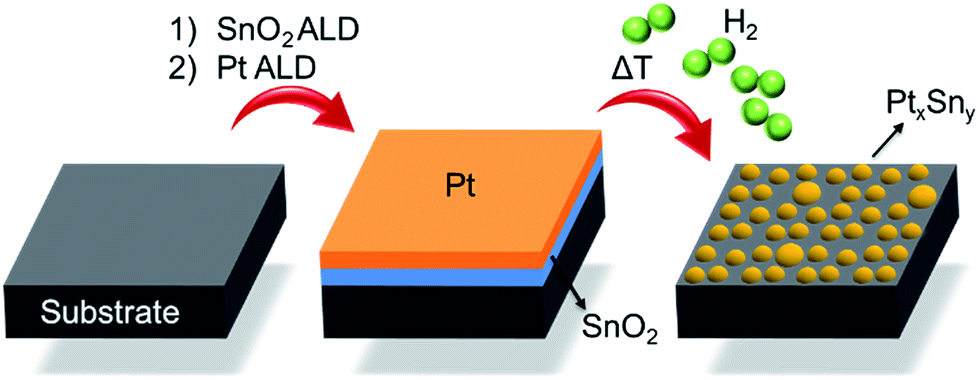 | ||
| Fig. 1 Schematic representation of the ALD-based Pt–Sn BMNPs synthesis. | ||
Experimental
Sample preparation
All depositions were performed at 150 °C in a home-built cold-wall ALD chamber16,23,24 with a base pressure of 10−6 mbar. Si wafers with a 100 nm thermally grown SiO2 layer on top were used as substrates. SnO2 and Pt were deposited using alternating exposures of TDMASn (99%, Strem Chemicals) and H2O for SnO2 ALD21 and (MeCp)PtMe3 (99%, Strem Chemicals) and O3 for Pt ALD.16 Ar with 99.999% purity was used as a carrier gas for all precursors. O3 was produced from a 99% O2/N2 mixture with an AC-2025 (USA Inc.) generator, resulting in an O3 concentration of 200 μg ml−1. A typical SnO2 ALD cycle lasted 20 s with 5 s pulse time for both the precursor and reactant, and 5 s pumping after each pulse. For Pt ALD, a static exposure mode was applied during both ALD half-cycles, resulting in a total cycle time of 50 s.Composition determination
Pt and SnO2 film thicknesses were determined by X-ray reflectivity measurements using a Bruker D8 Discover system with Cu Kα radiation. Alternatively, the thickness was extracted from calibrated X-ray fluorescence data obtained with a Bruker Artax system with Mo X-ray source. From the thickness, the specific weight of the Pt and Sn was calculated by assuming the densities of the films equal to that of bulk Pt and SnO2, respectively (see ESI† for the details).![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 564 eV) using a 35-element Ge detector (Canberra). XAS data reduction and analysis were executed with the Demeter 0.9.13 software package by following the methodology of Koningsberger et al.27
564 eV) using a 35-element Ge detector (Canberra). XAS data reduction and analysis were executed with the Demeter 0.9.13 software package by following the methodology of Koningsberger et al.27Results and discussion
A series of bilayer samples with different Pt/(Pt + Sn) atomic ratios was prepared by using the atomic level control of ALD to precisely define the thickness of SnO2 and Pt layers (Table 1, samples a–f). In situ XRD patterns collected during H2 TPR of the as-deposited bilayers a–d show similar phase evolution behavior (Fig. 2A). Initially, the patterns show a diffraction peak which corresponds to metallic Pt(111). No diffractions corresponding to SnO2 are observed, as a consequence of its amorphous structure. As the temperature increases, a gradual shift of the Pt(111) peak towards a lower 2θ angle occurs around 250 °C, resulting from the incorporation of reduced Sn into the Pt fcc lattice.28 The diffraction peaks at higher temperatures show that the uptake of reduced Sn results in the formation of PtSn intermetallic alloys. For sample e with a Pt/(Pt + Sn) atomic ratio of 47%, the formation of the crystalline alloy phase happens more abrupt at a temperature of 200 °C. In the case of the most Sn rich sample, sample f, there are no diffraction peaks visible initially, as the amount of Pt is under the detection limit of XRD. When the temperature reaches 500 °C, two diffractions corresponding to the PtSn4 phase appear and disappear as this phase is not stable above 522 °C. The absence of diffractions from SnO2 in the in situ XRD patterns indicates that the as-deposited amorphous SnO2 is reduced to Sn before being crystallized. Sn is also not visible as it is liquid at higher temperatures.29| Sample ID | Thickness (nm) | Pt/(Pt + Sn) atomic ratio | Phase(s) formed after TPR | Fractional Pt coordination numbers | ||
|---|---|---|---|---|---|---|
| SnO2 | Pt | nPt–Pt | nPt–Sn | |||
| a | 2.1 | 5.5 | 87 | Pt*, Pt3Sn | 0.81 | 0.19 |
| b | 4.4 | 5.5 | 75 | Pt3Sn | 0.62 | 0.38 |
| c | 7.9 | 5.5 | 63 | Pt3Sn*, PtSn | 0.58 | 0.42 |
| d | 7.9 | 3.8 | 54 | Pt3Sn, PtSn* | 0.45 | 0.55 |
| e | 4.4 | 1.7 | 47 | PtSn | 0.20 | 0.80 |
| f | 7.9 | 1.0 | 22 | PtSn4, Sn | — | — |
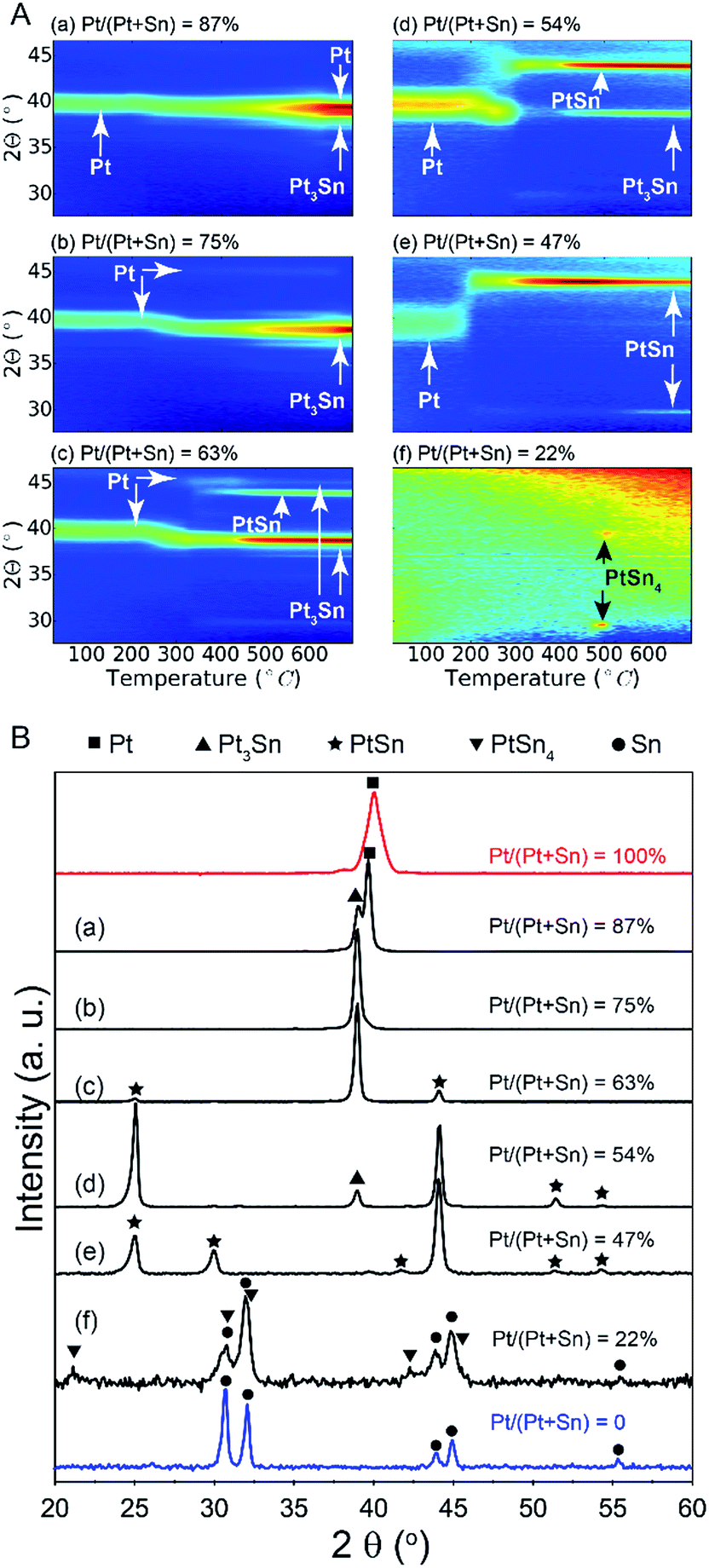 | ||
| Fig. 2 (A) In situ XRD patterns during TPR in 10% H2/N2 of Pt/SnO2 bilayer samples with Pt/(Pt + Sn) atomic ratios of (a) 87%, (b) 75%, (c) 63%, (d) 54%, (e) 47% and (f) 22%. (B) Ex situ XRD patterns measured after the TPR experiment. The red and blue patterns are measured after TPR of pure Pt and SnO2 layers, respectively. | ||
The ex situ XRD measurements after TPR and cool down (Fig. 2B) allow to pinpoint the exact Pt–Sn alloy phases for differently composed bilayers at room temperature (Table 1). There is a clear relation between the Pt/(Pt + Sn) atomic ratio of the as-deposited bilayers and their eventual Pt–Sn alloy composition/phase. A gradual transformation from Pt towards metallic Sn is observed, demonstrating the exceptional ability of ALD to tune the phase of bimetallic Pt–Sn alloys. For sample f, diffractions corresponding to metallic Sn are observed in addition to the PtSn4 peaks.
Ex situ Pt LIII edge extended X-ray absorption fine structure (EXAFS) data of the reduced samples (Fig. 3) furnish a detailed insight into the local atomic structure of the alloys, except for sample f for which the quality of the measured EXAFS data was too low for analysis. Qualitative analysis of the features in the higher R-range (R > 3 Å) reveals an fcc structure for samples a and b which gradually disappears for samples with lower Pt content. Because Pt and Pt3Sn have fcc lattice stacking, this result is in agreement with the long-range XRD data. Quantitative modeling of the EXAFS data with a Pt–Pt and Pt–Sn shell yields good agreement with the experimental signal for R in the 2–3 Å range. Pt–Sn alloying was assessed by deriving the fractional Pt coordination numbers (nPt–Pt = NPt–Pt/(NPt–Sn + NPt–Pt) and nPt–Sn = NPt–Sn/(NPt–Sn + NPt–Pt) in Table 1). A clear decrease [increase] in the fraction of Pt–Pt [Pt–Sn] bonds is observed with decreasing Pt content. More specifically, quasi phase pure Pt3Sn (sample b) and PtSn (sample e) display 62 [38] and 20 [80]% Pt [Sn] coordination around the Pt central absorber, respectively. This is in close accord with the theoretical 67 [33] and 25 [75]% Pt–Pt [Pt–Sn] bond abundance for Pt3Sn and PtSn alloy structures. The short-range information derived from EXAFS therefore fully corroborates with the long-range XRD data, proving the high phase-tuning potential of ALD for Pt–Sn alloys.
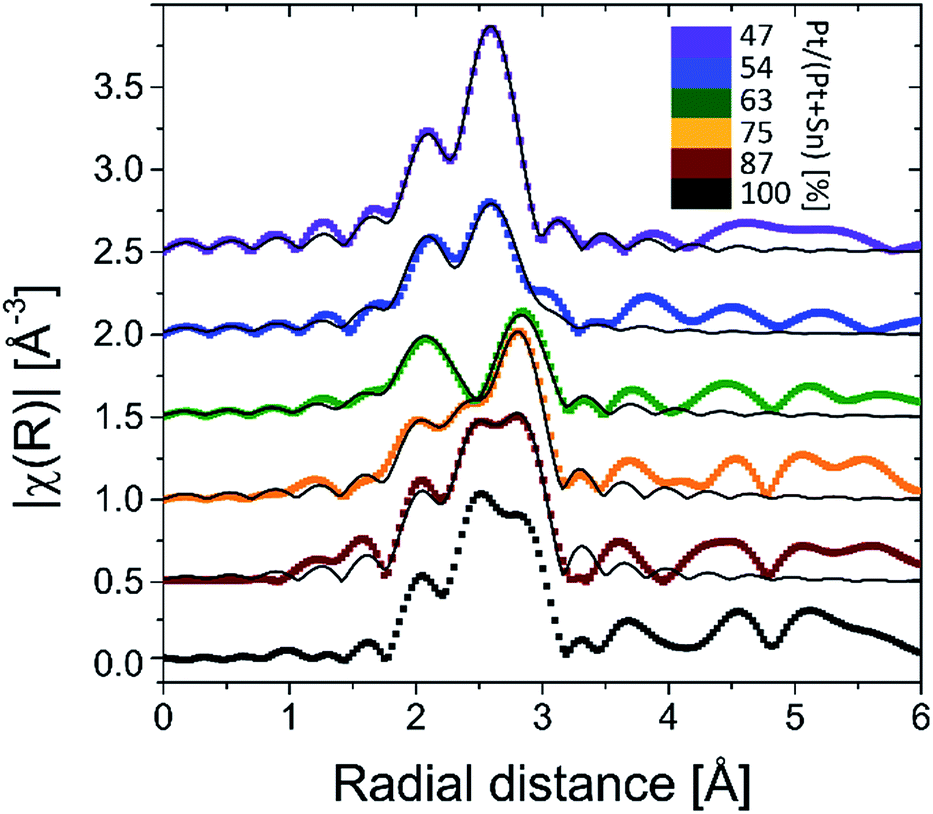 | ||
| Fig. 3 k2-Weighted Fourier transformed EXAFS magnitudes of the BMNPs obtained after TPR in 10% H2/N2 up to 700 °C. The thin black lines are the EXAFS modeling results (Pt–Pt + Pt–Sn shell). | ||
In addition to phase control, the presented method enables tuning of the Pt–Sn BMNP size. More particularly, particle size control is achieved by scaling the total bilayer film thickness, while keeping the Pt/(Pt + Sn) atomic ratio constant. This is demonstrated by the preparation of a series of bilayer films with a constant Pt/(Pt + Sn) atomic ratio of ca. 75%, but different total thickness, ranging from 1.4 to 5.8 nm (Table 2). The films were subjected to a TPR up to 700 °C in 10% H2/N2 and the formation of Pt3Sn was confirmed by XRD measurements (see ESI†). The morphology of the films was studied by scanning electron microscopy (SEM). The SEM images are shown in Fig. 4 together with a plot of the average particle size against the total deposited thickness. A linear relation is observed, proving that the average BMNP size can indeed be tuned via the total thickness of the bilayer ALD-film. While a wide distribution of nanoparticle sizes is observed for the largest nanoparticles in the SEM image of sample V, a narrow particle size distribution is obtained for the smallest nanoparticles that are of highest interest in the field of catalysis (see ESI†).
| Sample ID | Thickness (nm) | Total thickness (nm) | Average particle size (nm) | |
|---|---|---|---|---|
| SnO2 | Pt | |||
| I | 0.7 | 0.7 | 1.4 | 5 |
| II | 0.9 | 1.1 | 2.0 | 10 |
| III | 1.3 | 1.4 | 2.7 | 17 |
| IV | 1.5 | 1.7 | 3.2 | 25 |
| V | 2.8 | 3.0 | 5.8 | 58 |
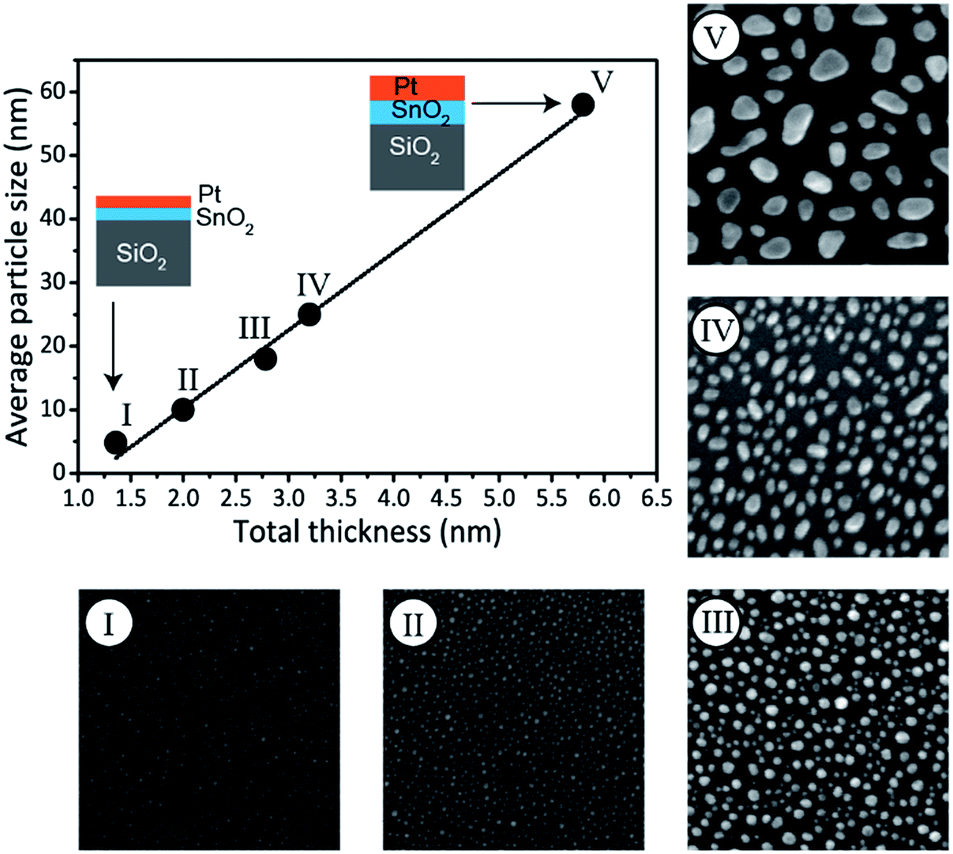 | ||
| Fig. 4 SEM images (full width of 500 nm) measured after TPR in 10% H2/N2 up to 700 °C of Pt/SnO2 bilayers with Pt/(Pt + Sn) atomic ratio of ca. 75%. The graph shows the average BMNP size formed after TPR as determined from SEM analysis versus the total deposited thickness. The line is a linear fit to the data points. The schematics show how the thickness of the Pt and SnO2 layers is scaled to tune the particle size. | ||
Conclusions
In summary, this work demonstrates that a reductive treatment of Pt/SnO2 bilayers deposited with ALD results in the formation of alloyed Pt–Sn particles for which the crystalline phase and average particle size can be controlled via the ALD process. The crystalline phase is determined by the as-deposited Pt/(Pt + Sn) atomic ratio, while the average particle size is defined by the total (SnO2 + Pt) thickness. Therefore, this approach highly takes advantage of the unique monolayer-level thickness control of ALD. Since the phase and size of the Pt–Sn bimetallic nanoparticles can be rationally tuned, we believe that the synthesis method we present here will open up a new way for constructing model systems that enable in-depth investigations of the relation between the Pt–Sn bimetallic particle structure and the catalytic efficiency and selectivity. In a first effort to validate the catalytic applicability of the presented Pt–Sn BMNP synthesis approach during propane dehydrogenation, activities comparable to conventionally synthesized Pt–Sn catalysts were observed. Detailed investigation towards optimization is ongoing.Acknowledgements
This research was supported by the Flemish Research Foundation (FWO-Vlaanderen), the Flemish Government (Long-term structural funding – Methusalem funding and Medium-scale research infrastructure funding-Hercules funding), the Special Research Fund BOF of Ghent University (GOA 01G01513), and the IAP7/05 Interuniversity Attraction Poles Programme, Belgian State, Belgian Science Policy. We are also grateful to the SAMBA-SOLEIL staff for smoothly running the facilities.Notes and references
- W. Yu, M. D. Porosoff and J. G. Chen, Chem. Rev., 2012, 112, 5780–5817 CrossRef CAS PubMed
.
- J. J. H. B. Sattler, J. Ruiz-Martinez, E. Santillan-Jimenez and B. M. Weckhuysen, Chem. Rev., 2014, 114, 10613–10653 CrossRef CAS PubMed
- J. Wu, Z. Peng and A. T. Bell, J. Catal., 2014, 311, 161–168 CrossRef CAS
- M. Filez, E. A. Redekop, H. Poelman, V. V. Galvita, M. Meledina, S. Turner, G. Van Tendeloo, C. Detavernier and G. B. Marin, Catal. Sci. Technol., 2016, 6, 1863–1869 CAS
- A. Sapi, F. Liu, X. Cai, C. M. Thompson, H. Wang, K. An, J. M. Krier and G. A. Somorjai, Nano Lett., 2014, 14, 6727–6730 CrossRef CAS PubMed
- C. Cui, M. Ahmadi, F. Behafarid, L. Gan, M. Neumann, M. Heggen, B. R. Cuenya and P. Strasser, Faraday Discuss., 2013, 162, 91–112 RSC
- X. Wang, L. Altmann, J. Stöver, V. Zielasek, M. Bäumer, K. Al-Shamery, H. Borchert, J. Parisi and J. Kolny-Olesiak, Chem. Mater., 2013, 25, 1400–1407 CrossRef CAS
- D. Y. Desario and F. J. Disalvo, Chem. Mater., 2014, 26, 2750–2757 CrossRef CAS
- S. M. George, Chem. Rev., 2010, 110, 111–131 CrossRef CAS PubMed
- V. Miikkulainen, M. Leskelä, M. Ritala and R. L. Puurunen, J. Appl. Phys., 2013, 113, 021301 CrossRef
- C. Detavernier, J. Dendooven, S. Pulinthanathu Sree, K. F. Ludwig and J. A. Martens, Chem. Soc. Rev., 2011, 40, 5242 RSC
- B. J. O'Neill, D. H. K. Jackson, J. Lee, C. Canlas, P. C. Stair, C. L. Marshall, J. W. Elam, T. F. Kuech, J. A. Dumesic and G. W. Huber, ACS Catal., 2015, 5, 1804–1825 CrossRef
- K. Cao, Q. Zhu, B. Shan and R. Chen, Sci. Rep., 2015, 5, 8470 CrossRef CAS PubMed
- J. Lu, K.-B. Low, Y. Lei, J. A. Libera, A. Nicholls, P. C. Stair and J. W. Elam, Nat. Commun., 2014, 5, 3264 Search PubMed
- S. T. Christensen, H. Feng, J. L. Libera, N. Guo, J. T. Miller, P. C. Stair and J. W. Elam, Nano Lett., 2010, 10, 3047–3051 CrossRef CAS PubMed
- J. Dendooven, R. K. Ramachandran, K. Devloo-Casier, G. Rampelberg, M. Filez, H. Poelman, G. B. Marin, E. Fonda and C. Detavernier, J. Phys. Chem. C, 2013, 117, 20557–20561 CAS
- J. Lu, J. W. Elam and P. C. Stair, Surf. Sci. Rep., 2016, 71, 410–472 CrossRef CAS
- M. J. Weber, M. A. Verheijen, A. A. Bol and W. M. M. Kessels, Nanotechnology, 2015, 26, 94002 CrossRef CAS PubMed
- M. J. Weber, A. J. M. MacKus, M. A. Verheijen, C. Van Der Marel and W. M. M. Kessels, Chem. Mater., 2012, 24, 2973–2977 CrossRef CAS
- R. K. Ramachandran, J. Dendooven, M. Filez, V. V. Galvita, H. Poelman, E. Solano, M. M. Minjauw, K. Devloo-Casier, E. Fonda, D. Hermida-Merino, W. Bras, G. B. Marin and C. Detavernier, ACS Nano, 2016, 10, 8770–8777 CrossRef CAS PubMed
- M. N. Mullings, C. Hägglund and S. F. Bent, J. Vac. Sci. Technol., A, 2013, 31, 61503 Search PubMed
- M. Filez, H. Poelman, R. K. Ramachandran, J. Dendooven, K. Devloo-Casier, E. Fonda, C. Detavernier and G. B. Marin, Catal. Today, 2014, 229, 2–13 CrossRef CAS
- Q. Xie, Y.-L. Jiang, C. Detavernier, D. Deduytsche, R. L. Van Meirhaeghe, G.-P. Ru, B.-Z. Li and X.-P. Qu, J. Appl. Phys., 2007, 102, 83521 CrossRef
- J. Musschoot, Q. Xie, D. Deduytsche, S. Van den Berghe, R. L. Van Meirhaeghe and C. Detavernier, Microelectron. Eng., 2009, 86, 72–77 CrossRef CAS
- W. Knaepen, C. Detavernier, R. L. Van Meirhaeghe, J. Jordan Sweet and C. Lavoie, Thin Solid Films, 2008, 516, 4946–4952 CrossRef CAS
- W. Knaepen, S. Gaudet, C. Detavernier, R. L. Van Meirhaeghe, J. J. Sweet and C. Lavoie, J. Appl. Phys., 2009, 105, 83532 CrossRef
- D. C. Koningsberger, B. L. Mojet, G. E. van Dorssen and D. E. Ramaker, Top. Catal., 2000, 10, 143–155 CrossRef CAS
- M. Filez, E. A. Redekop, V. V. Galvita, H. Poelman, M. Meledina, S. Turner, G. Van Tendeloo, A. T. Bell and G. B. Marin, Phys. Chem. Chem. Phys., 2016, 18, 3234–3243 RSC
- T. Biggs, S. S. Taylor and E. Van Der Lingen, Platinum Met. Rev., 2005, 49, 2–15 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental section; XRD patterns and particle size distributions of differently sized Pt3Sn nanoparticles. See DOI: 10.1039/c7ra01463e |
| ‡ Present address: Inorganic Chemistry and Catalysis group, Debye Institute for Nanomaterials Science, Utrecht University, Universiteitsweg 99, 3584 CG Utrecht, The Netherlands. |
| This journal is © The Royal Society of Chemistry 2017 |