
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAdamantyl derivatives and rearranged benzophenones from Garcinia xanthochymus fruits†
Yu Chen‡
a,
Fei Gan‡a,
Shan Jinb,
Hui Liua,
Shijun Wuc,
Wenting Yangc and
Guangzhong Yang *b
*b
aCollege of Chemistry and Material Sciences, South-Central University for Nationalities, Wuhan 430074, P. R. China
bSchool of Pharmaceutical Sciences, South-Central University for Nationalities, Wuhan 430074, P. R. China. E-mail: yanggz888@126.com; Fax: +86-27-67841196; Tel: +86-27-67841196
cCollege of Environmental and Biological Engineering, Wuhan Technology and Business University, Wuhan 430065, P. R. China
First published on 21st March 2017
Abstract
Garcinia xanthochymus is a tropical fruit yielding tree native to South East Asia. Its fruit is used to treat bilious conditions, diarrhea and dysentery. In this study, three new adamantyl derivatives, two new rearranged benzophenones, named as garcixanthochymones A–E (1–5), together with 12 known compounds including 7 xanthones (6–12) and 5 flavonoids (13–17) were isolated from the fruits of G. xanthochymus. Their structures were elucidated by detailed spectroscopic analyses. All isolated compounds were evaluated for their anti-proliferative activities against four human tumor cells (HepG2, A549, SGC7901, MCF-7). Adamantyl derivatives (1–3) and rearranged benzophenones (4–5) displayed potential inhibitory activity against four human cancer cell lines with IC50 values of 5.16–16.45 μM. These data suggested that the extracts of the fruits of G. xanthochymus are potent candidates for cancer prevention.
1. Introduction
Fruits contain high levels of biologically active components that impart health benefits beyond their basic nutritional value. Non-nutritive components in fruits such as phenolics, flavonoids, and in particular anthocyanins contribute in varying degrees to the antioxidant activity of individual fruits. Antioxidants play a crucial role in the prevention of chronic ailments such as heart attacks, cancer, diabetes, hypertension, stroke and Alzheimer's disease by combating oxidative stress. This suggests that a change in dietary behavior, such as increasing the consumption of fruits, may provide desirable health benefits for reducing the incidence of chronic diseases.1–3 The genus Garcinia belongs to Guttiferae (Clusiaceae) family and is a rich source of secondary metabolites including xanthones, flavonoids, benzophenones, lactones and phenolic acids. Several species have been documented as ethnobotanically important plants used traditionally as food and medicine.4 Garcinia xanthochymus is a fruit yielding tree native to South East Asia. Its fruits are edible and also used in jams preserves and vinegar. A preparation of the fruit of G. xanthochymus is given to treat bilious conditions, diarrhea and dysentery.5,6 Phytochemical studies have revealed that the fruits of G. xanthochymus contain various chemical constituents, including 7 polyisoprenylated benzophenones (PIBs): guttiferone H, gambogenone, aristophenone A, xanthochymol, guttiferone E, cycloxanthochymol, isoxanthochymol, 4 xanthones: alloathyriol, maclurin, euxanthone, 1,5-dihydroxyxanthone and 8 bifavonoids: amentoflavone, 3,8′′-biapigenin, (±)-fukugetin, (±)-fukugiside, 3,8′′-biflavanone, morelloflavone, GB 1a, (±)-volkensiflavone.6–9 The fruits of G. xanthochymus have been reported to show a variety of biological activities such as antioxidant,10 antidiabetic,11 antiplasmodial7 and cytotoxic activities.12 It has been reported that the G. xanthochymus fruits possess 31.31 mg g−1 and 5.313 mg g−1 of total phenol and flavonoid contents.2 PIBs are a major class of compounds isolated from the fruits of Garcinia plant, since fruits are the only edible plant part. PIBs with structural diversity have a wide range of biological activities.4 Much effort has been devoted to verify the effectiveness of the fruit of G. xanthochymus against cancer. A series of studies found that PIBs isolated from the fruit of G. xanthochymus showed inhibitory effects on human colon cancer cell lines by regulating the mammalian target of rapanmycin (mTOR) cell survival pathway.12As part of our continuing research on bioactive components from nutritional fruits, we have investigated the fruits of G. xanthochymus, isolated and identified three new adamantyl derivatives, two new rearranged benzophenones, named as garcixanthochymones A–E (1–5) and 12 known compounds. Cytotoxicities against four human tumor cell lines (HepG2, A549, SGC7901, MCF-7) of the 17 isolated compounds are also reported.
2. Materials and methods
2.1 General experimental procedures
Optical rotations were determined in MeOH on a P-1020 digital polarimeter. (JASCO Co., Tokyo, Japan). UV spectra were obtained on a 2401PC spectrophotometer (Shimadzu Co., Tokyo, Japan). 1D and 2D NMR spectra were recorded on a Bruker AVANCE III-500 MHz and a Bruker AV-400 MHz spectrometer (Bruker, Ettlingen, Germany) in DMSO-d6, C5D5N, acetone-d6 using tetramethylsilane (TMS) as an internal reference standard. Chemical shifts (δ) have been expressed in ppm and the coupling constants (J) have been given in Hz. High-resolution electrospray mass spectroscopy was performed on an Agilent G6230 TOF mass spectrometer (HR-ESI-MS) (Agilent Technologies Co. Ltd., California, America) and A waters Autospec Premier 776 mass spectrometer (HR-EI-MS) (Waters Technologies Co. Ltd., Massachusetts, America). High-performance liquid chromatography (HPLC) was conducted on an Ultimate 3000 HPLC system (Dionex Co., Sunnyvale, CA, USA) equipped with an Ultimate 3000 pump and Ultimate 3000 variable wavelength detector, as well as a semi-preparative YMC-Pack ODS-A column (250 × 10 mm, 5 μm) and a preparative YMC-Pack ODS-A column (250 × 20 mm, 5 μm) from YMC Co., Ltd (Kyoto, Japan), column chromatography (CC) was conducted over silica gel (200–300 mesh and 300–400 mesh, Qingdao Haiyang Chemical Industry Co., Ltd., Qingdao, China). Chromatographic grade acetonitrile was purchased from Chang Tech Enterprise Co., Ltd (Taiwan, China). The cell lines were purchased from the cell bank of Chinese Academy of Sciences (Shanghai, China). 3-(4,5-Dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide (MTT) was purchased from Biosharp Co. Ltd. (Hefei, Anhui, China). Doxorubicin hydrochloride was purchased from Aladdin Chemical Co. Ltd. (Shanghai, China). Dulbecco modified Eagle medium (DMEM), phosphate buffer saline (PBS) and penicillin–streptomycin solution were purchased from Hyclone Laboratories, GE healthcare life science (Logan, UT, USA). Fetal bovine serum (FBS) was purchased from Gibco, Life technologies (Grand Island, NY, USA). Reagent grade dimethyl sulfoxide (DMSO) was purchased from Vetec, Sigma Chemical Co. (St. Louis, MO, USA). The absorbance was read on a Multiskan GO microplate reader (Thermo Fisher Scientific Inc. Waltham, MA, USA).2.2 Plant material
The fruits of G. xanthochymus were purchased from Xishuangbanna Prefecture, Yunnan Province, P. R. China and identified by Prof. Ying-hong Zhao, Xishuangbanna Prefecture National Medicine Research Institute. The voucher specimen (2013![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 091201) was deposited in the herbarium of School of Pharmaceutical Sciences, South Central University for Nationalities.
091201) was deposited in the herbarium of School of Pharmaceutical Sciences, South Central University for Nationalities.
2.3 Extraction and isolation
The dried fruits of G. xanthochymus (6.18 kg) were powdered and extracted with 95% EtOH at room temperature for three times (each time for 24 h) to obtain EtOH extract 2.94 kg, and then successively partitioned with petroleum ether (P. E.), EtOAc and n-BuOH to get petroleum ether extract 267 g, EtOAc extract 711 g, n-BuOH extract 460 g. The EtOAc extract (332 g) was chromatographed on a silica gel column (200–300 mesh) eluted successively with P. E./EtOAc gradient (19:1, 9:1, 7:3, 6:4, 1:1, 4:6, 3:7, 0:1) to obtain 10 fractions (Fr. 1–Fr. 10). Fr. 3 (13.3 g) was subjected to octadecylsilane CC eluting with H2O–MeOH (7:3, 6:4, 1:1, 4:6, 3:7, 2:8, 0:1) to obtain 10 major fractions (Fr. 3.1–Fr. 3.10). Fr. 3.2 (145.4 mg) was purified by semipreparative HPLC (MeOH–H2O, 52:48) to afford 11 (7.4 mg) at tR 14.0 min. Fr. 3.4 (33 mg) was purified by semipreparative HPLC (MeOH–H2O, 55:45) to afford 13 (5.1 mg) at tR 13.9 min. Fr. 3.5 (287 mg) was purified by recrystallization to afford 10 (6.5 mg), then further subjected to a silica gel column (300–400 mesh) eluted successively with CHCl3/MeOH gradient (200:1, 100:1, 50:1, 19:1, 7:3, 1:1, 3:7, 0:1) to obtain 6 fractions (Fr. 3.5.1–Fr. 3.5.6). Fr. 3.5.3 (20 mg) was purified by semipreparative HPLC (CH3CN–H2O, 35:65) to afford 6 (1.5 mg) at tR 23.6 min. Fr. 3.5.4 (10.5 mg) was purified by semipreparative HPLC (MeOH–H2O, 50:50) to afford 9 (2.5 mg) at tR 37.0 min. Fr. 3.5.5 (50 mg) was purified by semipreparative HPLC (MeOH–H2O, 63:37) to afford 7 (4.3 mg) at tR 12.3 min, 8 (10.2 mg) at tR 14.1 min respectively. Fr. 3.6 (1.16 g) was subjected to a silica gel column (300–400 mesh) eluted successively with CHCl3/MeOH gradient (200:1, 100:1, 50:1, 20:1, 10:1, 8:2, 6:4, 1:1, 2:8, 0:1) to obtain 7 fractions (Fr. 3.6.1–Fr. 3.6.7). Fr. 3.6.3 (350 mg) was purified by semipreparative HPLC with gradient MeOH–H2O (73:27 to 98:2) to afford 14 (23.1 mg) at tR 25.9 min and the lower polar fractions (61.3 mg) at tR 41 min, which was further purified by semipreparative HPLC (CH3CN–H2O, 64:36) to afford 1 (6.6 mg) at tR 33.5 min, 2 (4.1 mg) at tR 42.3 min, 3 (4.0 mg) at tR 37.4 min. Fr. 3.7 (4.3 g) was subjected to a silica gel column (300–400 mesh) eluted successively with P. E./CHCl3/MeOH gradient (20:5:0.1, 10:5:0.1, 5:5:0.1, 5:5:1, 5:5:2, 0:9:1, 0:0:1) to obtain 3 fractions (Fr. 3.7.1–Fr. 3.7.3). Fr. 3.7.3 (1.58 g) was subjected to a silica gel column (300–400 mesh) eluted successively with P. E./CHCl3/MeOH gradient (20:5:0.1, 5:5:0.1, 5:5:0.2, 0:0:1) to obtain 3 fractions (Fr. 3.7.3.1–Fr. 3.7.3.3). Fr. 3.7.3.2 (620 mg) was purified by preparative HPLC (CH3CN–H2O, 93:7) to obtain 11 fractions (Fr. 3.7.3.2.1–Fr. 3.7.3.2.11). Fr. 3.7.3.2.1 (53.5 mg) was purified by semipreparative HPLC (CH3CN–H2O, 75:25) to afford 4 (23.2 mg) at tR 19.5 min. Fr. 3.7.3.2.2 (81.5 mg) was purified by semipreparative HPLC (CH3CN–H2O, 70:30) to afford 5 (24.9 mg) at tR 31.1 min. Fraction 5 (9.5 g) was subjected to octadecylsilane CC eluting with H2O–MeOH (7:3, 6:4, 1:1, 4:6, 3:7, 2:8, 0:1) to give 12 major fractions (Fr. 5.1–Fr. 5.12). Fr. 5.6 (30 mg) was purified by semipreparative HPLC (MeOH–H2O, 82:18) to afford 12 (5.4 mg) at tR 13.1 min. Fraction 8 (49.9 g) was purified by recrystallization to afford 16 (364 mg). Fraction 7 (32.4 g) was subjected to a silica gel column (200–300 mesh) eluted successively with CHCl3/MeOH gradient (500:1, 100:1, 20:1, 10:1, 7:3, 0:1) to obtain 14 fractions (Fr. 7.1–Fr. 7.14). Fr. 7.12 (2.3 g) was subjected to octadecylsilane CC eluting with H2O–MeOH (7:3, 1:1, 3:7, 1:9, 0:1) to obtain 15 (200 mg) and 17 (166 mg).
ε): 258 (4.07), 309 (3.79), 360 (3.86); 1H- and 13C-NMR: see Table 1. HR-EI-MS m/z: 618.3550 [M]+ (calcd for C38H50O7: 618.3557).
| No. | 13C-NMR | 1H-NMR | ||||
|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 1 | 2 | 3 | |
| a The chemical shifts have been expressed in δ ppm. The coupling constants (J) have been expressed in Hz. | ||||||
| 1 | 77.6 | 77.6 | 77.2 | |||
| 2 | 202.3 | 202.6 | 207.7 | |||
| 3 | 80.3 | 80.2 | 75.5 | |||
| 4 | 202.7 | 202.5 | 107.3 | |||
| 5 | 69.1 | 69.3 | 57.7 | |||
| 6 | 45.4 | 45.0 | 39.0 | 2.51 m 2H | 2.55 m 2H | 3.63 dd (14.8, 4.8) |
| 1.74 m | ||||||
| 7 | 48.3 | 48.3 | 43.4 | 1.81 m | 1.83 m | 2.23 m |
| 8 | 54.5 | 54.4 | 57.0 | |||
| 9 | 204.3 | 204.3 | 208.0 | |||
| 10 | 192.3 | 192.1 | 198.2 | |||
| 11 | 128.9 | 128.6 | 133.2 | |||
| 12 | 116.8 | 117.4 | 119.5 | 6.91 d (2.0) | 6.87 d (1.6) | 7.12 d (2.0) |
| 13 | 145.3 | 145.1 | 143.8 | |||
| 14 | 150.5 | 150.8 | 149.2 | |||
| 15 | 115.1 | 115.3 | 113.8 | 6.78 d (8.4) | 6.78 m | 6.70 d (8.4) |
| 16 | 124.0 | 124.1 | 125.2 | 6.72 dd (8.4, 2.0) | 6.78 m | 6.97 dd (8.4, 2.0) |
| 17 | 34.9 | 34.4 | 28.8 | 1.89 dd (14.4, 6.0) | 1.91 dd (14.8, 6.8) | 2.17 dd (13.2, 3.2) |
| 2.00 m | 1.98 dd (14.8, 6.8) | 1.30 (m) | ||||
| 18 | 43.8 | 39.9 | 41.7 | 2.73 m | 3.15 m | 1.56 m |
| 19 | 149.4 | 149.0 | 80.1 | |||
| 20 | 113.5 | 113.9 | 22.3 | 4.76 dd (2.4, 1.2) | 4.79 d (2.4) | 1.01 s 3H |
| 4.66 d (2.4) | 4.83 m | |||||
| 21 | 18.0 | 18.3 | 29.2 | 1.61 s 3H | 1.67 s 3H | 0.80 s 3H |
| 22 | 29.2 | 41.2 | 29.4 | 1.46 m | 1.62 m | 1.45 m |
| 1.30 m | 1.37 m | 1.02 m | ||||
| 23 | 42.4 | 73.4 | 36.2 | 1.30 m 2H | 3.89 d (10.0) | 2.25 m, 2.02 m |
| 24 | 70.5 | 150.1 | 146.7 | |||
| 25 | 29.5 | 109.7 | 110.7 | 1.17 s 3H | 4.72 s 4.93 s | 4.76 s 4.71 s |
| 26 | 29.5 | 18.4 | 22.8 | 1.17 s 3H | 1.72 s 3H | 1.74 s 3H |
| 27 | 24.0 | 24.0 | 24.2 | 2.39 dd (14.4, 6.2) | 2.49 dd (14.0, 6.4) | 2.34 m 2H |
| 2.50 m | 2.38 dd (14.0, 6.8) | |||||
| 28 | 121.5 | 121.4 | 122.8 | 4.92 t (6.2) | 4.89 t (6.4) | 5.06 t (7.0) |
| 29 | 133.8 | 133.9 | 131.9 | |||
| 30 | 18.3 | 18.5 | 18.2 | 1.66 s 3H | 1.65 s 3H | 1.64 s 3H |
| 31 | 26.1 | 26.2 | 26.2 | 1.60 s 3H | 1.58 s 3H | 1.64 s 3H |
| 32 | 23.1 | 23.1 | 24.6 | 1.17 s 3H | 1.17 s 3H | 1.05 s 3H |
| 33 | 22.7 | 22.7 | 23.5 | 1.22 s 3H | 1.22 s 3H | 1.02 s 3H |
| 34 | 52.0 | 52.1 | 52.2 | 4.15 dt (8.0, 2.4) | 4.14 d (7.8) | 3.68 t (3.2) |
| 35 | 122.4 | 122.4 | 88.8 | 4.98 dt (8.0, 1.2) | 5.01 d (7.8) | 4.26 d (3.2) |
| 36 | 134.2 | 134.2 | 71.7 | |||
| 37 | 26.3 | 26.2 | 29.5 | 1.69 s 3H | 1.69 s 3H | 1.34 s 3H |
| 38 | 18.5 | 18.7 | 28.0 | 1.74 s 3H | 1.75 s 3H | 1.27 s 3H |
ε): 258 (3.95), 310 (3.69), 363 (3.75); 1H- and 13C-NMR: see Table 1. HR-ESI-MS (positive ion mode) m/z: 639.3296 [M + Na]+ (calcd for C38H48O7Na: 639.3292).ε): 258 (3.93), 309 (3.77), 357 (3.73); 1H- and 13C-NMR: see Table 1. HR-ESI-MS (positive ion mode) m/z: 635.3577 [M + H]+ (calcd for C38H51O8: 635.3578).ε): 260 (3.95), 323 (3.67), 365 (3.77); 1H- and 13C-NMR: see Table 2. HR-ESI-MS (positive ion mode) m/z: 555.2718 [M + Na]+ (calcd for C33H40O6Na: 555.2723).
| No. | 4 | 5 | ||||
|---|---|---|---|---|---|---|
| 13C-NMR | 1H-NMR | HMBC | 13C-NMR | 1H-NMR | HMBC | |
| 1 | 64.4 | 64.3 | ||||
| 2 | 214.8 | 214.8 | ||||
| 3 | 71.2 | 71.2 | ||||
| 4 | 33.6 | 2.28, 1.91 m 2H | 33.8 | 1.97 dd (11, 8.5 Hz), 2.26 m 2H | ||
| 5 | 57.2 | 1.99 m | 57.7 | 2.07 m | ||
| 6 | 42.8 | 42.7 | ||||
| 7 | 93.1 | 93.2 | ||||
| 8 | 70.1 | 70.1 | ||||
| 9 | 206.6 | 206.5 | ||||
| 10 | 48.3 | 1.91, 2.10 m 2H | 48.3 | 2.09 m, 1.91 d (13.5) 2H | ||
| 11 | 26.7 | 2.66 m 2H | 9, 2, 12, 13, 1, 10 | 26.7 | 2.66 m 2H | 9, 2, 12, 13, 1 |
| 12 | 120.8 | 5.54 t (7.5) | 120.6 | 5.54 t (7.5) | 14, 15 | |
| 13 | 134.2 | 134.2 | ||||
| 14 | 26.3 | 1.62 s 3H | 12, 13 | 26.3 | 1.62 s 3H | 12, 13 |
| 15 | 18.3 | 1.63 s 3H | 12, 13 | 18.3 | 1.64 s 3H | 12, 13 |
| 16 | 29.9 | 2.51 dd (11.5, 9.2), 2.24 m 2H | 18, 17, 3, 2 | 30.0 | 2.50 dd (11.0, 9.5), 2.23 m 2H | 18, 17, 3, 2 |
| 17 | 57.6 | 3.07 dd (9.2, 8.5) | 7, 8, 18, 19, 20, 9 | 57.6 | 3.06 dd (9.5, 8.0) | 7, 8, 18, 19, 20, 9 |
| 18 | 37.4 | 37.4 | ||||
| 19 | 27.7 | 1.24 s 3H | 17, 29 | 27.8 | 1.22 s 3H | 17, 29, 18 |
| 20 | 31.0 | 1.42 s 3H | 17, 29 | 31.0 | 1.39 s 3H | 17, 29, 18 |
| 21 | 33.8 | 1.84, 1.91 m 2H | 34.1 | 2.40 m 2H | ||
| 22 | 37.1 | 2.09, 1.83 m | 124.6 | 5.14 t (7.0) | 24, 25 | |
| 23 | 146.5 | 132.1 | ||||
| 24 | 110.7 | 4.78 s, 4.80 s | 25, 22 | 26.3 | 1.68 s 3H | 22, 23 |
| 25 | 22.9 | 1.69 s 3H | 24, 23 | 18.4 | 1.60 s 3H | 22, 23 |
| 26 | 19.7 | 1.78 s 3H | 7, 6, 10 | 19.7 | 1.80 s 3H | 7, 6, 10, 5 |
| 27 | 196.7 | 196.7 | ||||
| 28 | 129.0 | 129.0 | ||||
| 29 | 146.1 | 146.1 | ||||
| 30 | 112.4 | 7.23 s | 18, 28, 31, 32 | 112.4 | 7.22 s | 18, 28, 31, 32 |
| 31 | 146.3 | 146.3 | ||||
| 32 | 154.0 | 154.0 | ||||
| 33 | 115.1 | 8.06 s | 27, 32, 31 | 115.1 | 8.06 s | 27, 32, 31 |
ε): 260 (3.97), 322 (3.71), 365 (3.77); 1H- and 13C-NMR: see Table 2. HR-ESI-MS (positive ion mode) m/z: 555.2726 [M + Na]+ (calcd for C33H40O6Na: 555.2723).2.4 Measurement of inhibition activity on tumor cell proliferation
The antiproliferative activities against four human tumor cell lines (HepG2, A549, SGC7901, MCF-7) of isolated compounds were measured by the MTT assay. The four human tumor cell lines were cultured in 96-well plates at a concentration of 104 cells per well in DMEM containing 10% FBS and 1% penicillin–streptomycin at 37 °C in 5% CO2 atmosphere. After 24 h of growth, washed the cells for three times by PBS, then added 200 μL sample solution or doxorubicin solution (dissolved in DMEM containing 1% penicillin–streptomycin) in each well at a gradient of 50 μM, 25 μM, 12.5 μM, 6.25 μM and 3.125 μM. After 24 h of incubation, removed the cell culture medium and washed the cells for three times by PBS, then added 100 μL MTT (0.5 mg mL−1) solution (dissolved in DMEM containing 1% penicillin–streptomycin) in each well incubated for 4 h. After the incubation, removed the MTT solution and add 100 μL DMSO, read the absorbance at a wavelength of 492 nm by using the microplate reader. The inhibition rate of the compound (%) was calculated with the formula [A]test/[A]control × 100%, where [A]test is the absorbance of the test group that the cells incubated in sample solution, and [A]control is the absorbance of the control group that the cells incubated only with the cell culture medium. The IC50 values was calculated according to the inhibition rate by using GraphPad Prism 6. All the 17 compounds have been tested the antiproliferative activities against four human tumor cell lines (HepG2, A549, SGC7901, MCF-7) as summarized in Table 3.| (IC50 in μM) | ||||
|---|---|---|---|---|
| Compoundsa | HepG2 | A549 | SGC7901 | MCF-7 |
| a Other isolates with IC50 > 50 μM for all cell lines are not listed.b Doxorubicin was used as positive control. | ||||
| Doxorubicinb | 6.52 ± 0.13 | 14.03 ± 0.21 | 7.54 ± 1.11 | 4.40 ± 1.17 |
| 1 | 8.34 ± 1.2 | 15.88 ± 0.48 | 7.32 ± 0.29 | 5.77 ± 0.17 |
| 2 | 5.77 ± 1.43 | 15.13 ± 5.99 | 5.16 ± 1.16 | 14.12 ± 1.29 |
| 3 | 6.64 ± 0.87 | 16.25 ± 3.11 | 6.80 ± 0.29 | 9.75 ± 0.62 |
| 4 | 9.08 ± 0.91 | 7.48 ± 0.03 | 10.71 ± 1.97 | 15.69 ± 0.87 |
| 5 | 9.03 ± 0.36 | 8.24 ± 0.16 | 15.25 ± 4.79 | 16.45 ± 0.23 |
| 14 | 12.82 ± 2.49 | 11.29 ± 1.40 | 19.30 ± 3.06 | 13.55 ± 1.97 |
3. Result and discussion
3.1 Structural elucidation of isolated compounds
Garcixanthochymone A (1) was isolated as a white powder and gave the molecular formula C38H50O7 with 14 degrees of unsaturation by its HR-EI-MS ([M]+ m/z 618.3550, calcd 618.3557). Its UV spectrum gave its maximum absorption wavelengths at 258 nm, 309 nm and 360 nm which indicated the presence of an aromatic moiety. The 1H-NMR spectrum revealed the presence of a 1,3,4-trisubstituted benzene ring signals [δH 6.72 (1H, dd, J = 8.4, 2.0 Hz), 6.78 (1H, d, J = 8.4 Hz) and 6.91 (1H, d, J = 2.0 Hz)], two olefinic proton signals [δH 4.92 (1H, t, J = 6.2 Hz) and 4.98 (1H, dt, J = 8.0, 1.2 Hz)], one terminal double bonds [δH 4.66 (1H, d, J = 2.4 Hz) and 4.76 (1H, dd, J = 2.4, 1.2 Hz)], nine methyl signals [δH 1.17 (3H, s), 1.17 (3H, s), 1.17 (3H, s), 1.22 (3H, s), 1.60 (3H, s), 1.61 (3H, s), 1.66 (3H, s), 1.69 (3H, s) and 1.74 (3H, s)], one methylene signal [δH 2.51 (2H, m)], two methine signals [δH 1.81 (1H, m), δH 4.15 (1H, dt, J = 8.0, 2.4 Hz)]. The 13C-NMR and DEPT spectrum indicated the presence of three nonconjugated carbonyl carbons [δC 202.3, 202.7, and 204.3], one conjugated carbonyl carbon [δC 192.3], a 3,4-dihydroxybenzoyl group [δC 115.1, 116.8, 124.0, 128.9, 145.3 and 150.5], two trisubstituted double bonds [δC 121.5, 122.4, 133.8, and 134.2], one terminal double bonds [δC 113.5, 149.4], five quaternary carbons [δC 54.5, 69.1, 70.5, 77.6 and 80.3], five methylenes [δC 24.0, 29.2, 34.9, 42.4 and 45.4], three methines [δC 43.8, 48.3 and 52.0], nine methyls [δC 18.0, 18.3, 18.5, 22.7, 23.1, 26.1, 26.3, 29.5 and 29.5]. On the basis of these data, 1 has an adamantyl phloroglucinol skeleton.13 Comparison of 1H and 13C NMR data of 1 with those of garcinialiptone A14 indicated that the two compounds were closely related, except for a 3-hydroxy-3-methylbutyl group at C-18 of 1 [δH 1.17 (6H, s), δH 1.30 (3H, m), δH 1.46 (1H, m); δC 29.2 (C-22), 29.5 (C-25, 26), 42.4 (C-23), 70.5 (C-24)] instead of a 3-methylbut-3-enyl group at C-18 of garcinialiptone A.14 This assumption was further confirmed by the 1H–1H COSY form H-18 to H2-22 and H2-22 to H2-23 and HMBC correlations (see Fig. 2) from H3-25, H3-26 to C-23 (δC 42.4) and C-24 (δC 70.5). Regarding the relative configuration of 1, the ROESY spectrum of 1 (see Fig. 3) showed correlations of H-34/H3-33, H-34/H-7, which indicated that H-34, H-7, H3-33 are on the same side of molecular plane which was the same as that of garcinialiptone A. Furthermore, the ROESY correlations between H-35/H2-6, H2-6/H3-32 revealed that the H-35, H3-32 and H2-6 were on the opposite face to H-34, H-7, H3-33. As a result of the above information, the structure of 1 was deduced completely as showed in the Fig. 1. | ||
| Fig. 1 Structures of compounds 1–5. | ||
 | ||
| Fig. 2 Selected HMBC for compound 1–3. | ||
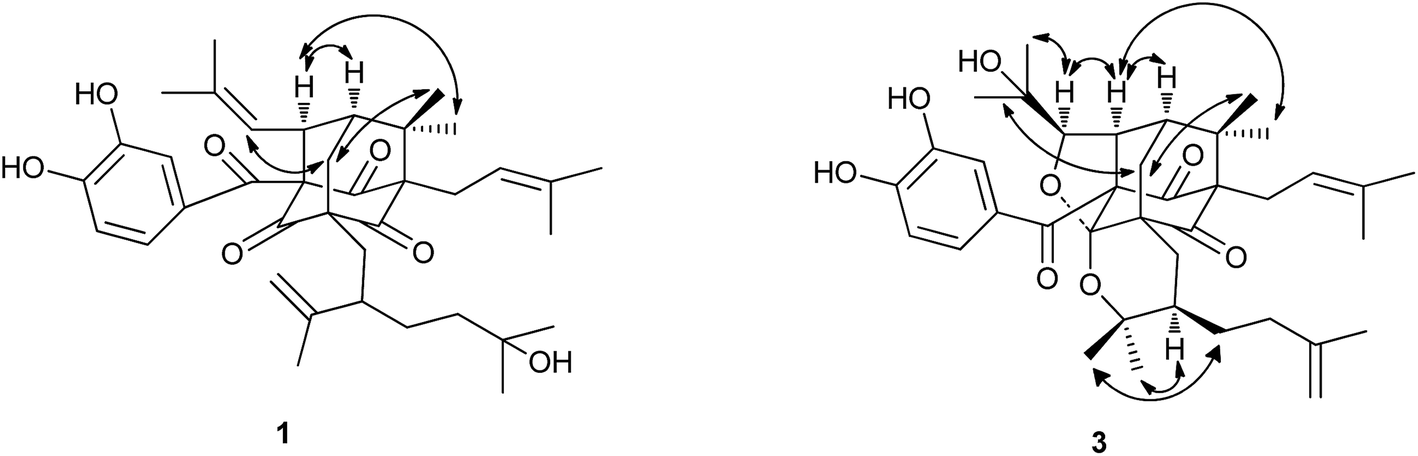 | ||
| Fig. 3 Selected ROESY of compounds 1 and 3. | ||
Garcixanthochymone B (2) was obtained as a white powder and gave the molecular formula C38H48O7 with 15 degrees of unsaturation by its HR-ESI-MS ([M + Na]+ m/z 639.3296, calcd 639.3292). Both 2 and 1 gave similar 1H and 13C NMR spectra data, except for the signals of a 2-hydroxy-3-methylbut-3-enyl group (C(22)–C(26)) at C-18 in the former rather than 3-hydroxy-3-methylbutyl group in the latter. This finding was also supported by the 13C NMR data of C-23 (δ 73.4), C-24 (δ 150.1), and C-25 (δ 109.7) and HMBC correlations (see Fig. 2) between H-23/C-26, 22, 25, 24. Thus, compound 2 was elucidated as 23-hydroxy garcinialiptone A, named as garcixanthochymone B.
Garcixanthochymone C (3) was obtained as a white powder and gave the molecular formula C38H50O8 with 14 degrees of unsaturation by its HR-ESI-MS ([M + H]+ m/z 635.3577, calcd 635.3578). In comparison of the NMR data of 3 with those of 1, revealed that 3 contained an adamantyl phloroglucinol skeleton as that of 1, but lacked resonances for an olefinic proton in 1H NMR and one trisubstituted double bond in 13C NMR in 1. In contrast, four additional carbon resonances, including one oxygenated methine (δC 88.8), two oxygenated quaternary carbon (δC 71.7, 80.1) and one ketal carbon (δC 107.3) were found in 3. It suggested that the 2-methyl-2-propenyl group at C-34 and 2-isopropenyl-5-methyl-5-hydroxyhexyl group at C-5 in 1 had undergone an alternative cyclization. HMBC correlations (see Fig. 2) of δH 1.27 (CH3-37) and 1.34 (CH3-38) to the carbons at δC 88.8 (C-35) and 71.7 (C-36), of δH 4.26 (H-35) to the carbons at δC 52.2 (C-34) and 43.4 (C-7) indicated that a 2,3-dioxygenated 2-methylpropyl group was attached to C-34 instead of 2-methyl-2-propenyl group at C-34 in 1. HMBC correlations of δH 1.01 (CH3-20) and 0.80 (CH3-21) to the carbons at δC 80.1 (C-19) and 41.7 (C-18) indicated that 2-oxygenated isopropyl group was attached to C-18 instead of isopropenyl group at C-18 in 1. The aforementioned groups accounted for 12 out of 14 degrees of unsaturation in compound 3, indicating the presence of two additional rings. The chemical shifts of C-35 (δC 88.8) and C-19 (δC 80.1) and one more ketal carbon (δC 107.3) implied the ether ring closure between C-35 and C-4, C-19 and C-4 to construct 2-(1-hydroxy-1-methylethyl)-tetrahydrofuran ring fused with the phloroglucinol moiety at C-3 and C-4 and 2,2-dimethyl-3-(3-methylbut-3-enyl)-tetrahydropyrano ring fused with the phloroglucinol moiety at C-5 and C-4. Regarding the relative configuration of 3, the ROESY spectrum of 3 (see Fig. 3) showed correlations of H-34/H3-33, H-34/H-7, H-34/H-35, H2-6a/H3-32 and H2-6b/H3-37 which indicated that H-34, H-35, H-7 and H3-33 are on the same side of molecular plane and H2-6 and H3-32 are on the other side of molecular plane. The ROESY correlations between Heq-17 at δH 2.17 (1H, dd, J = 13.2, 3.2)/H-18 and H-18/H3-21 revealed that the C-18 side chain on the tetrahydropyran ring should be in an equatorial configuration. Therefore, the structure of 3 was deduced completely as showed in the Fig. 1.
Garcixanthochymone D (4) was obtained as a white powder and gave the molecular formula C33H40O6 with 14 degrees of unsaturation by its HR-ESI-MS ([M + Na]+ m/z 555.2718, calcd 555.2723). The 1H NMR spectrum exhibited three olefinic protons [δH 4.78 (1H, s), 4.80 (1H, s), 5.54 (1H, t, J = 7.5 Hz)], a 1,2,4,5-tetrasubstituted benzene moiety [δH 7.23 (1H, s), 8.06 (1H, s)], and six singlet methyls [δH 1.24 (3H, s), 1.42 (3H, s), 1.62 (3H, s), 1.63 (3H, s), 1.69 (3H, s), 1.78 (3H, s)]. The 13C NMR spectrum showed 33 carbons attributable to six methyls, seven methylenes, of which one is a terminal olefinic carbon [δC 110.7 (C-24)], five methines, of which two are aromatic and one olefinic [δC 112.4 (C-30), 115.1 (C-33), 120.8 (C-12)], 15 quaternary carbons, of which three are keto groups [δC 196.7 (C-27), 206.6 (C-9), 214.8 (C-2)]. From the above analysis of the 1H and 13C-NMR spectroscopic data, it was implied that compound 4 is a tetracyclo [4.3.2.11,6.03,7] decane derivative similar to that of doitunggarcinone A.15 Further comparison of the NMR spectroscopic data of 4 with those of doitunggarcinone A revealed that a 1,2,4,5-tetrasubstituted benzene moiety fused at C-28 and C-29 in 4 instead of an ortho-disubstituted benzene moiety in doitunggarcinone A. This difference was evidenced by the HMBC correlations from H-30 (δH 7.23) to C-18 (δC 37.4) and C-28 (δC 129.0) and from H-33 (δH 8.06) to C-27 (δC 196.7). The relative configuration of doitunggarcinone A was previously deduced from ROESY correlations, and 3-methylbut-3-enyl at C-5 was determined as α-orientation which was opposite to CH3-26 configuration. However, the relative stereochemistry of doitunggarcinone A have been revised by synthesis and comparison of the 13C NMR spectral data with those of garcibracteatone and 5-epi-garcibracteatone. It was suggested that the stereochemistry at C-5 is a β-orientation.16 In order to determine the relative stereochemistry of 4, 1H, 13C-NMR and 2D-NMR of 4 were measured again in CDCl3. By comparison of the 13C NMR spectral data of 4 with those of doitunggarcinone A in CDCl3, it suggested that the relative stereochemistry of 4 is the same as that of doitunggarcinone A. Thus, the structure of 4 was deduced completely as showed in the Fig. 1.
Garcixanthochymone E (5) was obtained as a white powder and gave the molecular formula C33H40O6 with 14 degrees of unsaturation by its HR-ESI-MS ([M + Na]+ m/z 555.2726, calcd 555.2723), which was the same as that of 4. By comparing the NMR data of 5 with those of 4, it indicated that 5 was the isomer of 4. However, the main difference found was that compound 5 displays 1H NMR and 13C NMR data for a 3-methylbut-2-enyl group at C-5 instead of a 3-methylbut-3-enyl group in 4. This deduction was supported by 1H–1H COSY correlations of H2-4/H-5/H2-21/H-22. Thus, the structure of 5 was determined and named garcixanthochymone E.
Polycyclic polyprenylated acylphloroglucinols (PPAPs) are a class of secondary metabolites that usually possess bicyclo[3.3.1]nonane-2,4,9-trione, adamantyl, or homoadamantyl-like core structures.17 Up to now, 7 PPAPs have been isolated and identified from G. xanthochymus, such as xanthochymol and isoxanthochymol which belongs to bicycle [3.3.1] nonane-2,4,9-trione type.6 To the best of our knowledge, it was firstly time to isolate adamantly PPAPs from this plant. In addition, compound 3 featured two additional furan and pyrano rings incorporated into the tricyclo [3.3.1.13,7] decane system which are unique in natural adamantyl PPAPs. Few adamantyl-like PPAPs, such as sampsoniones A, B,18 hyperisampsins D19 and xerophenones A and B20 were found to possess a hemiketal carbon in the core skeleton, therefore, 3 represents the first example of adamantyl PPAPs with an unusual ketal carbon in the adamantyl skeleton.
Structurally, compound 4 and 5 was elucidated to possess an unprecedented tetracyclo-[4.3.2.11,6.03,7] decane core similar to that of garcibracteatone,21 doitunggarcinone A,15 hyperuralone B,22 hyphenrone R and S.23 Up to now, this type of caged PPAPs is the most structurally complex PPAP natural product with a highly compact polycyclic ring system containing seven stereocenters, five of which are quaternary. The biosynthetic pathway of 5 was proposed as shown in Scheme 1. An intramolecular Diels–Alder reaction of M would form the key intermediate A, then underwent radical cyclization, tautomerization and oxidative rearomatization to give 5.
 | ||
| Scheme 1 Plausible biogenetic pathway for 5. | ||
The twelve known compounds including 7 xanthones (6–12) and 5 flavonoids (13–17)were identified as 1,4,5-trihydroxyxanthone (6),24 1,3,7-trihydroxyxanthone (7),25 1,3,5-trihydroxyxanthone (8),26 1,5,6-trihydroxy-3-methoxyxanthone (9),26 1,3,6-trihydroxy-7-methoxyxanthone (10),27,28 2,5-dihydroxy-1-methoxylxanthone (11),29 1,3,5,6-tetrahydroxy-2-isoprenylxanthone (12),30 naringenin (13),31 6-prenyl-4′,5,7-trihydroxyflavone (14),32 GB-2a (15),33 (±)-fukugetin (16),34 volkensiflavone (17)34 by comparison of the spectroscopic data with those reported in the literature.
3.2 The antiproliferative activities of isolated compounds
The antiproliferative activities of 17 compounds were evaluated on four human tumor cell lines (HepG2, A549, SGC7901, MCF-7). As shown in Table 3, only compounds 1–5 and 12 could inhibit cancer proliferation derived from different tissues. PPAP derivatives 1–5 exhibited a stronger inhibitory effect on all four cancer cell, especially more effectively against HepG2 cell lines with IC50 values less than 10 μM. These results suggested that PPAPs might be responsible for the antitumor effect of G. xanthochymus.4. Conclusions
In this study, chemical investigation on the fruits of Garcinia xanthochymus yielded five new PPAPs (1–5) and 12 known compounds. Their structures were identified by extensive spectroscopic analyses. To the best of our knowledge, this study represents the first reported account of the isolation of adamantyl PPAPs from this plant. All isolated compounds were evaluated anti-proliferative activities against four human tumor cells (HepG2, A549, SGC7901, MCF-7). As a result, PPAPs derivatives (1–5) displayed potential inhibitory activity against four human cancer cell lines with IC50 values of 5.16–16.45 μM. These data suggest that the extracts of the fruits of G. xanthochymus are potent candidates for cancer prevention.Acknowledgements
This work was financially supported by the National Science Foundation of China (No. 31370379) and Key projects of technological innovation of Hubei province (No. 2016ACA138).References
- S. Karuppusamy, G. Muthuraja and K. M. Rajasekaran, Indian J. Nat. Prod. Resour., 2011, 2, 174–178 CAS.
- B. Sharma, P. J. Handique and H. S. Devi, J. Food Sci. Technol., 2015, 52, 894–902 CrossRef PubMed.
- K. U. Yilmaz, S. Ercisli, Y. Zengin, M. Sengul and E. Y. Kafkas, Food Chem., 2009, 114, 408–412 CrossRef CAS.
- S. Kumar, S. Sharma and S. K. Chattopadhyay, Fitoterapia, 2013, 89, 86–125 CrossRef CAS PubMed.
- K. Trisuwan, S. Boonyaketgoson, V. Rukachaisiriku and S. Phongpaichit, Tetrahedron Lett., 2014, 55, 3600–3602 CrossRef CAS.
- S. Baggett, P. Protiva, E. P. Mazzola, H. Yang, E. T. Ressler, M. J. Basile, I. B. Weinstein and E. J. Kennelly, J. Nat. Prod., 2005, 68, 354–360 CrossRef CAS PubMed.
- J. T. Lyles, A. Negrin, S. I. Khan, K. He and E. J. Kennelly, Planta Med., 2014, 80, 676–681 CrossRef CAS PubMed.
- R. K. Baslas and P. Kumar, Acta Cienc. Indica, Chem., 1981, 7, 31–34 CAS.
- R. K. Baslas and P. Kumar, Curr. Sci., 1979, 48, 814–815 CAS.
- M. Fu, H. J. Feng, Y. Chen, D. B. Wang and G. Z. Yang, Chin. J. Nat. Med., 2012, 10, 129–134 CrossRef CAS.
- M. Fu, X. Hu, J. Xu, Y. Chen and G. Z. Yang, Nat. Prod. Res. Dev., 2014, 26, 255–259 Search PubMed.
- P. Protiva, M. E. Hopkins, S. Baggett, H. Yang, M. Lipkin, P. R. Holt, E. J. Kennelly and I. B. Weinstein, Int. J. Cancer, 2008, 123, 687–694 CrossRef CAS PubMed.
- N. Tanaka, Y. Takaishi, Y. Shikishima, Y. Nakanishi, K. Bastow, K. H. Lee, G. Honda, M. Ito, Y. Takeda, O. K. Kodzhimatov and O. Ashurmetov, J. Nat. Prod., 2004, 67, 1870–1875 CrossRef CAS PubMed.
- L. J. Zhang, C. T. Chiou, J. J. Cheng, H. C. Huang, L. M. Yang Kuo, C. C. Liao, K. F. Bastow, K. H. Lee and Y. H. Kuo, J. Nat. Prod., 2010, 73, 557–562 CrossRef CAS PubMed.
- C. Tantapakul, W. Phakhodee, T. Ritthiwigrom, S. Cheenpracha, U. Prawat, S. Deachathai and S. Laphookhieo, J. Nat. Prod., 2012, 75, 1660–1664 CrossRef CAS PubMed.
- H. P. Pepper, H. C. Lam, W. M. Bloch and J. H. George, Org. Lett., 2012, 14, 5162–5164 CrossRef CAS PubMed.
- R. Ciochina and R. B. Grossman, Chem. Rev., 2006, 106, 3963–3986 CrossRef CAS PubMed.
- L. H. Hu and K. Y. Sim, Tetrahedron Lett., 1998, 39, 7999–8002 CrossRef CAS.
- H. Zhu, C. Chen, J. Yang, X. Li, J. Liu, B. Sun, S. Huang, D. Li, G. Yao, Z. Luo, Y. Li, J. Zhang, Y. Xue and Y. Zhang, Org. Lett., 2014, 16, 6322–6325 CrossRef CAS PubMed.
- G. E. Henry, H. Jacobs, S. McLean, W. F. Reynolds and J. Yang, Tetrahedron Lett., 1995, 36, 4575–4578 CrossRef CAS.
- O. Thoison, D. D. Cuong, A. Gramain, A. Chiaroni, N. V. Hung and T. Séveneta, Tetrahedron, 2005, 61, 8529–8535 CrossRef CAS.
- J. Zhang, J. Yang, Y. Liao, X. Yang, J. Ma, Q. Xiao, L. Yang and G. Xu, Org. Lett., 2014, 16, 4912–4915 CrossRef CAS PubMed.
- Y. Liao, S. Yang, X. Li, X. Yang and G. Xu, Sci. China: Chem., 2016, 59, 1216–1223 CrossRef CAS.
- M. Iin uma, H. Tosa, T. Tanaka, F. Asai and R. Shimano, Phytochemistry, 1995, 38, 247–249 CrossRef CAS.
- C. N. Lin, M. I. Chung, S. J. Liou, T. H. Lee and J. P. Wang, J. Pharm. Pharmacol., 1996, 48, 532–538 CrossRef CAS PubMed.
- W. Li, Y. N. Sun, X. T. Yan, S. Y. Yang, C. W. Choi, J. W. Hyun, H. K. Kang, K. Y. Paek and Y. H. Kim, Food Sci. Biotechnol., 2013, 22, 945–949 CrossRef CAS.
- M. Linuma, H. Tosa, T. Ito, T. Tanaka and S. Riswan, Chem. Pharm. Bull., 1996, 44, 1744–1747 CrossRef.
- H. Tosa, M. Linuma, K. I. Murakami, T. Ito, T. Tanaka, V. Chelladurai and S. Riswan, Phytochemistry, 1997, 45, 133–136 CrossRef CAS.
- H. Minami, E. Takahashi, M. Kodama and Y. Fukuyama, Phytochemistry, 1996, 41, 629–633 CrossRef CAS.
- K. Panthong, W. Pongcharoen, S. Phongpaichit and W. C. Taylor, Phytochemistry, 2006, 67, 999–1004 CrossRef CAS PubMed.
- W. Gao, L. Yang, H. H. Li, C. Q. Liu, S. J. Liu and J. M. Hu, Mod. Chin. Med., 2015, 17, 311–314 CAS.
- S. J. Lee, A. R. Wood, C. G. A. Maier, R. A. Dixon and T. J. Mabry, Phytochemistry, 1998, 49, 2573–2577 CrossRef CAS.
- B. M. Abegaz, B. T. Ngadjui, E. Dongo and H. Tamboue, Phytochemistry, 1998, 49, 1147–1150 CrossRef CAS.
- R. S. Compagnone, A. C. Suarez, S. G. Leitao and F. D. Monache, Rev. Bras. Farmacogn., 2008, 18, 6–10 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra01543g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |