
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ferrous complexes supported by sterically encumbered asymmetric bis(arylimino)acenaphthene (BIAN) ligands: synthesis, characterization and screening for catalytic hydrosilylation of carbonyl compounds†
Xun Yu‡
,
Feifeng Zhu‡,
Donglei Bu and
Hao Lei *
*
Department of Chemistry, College of Chemistry and Materials Science, Jinan University, Guangzhou 510632, China. E-mail: tleihao@jnu.edu.cn
First published on 9th March 2017
Abstract
Six ferrous chloride complexes ((Ar-BIANX)FeCl2: Ar = 2,6-diisopropylphenyl (Dipp), X = F (1), Cl (2), Me (3); Ar = mesityl (Mes), X = F (4), Cl (5), Me (6)) supported by sterically bulky asymmetric bis(arylimino)acenaphthene (BIAN) ligands were prepared through the treatment of anhydrous FeCl2 with the corresponding ligands in a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. The compounds were characterized by X-ray crystallography, IR, NMR and electrochemical methods. This series of complexes represents rare examples of structurally characterized iron compounds bearing asymmetric bidentate BIAN ligands. The complexes were tested for catalytic hydrosilylation of aldehydes and ketones at room temperature, and moderate to good yields of alcohol derivatives were obtained after hydrolysis workup.
:1. The compounds were characterized by X-ray crystallography, IR, NMR and electrochemical methods. This series of complexes represents rare examples of structurally characterized iron compounds bearing asymmetric bidentate BIAN ligands. The complexes were tested for catalytic hydrosilylation of aldehydes and ketones at room temperature, and moderate to good yields of alcohol derivatives were obtained after hydrolysis workup.
Introduction
Within the last decade, considerable attention has been directed towards metal complexes supported by redox-active ligands,1,2 owing to their intriguing electronic structures and potential applications in catalysis.3–7 Due to the multiple consecutive oxidation states of redox-active ligands, their coordination to metal centers usually induces radical reactivity and electron reservoir behavior. α-Diimines comprise a class of the most widely investigated redox-active ligands, demonstrating three different oxidation states in their coordination chemistry, i.e. the neutral ligand, radical anion and the dianionic form.8,9 The electronic and steric properties of their metal compounds can be regulated by variation of the substituents on the N–C–C–N backbone. Specifically, when the backbone C–C bond is fused with a conjugated naphthalene ring, a rigid and robust ligand framework is derived, i.e. bis(arylimino)acenaphthene (BIAN). Similar to the parent diimine ligand sets, BIAN ligands can also undergo successive facile single-electron reductions. Furthermore, the neutral form of BIAN ligands can accept up to four electrons, due to the incorporation of naphthalene fragment. In addition, the rigid and planar ligand backbone ensures the two nitrogen donor atoms chelating to the same metal center in a syn conformation, which are crucial for the preparation of certain single-metal catalysts.10Synthesis and reactivity of metal BIAN compounds are well documented in the literature. For instance, metal-to-ligand charge transfer was commonly observed in a series of heavier p-block metal compounds supported by BIAN ligands.11–15 In recent years, Clark and co-authors explored the reactivity and redox capability of BIAN ligands in early transition metal complexes, revealing very rich electronic behavior for such metal/ligand combination.16,17 In contrast, iron compounds bearing BIAN ligand framework are relatively less investigated. A search in Cambridge Structural Database (CSD) showed that only several structurally characterized BIAN-Fe complexes have been reported, including those with tridentate BIAN ligands with an additional donor arm other than the two N donors.18 It was discovered that the solid-state structures of these tridentate BIAN metal complexes exhibited a range of coordination modes depending on the crystallization solvent as well as the nucleophilicity of the third donor atoms.19 Other known BIAN-Fe compounds usually involve identical aryl N-substituents (Mes or Dipp), featuring mononuclear,20,21 dinuclear22 or multinuclear structural motifs.23 It was discovered that the radical anionic form of BIAN ligands could be installed to Fe(II) centers through the treatment of FeI2 with various molar ratio of potassium salt of the radical anion.22 Findlater and co-workers reported the structure of a toluene-capped low-valent iron complex supported by a partially reduced BIAN ligand, i.e. (BIAN)Fe(η6-C7H8), synthesized via reduction of the ferrous precursor with sodium amalgam.20 This compound was shown to be an efficient precatalyst for the hydrosilylation of aldehydes and ketones using Ph2SiH2 at 70 °C. Very recently, similar (ArBIAN)FeBr2 complexes (Ar = Mes or Dipp) were reported by Hoyt and co-workers, and the catalytic hydrosilylation of 1-hexene was investigated.21
Reduction of carbonyl compounds is among the most valuable synthetic routes in both academia and industry. Transition-metal-catalyzed hydrosilylation followed by hydrolysis workup has emerged as a useful protocol for the production of alcohols.24 Although the platinum group metals have long been proved to be efficient for such transformations, their high cost and toxicity prompts the search for more sustainable homogeneous catalytic systems. Complexes with earth-abundant metals (in particular Fe) have attracted increasing attention in recent years, due to their potential as alternative cheap catalysts for various organic transformations.25–28 During the last decade, rapid developments have occurred in the area of Fe-catalyzed hydrosilylation of aldehydes and ketones, as a result of the efforts from the research groups of Beller,29 Nishiyama,30 Guan,31 Chirik,32,33 Tilley,34 Gade35,36 and others.37,38 In spite of such considerable development in this area, examples of Fe-catalyzed hydrosilylation of carbonyls under mild conditions (e.g. at room temperature) are still limited to several recent reports.32–38 These include iron compounds bearing tridentate NNN32,33,35 or NNO37 pincer ligands, as well as those featuring two-34 or three-coordinate38 Fe centers, the preparation of which usually involves sophisticated ligand design and multiple synthetic steps. Very recently, Gade and co-workers reported highly active iron alkyl and alkoxide catalysts with turnover frequency (TOF) up to 240 h−1 at −40 °C for hydrosilylation reactions.35,36 In this context, discovery of easily-prepared Fe-containing hydrosilylation catalysts that function at room temperature (or lower) still remains a current challenge in this research area.
As mentioned earlier, a BIAN supported Fe complex has been reported to efficiently catalyze the hydrosilylation of aldehydes and ketones at 70 °C under solvent-free condition.20 It is reasonable to assume that increasing the steric bulk at N atoms on the BIAN backbone, might further stabilize the proposed Fe hydride catalytic intermediate, thus improves the efficiency of this catalytic system. In recent years, 2,6-dibenzhydryl phenyl substituents (abbreviated as Ar*) were introduced to the BIAN ligand framework by Sun and others, and their nickel and palladium complexes were demonstrated to be effective catalysts for either ethylene polymerization or C–C cross coupling reactions.39–45 Following our previous assumption, herein we report the synthesis of a series of Fe(II) complexes supported by sterically hindered BIAN ligands (Ar-BIANX)FeCl2 (Ar = Dipp, X = F (1), Cl (2), Me (3); Ar = Mes, X = F (4), Cl (5), Me (6)). The solid-state structures of the six compounds were determined by single crystal X-ray diffraction, rendering them rare examples of structurally characterized iron complexes with asymmetric BIAN ligands. The new compounds were further demonstrated to be efficient precatalysts for room temperature hydrosilylation of various aldehyde and ketone substrates.
Results and discussion
The asymmetric bulky BIAN ligands are facile to prepare via stepwise condensation of acenaphthaquinone with anilines under acid-catalyzed conditions (Scheme 1).42–45 In the current study, we chose to employ BIAN ligands that have two different substituted aryl groups on the N donor atoms. In a certain ligand framework, one of the nitrogen atoms is substituted with Mes or Dipp group, while the other N atom is bonded to a sterically encumbered 2,6-dibenzhydryl phenyl group (Ar*), whose para-position is occupied by different electron donating or withdrawing groups (–F, –Cl or –Me). Thus, such ligand design strategy results in a series of asymmetric bulky BIAN ligands with tunable electronic property and steric bulk.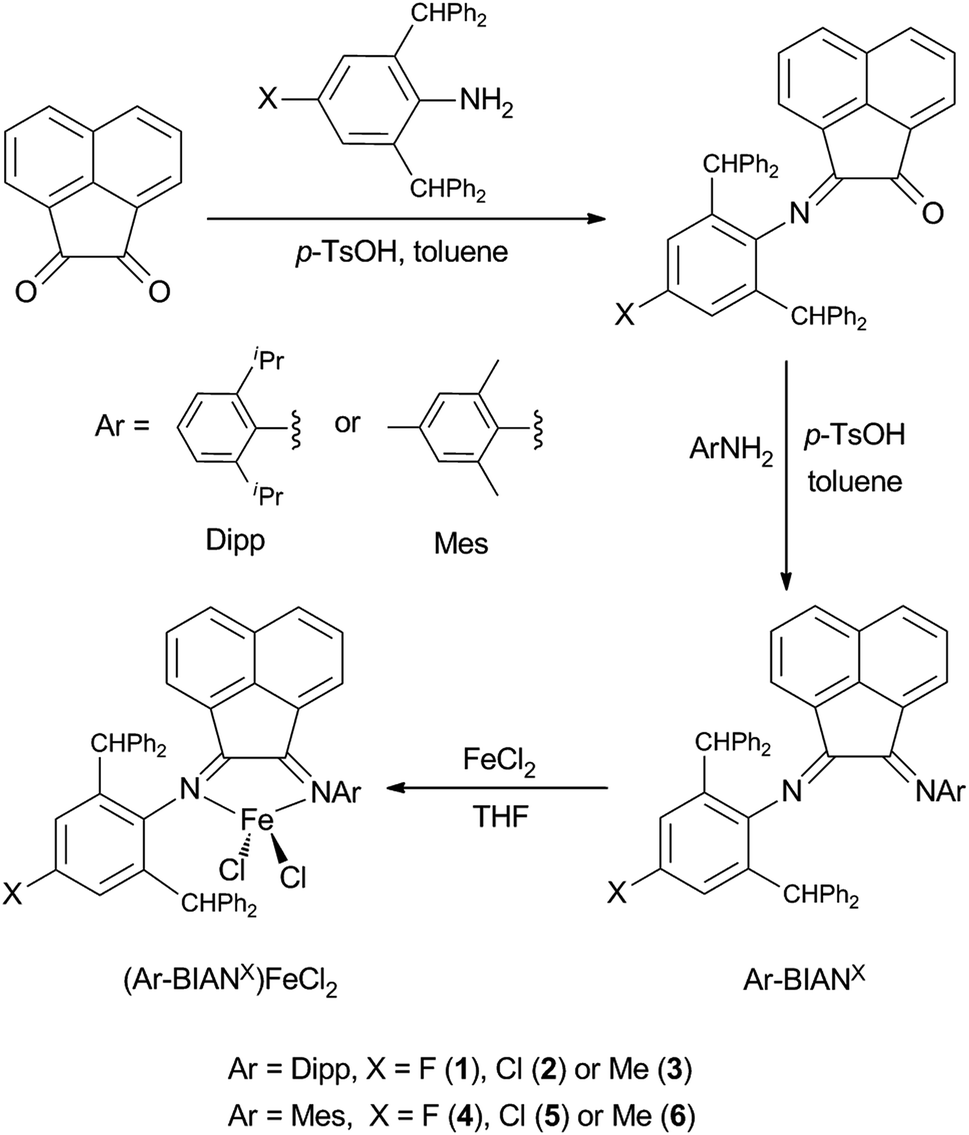 | ||
| Scheme 1 Synthetic routes to complexes 1–6. | ||
Treatment of the bulky BIAN ligands with anhydrous FeCl2 in THF at room temperature afforded the corresponding ferrous complexes 1–6 in good yields (Scheme 1). Although crystal structures of 1–6 revealed different extent of solvation with THF molecules (either coordinated or non-coordinated, vide infra), the elemental analysis data confirmed their formula composition as (Ar-BIANX)FeCl2. All six compounds showed very high thermal stability under inert atmosphere, with melting points higher than 320 °C. The 1H NMR spectra of these compounds were featured with broadened and paramagnetically shifted resonances within the range of −16 to +18 ppm (see ESI†), suggesting high-spin electron configuration for the iron centers. This is similar to the observation for previously reported analogous BIAN Fe(II) halide complexes.20,21
The new ferrous complexes were further characterized by IR spectroscopy. As summarized in Table 1, the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretching frequencies of the iron compounds are lower than those of the corresponding neutral BIAN ligands, indicating the presence of bonding interactions between imine groups and the metal centers. In addition, the relatively small shifts of the stretching frequencies between the ligands and corresponding iron complexes (Δν(CN) < 25 cm−1) imply that no significant metal-to-ligand electron transfer has occurred, and the coordinated BIAN ligands all remain in their neutral oxidation states. A closer look at the IR data revealed that all the six Fe compounds showed comparable CN vibration frequencies (Δν(CN) < 10 cm−1), indicating the presence of similar electronic structures across this series of metal complexes. Furthermore, the CN stretching frequencies of these Fe species were consistent with the reported values for the Ni and Pd complexes bearing the same ligand sets,42–45 but lower than that reported for analogous Fe compounds with symmetric BIAN ligands.21
N stretching frequencies of the iron compounds are lower than those of the corresponding neutral BIAN ligands, indicating the presence of bonding interactions between imine groups and the metal centers. In addition, the relatively small shifts of the stretching frequencies between the ligands and corresponding iron complexes (Δν(CN) < 25 cm−1) imply that no significant metal-to-ligand electron transfer has occurred, and the coordinated BIAN ligands all remain in their neutral oxidation states. A closer look at the IR data revealed that all the six Fe compounds showed comparable CN vibration frequencies (Δν(CN) < 10 cm−1), indicating the presence of similar electronic structures across this series of metal complexes. Furthermore, the CN stretching frequencies of these Fe species were consistent with the reported values for the Ni and Pd complexes bearing the same ligand sets,42–45 but lower than that reported for analogous Fe compounds with symmetric BIAN ligands.21
N stretching frequencies for BIAN ligands and Fe complexes 1–6
| ν(CN)/cm−1 |
||
|---|---|---|
| BIAN ligands | Fe complexes | |
| 1 | 1639, 1593 | 1619, 1582 |
| 2 | 1640, 1595 | 1621, 1584 |
| 3 | 1631, 1590 | 1622, 1585 |
| 4 | 1643, 1593 | 1619, 1587 |
| 5 | 1642, 1598 | 1623, 1588 |
| 6 | 1640, 1594 | 1621, 1586 |
The electronic structures of the BIAN supported Fe complexes were also probed by UV-vis spectroscopy. In the spectra (see ESI†), each of the complexes showed a shoulder absorption band around ca. 420 nm, which is probably a result of the n → π* transition from the diimine to the acenaphthene. In contrast, the high energy absorption bands around ca. 320 nm for the complexes were tentatively assigned to the π → π* transition within the BIAN ligand framework. This is in good comparison to those reported for related BIAN indium compounds.14 Again, the similarity of the electronic transition energy for the six ferrous complexes confirmed their comparable electronic structures.
The compositions and solid-state structures of complexes 1–6 were further corroborated by X-ray crystallography. Vapor diffusion of n-hexane into the THF solution of these complexes afforded bronze crystals of high quality. Compound 1 crystallizes in the orthorhombic space group P212121, while compound 3 crystallizes in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . The other four iron complexes all crystallizes in monoclinic space groups, i.e. P21/n for compounds 2 and 4, and P21/c for compounds 5 and 6. The crystallographic data and collection parameters are presented in Table 2. In general, all six structures showed expected bonding environment and atom connectivity, with two N atoms coordinated to the iron center of a ferrous dichloride unit (Fig. 1). However, to our surprise, two different types of coordination environment for the Fe centers were observed, i.e. four-coordinate (as in complexes 2, 3, 4 and 6) and five-coordinate (as in complexes 1 and 5). The major difference between these two bonding situations is whether a THF molecule is directly coordinated to the iron through its oxygen atom. The four-coordinate compounds showed a distorted tetrahedral geometry with calculated τ4 values in the range of 0.83 to 0.88.46 In contrast, the five-coordinate complexes demonstrated a more distorted trigonal bipyramidal geometry (τ5 = 0.61–0.65),47 with the equatorial positions occupied by two chlorine atoms and one nitrogen atom, while the axial positions occupied by the other nitrogen atom and the oxygen atom of coordinated THF molecule. The fact that some of this series of Fe compounds could adopt a five-coordinate environment indicates that in this case the effective steric bulk of the Ar* substituent is not as great as originally assumed. This is further supported by the observation that in the structure of 1, the THF molecule coordinates to Fe from the same side as the Ar* group, while in the structure of 5, the coordinated THF molecule lies on the same side of the Mes group. This implies that within the BIAN ligand scaffold, the steric bulk follows the sequence of Dipp > Ar* > Mes. It is reasonable to deduce that using Ar* groups on both coordinating N atoms of the BIAN ligands would enforce a much more hindered coordination environment and ensure low-coordination at the iron center. However, such symmetric doubly Ar*-substituted BIAN ligands could not be prepared using conventional acid-catalyzed condensation methods, in accordance with previous literature reports.39 It is also noteworthy that for this series of compounds, whether or not to have an additional coordinated THF in the structures does not seem to be affected by the electronic property of remote ligand substituents (X = F, Cl or Me), nor by the difference in steric bulk of the other N substituent (Dipp vs. Mes). This implies that the isolation of both 4- and 5-coordinate Fe complexes across this series might be a result of intermolecular forces during crystallization (e.g. packing effect).
. The other four iron complexes all crystallizes in monoclinic space groups, i.e. P21/n for compounds 2 and 4, and P21/c for compounds 5 and 6. The crystallographic data and collection parameters are presented in Table 2. In general, all six structures showed expected bonding environment and atom connectivity, with two N atoms coordinated to the iron center of a ferrous dichloride unit (Fig. 1). However, to our surprise, two different types of coordination environment for the Fe centers were observed, i.e. four-coordinate (as in complexes 2, 3, 4 and 6) and five-coordinate (as in complexes 1 and 5). The major difference between these two bonding situations is whether a THF molecule is directly coordinated to the iron through its oxygen atom. The four-coordinate compounds showed a distorted tetrahedral geometry with calculated τ4 values in the range of 0.83 to 0.88.46 In contrast, the five-coordinate complexes demonstrated a more distorted trigonal bipyramidal geometry (τ5 = 0.61–0.65),47 with the equatorial positions occupied by two chlorine atoms and one nitrogen atom, while the axial positions occupied by the other nitrogen atom and the oxygen atom of coordinated THF molecule. The fact that some of this series of Fe compounds could adopt a five-coordinate environment indicates that in this case the effective steric bulk of the Ar* substituent is not as great as originally assumed. This is further supported by the observation that in the structure of 1, the THF molecule coordinates to Fe from the same side as the Ar* group, while in the structure of 5, the coordinated THF molecule lies on the same side of the Mes group. This implies that within the BIAN ligand scaffold, the steric bulk follows the sequence of Dipp > Ar* > Mes. It is reasonable to deduce that using Ar* groups on both coordinating N atoms of the BIAN ligands would enforce a much more hindered coordination environment and ensure low-coordination at the iron center. However, such symmetric doubly Ar*-substituted BIAN ligands could not be prepared using conventional acid-catalyzed condensation methods, in accordance with previous literature reports.39 It is also noteworthy that for this series of compounds, whether or not to have an additional coordinated THF in the structures does not seem to be affected by the electronic property of remote ligand substituents (X = F, Cl or Me), nor by the difference in steric bulk of the other N substituent (Dipp vs. Mes). This implies that the isolation of both 4- and 5-coordinate Fe complexes across this series might be a result of intermolecular forces during crystallization (e.g. packing effect).
| 1(THF)·2THF | 2·THF | 3 | 4 | 5·(THF) | (6)2·THF | |
|---|---|---|---|---|---|---|
| a R1 = ∑||Fo| − |Fc||/∑|Fo|.b wR2 = [∑[w(Fo2 − Fc2)2]/∑[w(Fo2)2]]1/2. | ||||||
| Formula | C68H71Cl2FFeN2O3 | C60H55Cl3FeN2O | C57H50Cl2FeN2 | C53H41Cl2FFeN2 | C57H49Cl3FeN2O | C112H96Cl4Fe2N4O |
| fw | 1110.01 | 982.26 | 889.74 | 851.63 | 940.18 | 1767.42 |
| Space group | P212121 | P21/n | P |
P21/n | P21/c | P21/c |
| a (Å) | 10.38300(14) | 10.51517(14) | 10.5987(3) | 13.87969(13) | 19.01587(18) | 19.7651(4) |
| b (Å) | 21.4869(4) | 18.6075(3) | 12.0841(3) | 16.89054(15) | 14.80189(15) | 14.8041(3) |
| c (Å) | 25.4145(3) | 26.0044(3) | 18.4864(5) | 18.35276(16) | 16.80964(17) | 34.0600(7) |
| α (deg) | 90 | 90 | 98.172(2) | 90 | 90 | 90 |
| β (deg) | 90 | 96.4438(11) | 99.125(2) | 103.3046(9) | 103.5203(10) | 102.115(2) |
| γ (deg) | 90 | 90 | 99.657(2) | 90 | 90 | 90 |
| V (Å3) | 5669.94(14) | 5055.91(12) | 2269.69(10) | 4187.06(7) | 4600.30(8) | 9744.1(3) |
| Z | 4 | 4 | 2 | 4 | 4 | 4 |
| T (K) | 93(2) | 93(2) | 150(10) | 150(10) | 150(10) | 150(10) |
| dcalcd (g cm−3) | 1.300 | 1.290 | 1.302 | 1.351 | 1.357 | 1.205 |
| μ (mm−1) | 3.414 | 4.177 | 4.052 | 4.406 | 4.567 | 0.458 |
| R1a | 0.0361 | 0.0439 | 0.0394 | 0.0396 | 0.0771 | 0.0646 |
| wR2b | 0.0972 | 0.1284 | 0.1041 | 0.1071 | 0.2012 | 0.1803 |
| Goodness of fit on F2 | 1.026 | 1.124 | 1.023 | 1.030 | 1.084 | 1.033 |
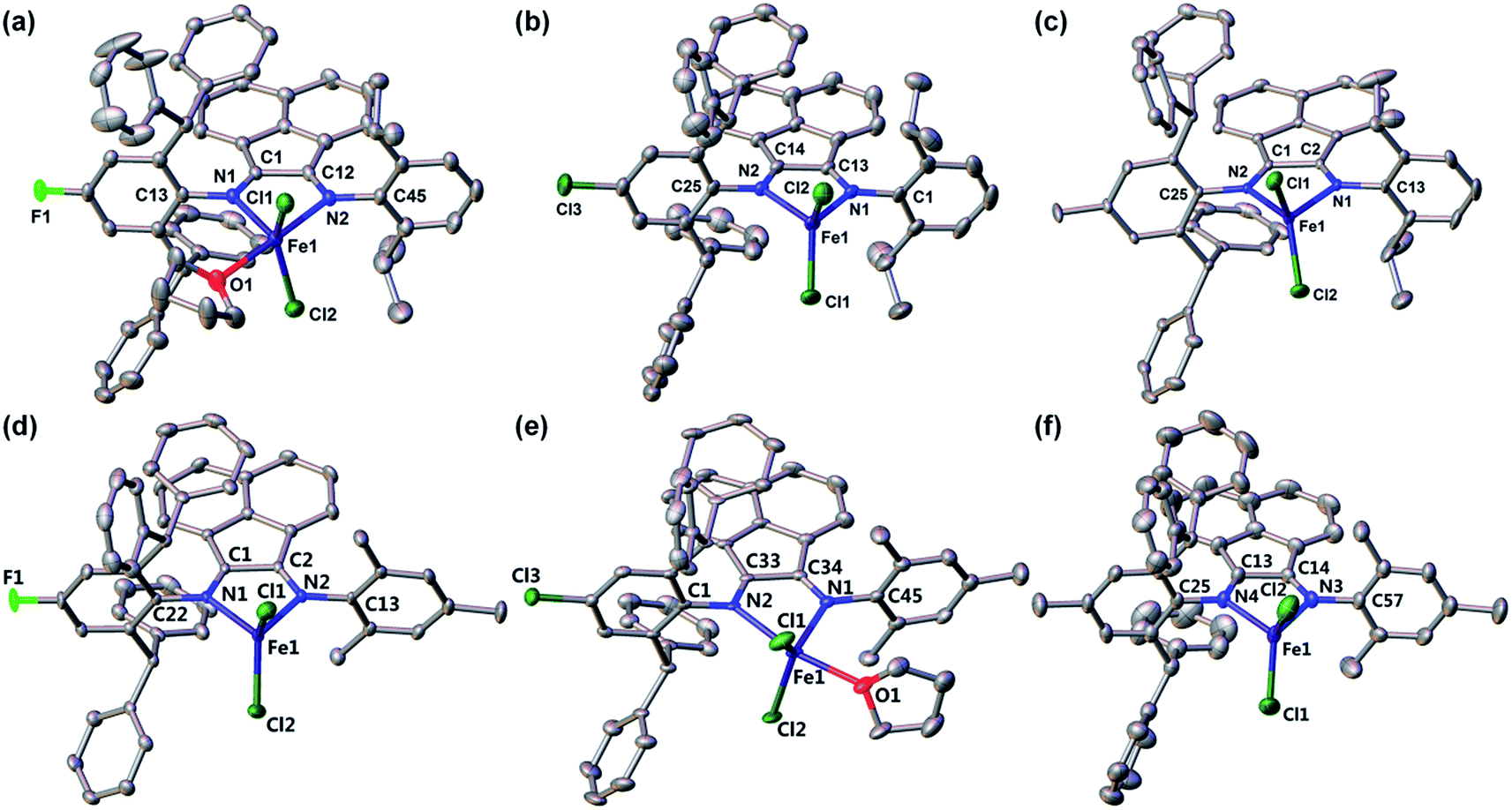 | ||
| Fig. 1 Solid-state structures of complexes 1–6 (from (a) to (f)) with hydrogen atoms omitted for clarity. The thermal ellipsoids are shown at 30% probability level. | ||
Selected bond distances for the solid-state structures of 1–6 are summarized in Table 3. For metal complexes with redox-active α-diimine or BIAN ligands, inspections of the bond lengths of their Nim–Cim–Cim–Nim backbones could establish the oxidation state of the ligands. The Cim–Cim distances in the structures of 1–6 (1.516–1.531 Å) are consistent with the values for typical Csp2–Csp2 single bond. The Cim–Nim bond lengths (1.274–1.289 Å) unambiguously indicate CN double bond character. Thus, the bonding scheme clearly shows that the BIAN ligands remain in their neutral diimine form in this series of Fe compounds. The molecular structures of the 4-coordinate complexes (2, 3, 4 and 6) feature almost identical Fe–Nim bond distances (2.13–2.15 Å). In contrast, one of the Fe–Nim bonds in each 5-coordinate complexes are significantly elongated (2.294(2) Å for 1 and 2.343(2) Å for 5), as a result of the coordination of a THF molecule to the metal center. The THF coordination also leads to slightly longer Fe–Cl bonds in 1 and 5 (2.26–2.29 Å) than those in 2, 3, 4 and 6 (2.21–2.23 Å).
| 1 | 2 | 3 | 4 | 5 | 6 | |
|---|---|---|---|---|---|---|
| a For four-coordinate complexes, τ4 = (360° − α − β)/(360° − 2θ); for five-coordinate complexes, τ5 = (β − α)/60°. α and β are the two largest bond angles around the metal center (α < β) and θ = 109.5°.46,47 | ||||||
| Cim–Cim | 1.521(4) | 1.516(3) | 1.531(2) | 1.522(2) | 1.520(4) | 1.523(4) |
| Cim–Nim | 1.289(4), 1.274(4) | 1.286(3), 1.279(3) | 1.282(2), 1.275(2) | 1.284(2), 1.279(2) | 1.281(4), 1.274(4) | 1.284(4), 1.278(4) |
| Fe–Nim | 2.294(2), 2.144(2) | 2.1455(18), 2.1290(18) | 2.1386(14), 2.1371(15) | 2.1501(15), 2.1367(15) | 2.343(2), 2.160(3) | 2.143(2), 2.131(3) |
| Fe–Cl | 2.2870(10), 2.2769(9) | 2.2179(7), 2.2138(7) | 2.2209(6), 2.2173(6) | 2.2312(6), 2.2227(6) | 2.2864(9), 2.2611(9) | 2.2225(11), 2.2219(11) |
| Fe–OTHF | 2.288(2) | N/A | N/A | N/A | 2.215(2) | N/A |
| τn (n = 4 or 5)a | 0.61 | 0.88 | 0.83 | 0.88 | 0.65 | 0.87 |
The electrochemical behavior of the iron complexes 1–6 was investigated by cyclic voltammetry (CV) as shown in Table 4. All six compounds show similar redox properties and the voltammogram of 2 in dichloromethane was illustrated in Fig. 2 as a representative example. The quasi-reversible redox wave centered at ca. −0.5 V (vs. Ag/AgCl) are tentatively assigned to a two-electron process involving the partial reduction of both Fe(II) center and neutral BIAN ligand. The detection of this 2e reduction process rather than two separate 1e ligand reductions, is consistent with the previously reported chemical reduction and isolation of (BIAN)Fe(η6-C7H8),20 which is a formally 2e reduction product. Furthermore, the half wave potentials (E1/2 = 1/2(Ep(ox) + Ep(red))) for the two fluorinated derivatives 1 (−0.493 V vs. Ag/AgCl) and 4 (−0.480 V vs. Ag/AgCl) are the most positive ones among this series of Fe compounds, in accordance with the strong electron-withdrawing ability of fluorine atoms. Consistently, complexes 3 and 6, whose supporting BIAN ligands contain electron-donating methyl substituents, show the most negative E1/2 values at −0.592 V and −0.616 V (vs. Ag/AgCl), respectively. This demonstrates that subtle modification at the N-aryl group of BIAN skeleton can effectively modulate the redox potential of the corresponding BIAN metal complexes within a considerably wide range (ca. 136 mV). As shown in Fig. 2, another quasi-reversible redox wave at a more positive potential (ca. 0.1 V vs. Ag/AgCl) was observed, which are attributed to the 1e process of Fe(II)/Fe(III) couple. In addition, an irreversible reduction wave could be observed at ca. −1.6 V vs. Ag/AgCl for complexes 1 and 4 (see ESI†), which can be ascribed to a further 2e reduction of the BIAN ligand.
| Ep(ox) | Ep(red) | E1/2 | |
|---|---|---|---|
| a Potentials are reported in volts vs. Ag/AgCl in CH2Cl2. | |||
| 1 | −0.435 | −0.550 | −0.493 |
| 2 | −0.448 | −0.616 | −0.532 |
| 3 | −0.543 | −0.640 | −0.592 |
| 4 | −0.414 | −0.545 | −0.480 |
| 5 | −0.477 | −0.575 | −0.526 |
| 6 | −0.566 | −0.666 | −0.616 |
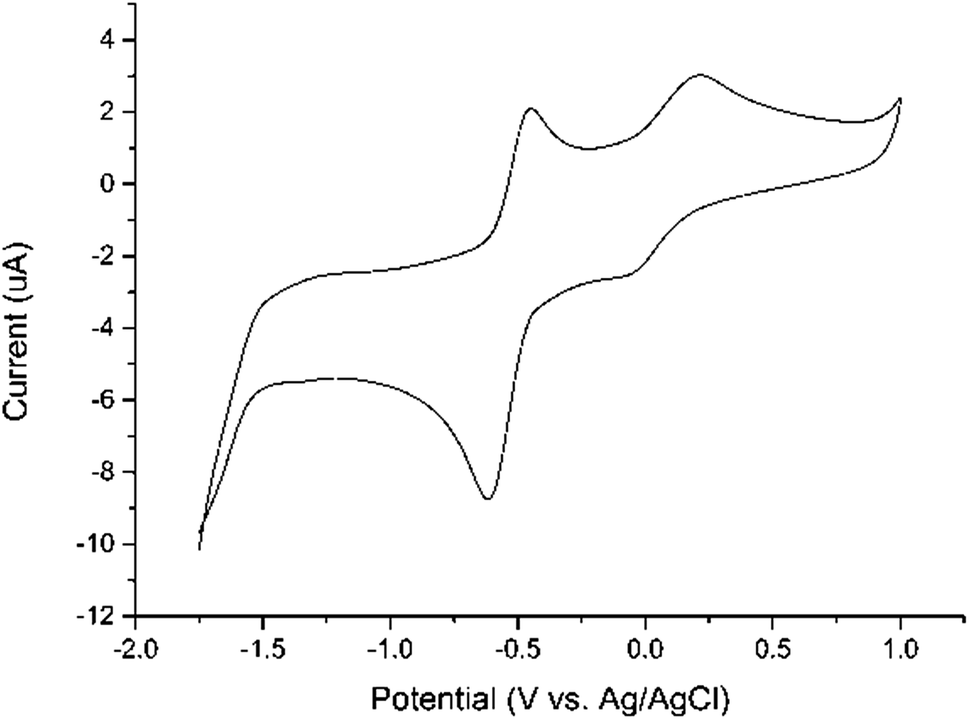 | ||
| Fig. 2 Cyclic voltammogram of 1.0 mM of 2 in CH2Cl2 with 0.10 M nBu4NPF6 as the supporting electrolyte at a scan rate of 100 mV s−1. The potentials are reported relative to Ag/AgCl. | ||
With complexes 1–6 synthesized and fully characterized, we then turned our attention to evaluating the performance of these compounds as precatalysts for room-temperature hydrosilylation of aldehydes and ketones. The initial screening was conducted by the reaction of equimolar of Ph2SiH2 and 4-methylbenzaldehyde, and the results are summarized in Table 5. The active catalysts were generated in situ upon the addition of LiCH2SiMe3 (2 equiv.) to the precursors 1–6. The reaction mixture containing silyl ether products were further converted to the corresponding alcohols by treatment of tetra-n-butylammonium fluoride (TBAF) solution. Such precatalyst activation strategy and product workup process have been employed in similar catalytic systems.21 The results were analyzed by 1H NMR spectroscopy using 1,3,5-trimethoxybenzene as an internal standard. As can be seen in Table 5, all six BIAN-supported ferrous complexes can serve as efficient precatalysts for the room-temperature hydrosilylation reaction. The difference in reactivity of 1–6 in the test reaction seems to be not very significant, judged by the yield of alcohol products, with compound 6 showing most promising performance. This implies that the differences in electronic property and steric bulk for 1–6 do not affect their catalytic reactivity very much, which is in sharp contrast to a previously reported series of palladium diimine complexes for catalytic ethylene polymerization.48 Thus, further optimization of catalytic reaction conditions and result discussions will be focused on complex 6.
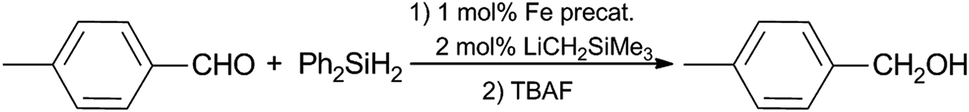
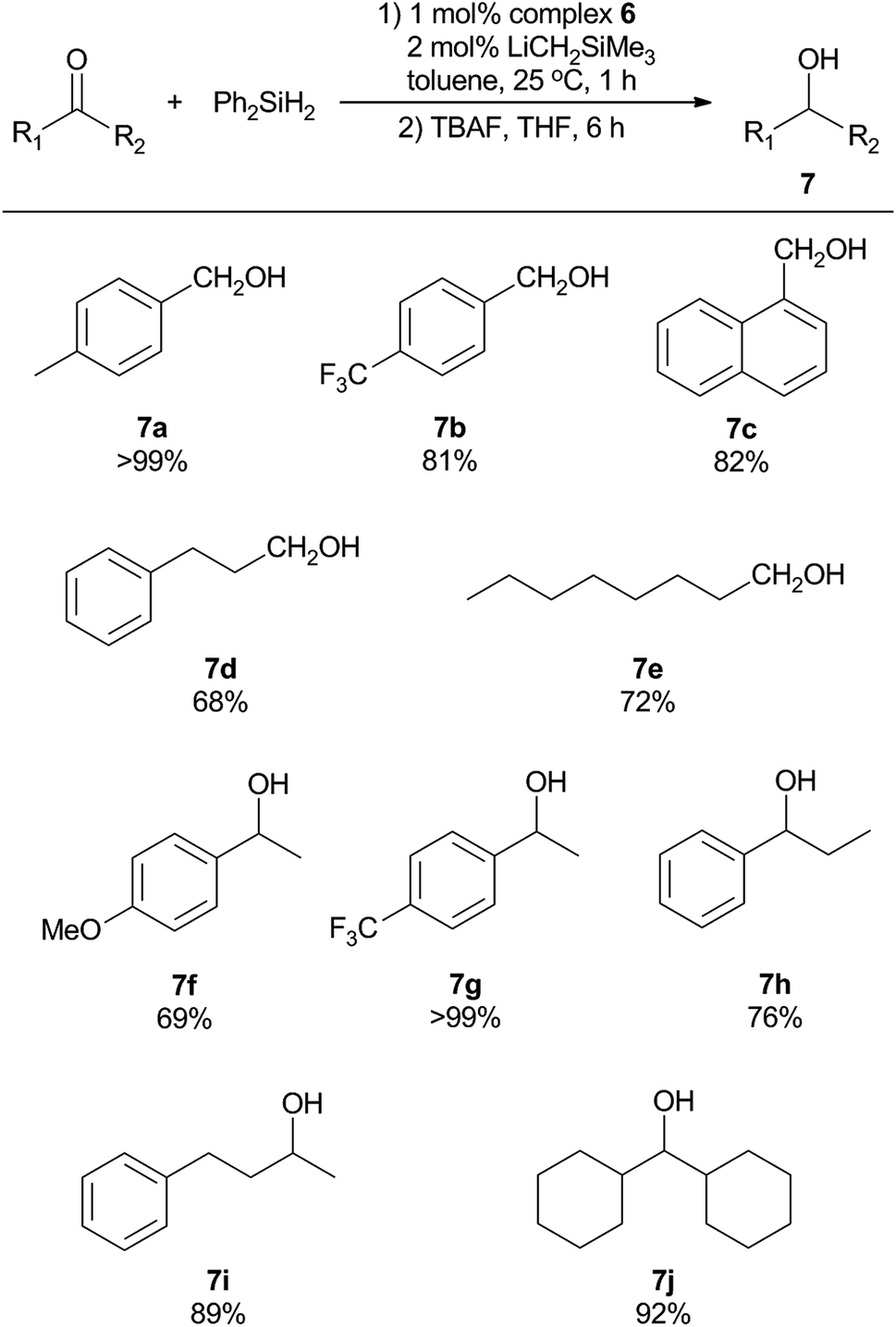
Further screening of the reaction was performed by variation of the type and amount of silane used, as well as the reaction time (Table 6). Employment of alternative silanes resulted in lower yields of alcohols in comparison to that with Ph2SiH2 (entry 1–4). In addition, increasing the amount of Ph2SiH2 to 1.5 or 2.0 equivalent led to lower yields of the product (entry 5 and 6). The reaction time was also optimized using conditions listed in Table 6 (entry 7–9), which demonstrates that the catalytic reaction reaches completion by 1 hour and the determined yield of alcohol product further decreases as the reaction time is extended. On this basis, we opted to use 1.0 equiv. of Ph2SiH2 and 1 hour reaction time as our so-far optimized condition for further exploration of reaction scope for other functionalized aldehydes and ketones. It was found that a number of substrates could be successfully accommodated, as summarized in Scheme 2. para-Substituted benzaldehydes bearing either electron-donating or electron-withdrawing substituents were successfully converted to the corresponding primary alcohols (7a and 7b). In addition, sterically bulkier aromatic aldehyde was reduced in good yield (7c). In contrast, the transformation of aliphatic aldehydes into the primary alcohol counterparts (7d and 7e) was not as efficient using the same catalytic protocol. The production of secondary alcohols using various ketones was also investigated. Substituted acetophenone derivatives were reduced to the related alcohols in moderate to good yields (7f and 7g), demonstrating that an electron-withdrawing group is more favorable for the reaction. Propiophenone was converted to the corresponding secondary alcohol 7h in a lower yield, possibly due to the increased steric bulk of the alkyl group. Unlike the aldehyde counterparts, structurally diverse dialkyl ketones were suitable substrates for this catalytic system, resulting in high yields of dialkyl alcohols (7i and 7j). Thus, this preliminary catalytic reactivity investigation showed that the sterically hindered asymmetric (BIAN)-FeCl2 complexes are versatile precatalysts for room-temperature hydrosilylation of aldehydes and ketones. The detailed mechanism of this catalytic system is not fully elucidated yet. Other than the more obvious Fe-mediated process, a recent study showed that BIAN ligands could be dearomatized by reactive s-block alkyls such as LiCH2SiMe3, and the resulting species are catalytically active towards hydroamination reactions.49
|
||||
|---|---|---|---|---|
| Entry | Silane | n | Time (h) | Yieldb (%) |
| a Reaction conditions: (1) 4-methylbenzaldehyde (1.0 mmol), silane (n mmol), 1 mol% complex 6, 2 mol% LiCH2SiMe3, toluene, 25 °C; (2) TBAF, THF, 6 h.b Yields were determined by 1H NMR with an internal standard. | ||||
| 1 | PhSiH3 | 1.0 | 1.0 | 72 |
| 2 | Ph3SiH | 1.0 | 1.0 | 70 |
| 3 | Et3SiH | 1.0 | 1.0 | 45 |
| 4 | (EtO)3SiH | 1.0 | 1.0 | 68 |
| 5 | Ph2SiH2 | 1.5 | 1.0 | 73 |
| 6 | Ph2SiH2 | 2.0 | 1.0 | 60 |
| 7 | Ph2SiH2 | 1.0 | 0.5 | 71 |
| 8 | Ph2SiH2 | 1.0 | 3.0 | 95 |
| 9 | Ph2SiH2 | 1.0 | 12.0 | 74 |
 | ||
| Scheme 2 Hydrosilylation of functionalized aldehydes and ketones by complex 6. | ||
Conclusions
In summary, we have prepared a series of iron complexes ligated by asymmetric bulky BIAN ligands. The solid-state structures of these complexes were determined by X-ray crystallography, showing either 4- or 5-coordinate iron centers. These compounds represent very rare examples of structurally characterized iron complexes with asymmetric bidentate BIAN ligands. The structural parameters of the BIAN ligand backbones reveal that the ligands remain in their neutral form in these complexes. The compounds were also characterized by 1H NMR, IR, UV-vis spectroscopy and cyclic voltammetry. The electrochemical measurements showed that this series of compounds undergo quasi-reversible 2-electron reduction of the complexes, rather than two successive 1e reduction on the ligand. Such synergistic reactivity of both ligand and metal center makes them possible precatalysts for 2e reduction of carbonyl compounds. Preliminary catalytic performance screening found that these compounds exhibited good catalytic reactivity for the room-temperature hydrosilylation of various aldehydes and ketones. Mechanistic investigation and further improvement of this catalytic system via ligand modification are ongoing and will be reported in due course.Experimental
Materials and methods
All reactions were performed under a nitrogen atmosphere with rigorous exclusion of oxygen and water using standard Schlenk or glovebox techniques. All solvents were dried and purified using an Innovative Technology PureSolv MD5 solvent purification system. Ligands Ar-BIANX were prepared following literature procedures.42,44,45 All other reagents were obtained commercially and used as received. UV-vis spectra were recorded on a Shimadzu UV-3600 UV-vis-NIR spectrophotometer in THF solution. Infrared spectra were measured as Nujol mulls between KBr plates on a Nicolet 6700 spectrometer. 1H NMR (300 MHz) spectra were collected using a Bruker Avance III 300 spectrometer and referenced internally to the residual solvent resonances. Cyclic voltammograms were obtained using a CH Instruments CHI-660D electrochemical analyzer in 0.10 M CH2Cl2 solution of nBu4NPF6, with Pt working and auxiliary electrodes, Ag/AgCl reference electrode, and a scan rate of 100 mV s−1. Melting points were measured on a Mel-Temp II apparatus and uncorrected, using glass capillaries sealed under inert atmosphere. Elemental analyses were performed by an Elementar Vario EL elemental analyzer.Synthesis of (Dipp-BIANF)FeCl2 (1)
To a stirred THF suspension of anhydrous FeCl2 (0.412 g, 3.25 mmol), a THF solution of Dipp-BIANF ligand (3.009 g, 3.92 mmol) was added dropwise at ambient temperature. The solution gradually turned to dark reddish brown and the resulting mixture was stirred overnight. Solvents were partially removed under reduced pressure, and the precipitated product was isolated by decantation, washed with toluene, and dried under vacuum. Yield: 2.72 g (94%). Mp: >320 °C. 1H NMR (300 MHz, CDCl3, 23 °C, δ in ppm): 12.62, 9.38, 8.14, 6.36, 5.79, 3.82, 2.91, 1.95, 1.34, −0.84, −3.27, −4.70, −15.62. UV-vis, λmax in nm (ε, M−1 cm−1): 316 (7.8 × 103), 425 (sh). IR (Nujol, ν(CN) in cm−1): 1619, 1582. Anal calcd for C56H47Cl2FFeN2 (found): C, 75.26 (75.09); H, 5.30 (5.43); N, 3.13 (2.86).
Synthesis of (Dipp-BIANCl)FeCl2 (2)
To a stirred THF suspension of anhydrous FeCl2 (0.444 g, 3.50 mmol), a THF solution of Dipp-BIANCl ligand (2.623 g, 3.35 mmol) was added dropwise at ambient temperature. The solution gradually turned to dark yellowish brown and the resulting mixture was stirred overnight. The suspension was filtered and all volatiles were removed under reduced pressure. The yellowish brown solid product was washed with toluene and dried under vacuum. Yield: 2.44 g (80%). Mp: >320 °C. 1H NMR (300 MHz, CDCl3, 23 °C, δ in ppm): 12.51, 9.32, 8.17, 6.41, 5.64, 3.82, 2.87, 1.91, 1.28, −0.70, −3.37, −4.78, −5.81, −15.50. UV-vis, λmax in nm (ε, M−1 cm−1): 313 (8.5 × 103), 425 (sh). IR (Nujol, ν(CN) in cm−1): 1621, 1584. Anal calcd for C56H47Cl3FeN2 (found): C, 73.90 (73.61); H, 5.20 (5.39); N, 3.08 (3.38).
Synthesis of (Dipp-BIANMe)FeCl2 (3)
To a stirred THF suspension of anhydrous FeCl2 (0.124 g, 0.98 mmol), a THF solution of Dipp-BIANMe ligand (0.730 g, 0.96 mmol) was added dropwise at ambient temperature. The solution gradually turned to dark brown and the resulting mixture was stirred overnight. The suspension was filtered and all volatiles were removed under reduced pressure. The greenish yellow solid product was washed with toluene and dried under vacuum. Yield: 0.854 g (85%). Mp: >320 °C. 1H NMR (300 MHz, CDCl3, 23 °C, δ in ppm): 16.45, 12.98, 12.64, 9.30, 8.16, 6.49, 6.32, 5.82, 5.07, 3.78, 2.96, 2.39, 1.88, 1.28, −1.02, −2.97, −5.14, −15.74. UV-vis, λmax in nm (ε, M−1 cm−1): 312 (7.0 × 103), 422 (sh). IR (Nujol, ν(CN) in cm−1): 1622, 1585. Anal calcd for C57H50Cl2FeN2 (found): C, 76.94 (75.54); H, 5.66 (5.89); N, 3.15 (3.02).
Synthesis of (Mes-BIANF)FeCl2 (4)
To a stirred THF suspension of anhydrous FeCl2 (0.210 g, 1.65 mmol), a THF solution of Mes-BIANF ligand (1.171 g, 1.61 mmol) was added dropwise at ambient temperature. The solution gradually turned to dark brown and the resulting mixture was stirred overnight. Solvents were partially removed under reduced pressure, and the precipitated yellowish brown product was isolated by decantation, washed with toluene, and dried under vacuum. Yield: 0.923 g (62%). Mp: >320 °C. 1H NMR (300 MHz, CDCl3, 23 °C, δ in ppm): 15.71, 14.74, 13.25, 12.52, 9.31, 8.25, 6.41, 6.23, 5.07, 3.79, 2.39, 1.88, 1.32, −3.60, −5.89. UV-vis, λmax in nm (ε, M−1 cm−1): 323 (9.0 × 103), 418 (sh). IR (Nujol, ν(CN) in cm−1): 1619, 1587. Anal calcd for C53H41Cl2FFeN2 (found): C, 74.74 (75.69); H, 4.85 (5.00); N, 3.29 (2.83).
Synthesis of (Mes-BIANCl)FeCl2 (5)
To a stirred THF suspension of anhydrous FeCl2 (0.221 g, 1.74 mmol), a THF solution of Mes-BIANCl ligand (1.283 g, 1.73 mmol) was added dropwise at ambient temperature. The solution gradually turned to dark brown and the resulting mixture was stirred overnight. The solution was concentrated under reduced pressure and 50 mL of hexanes was added. The precipitated greenish yellow product was isolated by decantation and dried under vacuum. Yield: 1.42 g (87%). Mp: >320 °C. 1H NMR (300 MHz, CDCl3, 23 °C, δ in ppm): 15.77, 14.67, 13.19, 12.48, 9.31, 8.24, 6.73, 6.47, 6.29, 5.07, 3.83, 2.34, 1.91, −3.70, −6.03. UV-vis, λmax in nm (ε, M−1 cm−1): 322 (8.2 × 103), 418 (sh). IR (Nujol, ν(CN) in cm−1): 1623, 1588. Anal calcd for C53H41Cl3FeN2 (found): C, 73.33 (72.71); H, 4.76 (4.68); N, 3.23 (2.92).
Synthesis of (Mes-BIANMe)FeCl2 (6)
To a stirred THF suspension of anhydrous FeCl2 (0.343 g, 2.71 mmol), a THF solution of Mes-BIANMe ligand (1.918 g, 2.67 mmol) was added dropwise at ambient temperature. The solution gradually turned to dark reddish brown and the resulting mixture was stirred overnight. The suspension was filtered and all volatiles were removed under reduced pressure. The dark brown solid product was washed with toluene and dried under vacuum. Yield: 1.972 g (80%). Mp: >320 °C. 1H NMR (300 MHz, CDCl3, 23 °C, δ in ppm): 16.81, 15.82, 14.89, 13.39, 12.60, 9.35, 8.28, 6.25, 5.54, 5.27, 2.40, 1.30, −3.26, −4.08, −5.39. UV-vis, λmax in nm (ε, M−1 cm−1): 324 (8.1 × 103), 422 (sh). IR (Nujol, ν(CN) in cm−1): 1621, 1586. Anal calcd for C54H44Cl2FeN2 (found): C, 76.51 (76.64); H, 5.23 (5.09); N, 3.33 (2.98).
X-ray crystallography
Single crystals of 1–6 suitable for X-ray crystallography were grown by vapor diffusion of hexanes into a THF solution of the compound. Data for 1 and 2 were collected on a Rigaku XtaLAB P300DS diffractometer with Cu Kα radiation (λ = 1.54178 Å) at 93 K, data for 3–5 were collected on a Rigaku Supernova diffractometer with Cu Kα radiation (λ = 1.54178 Å) at 150 K, and data for 6 were collected on a Rigaku Supernova diffractometer with Mo Kα radiation (λ = 0.71073 Å) at 150 K. The empirical absorption corrections were applied using spherical harmonics, implemented in the SCALE3 ABSPACK scaling algorithm.50 All the structures were solved using direct methods, which yielded the positions of all non-hydrogen atoms. Hydrogen atoms were placed in calculated positions in the final structure refinement. Structure determination and refinement were carried out using SHELXS and SHELXL programs, respectively.51 The PLATON SQUEEZE treatment was applied to all guest molecules in the structure of 6 because the guest molecules are extremely disordered and cannot be modeled. CCDC-1530840 (1), 1530844 (2), 1530839 (3), 1530842 (4), 1530841 (5), 1530843 (6) contain the supplementary crystallographic data for this paper.†Typical procedure for catalytic hydrosilylation of aldehydes and ketones
In a glove box, a 20 mL vial containing a stir bar was charged with complex 6 (8.5 mg, 1 mol%) and cooled to −35 °C. A toluene solution of LiCH2SiMe3 (0.01 M, 2.0 mL, 2 mol%) was then added to the vial, and the reaction mixture was allowed to warm up under stirring for 0.5 h. Subsequently, substrate (1.0 mmol) and Ph2SiH2 (186 μL, 1.0 mmol) were added. The resulting mixture was stirred for 1 h at ambient temperature. After that, the vial was removed from the glovebox, and the reaction mixture was treated with 2 mL of THF and 1 mL of 1.0 M THF solution of tetra-n-butylammonium fluoride. After the mixture was stirred for 6 h, all volatiles were removed under vacuum and the residue was extracted with CH2Cl2. The organic phase was separated, dried with MgSO4, filtered and concentrated. The obtained residue was analyzed by 1H NMR method using 1,3,5-trimethoxybenzene as an internal standard.Acknowledgements
We gratefully acknowledge financial support from the National Natural Science Foundation of China (No. 21301070), Pearl River S&T Nova Program of Guangzhou (No. 201610010118), the Fundamental Research Funds for the Central Universities (No. 21615401), Natural Science Foundation of Guangdong Province (No. 2016A030313093), China Postdoctoral Science Foundation (No. 2016M592598) and Jinan University.Notes and references
- W. Kaim, Inorg. Chem., 2011, 50, 9752–9765 CrossRef CAS PubMed.
- W. Kaim, Eur. J. Inorg. Chem., 2012, 343–348 CrossRef CAS.
- V. K. K. Praneeth, M. R. Ringenberg and T. R. Ward, Angew. Chem., Int. Ed., 2012, 51, 10228–10234 CrossRef CAS PubMed.
- O. R. Luca and R. H. Crabtree, Chem. Soc. Rev., 2013, 42, 1440–1459 RSC.
- D. L. J. Broere, R. Plessius and J. I. van der Vlugt, Chem. Soc. Rev., 2015, 44, 6886–6915 RSC.
- V. Lyaskovskyy and B. de Bruin, ACS Catal., 2012, 2, 270–279 CrossRef CAS.
- L. A. Berben, Chem.–Eur. J., 2015, 21, 2734–2742 CrossRef CAS PubMed.
- G. V. Koten and K. Vrieze, Adv. Organomet. Chem., 1982, 21, 151–239 CrossRef.
- A. Raghavan and A. Venugopal, J. Coord. Chem., 2014, 67, 2530–2549 CrossRef CAS.
- N. J. Hill, I. Vargas-Baca and A. H. Cowley, Dalton Trans., 2009, 240–253 RSC.
- C. J. Allan, B. F. T. Cooper, H. J. Cowley, J. M. Rawson and C. L. B. Macdonald, Chem.–Eur. J., 2013, 19, 14470–14483 CrossRef CAS PubMed.
- A. L. Brazeau, N. D. Jones and P. J. Ragogna, Dalton Trans., 2012, 41, 7890–7896 RSC.
- N. J. Hill, G. Reeske, J. A. Moore and A. H. Cowley, Dalton Trans., 2006, 4838–4844 RSC.
- J. Wang, R. Ganguly, L. Yongxin, J. Díaz, H. S. Soo and F. García, Dalton Trans., 2016, 45, 7941–7946 RSC.
- R. A. Zarkesh, A. S. Ichimura, T. C. Monson, N. C. Tomson and M. R. Anstey, Dalton Trans., 2016, 45, 9962–9969 RSC.
- K. M. Clark, Inorg. Chem., 2016, 55, 6443–6448 CrossRef CAS PubMed.
- J. Bendix and K. M. Clark, Angew. Chem., Int. Ed., 2016, 55, 2748–2752 CrossRef CAS PubMed.
- B. A. Schaefer, G. W. Margulieux, B. L. Small and P. J. Chirik, Organometallics, 2015, 34, 1307–1320 CrossRef CAS.
- B. M. Schmiege, M. J. Carney, B. L. Small, D. L. Gerlach and J. A. Halfen, Dalton Trans., 2007, 2547–2562 RSC.
- F. S. Wekesa, R. Arias-Ugarte, L. Kong, Z. Sumner, G. P. McGovern and M. Findlater, Organometallics, 2015, 34, 5051–5056 CrossRef CAS.
- M. J. Supej, A. Volkov, L. Darko, R. A. West, J. M. Darmon, C. E. Schulz, K. A. Wheeler and H. M. Hoyt, Polyhedron, 2016, 114, 403–414 CrossRef CAS.
- I. L. Fedushkin, A. A. Skatova, N. M. Khvoinova, A. N. Lukoyanov, G. K. Fukin, S. Y. Ketkov, M. O. Maslov, A. S. Bogomyakov and V. M. Makarov, Russ. Chem. Bull., 2013, 62, 2122–2131 CrossRef CAS.
- D. A. Piryazev, M. A. Ogienko, A. V. Virovets, N. A. Pushkarevsky and S. N. Konchenko, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2012, 68, m320–m322 CAS.
- B. Marciniec, Hydrosilylation: A Comprehensive Review on Recent Advances, Springer Science, Berlin, 2009 Search PubMed.
- K. Junge, K. Schröder and M. Beller, Chem. Commun., 2011, 47, 4849–4859 RSC.
- S. Enthaler, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2008, 47, 3317–3321 CrossRef CAS PubMed.
- B. Su, Z.-C. Cao and Z.-J. Shi, Acc. Chem. Res., 2015, 48, 886–896 CrossRef CAS PubMed.
- I. Bauer and H.-J. Knölker, Chem. Rev., 2015, 115, 3170–3387 CrossRef CAS PubMed.
- N. S. Shaikh, K. Junge and M. Beller, Org. Lett., 2007, 9, 5429–5432 CrossRef CAS PubMed.
- H. Nishiyama and A. Furuta, Chem. Commun., 2007, 760–762 RSC.
- P. Bhattacharya, J. A. Krause and H. Guan, Organometallics, 2011, 30, 4720–4729 CrossRef CAS.
- A. M. Tondreau, E. Lobkovsky and P. J. Chirik, Org. Lett., 2008, 10, 2789–2792 CrossRef CAS PubMed.
- A. M. Tondreau, J. M. Darmon, B. M. Wile, S. K. Floyd, E. Lobkovsky and P. J. Chirik, Organometallics, 2009, 28, 3928–3940 CrossRef CAS.
- J. Yang and T. D. Tilley, Angew. Chem., Int. Ed., 2010, 49, 10186–10188 CrossRef CAS PubMed.
- T. Bleith, H. Wadepohl and L. H. Gade, J. Am. Chem. Soc., 2015, 137, 2456–2459 CrossRef CAS PubMed.
- T. Bleith and L. H. Gade, J. Am. Chem. Soc., 2016, 138, 4972–4983 CrossRef CAS PubMed.
- Á. Raya-Barón, M. A. Ortuño, P. Oña-Burgos, A. Rodríguez-Diéguez, R. Langer, C. J. Cramer, I. Kuzu and I. Fernández, Organometallics, 2016, 35, 4083–4089 CrossRef.
- A. J. Ruddy, C. M. Kelly, S. M. Crawford, C. A. Wheaton, O. L. Sydora, B. L. Small, M. Stradiotto and L. Turculet, Organometallics, 2013, 32, 5581–5588 CrossRef CAS.
- J. L. Rhinehart, N. E. Mitchell and B. K. Long, ACS Catal., 2014, 4, 2501–2504 CrossRef CAS.
- S. Du, S. Kong, Q. Shi, J. Mao, C. Guo, J. Yi, T. Liang and W.-H. Sun, Organometallics, 2015, 34, 582–590 CrossRef CAS.
- X. Wang, L. Fan, Y. Yuan, S. Du, Y. Sun, G. A. Solan, C.-Y. Guo and W.-H. Sun, Dalton Trans., 2016, 45, 18313–18323 RSC.
- H. Liu, W. Zhao, X. Hao, C. Redshaw, W. Huang and W.-H. Sun, Organometallics, 2011, 30, 2418–2424 CrossRef CAS.
- Q. Ban, J. Zhang, T. Liang, C. Redshaw and W.-H. Sun, J. Organomet. Chem., 2012, 713, 151–156 CrossRef CAS.
- S. Kong, C.-Y. Guo, W. Yang, L. Wang, W.-H. Sun and R. Glaser, J. Organomet. Chem., 2013, 725, 37–45 CrossRef CAS.
- L. Fan, S. Du, C.-Y. Guo, X. Hao and W.-H. Sun, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 1369–1378 CrossRef CAS.
- L. Yang, D. R. Powell and R. P. Houser, Dalton Trans., 2007, 955–964 RSC.
- A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 RSC.
- S. Dai, X. Sui and C. Chen, Angew. Chem., Int. Ed., 2015, 54, 9948–9953 CrossRef CAS PubMed.
- M. Arrowsmith, M. S. Hill and G. Kociok-Köhn, Organometallics, 2014, 33, 206–216 CrossRef CAS.
- CrysAlis Pro, Version 1.171.38.43, Rigaku OD, 2015 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: NMR spectra, UV-vis spectra, cyclic voltammograms and CIFs for compounds 1–6. CCDC 1530839–1530844. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra01511a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |