
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
High ion-conducting solid polymer electrolytes based on blending hybrids derived from monoamine and diamine polyethers for lithium solid-state batteries†
Ta-Ming Liu,
Diganta Saikia ,
Sze-Yuan Ho,
Ming-Chou Chen* and
Hsien-Ming Kao*
,
Sze-Yuan Ho,
Ming-Chou Chen* and
Hsien-Ming Kao*
Department of Chemistry, National Central University, Chung-Li, 32054, Taiwan, Republic of China. E-mail: hmkao@cc.ncu.edu.tw; mcchen@ncu.edu.tw; Fax: +886-3-4227664; Tel: +886-3-4275054
First published on 7th April 2017
Abstract
In this study, polyetheramine and polyetherdiamine were cross-linked separately with (3-glycidyloxypropyl)trimethoxysilane (GLYMO) and poly(ethylene glycol) diglycidyl ether (PEGDGE) and then blended in different ratios to obtain blended hybrid solid polymer electrolytes (SPEs). The blend network architecture improved the LiClO4 salt dissociation, which led to a higher ionic conductivity of the SPEs (4.5 × 10−4 S cm−1 at 60 °C and 1.2 × 10−4 S cm−1 at 30 °C for the [O]/[Li] ratio of 16). When the salt concentration was increased, the phase of the SPEs changed from semi-crystalline to an amorphous state, as revealed by differential scanning calorimetry. Complexation with different constituents of the SPEs was evidenced from the Fourier transform infrared measurements, while 13C and 29Si solid-state NMR spectroscopy provided information about the successful formation of the blend organic–inorganic hybrid with a silicate architecture, and 7Li NMR static linewidth measurements gave an insight into the dynamic behaviour of the lithium ions inside the SPEs. The blend hybrid SPEs demonstrated an electrochemical stability window of over 5 V. The electrochemical performance of the blend hybrid SPE assembled in a battery with lithium as the anode and LiFePO4 as the cathode exhibited a good cycle life of over 100 cycles, with an initial discharge capacity of 110 mA h g−1 and a coulombic efficiency of over 99%. With these good characteristic properties, the present blend hybrid SPE has the potential to be used in lithium solid-state batteries and other electrochemical devices.
Introduction
With the technological prowess and advent of new electronic devices, the need for efficient energy storage devices is increasing urgently. Meanwhile, environmental concerns related to global warming and rising sea levels expedite the requirement for the use of renewable energy resources and effective storage systems. Among the storage devices, lithium-ion batteries have more market share, from consumer electronics to electric/hybrid electric vehicles.1,2 However, safety issues, such as the flammability of organic liquid electrolytes, poor thermal stability, electrolyte leakage and the formation of lithium dendrites that may cause internal short circuits, are the major problems associated with these Li-ion batteries.3 Thus, finding a suitable electrolyte with higher ionic conductivity and free from these drawbacks still remains a challenge. Over the last few decades, solid electrolytes, such as ceramics and polymer electrolytes, have been investigated for applications in Li-ion batteries as possible replacements for liquid electrolytes.4–6 In particular, the advantages, such as flame resistance, leakage proof, flexible and versatile, easily processable and cost-effectiveness, associated with solid polymer electrolytes (SPEs) have made them become the electrolytes of choice.7–9 Work on SPEs was started after Wright's group's report on solid-state electrolytes with a Li-ion doped poly(ethylene oxide) (PEO) system10 and when Armand recognized its technological importance in lithium batteries.11 However, it was soon recognized that the main drawback of SPEs was their low ionic conductivities at ambient temperature.12 In order to address this key issue, a number of strategies and/or modification procedures have been designed and employed to improve the ionic conductivity of SPEs. For example, various polymeric matrices with high dielectric constants, such as PEO, poly(propylene)oxide (PPO), poly(methyl methacrylate) (PMMA), polyacrylonitrile (PAN), poly(vinylidene fluoride) (PVdF) and their copolymers, have been tested by doping with different lithium salts.13–17 In view of the good solvation property of PEO with lithium salts, PEO-based SPEs are the most widely investigated system. Still, not much improvement in ionic conductivity values for PEO-based SPEs has yet been achieved. Another important modification of SPEs is the addition of ceramic fillers to improve the ionic conductivity and mechanical stability. Fillers like TiO2, Al2O3, SiO2, ZrO2, etc. have been successfully added to different polymeric matrices to give an enhancement in their ionic conductivity values.18–21 It is believed that the presence of fillers in SPEs helps to reduce the degree of crystallinity of polymers and polymer-filler grain boundaries, which generate conducting pathways for the rapid movement of ions, and thus results in an improvement in ionic conductivity. However, particle aggregation still remains a concern for the use of these electrolytes in lithium solid-state batteries. Alternatively, hybrid SPEs of organic and inorganic components have recently received much attention due to their higher ionic conductivities as compared to PEO-based SPEs and due to their flexibility in design.22–24 As the presence of organic and inorganic groups in the SPEs can be controlled in a desirable way, according to the requirements and structural design, these hybrid SPEs can hold unique chemical and physical properties that are different from the parent ones. Furthermore, the use of the sol–gel synthesis technique makes it easier to regulate the functionalization level of the material as well as the connectivity between interfaces. Alternately, an easy and convenient approach to synthesize SPEs with high ionic conductivity and mechanical stability is by polymer blending. SPEs based on polymer blending are simple to be processed and their physical properties can be easily adjusted when one part is softer and the other part is comparatively harder.25–27 Normally, intermolecular interactions, such as hydrogen bonding, ionic and dipole–dipole, play a role in the physico-chemical and electrochemical behaviours of the blended polymer systems.In this work, the blending of two organic–inorganic hybrids to form a new type of SPE was developed. The first hybrid was formed by cross-linking (3-glycidyloxypropyl)trimethoxysilane (GLYMO), an organosilane, with a monoamine-based polyether, while the second hybrid was prepared based on the reactions of GLYMO and poly(ethylene glycol) diglycidyl ether (PEGDGE) with a diamine-based polyether. These two hybrids were then blended together to obtain the final blended hybrid system. The present type of the hybrids has the advantage to offer the formation of in situ sub-micron silica particles, which gives a composite nature to the membrane for use. By varying the weight percentages of the blending parts and the [O]/[Li] ratios with LiClO4 salt, a series of blended hybrid SPEs with the optimum conductivity value and mechanical property can be obtained. Characterization of the blended SPEs was systematically performed to investigate their physical and electrochemical behaviours. DSC/TGA and FTIR analyses gave insights into the phase behaviour, thermal stability and interactions among the constituents of the blended system. Furthermore, multinuclear solid-state NMR spectroscopy (13C, 29Si, 7Li) was utilized as it is recognized to be a powerful technique to verify the successful synthesis of blended hybrid structure as well as good lithium-ion mobility within the blended SPEs. The present work focused on the ionic conductivity and electrochemical behaviours, such as the transference number of lithium cations, electrochemical stability window and cycle performance of the blended hybrid SPEs.
Experimental
Synthesis of the blended SPE membranes
The synthesis procedures of the blended hybrid SPEs are briefly illustrated in Scheme 1. The blended hybrid membranes were prepared by mixing two precursor materials in different weight percentages. The first precursor (precursor I) was synthesized as follows: a solution was made by dissolving 1 mol of polyetheramine (Jeffamine M-2070 with Mw = 2000 g mol−1, Huntsman) into 16 mL of THF and then adding 1 mol of GLYMO (Aldrich), which was stirred for 24 h at 50 °C. Afterwards, the dropwise addition of 0.3 mL of 1 M HCl to the above solution was conducted to ensure the hydrolysis and condensation reactions of the organosilane to form the silicate architecture. The hydrolysis and condensation reactions of GLYMO that led to the formation of silicate particles can be described by the following equations.
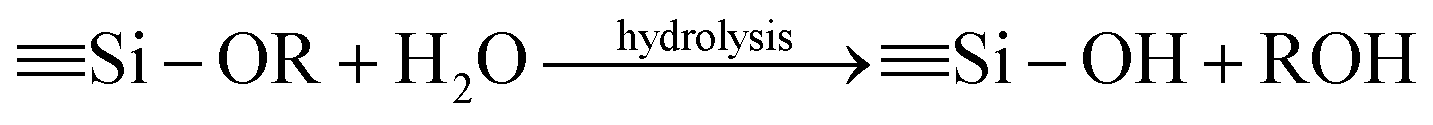 | (1) |
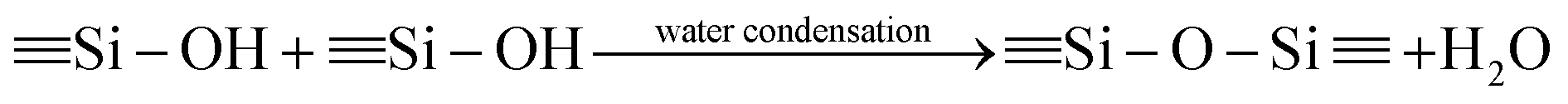 | (2) |
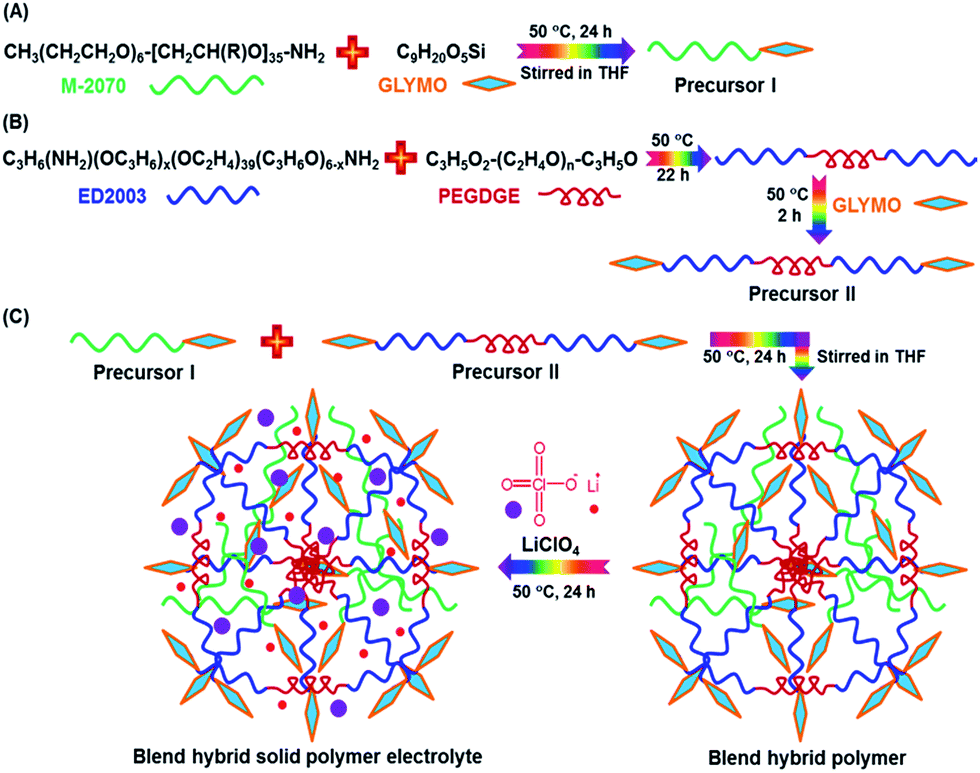 | ||
| Scheme 1 Schematic representation of the synthesis procedures for the blended organic–inorganic hybrid electrolytes. | ||
The hydrolysis of the GLYMO molecules forms silanol groups. The condensation between the silanol groups generates the siloxane bridges (Si–O–Si) that form the entire silicate structure. The GLYMO molecule has an organic linker covalently bonded with the silicon atom. The other end of the organic linker can react with the polyetheramine to form the hybrid structure, as shown in Scheme 1. Therefore, these silicate particles are attached to the polymeric sub-units and are uniformly dispersed in the hybrid structure. To synthesize the second precursor (precursor II), on the other hand, another solution consisting of polyetherdiamine (H2N-(PPG)x(PEG)y(PPG)z-NH2, Jeffamine ED2003 with Mw = 2000 g mol−1 containing x + z = 6; y = 39, Huntsman) and PEGDGE (Mw = 526 g mol−1, Aldrich), which were dissolved in 16 mL of THF in 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mol ratio, was kept at 50 °C for 22 h under vigorous stirring. Then, the resulting solution was further stirred for 2 h at 50 °C after the addition of 2 mol of GLYMO. As in the previous case, the same amount of HCl was added and GLYMO went through the hydrolysis and condensation process in a similar way. Both the precursor I and precursor II solutions were then mixed in different weight ratios, under conditions of stirring at 50 °C for 24 h, to form a series of the blended hybrid. Finally, a certain amount of LiClO4 salt was added to the blended hybrid solution for the desired [O]/[Li] ratios at the same temperature. After that, the resulting viscous solution was cast onto Teflon dishes and dried at 70 °C under vacuum for 2 days. The blended hybrid SPE membrane with a thickness ranging from 70 to 90 μm was finally formed. The blended hybrid SPEs were abbreviated as MP(x:y)-Z, where M and P correspond to precursors I and II with the weight ratio of x and y, respectively, and Z stands for the [O]/[Li] ratio calculated from the ether oxygen atoms from both the mono and diamine polyethers and PEGDGE. The blended hybrid SPEs were stored in a glove box for further use in the different characterizations.
:1 mol ratio, was kept at 50 °C for 22 h under vigorous stirring. Then, the resulting solution was further stirred for 2 h at 50 °C after the addition of 2 mol of GLYMO. As in the previous case, the same amount of HCl was added and GLYMO went through the hydrolysis and condensation process in a similar way. Both the precursor I and precursor II solutions were then mixed in different weight ratios, under conditions of stirring at 50 °C for 24 h, to form a series of the blended hybrid. Finally, a certain amount of LiClO4 salt was added to the blended hybrid solution for the desired [O]/[Li] ratios at the same temperature. After that, the resulting viscous solution was cast onto Teflon dishes and dried at 70 °C under vacuum for 2 days. The blended hybrid SPE membrane with a thickness ranging from 70 to 90 μm was finally formed. The blended hybrid SPEs were abbreviated as MP(x:y)-Z, where M and P correspond to precursors I and II with the weight ratio of x and y, respectively, and Z stands for the [O]/[Li] ratio calculated from the ether oxygen atoms from both the mono and diamine polyethers and PEGDGE. The blended hybrid SPEs were stored in a glove box for further use in the different characterizations.
Characterization methods
The thermal behaviours of the present hybrid electrolytes were investigated by differential scanning calorimetry (DSC) and thermogravimetric analysis (TGA) by using a Perkin-Elmer Pyris 6 and TA instrument Q50, respectively. The samples (around 6 to 8 mg) were heated at a heating rate of 10 °C min−1 under a nitrogen environment for the measurements. Fourier transform infrared (FTIR) spectroscopy was recorded in the range of 4000–500 cm−1 (resolution of 4 cm−1) on a JASCO 4200 spectrometer using the KBr wafer technique. The degree of ionic dissociation was further analyzed by conducting band deconvolution in the spectral range of 650–600 cm−1 for the ClO4− vibrations.Multinuclear solid-state NMR spectra were recorded by using a Varian Infinityplus-500 NMR spectrometer at the Larmor frequencies of 194.3, 125.36 and 99.03 MHz, respectively, for 7Li, 13C and 29Si spins. Adamantane and octakis(trimethylsiloxy)-silsesquioxane (Q8M8) were used as the standards to establish the set-up of the Hartmann–Hahn matching conditions for the 13C and 29Si cross-polarization magic angle spinning (CPMAS) conditions, respectively. 7Li chemical shifts were referred to 1 M LiCl (aq) at 0 ppm, while both the 13C and 29Si chemical shifts were referenced to a single standard tetramethylsilane (TMS) at 0 ppm.
The AC impedance of the blended hybrid SPEs was measured by using the Autolab/PGSTAT302 (Metrohm Autolab B.V., Netherlands) frequency response analyzer. The frequency range was between 100 kHz and 1 Hz and an amplitude of 10 mV was employed. The temperature range of 15–80 °C was chosen to carry out the measurements. Before recording the data, the samples were thermally equilibrated for at least 30 min at each chosen temperature. The bulk electrolyte resistance (Rb) can be deduced from the intercept at the real impedance axis in the complex impedance plot. Since both the thickness (t) of the blended SPE membrane and the electrode area (A) are known, the ionic conductivity (σ) can be calculated from the equation σ = (1/Rb)(t/A).
The method of Evans et al. was employed in this study to measure the Li transference numbers (t+) of the blended hybrid SPEs.28 The present blended SPEs with non-blocking lithium (Alfa Aesar) electrodes were assembled in an argon environment using a standard 2032 coin-cell holder. The current and resistance values were measured at 70 °C on an Autolab/PGSTAT302 frequency response analyzer with an applied dc voltage of 10 mV.
A coin-cell holder was used to fabricate the cell for measuring the electrochemical stability window of the blended hybrid SPEs by the linear sweep voltammetry technique. The cell was made of the blended SPE sandwiched in-between a stainless steel (SS) working electrode and lithium as the counter and reference electrodes. The measurement was performed from 0 to 6 V vs. Li/Li+ at a scan rate of 1 mV s−1.
A LANHE CT2001A battery testing system was employed to study the charge–discharge behaviour of the cell with the blended hybrid SPE. Lithium metal (Alfa product) and LiFePO4 were used as the anode and cathode, respectively, to fabricate the battery and the test was carried out at a current rate of 0.2C. The LiFePO4 cathode was prepared by mixing 80 wt% of the active material, 10 wt% super P carbon and 10 wt% poly(vinylidene fluoride) (PVdF) binder in NMP (N-methyl-2-pyrrolidone) solution. To achieve good contact with the hybrid electrolyte membrane, 2–3 drops of liquid electrolyte were poured over the cathode surface.
The cycled cell was disassembled and the hybrid SPE membrane was recovered to be characterized by SEM to analyse the morphology after prolonged cycles.
Results and discussion
Optimization of the blending ratio of precursor I and precursor II, with a fixed [O]/[Li] ratio of 16, was the first priority to maximize the ionic conductivity as well as the mechanical integrity of the present SPEs system. The temperature dependence of ionic conductivity as a function of the weight ratios of precursors I and II was investigated and the results are shown in Fig. S1 (ESI†). It was found that the blended SPE sample, MP(70:30)-16 with 70 wt% precursor I and 30 wt% precursor II, possessed the maximum ionic conductivity at 30 °C among the compositions investigated. When the amount of precursor I was increased to 80 wt%, the MP(80:20)-16 sample became mechanically unstable and the ionic conductivity could not be measured. Hence, a decision was made to prepare a 70:30 weight ratio of precursor I and precursor II, which allowed the blended hybrid membrane to be sufficiently flexible and mechanically strong enough to consider it further as a potential electrolyte matrix for electrochemical applications. Based on the 70:30 weight ratio of the two precursors, the [O]/[Li] ratios were further varied to optimize the final blended hybrid SPE system with the highest ionic conductivity. Detailed characterizations on the MP(70:30)-Z blended SPEs system were therefore performed in this study.
DSC and TGA of the blended hybrid SPEs
The melting (Tm) and glass transition (Tg) temperatures of the MP(70:30)-Z blended hybrid SPEs was measured by DSC in order to evaluate the degree of crystallinity and flexibility of the blended SPEs. The DSC thermograms of pure starting materials and the blended hybrid SPEs without and with various [O]/[Li] ratios are depicted in Fig. 1. While the pure polymers polyetherdiamine (ED2003) and polyetheramine (M2070) displayed crystalline melting peaks (Tm) around 35.4 °C and 1 °C, respectively, the blended hybrid MP(70:30) showed the melting transition at 18.5° (Table 1). In the blended hybrid electrolytes, the melting transition shifted to lower temperature starting with MP(70:30)-32 and completely disappeared from MP(70:30)-16 onwards, suggesting a semi-crystalline to amorphous phase transition of the synthesized blended hybrid electrolytes with increasing salt concentrations. Therefore, it is clear that the effect of the salt transforms the semi-crystalline hybrid electrolytes to an amorphous one. Normally, the degree of crystallinity (χc) of SPEs can be estimated from the area under the melting endotherm curve based on the equation:
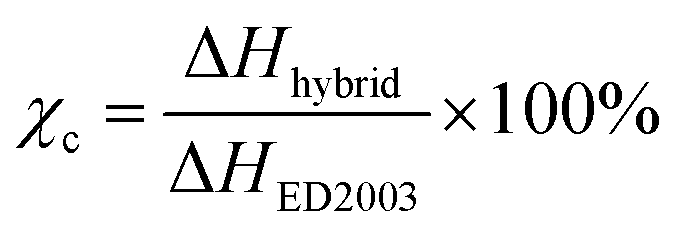 | (3) |
:30)-8 sample became completely amorphous. It is generally accepted that the increased interactions of ether oxygen atoms from the polyethers and Li+ cations restrict the reorganization of polymer chains or destroy the regular crystallized structure, leading to a reduction in the degree of crystallinity.29 An exothermic peak at −27.6 °C was observed for the MP(70:30)-32 sample, which shifted to −9.9 °C for the MP(70:30)-24 sample and became less intense. This could be due to the recrystallization of polyetherdiamine, but such peaks were not observed for the MP(70:30)-Z (Z = 16–8) samples with higher salt concentrations. This observation suggested that a higher salt amount in the MP(70:30)-Z (Z = 16–8) samples suppresses the recrystallization process.30
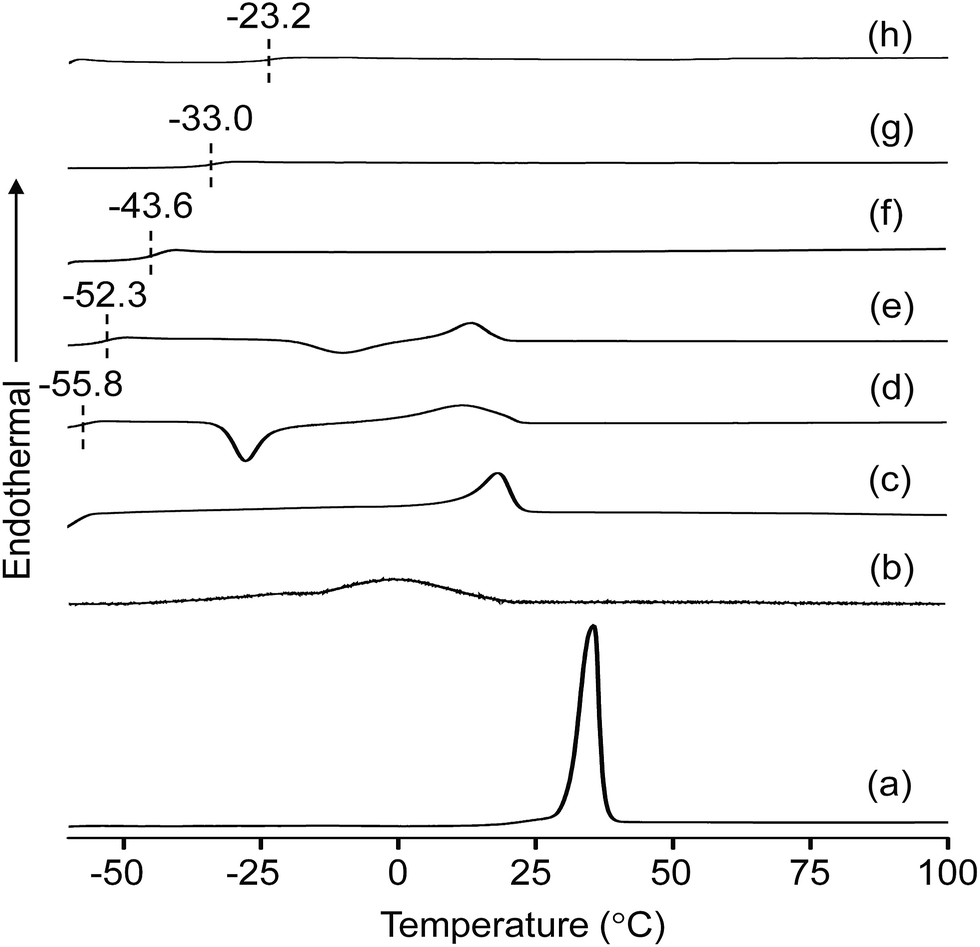 | ||
| Fig. 1 DSC thermograms of: (a) ED2003, (b) M-2070 and MP(70:30)-Z blended hybrid SPEs with Z = (c) ∞, (d) 32, (e) 24, (f) 16, (g) 12 and (h) 8. Z indicates the [O]/[Li] ratio. | ||
:30)-Z blended hybrid electrolytes
| Sample | Tg (°C) | Tm (°C) | ΔHhybrid (J g−1) | χc (%) | A (S cm−1 K−0.5) | B (kJ mol−1) | t+ |
|---|---|---|---|---|---|---|---|
| a Enthalpy of fusion for pure ED2003 (ΔHED2003). | |||||||
| ED2003 | — | 35.4 | 96.3a | 100 | — | — | — |
| M-2070 | — | 1.0 | 21.9 | 22.7 | — | — | — |
| MP(70:30)-∞ |
— | 18.5 | 31.5 | 32.7 | — | — | — |
| MP(70:30)-32 |
−55.8 | 12.8 | 24.4 | 25.3 | 2.7 | 8.4 | 0.23 |
| MP(70:30)-24 |
−52.3 | 13.7 | 16.9 | 17.5 | 2.4 | 8.0 | 0.24 |
| MP(70:30)-16 |
−43.6 | — | — | — | 2.3 | 7.2 | 0.28 |
| MP(70:30)-12 |
−33.0 | — | — | — | 4.5 | 8.1 | 0.20 |
| MP(70:30)-8 |
−23.2 | — | — | — | — | — | — |
Measurements of the Tg values of the blended hybrid SPEs provide information about the flexibility of the polymer chains. Both the results presented in Fig. 1 and the data in Table 1 indicated that the Tg values increased with the increase in salt concentration. The MP(70:30)-32 blended SPE sample showed a Tg value of −55.8 °C, which increased to −23.2 °C for the MP(70:30)-8 sample. It is believed that the interaction of polyether units (from precursor I and precursor II) with the Li+ cations enhances the Tg values gradually, since the increased salt concentration causes an increase in the intermolecular interaction, which effectively results in less movement of the polymer chains.31
TGA experiments were conducted to determine the thermal stability of the blended hybrid SPEs, and the results are presented in Fig. S2 (ESI†). The physisorbed water on the surface of the MP(70:30)-Z blended hybrid SPEs corresponds to the initial 1–2% weight loss observed at 100 °C. The decomposition of EO units of M-2070 and ED2003 started, accounting for the weight loss up to 3–4%, when the temperature was increased to 270 °C. Between 270 °C and 430 °C, the maximum weight loss occurred due to the complete decomposition of the EO and PO units from the mono and diamine polyethers. The remaining weight (varying from 3% to 9%) at 900 °C was mainly attributed to the silicon, which formed the silicate architecture in the blended hybrid matrix. From the above results, it is clear that the present blended hybrid SPEs are thermally stable up to 260 °C and can be used to that temperature without any adverse effect on the electrochemical devices.
Infrared spectroscopic investigations
FTIR investigation is useful to monitor the molecular interactions and formation of the organic–inorganic structure. Fig. 2 depicts the FTIR spectra of the blended hybrid SPEs without and with various [O]/[Li] ratios. Different fingerprint bands can be observed in the IR spectra for particular functional groups. A broad band due to the hydrogen bonded N–H stretching mode was observed around 3445 cm−1.32 The asymmetric and symmetric –CH2 stretching vibrations gave rise to bands at 2917 and 2878 cm−1, respectively,33 while the bands around 1653 and 1300 cm−1 can be ascribed to the C–N stretching vibration,34,35 and the band at 1472 cm−1 was due to the CH2 scissoring.36 The 1375 cm−1 band was assigned to the CH3 bending vibration of the PPG unit.35,36 Other bands around 1457, 1350, 1251 and 842 cm−1 correspond to CH2 vibrational modes, such as bending, wagging, twisting and rocking, respectively.35,37 The C–O–C stretching vibrations of the blended hybrids gave rise to the major bands at 1108 and 1091 cm−1.38 Unfortunately, the Si–O–C and Si–O–Si bands due to the hydrolysis of GLYMO also appeared in the region of 1000–1200 cm−1, and therefore could be possibly seriously overlapped with the bands of the C–O–C stretching vibrations. Therefore, it was not possible to ascertain separately the effect of the salt concentration on these interactions.37,39 The band found at 952 cm−1 was ascribed to CH2 rocking vibration and/or the non-condensed Si–OH from GLYMO.40 The absorption bands in the range of 635–625 cm−1 were attributed to the characteristic ν(ClO4−) modes of the LiClO4 salt that was doped within the blended hybrid SPEs.35,37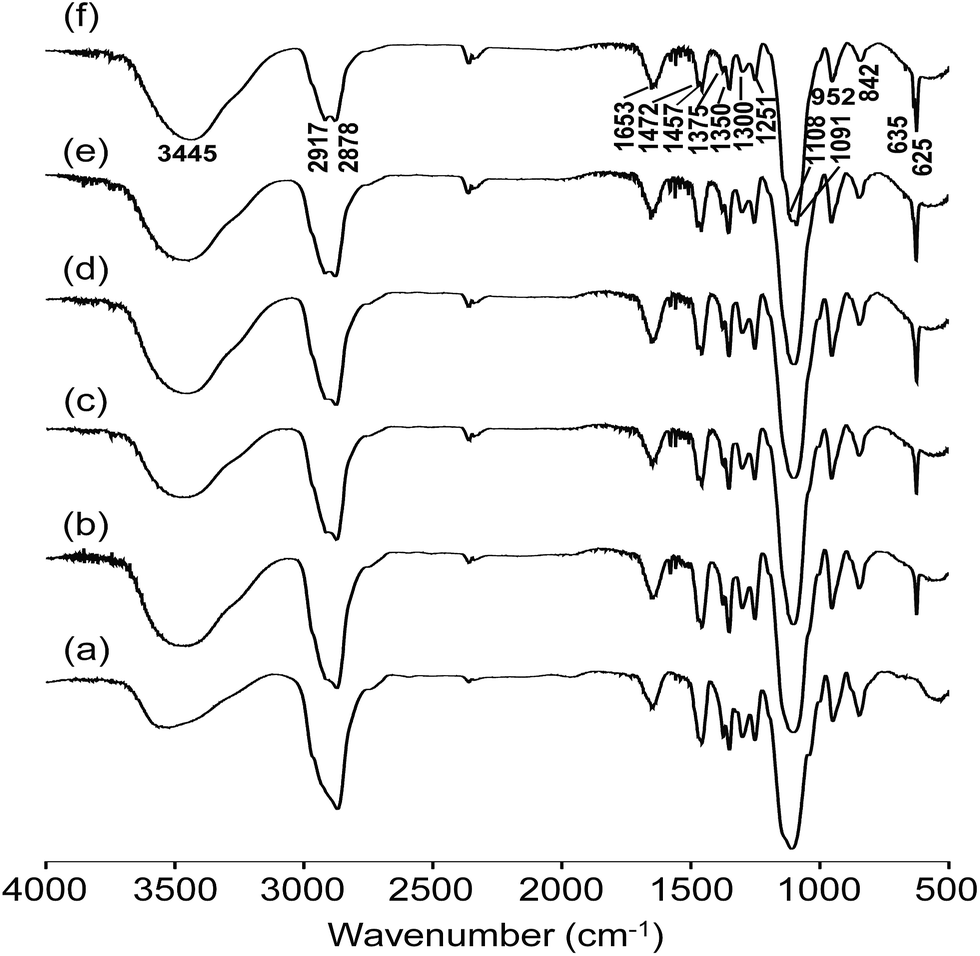 | ||
| Fig. 2 FTIR spectra of the MP(70:30)-Z blended hybrid SPEs with Z = (a) ∞, (b) 32, (c) 24, (d) 16, (e) 12 and (f) 8. | ||
The information about the dissociation of lithium salt in SPEs can be provided by the characteristic ν(ClO4−) mode of the LiClO4 salt in the frequency region of 650–600 cm−1. When Gaussian–Lorentzian functions were applied to deconvolute the IR spectra in the frequency region of 650–600 cm−1, one band centred at 635 cm−1 and another at 625 cm−1 were generated. The band around 625 cm−1 was assigned to the vibration mode of free ClO4− anions, whereas the band at 635 cm−1 was related to the vibration of Li+ClO4− contact-ion pairs.41,42 It can be observed from Fig. S3 (ESI†) that around 86–90% of free ClO4− anions are available for the MP(70:30)-Z (Z = 32–12) blended hybrid electrolytes, which means that the same amount of free Li+ cations are available for conduction. When a higher salt concentration was doped in the MP(70:30)-8 sample, the amount of free ClO4− anions was reduced to around 78% due to the formation of more contact-ion pairs. It is possible that the ClO4− anion and Li+ cation are not far apart from each other at higher salt concentrations due to the presence of a large number of ions in the electrolytes. As a result, the opposite ions can easily combine and thus increase the number of contact-ion pairs but this reduces the free ions availability.
Structural confirmation from the 13C and 29Si solid-state NMR
The architectural design of the present polymer matrices can be explored by 13C solid-state NMR spectroscopy. As observed in Fig. 3(A), the 13C CPMAS NMR spectra of the MP(70:30)-Z blended hybrid SPEs exhibited a prominent peak at 70 ppm, which can be easily assigned to the ether carbons in the ethylene oxide segments of polymers ED2003, M-2070 and PEGDGE.38 On the other hand, the ether carbons in the propylene oxide segments of ED2003, M-2070, and PEGDGE accounted for the small peak at 75 ppm. Although the carbon atoms of the epoxy ring of GLYMO were expected to resonate at 42 and 49 ppm,43 they were not observable in the present case. Instead, a small peak at 55 ppm was observed, which indicated the opening of the epoxide ring of GLYMO after the hydrolysis reaction. Two other peaks at 22 and 9 ppm can be ascribed to the methylene carbons in the α and β positions to the Si atom of GLYMO, respectively. The carbons from the –CH3 groups of the propylene oxide units appeared at 17 ppm. The presence of these characteristics peaks of carbons from different groups suggested the successful synthesis of the blended hybrid structure.
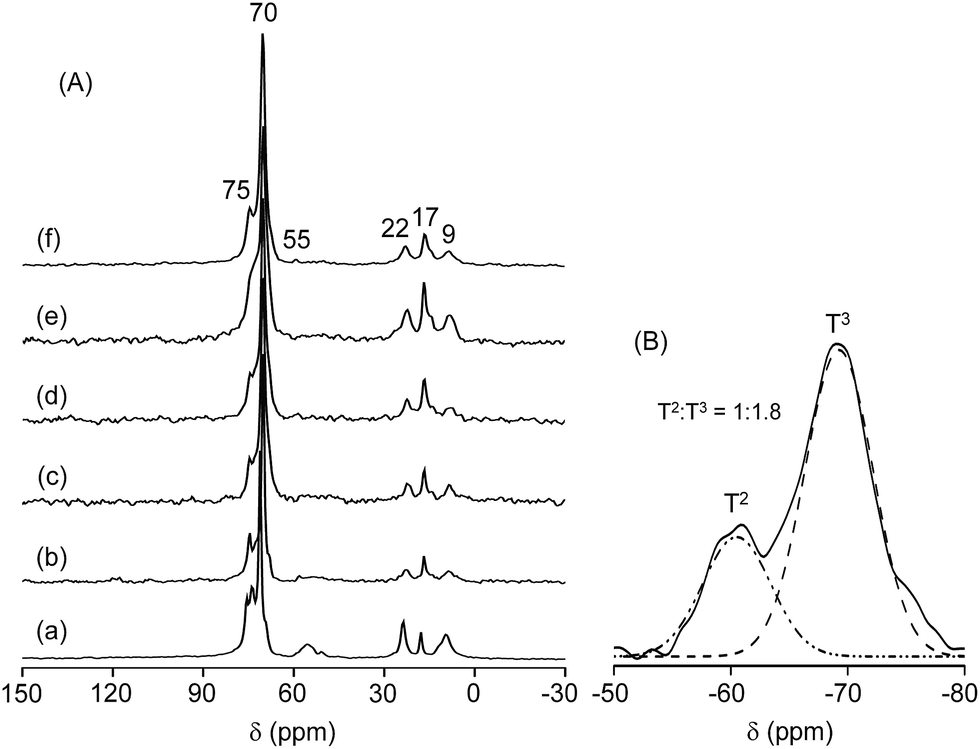 | ||
| Fig. 3 (A) 13C CPMAS NMR spectra of the MP(70:30)-Z blended hybrid SPEs with Z = (a) ∞, (b) 32, (c) 24, (d) 16, (e) 12 and (f) 8. (B) Deconvoluted 29Si CPMAS NMR spectra of the MP(70:30)-16 blended hybrid SPE. | ||
29Si CPMAS NMR spectroscopy can provide information about the silicate structure in the blended hybrid system. The deconvoluted 29Si CPMAS NMR spectrum of the MP(70:30)-16 blended hybrid SPE is displayed in Fig. 3(B). Two peaks related to the T2 (RSi(OSi)2(OH), R = alkyl group) and T3 (RSi(OSi)3) silicon species were observed around −66.9 and −58.0 ppm, respectively. The presence of T groups in the 29Si NMR spectrum suggested the formation of silicon domains or silica particles within the blended hybrid structure. Moreover, silica condensation in the present system was relatively high since the intensity of T3 was almost twice that of T2 (T3:T2 = 1.8:1).
Ionic conductivity as a function of the salt concentration
The temperature-dependent ionic conductivities of the MP(70:30)-Z blended hybrid SPEs as a function of the [O]/[Li] ratio are shown in Fig. 4(A). For all the electrolyte samples, it was observed that ionic conductivity increased continuously with the increase in temperature. The MP(70:30)-16 sample exhibited the maximum ionic conductivity values of 1.2 × 10−4 S cm−1 and 8.3 × 10−4 S cm−1 at 30 °C and 80 °C, respectively, among the salt concentrations investigated. The present ionic conductivity value is comparatively higher since most of the ionic conductivity values reported for other organic–inorganic hybrid SPEs are well below 10−4 S cm−1 at 30 °C.23,44–46 The observed curve-like Arrhenius plots suggested that ionic movements followed the Vogel–Tamman–Fulcher (VTF) behaviour, indicative of ion transport through the segmental motion of polymers.
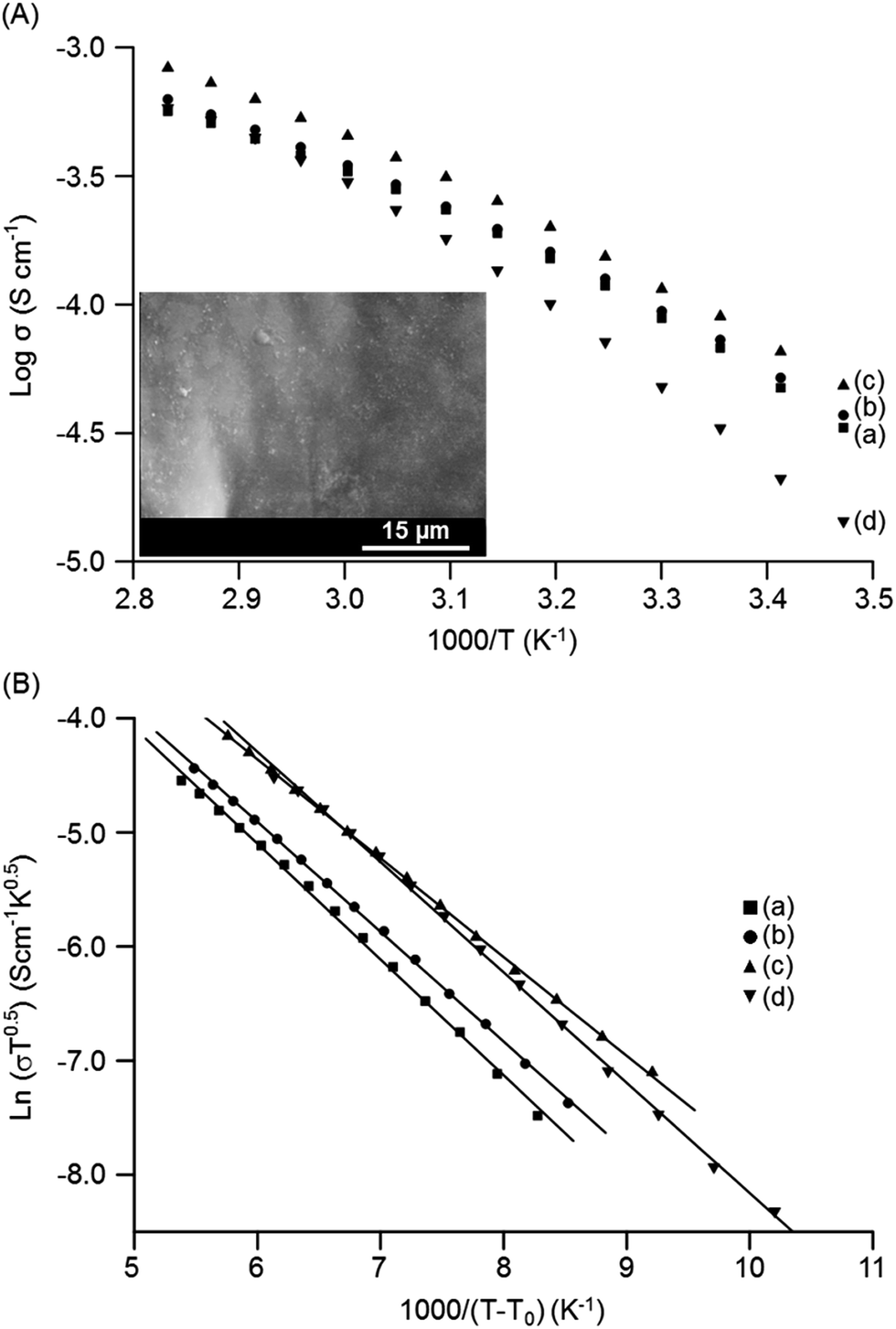 | ||
| Fig. 4 Temperature dependence of: (A) ionic conductivity and (B) the corresponding VTF fitting curves of the MP(70:30)-Z blended hybrid SPEs with Z = (a) 32, (b) 24, (c) 16 and (d) 12. Inset of (A) shows the SEM image of the MP(70:30)-16 blended hybrid SPE. | ||
Generally, several factors can contribute to the total ionic conductivity of an electrolyte, and these can be summarized by the equation:
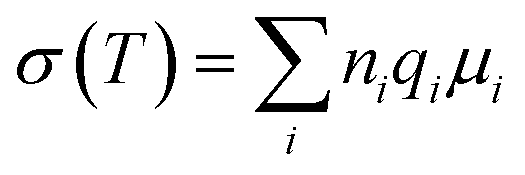 | (4) |
:30)-12 blended SPE with a higher salt amount. Another important factor that can influence the ionic conductivity is the Tg value of the electrolytes, which is an indication of segmental motion of the polymers. As seen in Table 1, the Tg values increased with the increase in salt concentration, suggesting a reduction of the flexibility of the blended SPEs. The increased Tg values of the blended hybrid SPEs suggested that the transient coordination of ether dipoles with Li+ cations restricted segmental movements of the polymers, and thus caused a decrease in the free volume and ionic mobility.47 Thus, the factors, such as the number of total charge carriers, free ions, free volume and segmental movement of the polymers, collaborate in a well suited way to make the MP(70:30)-16 sample the most ion conductive sample in the present blended hybrid SPE system.
The use of organosilanes in the synthesis mixture also produces some sub-micron-sized in situ SiO2 particles in the present organic–inorganic hybrid blended electrolytes, as observed in the inset SEM image of Fig. 4(A). The ionic conductivity behaviour can be explained by the effective-medium theory (EMT).48,49 The present hybrid blended SPE can be viewed as a composite system that is composed of ion-conducting polymer matrices accompanied with dispersed silicate particles. The presence of these sub-micron SiO2 particles reduce the polymer–cation interactions and suppress the crystallinity, resulting in an enhancement in the ionic conductivity.50 Moreover, a highly conducting interface layer (grain boundary) that can assist in the enhancement of the ionic conductivity of the blended hybrid SPEs might be generated by the polymer–SiO2 interactions.51 Grain boundaries are sites of high defect regions that may allow rapid ion movement. Such grain boundaries between polymers and sub-micron SiO2 particles can act as channels for the free ions to pass through, and thus facilitate rapid ion movement and increase the ionic conductivity.52 Therefore, the in situ formation of sub-micron-sized SiO2 particles during the synthesis of organic–inorganic hybrids plays a key role in achieving such a high conductivity value for this blended hybrid SPE system. Furthermore, the Lewis-acid-based interactions among the surface groups of silicate particles, PEO/PPO segments and lithium salt anions enhance the ionic conductivity.53,54 The Lewis acid –OH groups on the silicate surface may interact with the ether oxygen atoms of the PEO/PPO segments and ClO4− anion of the salt. This results in a structural modification with a decrease in the PEO/PPO chains reorganization and an increase in the amorphous phase fraction, thereby creating Li+-conducting pathways at the surfaces of silicate particles. The interactions of –OH groups with the ClO4− ions reduce the ionic coupling and promote the salt dissociation by forming ion-silicate-type complexes.54 These interactions facilitate the formation of more free ions, and thus support the enhancement in ionic conductivity.
As the Arrhenius conductivity plots showed a VTF-like character, the conductivity data were fitted with the VTF equation:
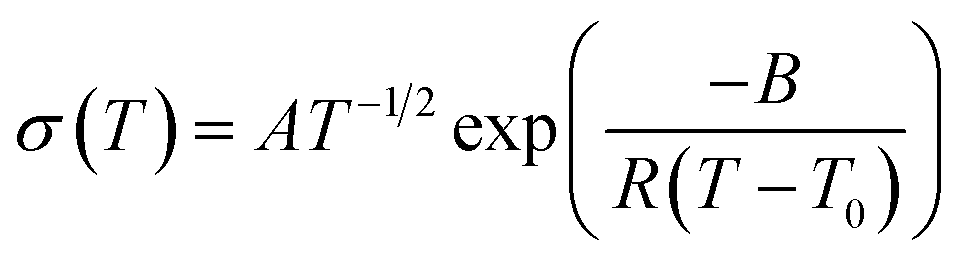 | (5) |
Transference number measurements
The cationic contribution to the ionic conductivity can be evaluated by measuring the Li+ transference number, which is derived by the following equation from Evans et al.:28
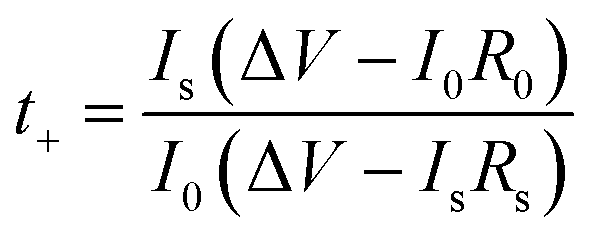 | (6) |
:30)-16 is shown in Fig. 5 and the values of t+ for the blended hybrid SPEs are presented in Table 1. As observed in Fig. 5, the low frequency semi-circle is extended in the steady-state condition in comparison to the initial state, suggesting that a passivation layer on the lithium electrode was formed that led to an enhancement in the interfacial resistance.59,60 This passive layer mainly consists of the lithium electrode and the polymer, salt and other impurities derived from the corrosion reaction products. Therefore, the establishment of a concentration polarization of the anion and development of the passivating layer resulted in the steady-state current, as observed in Fig. 5. The interactions of the Li+ cations with the ether oxygen atoms of the different polymeric groups (EO and PO segments) and the oxygen atoms in ClO4− limited the movement of Li+ ions. As a result, low t+ values (<0.5) were obtained in the present blended hybrid SPEs.18 It can be concluded that the main contribution to the ionic conductivity came from the anionic species of the blended hybrid SPEs, since the transference number values of the lithium cations in the MP(70:30)-Z blended hybrid SPEs were all lower than 0.5.
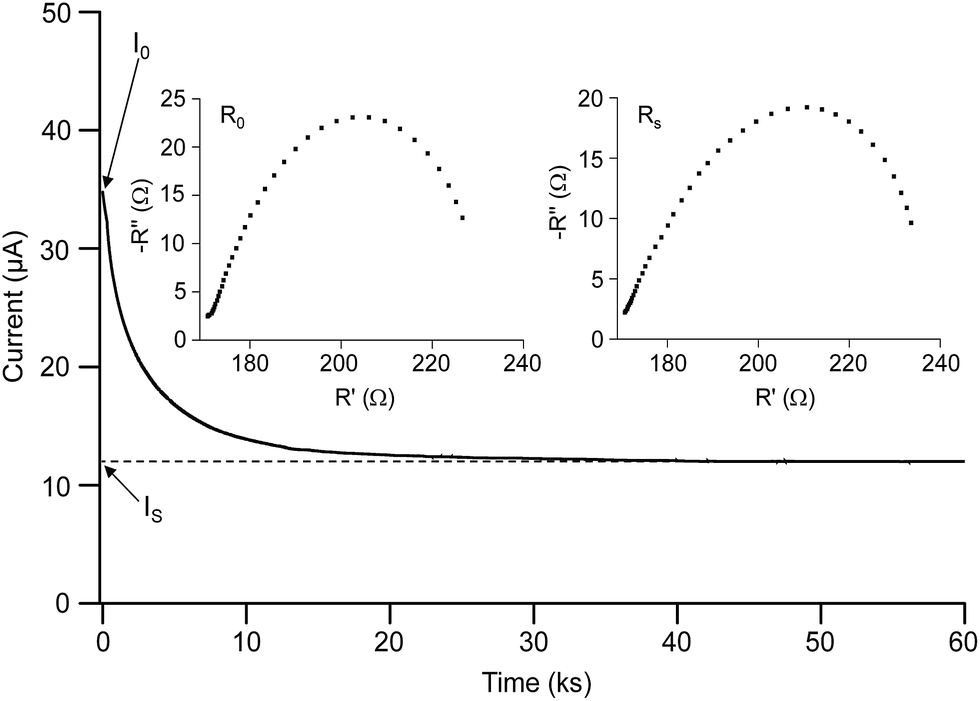 | ||
| Fig. 5 Typical depolarization curve of the MP(70:30)-16 blended hybrid SPE. | ||
Determination of Li+ ion mobility from the 7Li static linewidths
The temperature-dependent 7Li static linewidths measured from 7Li solid-state NMR provides information about the lithium-ion mobility in the blended hybrid SPEs. Fig. 6 depicts the temperature dependence of the 7Li static linewidths for the MP(70:30)-32 and MP(70:30)-16 blended hybrid SPEs, acquired under conditions of with and without 1H decoupling. As observed in Fig. 6, the linewidth evolution could be described by two flat regions separated by a temperature range, in which the linewidths changed dramatically. A broad linewidth with a full width half height (FWHH) of around 5 kHz was observed at a low sample temperature (e.g. <−60 °C), which was well below its Tg value. At this stage, the mobility of lithium ions was much hindered by the low temperatures, while the interactions from either quadrupolar or internuclear dipole–dipole interactions caused such a broad linewidth. With increasing the sample temperature, the linewidth started to sharpen approaching the Tg of the samples. At −50 °C, the linewidths of MP(70:30)-32 and MP(70:30)-16 were measured to be 4.6 and 5.1 kHz, respectively. This observed linewidth might come from two interactions, that is, 7Li–1H dipolar interactions and 7Li–7Li homonuclear interactions. When the proton decoupling technique was employed to eliminate the 7Li–1H dipolar interaction, linewidths of 0.95 and 1.0 kHz were obtained for the MP(70:30)-32 and MP(70:30)-16 samples, respectively. Therefore, the 7Li–1H dipolar interactions contributed 3.65 kHz for MP(70:30)-32 and 4.1 kHz for MP(70:30)-16 to the total lithium linewidths. Approximately 80% of interaction in the MP(70:30)-32 and MP(70:30)-16 SPE samples was because of the 7Li–1H dipolar interactions. These 7Li static NMR results are comparable to those for the other reported SPEs.61,62
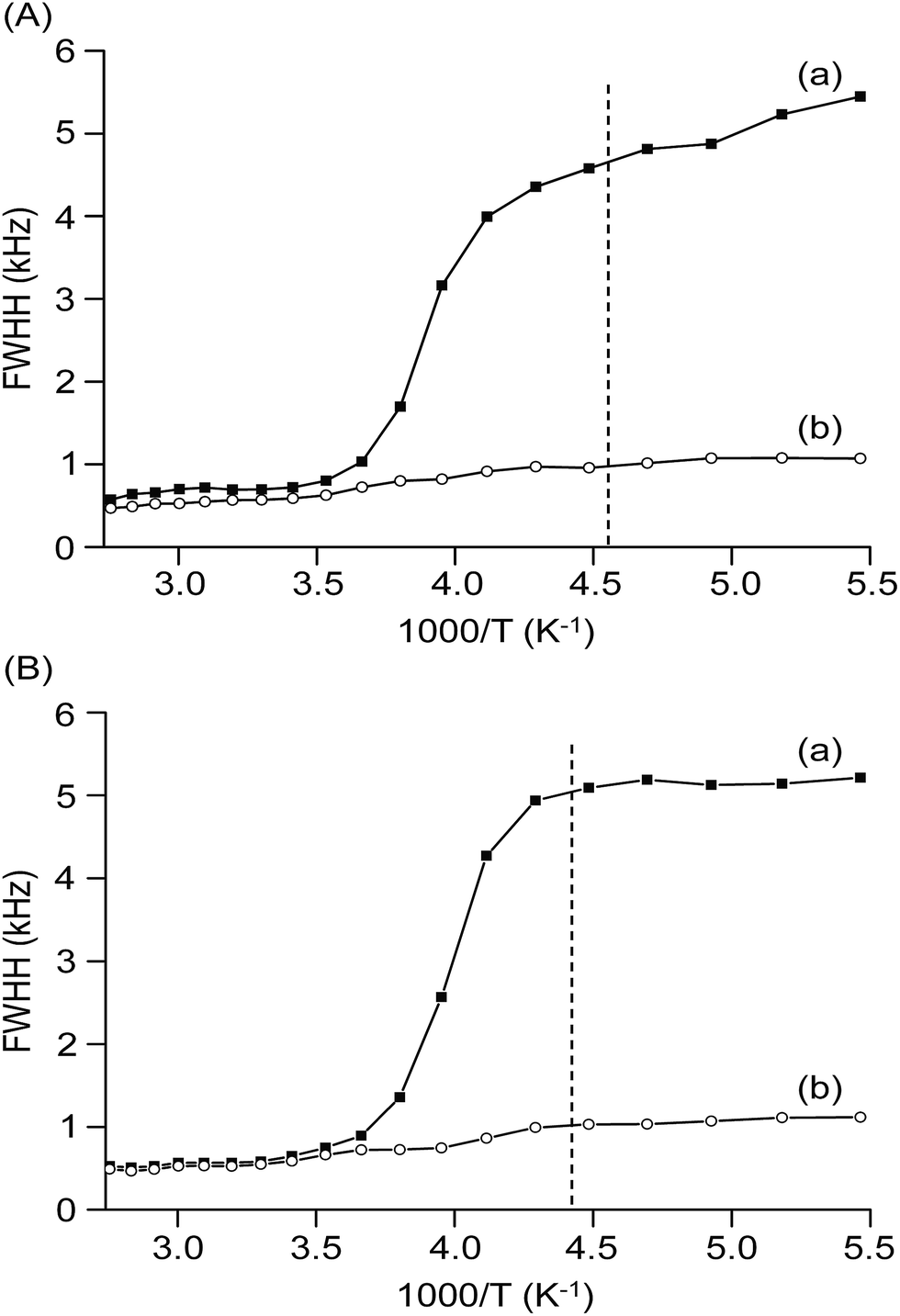 | ||
| Fig. 6 Temperature dependence of the 7Li static linewidths of (A) MP(70:30)-32 and (B) MP(70:30)-16 blended hybrid SPEs, measured (a) without and (b) with proton decoupling. | ||
From the NMR line narrowing data, it is also possible to estimate the activation energy of the blended hybrid SPEs. The activation energy (Ea) can be obtained from the following equation:63
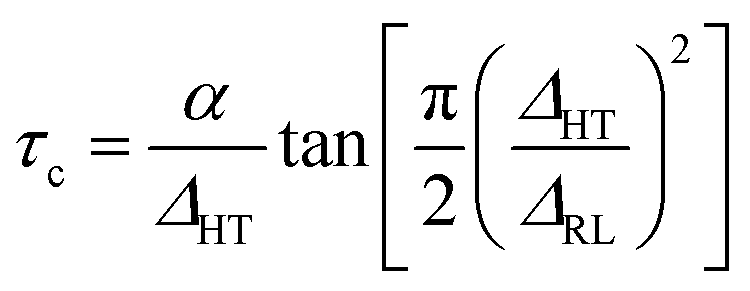 | (7) |
:30)-32 and MP(70:30)-16 samples took place around −50 °C. Considering that τc is thermally activated, the relationship becomes:|
τc = τ0exp(Ea/kT)
| (8) |
By fitting eqn (7) and (8), the Ea values could be estimated from the linewidth and were found to be 0.15 and 0.14 eV for MP(70:30)-32 and MP(70:30)-16, respectively. The current Ea values are lower or comparable to other reported literature.62,64,65
7Li–{1H} MAS NMR study
The local environments of Li+ cations in the MP(70:30)-32 and MP(70:30)-16 SPEs were explored with the variable temperature 7Li–{1H} (i.e. proton decoupled) MAS NMR measurements in the temperature range of −90 to +90 °C. The most dominant coordination sites for Li+ cations are the ether oxygen atoms of polyethers from the M-2070, ED2003 and PEGDGE, which correspond to the peak observed at −1.0 ppm, as shown in Fig. S4 (ESI†). The Li+ cations coordinated with the oxygen atoms of GLYMO were difficult to be detected due to the smaller amount of GLYMO in the blended hybrids, while the lithium linewidths were broad at −90 °C with FWHH values of 0.79 and 0.64 kHz for MP(70:30)-32 and MP(70:30)-16, respectively, and then became narrower to 0.51 and 0.47 kHz at +90 °C, suggesting that the mobility of lithium ions was enhanced with increasing the sample temperature.
Electrochemical stability window of the blended hybrid SPEs
The electrochemical stability window of an electrolyte is an important parameter that can be used to evaluate whether the electrolyte can perform safely up to certain voltage limit in a battery. The linear sweep voltammograms of the cell consisting of the MP(70:30)-Z blended hybrid electrolytes sandwiched in-between the lithium and SS electrodes are presented in Fig. 7. The measurement was carried out from 0 to 6 V and at a scan rate of 1 mV s−1. In the potential regions of 2 to 5 V, a small background current flowed through the cell. The changes in the stainless steel surface might contribute to the observed lower current.66 When the voltage crossed over 5 V, the current increased considerably, suggesting that the decomposition process of the electrolyte had started. The oxidation of some trace species generated a hump between 2 and 3.5 V for the blended hybrid SPEs.67 One hump was also observed at a higher voltage of 4.3 V for the MP(70:30)-32 blended hybrid electrolyte. Overall, the attainment of an electrochemical stability window of over 5 V opens the opportunity for the presented blend hybrid SPEs to be utilized in applications in high voltage lithium-ion batteries.
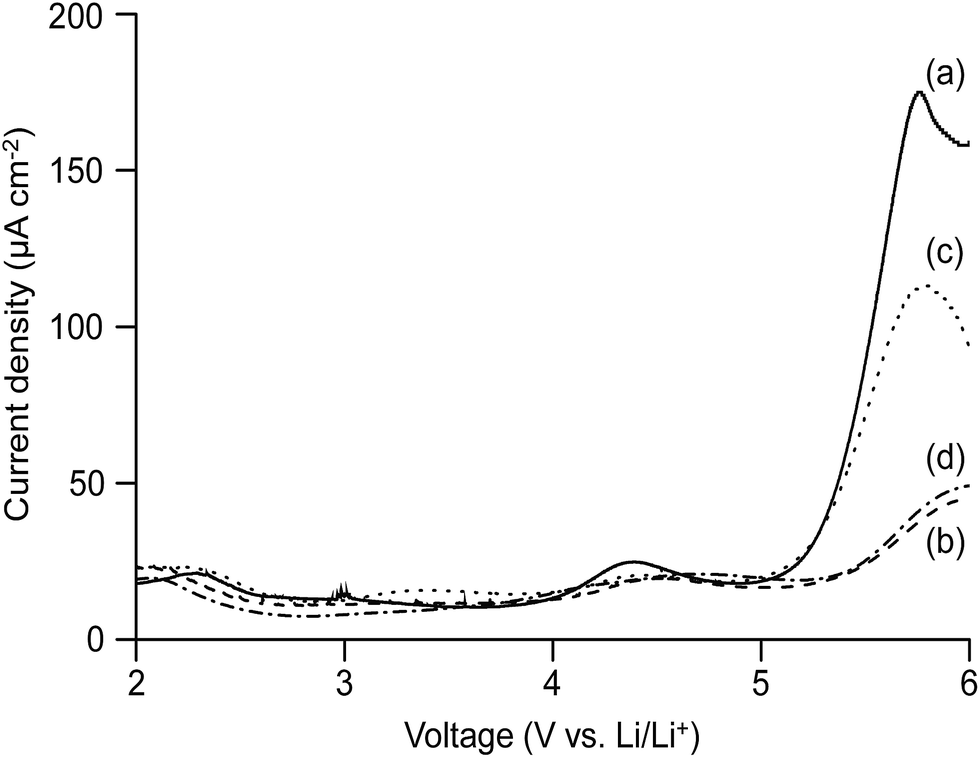 | ||
| Fig. 7 Linear sweep voltammetry curves of the cell prepared with the MP(70:30)-Z blended hybrid SPEs with Z = (a) 32, (b) 24, (c) 16 and (d) 12. | ||
Charge–discharge behaviour of the blended hybrid SPE
The highest ion-conducting sample MP(70:30)-16 was chosen to fabricate the lithium-ion battery with upper and lower cut off voltages of 3.6 and 2.2 V, respectively, at a current rate of 0.2C. Fig. 8 shows the discharge capacity and coulombic efficiency of the test cell for 100 cycles. As shown in Fig. 8, the cell delivered an initial discharge capacity value of 110 mA h g−1. However, the capacity declined continuously and it delivered lower values of 79 mA h g−1 after 50 cycles and 61 mA h g−1 after 100 cycles. The reduction in capacity was mainly attributed to the formation of a passivation layer at the lithium electrode/electrolyte interface.68 Although the capacity retention was almost 72% after 50 cycles, it gradually decreased and the cell finally retained only 55% of capacity after 100 cycles. This suggested that the formation of a passivation layer on the electrode surface and the physical changes in the active materials gradually enhance the cell's internal resistance and resist the charge transfer between the hybrid SPE and the lithium anode, thus resulting in the loss in discharge capacity with the increase in cycle numbers. However, the capacity fading slowed down between the 50th and 100th cycles, suggesting an improved stability of the hybrid SPE with the electrodes.
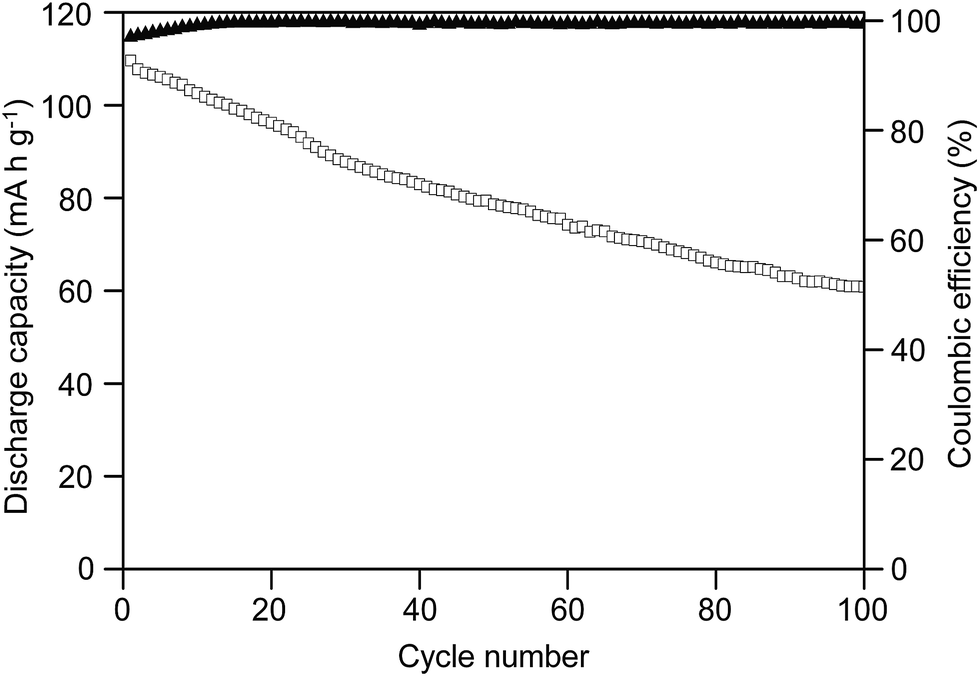 | ||
| Fig. 8 Cycle performance and coulombic efficiency of the cell fabricated with the MP(70:30)-16 blended hybrid SPE. | ||
As observed in Fig. 8, the first cycle coulombic efficiency value was 97%, which slowly increased to above 99% at the 10th cycle and then the value was continuously maintained above 99% up to the 100th cycle. The formation of the passivation layer on the surface of the lithium electrode due to decomposition of the electrolyte caused the irreversible capacity loss. The formed passivation layer consumed part of the anode capacity and resulted in an irreversible capacity loss, which led to a lower coulombic efficiency value for the initial few cycles. On the other hand, the passivation layer shielded the hybrid SPE from further reduction by the lithium and helped to maintain stable coulombic efficiency values of over 99% up to 100 cycles, excluding the first 9 cycles. The high coulombic efficiency values suggested good reversibility of charge transfer at the electrode/electrolyte interface.
Morphological study of the blended hybrid SPE after the cycle tests
The hybrid SPE membrane was recovered from the cell after the charge–discharge cycling and subjected to a morphological study by SEM to analyse whether the silicate particles were uniformly dispersed or aggregated after the cycle tests. The SEM images of the MP(70:30)-16 hybrid SPE membrane before and after the charge–discharge cycle test are displayed in Fig. S5(a and b) (ESI†). As observed in the Fig. S5(a),† the hybrid SPE membrane was quite smooth with uniformly dispersed sub-micron silicate particles. After the charge–discharge cycles, there was no obvious change related to particle aggregation or cracking of the hybrid SPE membrane, suggesting good stability of the hybrid SPE membrane (Fig. S5(b)†). It also established that the present hybrid membrane is able to prevent aggregation and delivers a suitable capacity in lithium-ion batteries. The presence of silicate particles was confirmed from the EDS spectrum presented in Fig. S5(c).† From the results, it can be inferred that the present synthesis strategy successfully prevents particle aggregation in the hybrid SPE membrane after prolonged charge–discharge cycles.
Conclusions
In summary, a highly conductive hybrid SPE based on the blending of two organic–inorganic hybrids was synthesized successfully. The alkoxysilane (3-glycidyloxypropyl)trimethoxysilane was used to modify the amine ends of the polyethers and poly(ethylene glycol) diglycidyl ether to design a silicate network architecture along with the polymer networks. The blended hybrid SPE possessed the maximum ionic conductivity value of 1.2 × 10−4 S cm−1 at 30 °C for the [O]/[Li] ratio of 16 and an electrochemical stability window of over 5 V. The almost amorphous structure of the blended hybrid SPEs at higher salt concentrations was believed to make an important contribution to the reported ionic conductivity as the amorphous networks mainly contributed to the movement of ions in the SPEs. Multinuclear solid-state NMR spectroscopy confirmed the successful synthesis of the blended hybrids as well as the interactions of Li+ cations with the polymer chains. 7Li solid-state NMR further established that the main ion conduction paths resulted from the segmental motions of polymer chains, which was also verified through the VTF fitting results of the ionic conductivity data. The good thermal stability of the blended hybrid SPE is promising for enhancing the safety of electrochemical devices if used. A test cell with the blended hybrid SPE, lithium anode and LiFePO4 cathode possessed an initial discharge capacity value of 110 mA h g−1 at 0.2C and a coulombic efficiency of about 99% up to 100 cycles. These characteristic features of the present blend hybrid SPEs suggest the good possibility of their use in high voltage lithium solid-state batteries.Acknowledgements
The financial support of this work by the Ministry of Science and Technology of Taiwan (Grant number: MOST 105-2119-M-008-012) is gratefully acknowledged.Notes and references
- N. Nitta, F. Wu, J. T. Lee and G. Yushin, Mater. Today, 2015, 18, 252–264 CrossRef CAS.
- J. B. Goodenough and K.-S. Park, J. Am. Chem. Soc., 2013, 135, 1167–1176 CrossRef CAS PubMed.
- K. Xu, Chem. Rev., 2004, 104, 4303–4418 CrossRef CAS PubMed.
- D. Golodnitsky, E. Strauss, E. Peled and S. Greenbaum, J. Electrochem. Soc., 2015, 162, A2551–A2566 CrossRef CAS.
- L. Long, S. Wang, M. Xiao and Y. Meng, J. Mater. Chem. A, 2016, 4, 10038–10069 CAS.
- K. S. Ngai, S. Ramesh, K. Ramesh and J. C. Juan, Ionics, 2016, 22, 1259–1279 CrossRef CAS.
- K. Xu, Chem. Rev., 2014, 114, 11503–11618 CrossRef CAS PubMed.
- J. B. Goodenough and Y. Kim, Chem. Mater., 2010, 22, 587–603 CrossRef CAS.
- J. W. Fergus, J. Power Sources, 2010, 195, 4554–4569 CrossRef CAS.
- D. E. Fenton, J. M. Parker and P. V. Wright, Polymer, 1973, 14, 589 CrossRef CAS.
- M. B. Armand, J. M. Chabagno and M. Duclot, in Fast Ion Transport in Solids, ed. P. Vashisha, J. N. Mundy and G. K. Shenoy, North Holland, Amsterdam, 1979 Search PubMed.
- J. G. Kim, B. Son, S. Mukherjee, N. Schuppert, A. Bates, O. Kwon, M. J. Choi, H. Y. Chung and S. Park, J. Power Sources, 2015, 282, 299–322 CrossRef CAS.
- S. Lascaud, M. Perrier, A. Vallee, S. Besner, J. Prud'homme and M. Armand, Macromolecules, 1994, 27, 7469–7477 CrossRef CAS.
- M. Kono, E. Hayashi and M. Watanabe, J. Electrochem. Soc., 1998, 145, 1521–1527 CrossRef CAS.
- A. Ferry, L. Edman, M. Forsyth, D. R. MacFarlane and J. Sun, J. Appl. Phys., 1999, 86, 2346–2348 CrossRef CAS.
- M. A. Gebreyesus, Y. Purushotham and J. Siva Kumar, Heliyon, 2016, 2, e00134 CrossRef PubMed.
- A. I. Gopalan, P. Santhosh, K. M. Manesh, J. H. Nho, S. H. Kim, C.-G. Hwang and K.-P. Lee, J. Membr. Sci., 2008, 325, 683–690 CrossRef CAS.
- F. Croce, G. B. Appetecchi, L. Persi and B. Scrosati, Nature, 1998, 394, 456–458 CrossRef CAS.
- N. Kaskhedikar, J. Paulsdorf, A. Burjanadze, Y. Karatas, B. Roling and H. D. Wiemhofer, Solid State Ionics, 2006, 177, 2699–2704 CrossRef CAS.
- L. Fan, C.-W. Nan and S. Zhao, Solid State Ionics, 2003, 164, 81–86 CrossRef CAS.
- F. Croce, L. Settimi and B. Scrosati, Electrochem. Commun., 2006, 8, 364–368 CrossRef CAS.
- P. Judeinstein, J. Titman, M. Stamm and H. Schmidt, Chem. Mater., 1994, 6, 127–134 CrossRef CAS.
- J. Shim, D.-G. Kim, J. H. Lee, J. H. Baik and J.-C. Lee, Polym. Chem., 2014, 5, 3432–3442 RSC.
- D. Saikia, Y.-C. Pan, C.-G. Wu, J. Fang, L.-D. Tsai and H.-M. Kao, J. Mater. Chem. C, 2014, 2, 331–343 RSC.
- J. Nunes-Pereira, C. M. Costa and S. Lanceros-Méndez, J. Power Sources, 2015, 281, 378–398 CrossRef CAS.
- C. A. Nguyen, S. Xiong, J. Ma, X. Lu and P. S. Lee, Phys. Chem. Chem. Phys., 2011, 13, 13319–13326 RSC.
- S. A. M. Noor, A. Ahmad, I. A. Talib and M. Y. A. Rahman, Ionics, 2010, 16, 161–170 CrossRef CAS.
- J. Evans, C. A. Vincent and P. G. Bruce, Polymer, 1987, 28, 2324–2328 CrossRef CAS.
- J. Xi, X. Qiu, X. Ma, M. Cui, J. Yang, X. Tang, W. Zhu and L. Chen, Solid State Ionics, 2005, 176, 1249–1260 CrossRef CAS.
- W. A. Henderson, Macromolecules, 2007, 40, 4963–4971 CrossRef CAS.
- J. F. Le Nest, A. Gandini and C. Schoenenberger, Trends Polym. Sci., 1994, 2, 432–437 CAS.
- M. M. Coleman, K. H. Lee, D. J. Skrovanek and P. C. Painter, Macromolecules, 1986, 19, 2149–2157 CrossRef CAS.
- L. S. Teo, C. Y. Chen and J. F. Kuo, Macromolecules, 1997, 30, 1793–1799 CrossRef CAS.
- T. Miyazawa, T. Shimanouchi and S.-I. Mizushima, J. Chem. Phys., 1956, 24, 408–418 CrossRef CAS.
- D. L. Pavia, G. M. Lampman and G. S. Kriz, Introduction to Spectroscopy, Harcourt College Publication, USA, 2001, pp. 15–84 Search PubMed.
- B. H. Stuart, Infrared Spectroscopy: Fundamentals and Applications, John Wiley & Sons Ltd., 2004, p. 139 Search PubMed.
- P. Patnaik, Dean's Analytical Chemistry Handbook, McGraw-Hill, New York, 2nd edn, 2004, pp. 7.1–7.43 Search PubMed.
- H.-M. Kao, T. T. Hung and G. T. K. Fey, Macromolecules, 2007, 40, 8673–8683 CrossRef CAS.
- A. Kioul and L. Mascia, J. Non-Cryst. Solids, 1994, 175, 169–186 CrossRef CAS.
- M. M. Silva, V. de Zea Bermudez, L. D. Carlos, A. P. P. de Almeida and M. J. Smith, J. Mater. Chem., 1999, 9, 1735–1740 RSC.
- M. Salomon, M. Xu, E. M. Eyring and S. Petrucci, J. Phys. Chem., 1994, 98, 8234–8244 CrossRef CAS.
- H.-W. Chen, C.-Y. Chiu, H.-D. Wu, I.-W. Shen and F.-C. Chang, Polymer, 2002, 43, 5011–5016 CrossRef CAS.
- M. Templin, U. Wiesner and H. W. Spiess, Adv. Mater., 1997, 9, 814–817 CrossRef CAS.
- P. C. Barbosa, M. M. Silva, M. J. Smith, A. Gonçalves, E. Fortunato, S. C. Nunes and V. de Zea Bermudez, Electrochim. Acta, 2009, 54, 1002–1009 CrossRef CAS.
- S. C. Nunes, V. de Zea Bermudez, D. Ostrovskii, M. M. Silva, S. Barros, M. J. Smith, L. D. Carlos, J. Rocha and E. Morales, J. Electrochem. Soc., 2005, 152, A429–A438 CrossRef CAS.
- D. Saikia, H.-Y. Wu, C.-P. Lin, Y.-C. Pan, J. Fang, L.-D. Tsai, G. T. K. Fey and H.-M. Kao, Polymer, 2012, 53, 6008–6020 CrossRef CAS.
- S. Ahmad, H. B. Bohidar, S. Ahmad and S. A. Agnihotry, Polymer, 2006, 47, 3583–3590 CrossRef CAS.
- C. W. Nan and D. M. Smith, Mater. Sci. Eng., B, 1991, 10, 99–106 CrossRef.
- W. Wieczorek, D. Raducha, A. Zalewska and J. R. Stevens, J. Phys. Chem. B, 1998, 102, 8725–8731 CrossRef CAS.
- Y. Dai, S. Greenbaum, D. Golodnitsky, G. Ardel, E. Strauss, E. Peled and Y. Rosenberg, Solid State Ionics, 1998, 106, 25–32 CrossRef CAS.
- C. W. Nan and D. M. Smith, Mater. Sci. Eng., B, 1991, 10, 99–106 CrossRef.
- J. Maier, Prog. Solid State Chem., 1995, 23, 171–263 CrossRef CAS.
- W. Wieczorek, J. R. Stevens and Z. Florjańczyk, Solid State Ionics, 1996, 85, 67–72 CrossRef CAS.
- F. Croce, L. Persi, B. Scrosati, F. Serraino-Fiory, E. Plichta and M. A. Hendrickson, Electrochim. Acta, 2001, 46, 2457–2461 CrossRef CAS.
- J. L. Souquet, M. Duclot and M. Levy, Solid State Ionics, 1996, 85, 149–157 CrossRef CAS.
- J. Zhang, X. Huang, H. Wei, J. Fu, Y. Huang and X. Tang, Electrochim. Acta, 2010, 55, 5966–5974 CrossRef CAS.
- Y.-Y. Chu, Z.-Y. Liu, D. Saikia, C.-J. Chou, H.-M. Kao and M.-C. Chen, Ionics, 2015, 21, 2523–2534 CrossRef CAS.
- Y. H. Liang, C. C. Wang and C. Y. Chen, Eur. Polym. J., 2008, 44, 2376–2384 CrossRef CAS.
- D. Fauteux, Solid State Ionics, 1985, 17, 133–138 CrossRef CAS.
- S. Morzilli, F. Bonino and B. Scrosati, Electrochim. Acta, 1987, 32, 961–964 CrossRef CAS.
- A. C. Bloise, C. C. Tambelli, R. W. A. Franco, J. P. Donoso, C. J. Magon, M. F. Souza, A. V. Rosario and E. C. Pereira, Electrochim. Acta, 2001, 46, 1571–1579 CrossRef CAS.
- P. H. de Souza, R. F. Bianchi, K. Dahmouche, P. Judeinstein, R. M. Faria and T. J. Bonagamba, Chem. Mater., 2001, 13, 3685–3692 CrossRef CAS.
- S. H. Chung, K. R. Jeffrey and J. R. Stevens, J. Chem. Phys., 1991, 94, 1803–1811 CrossRef CAS.
- P. Mustarelli, C. Capiglia, E. Quartarone, C. Tomasi, P. Ferloni and L. Linati, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 60, 7228–7233 CrossRef CAS.
- S. T. C. Ng, M. Forsyth, M. Garcia, D. R. MacFarlane and M. E. Smith, Electrochim. Acta, 1998, 43, 1551–1556 CrossRef CAS.
- A. C. Bloise, J. P. Donoso, C. J. Magon, A. V. Rosario and E. C. Pereira, Electrochim. Acta, 2003, 48, 2239–2246 CrossRef CAS.
- Y. X. Jiang, Z. F. Chen, Q. C. Zhuang, J. M. Xu, Q. F. Dong, L. Huang and S. G. Sun, J. Power Sources, 2006, 160, 1320–1328 CrossRef CAS.
- H.-H. Kuo, W.-C. Chen, T.-C. Wen and A. Gopalan, J. Power Sources, 2002, 110, 27–33 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Temperature dependence of ionic conductivity of MP(x:y)-16 blend hybrid SPEs with x:y = (a) 0:100 (b) 30:70 (c) 50:50 (d) 70:30 (Fig. S1); TGA curves of MP(70:30)-Z blend hybrid SPEs with Z = (a) 32, (b) 24, (c) 16, (d) 12, and (e) 8 (Fig. S2); deconvoluted FTIR spectra of MP(70:30)-Z blend hybrid SPEs with Z = (a) 32, (b) 24, (c) 16, (d) 12, and (e) 8 (Fig. S3); 7Li–{1H} MAS NMR spectra of MP(70:30)-Z blend hybrid SPEs with Z = (A) 32 and (B) 16, acquired at a spinning speed of 3 kHz (Fig. S4); SEM images of MP(70:30)-16 blend hybrid SPE (a) before and (b) after charge–discharge cycle testing with EDS spectrum (c) (Fig. S5). See DOI: 10.1039/c7ra01542a |
| This journal is © The Royal Society of Chemistry 2017 |