
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Perfect symmetrical cyclic aromatic trimer motif in tripodal molecule†
Shankar Deval Sathiyashivana,
Chakka Kiran Kumar b,
Bhaskaran Shankarb,
Malaichamy Sathiyendiran*b and
Dhanraj T. Masram*a
b,
Bhaskaran Shankarb,
Malaichamy Sathiyendiran*b and
Dhanraj T. Masram*a
aDepartment of Chemistry, University of Delhi, India. E-mail: dtmasram@chemistry.du.ac.in
bSchool of Chemistry, University of Hyderabad, India. E-mail: msathi@uohyd.ac.in
First published on 20th March 2017
Abstract
A noncovalent perfect symmetrical cyclic aromatic trimer motif stabilized through edge-to-face C–H⋯π interactions was found in substituted benzimidazolyl-based tripodal molecule. The distance 4.8 Å, centroid to centroid, and interplanar angle 60° between any two aromatic units in the trimer motif are in excellent agreement with the theoretically predicted symmetrical cyclic benzene trimer cluster.
Edge-to-face C–H⋯π and face-to-face π⋯π interactions between aromatic units in biomolecules result in dimers, trimers, tetramers and oligomers, and are vital to the structural stabilization of DNA, proteins, protein folding, and protein–protein recognition.1–3 Theoretical as well as synthetic investigations of the benzene dimer that is prevalent both in bio- and man-made molecules are now well-documented.1–3 Theoretical studies of aromatic clusters beyond the benzene dimer, in particular cyclic aromatic trimers (CATs), have been intensively carried out by several groups due to their presence in half of all proteins and CATs are considered as a basic unit for higher-order aromatic clusters (Fig. 1).1–3 However, the design and synthesis of most stable CATs are very limited.1–4 Assembling CAT motifs from small biomolecules is challenging because three aromatic units have to be arranged in a cyclic manner with an intermonomer distance of 5.8 ± 1 Å (dCOM⋯COM, centre of mass (COM) to COM) and dihedral angle (τ) of ∼60°. Furuta et al. have reported the cyclic benzene trimer (CBT) motif in the supramolecular structure I with metric parameters (dCOM⋯COM = 4.97/4.62/5.00 Å; τ = 68/33/59°; Fig. S1 in the ESI†) which are close to the calculated distance for the optimized geometry of the C3h-symmetric CBT motif (dCOM⋯COM = 4.8 Å).5a We have recently reported a method to assemble CAT motifs in organic molecules.6 The above approach provides close to symmetrical CAT motifs with average intermonomer distances of 5.06 to 5.43 Å with the average dihedral angle of 60° (Table S1 in the ESI†). Though both methods offer excellent approaches for making CAT motifs in metal complexes and organic molecules, to the best of our knowledge, synthetic complexes or organic molecules possessing perfect CAT motifs are scarce.1–6 Herein, we report the first perfect symmetrical CAT motif in an organic molecule as theoretically predicted for the CBT. A family of mono-/di-/tri-methoxyphenyl substituted benzimidazolyl based molecules (1–9) with/without alkyl substituted spacers is synthesized (Fig. 2). Molecules 2–3, 5–6 and 8–9 with alkyl substituted spacers show a CAT motif in solution. Molecules 3 and 6 were further characterized using single crystal X-ray diffraction analysis and were found to possess a CAT motif in the solid state; in particular molecule 6 has a perfect CAT motif with a distance of 4.88 Å (dCOM⋯COM) and dihedral angle (τ) of 60°.
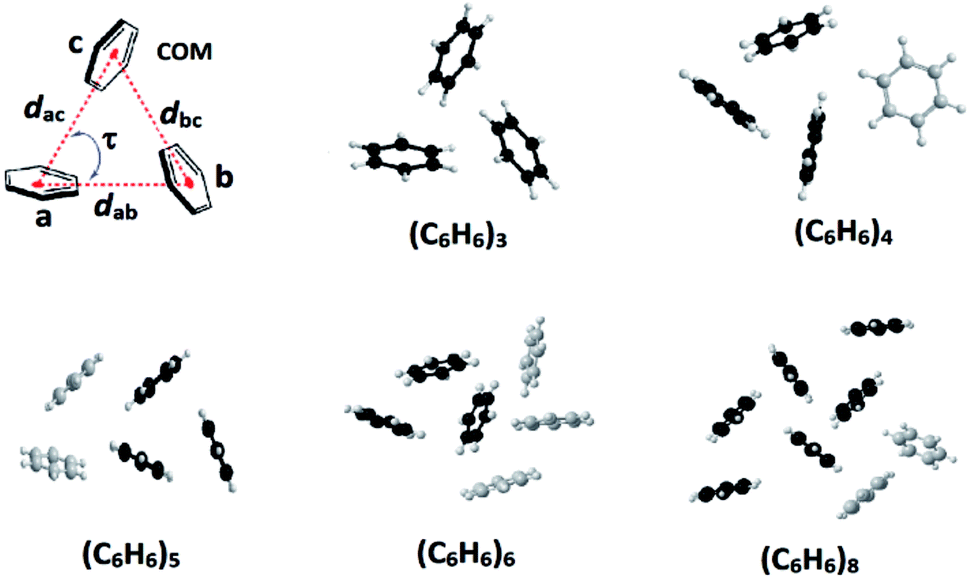 | ||
| Fig. 1 Cyclic benzene trimer (CBT) motif (d = dCOM⋯COM; COM = centre of mass; τ = dihedral angle) and low-lying optimized structures of the trimer, and higher order benzene clusters possessing the CBT basic unit(s).1,2,3f | ||
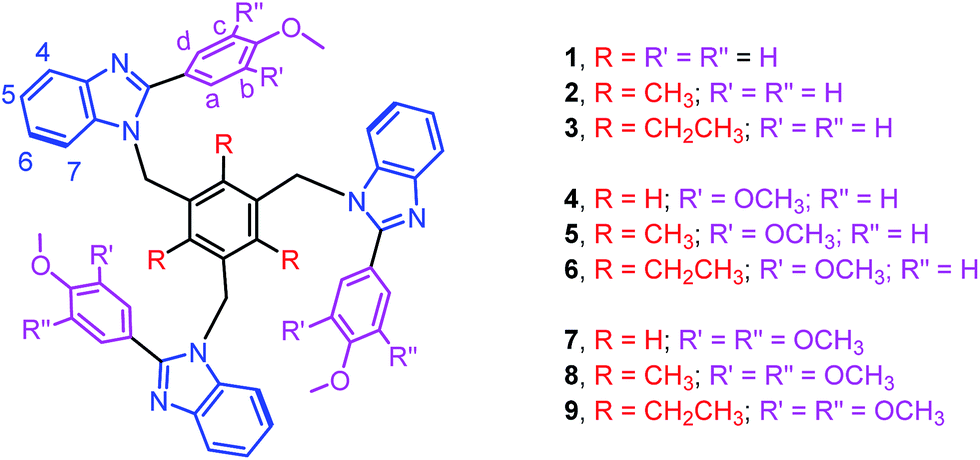 | ||
| Fig. 2 Chemdraw structures of molecules 1–9. | ||
The treatment of 2-substituted benzimidazoles (L1–L3) with 1,3,5-tris(bromomethyl)benzene, 2,4,6-tris(bromomethyl)mesitylene or 1,3,5-tris(bromomethyl)-2,4,6-triethylbenzene in the presence of NaH yielded the corresponding tripodal molecules 1–9 (Fig. 2).6,7 The molecules are air and moisture stable and soluble in organic solvents. All molecules are characterized using elemental analysis and high resolution mass and 1H NMR spectroscopic methods. The electrospray ionization mass spectra of 1–9 displayed molecular ion peaks (m/z for [M + H]+) that match the theoretical values.
Molecule 6 crystallized in the P![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) c1 space group. Each asymmetric unit contains one third of molecule 6 and adopts a syn-conformation with three benzimidazolyl units above the central benzene core, and three dimethoxyphenyl units and three ethyl units below it (Fig. 3). The dihedral angle (τ) between the benzimidazolyl units is 60°. The distance between the centre of mass (COM) of one benzene unit of benzimidazolyl to the COM of the adjacent benzene unit of benzimidazolyl unit is 4.88 Å. These bonding parameters suggest that three benzimidazolyl units are arranged in a cyclic manner similar to the optimized perfect symmetrical CBT cluster.
c1 space group. Each asymmetric unit contains one third of molecule 6 and adopts a syn-conformation with three benzimidazolyl units above the central benzene core, and three dimethoxyphenyl units and three ethyl units below it (Fig. 3). The dihedral angle (τ) between the benzimidazolyl units is 60°. The distance between the centre of mass (COM) of one benzene unit of benzimidazolyl to the COM of the adjacent benzene unit of benzimidazolyl unit is 4.88 Å. These bonding parameters suggest that three benzimidazolyl units are arranged in a cyclic manner similar to the optimized perfect symmetrical CBT cluster.
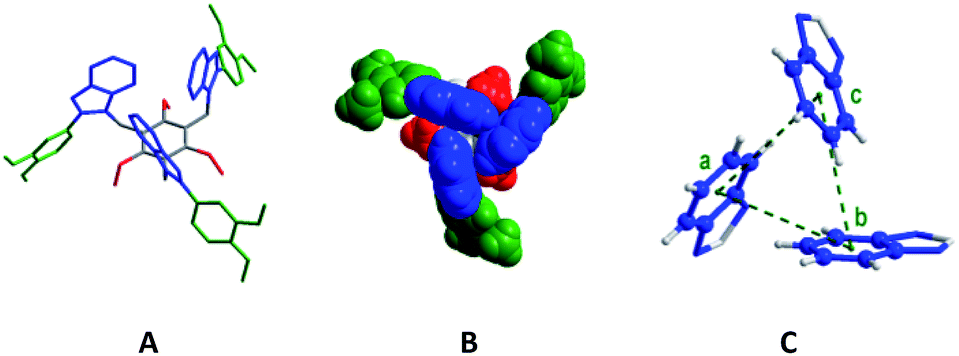 | ||
| Fig. 3 Molecular structures of 6 (A: stick model; H-atoms are removed; B: space-filling model; C: three benzimidazolyl units of 6), and a, b and c are the COM points of the benzene ring. (Color code: dimethoxyphenyl = green, ethyl = red, benzene = grey, benzimidazolyl = blue, and H-atoms in C = grey). | ||
The monomethoxyphenyl-substituted molecule 3 also adopts the syn-conformation similar to molecule 6 (Fig. S1 in the ESI†). The bonding parameters (dCOM⋯COM = 5.32/4.92/5.24 Å and τ = 72/74/34°) in the three benzimidazolyl units of 3 suggest that it possesses the unsymmetrical CAT motif which is stabilized through edge-to-face C–H⋯π interactions. Molecule 3 and tripodal molecules with furan-/thiophene-/2-pyridyl-substituted benzimidazolyl and alkyl substituted spacers (II–IV, Table S1 in the ESI†) adopt the CAT motif with a significant deviation from the perfect CBT motif, i.e. three different distances between the benzene units (dCOM⋯COM) and three dihedral angles between these three units (Table S1 in the ESI†).6
The crystal structures of 3 and 6 are stabilized by extensive inter-ligand C(alkyl)–H⋯π, and edge-to-face C–H⋯π interactions (Fig. S2–S5 in the ESI†). Among the three alkyl chains in molecule 3, two alkyl chains are involved in the CH⋯π interactions with the neighbouring methoxyphenyl unit (C(CH2)–H⋯π(COM), d, C27–H/C16–H⋯COM = 2.82/2.71 Å; ∠C–H⋯COM = 149/129°). Intermolecular edge-to-face C–H⋯π interactions were found between the two anti-cofacially arranged methoxyphenylbenzimidazolyl units (τ = 35/37°; d, C53–H/C52–H ⋯COM(benzene)/COM(imidazole) = 3.01/2.71 Å; ∠C–H⋯COM = 121/125°). The third methoxyphenylbenzimidazolyl unit interacts with the neighbouring benzimidazolyl unit via offset π⋯π stacking interactions (d, C⋯C = 3.48–3.64 Å). One benzene in the CBT unit is in contact with the neighbouring benzene spacer via edge-to-face CH/π interactions (d, C52–H⋯COM = 2.71 Å; ∠C–H⋯COM = 125°) (Fig. S3 in the ESI†).
The three neighbouring molecules in the crystal structure of 6 are arranged in a circular shape and are in contact with the central molecule. Both C(alkyl)–H⋯π, and edge-to-face C–H⋯π interactions are observed between the two anti-cofacially arranged methoxyphenylbenzimidazolyl units (d, C19–H⋯COM (methoxyphenyl unit) = 2.96 Å; ∠C–H⋯COM = 135° and C12–H/C13–H⋯COM = 3.03/2.82 Å; ∠C–H⋯COM = 123/127°). In contrast to the solid state structure of 3, all of the benzene rings of the CBT unit of 6 are in contact with the neighbouring benzene spacer with edge-to-face C–H⋯π interactions (d, C3–H⋯C17/COM (benzene) = 3.08/3.85 Å; ∠C–H⋯C17/COM = 156/139°) (Fig. S4–S5 in the ESI†).
The 1H and 13C NMR spectra of all of the molecules (1–9) were recorded in d6-DMSO (Fig. S6–S29 in the ESI†). Molecule 2 displays a well-separated and single set of chemical resonances for all of the protons (Fig. S6 in the ESI†). H6 and H7 of the benzimidazolyl unit of 2 appeared as a triplet and doublet with a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 proton ratio (Fig. 4A and S6 in the ESI†). These protons were significantly upfield shifted by 0.54 and 1.18 ppm compared to 4-methoxyphenylbenzimidazole (L1). The remaining two protons (H4 and H5) of the benzimidazolyl unit of 2 were slightly downfield shifted. These data reveal that molecule 2 adopts the syn-conformation with an edge-to-face arrangement of the three benzimidazolyl units, i.e. a ‘cyclic benzene trimer’ arrangement similar to the solid state arrangement of the three benzimidazolyl units in 3 and 6 with the C6(H)–C7(H) edge of the benzene ring directed over the face of the neighbouring benzene ring (Fig. 3). Similarly, molecules 5 and 8, consisting of 2 and a methoxy group(s) (–OCH3 for 5; two –OCH3 groups for 8), also display a similar 1H NMR pattern for the protons of the benzimidazolyl units (Fig. S9 in the ESI†).
:3 proton ratio (Fig. 4A and S6 in the ESI†). These protons were significantly upfield shifted by 0.54 and 1.18 ppm compared to 4-methoxyphenylbenzimidazole (L1). The remaining two protons (H4 and H5) of the benzimidazolyl unit of 2 were slightly downfield shifted. These data reveal that molecule 2 adopts the syn-conformation with an edge-to-face arrangement of the three benzimidazolyl units, i.e. a ‘cyclic benzene trimer’ arrangement similar to the solid state arrangement of the three benzimidazolyl units in 3 and 6 with the C6(H)–C7(H) edge of the benzene ring directed over the face of the neighbouring benzene ring (Fig. 3). Similarly, molecules 5 and 8, consisting of 2 and a methoxy group(s) (–OCH3 for 5; two –OCH3 groups for 8), also display a similar 1H NMR pattern for the protons of the benzimidazolyl units (Fig. S9 in the ESI†).
The 1H NMR spectra of molecules 3, 6 and 9, possessing three ethyl groups on the central benzene scaffold, show a similar upfield shift for H6 and H7 of the benzimidazolyl protons (Fig. 4B and S6–S8 in the ESI†). However, these two protons were further upfield shifted in comparison to molecules 2, 5, and 8 (Fig. 4). In particular, the separation between H6 and H7 in 3, 6 and 9 is small compared to in 2, 5, and 8. Though these two protons appeared as a triplet and doublet, their signals started to broaden in 3. In the case of 6, a broad singlet was observed for H6. Both H6 and H7 were broadened in 9. The peak positions of H6 and H7 of molecules 3, 6 and 9 are almost similar, indicating the very similar arrangement of the benzimidazolyl units. A similar kind of broadening was also observed in the case of the tripodal molecules with furan-/thiophene-/2-pyridyl-substituted benzimidazolyl and ethyl substituted benzene spacers. The broadening of H6 and H7 of the benzimidazolyl unit in the molecules is attributed to the three types of syn-conformers existing in solution, which undergo rapid equilibrium in the NMR time scale.6b This may also occur presumably on account of some slowed partial rotation of the three benzimidazolyl units. The higher upfield shift of these two protons reveals that the CBT interactions in solution are stronger in 3, 6 and 9 than in 2, 5, and 8.
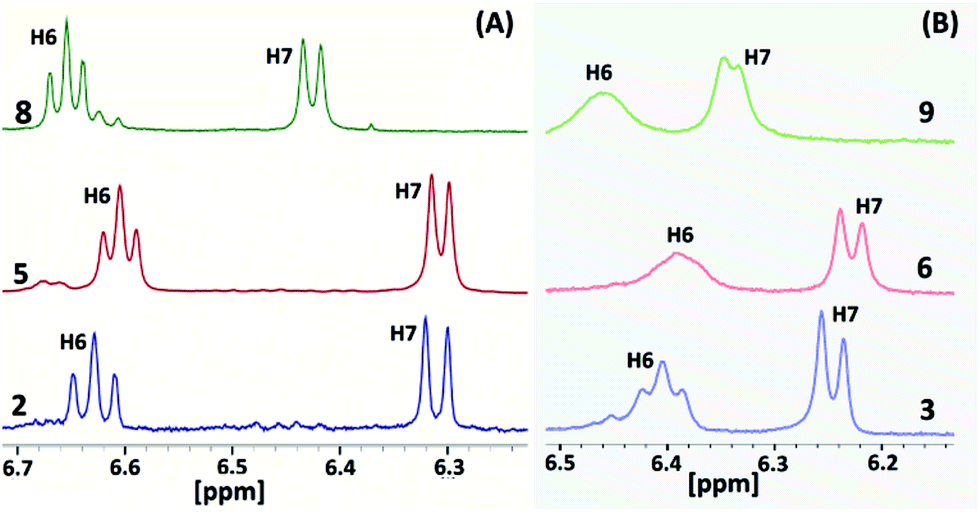 | ||
| Fig. 4 Partial 1H NMR spectra of 2, 5 and 8 (A) and 3, 6 and 9 (B) in d6-DMSO. | ||
These parameters clearly suggest that the ethyl substituted centre benzene scaffold based molecules 3, 6 and 9 possess the CBT motif with stronger edge-to-face C–H⋯π interactions in solution than the methyl substituted molecules 2, 5, and 8. Among the ethyl/methyl substituted benzene centre scaffold molecules, the mono-/di-methoxyphenyl benzimidazolyl based molecules (2–3 and 5–6) display stronger CBT interactions than the trimethoxyphenyl substituted molecules (8–9).
It is well known that the benzimidazolyl-based ligand coordinates to several metal ions.8 The conformation of the ligand in the final product, either a discrete or polymer complex, depends on the geometric requirements of the metal ions and the packing interactions, steric group, and the crystallization of the solvent.8 To check whether the molecules 2–3, 5–6 and 8–9 keep the CBT motif intact in the presence of metal ions in solution, silver(I) ion is added to the DMSO-d6 solution of molecule 5 (Fig. 5). A significant downfield shift was observed for the H4 proton, adjacent to a tertiary nitrogen, due to coordination of the ligand to silver ions. However, H6 and H7 are significantly upfield shifted in comparison to 3,4-dimethoxyphenylbenzimidazole. In addition, the upfield shift value for H6 is slightly more than that in molecule 5. These observations suggest that the CBT cluster remains intact even after 5 coordinates with the silver ion. However, the CAT is stabilized by weak edge-to-face C–H⋯π interactions in comparison to molecule 5.
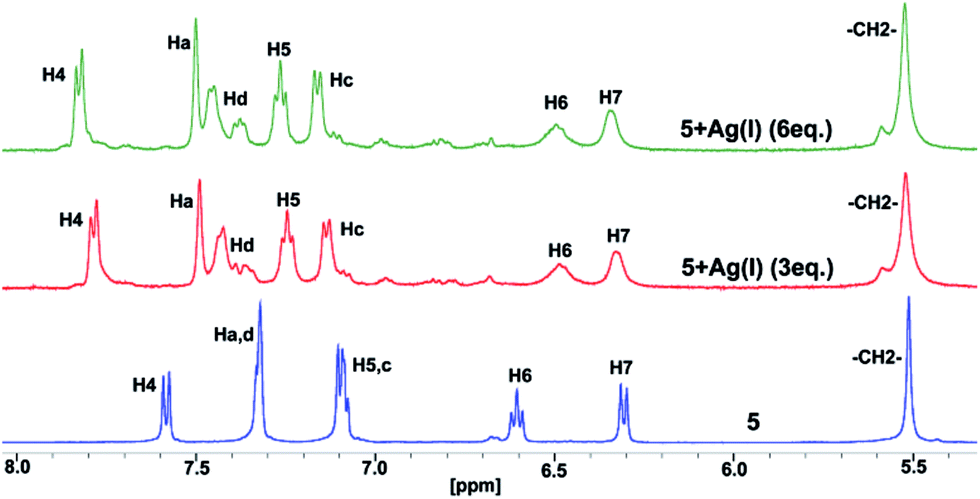 | ||
| Fig. 5 Partial 1H NMR spectra of 5 and 5 + Ag(I) in d6-DMSO. | ||
In conclusion, a family of organic molecules possessing CAT motifs was prepared. The study also provides the first synthetic molecule possessing a perfect symmetrical CAT as predicted theoretically for the low-energy CBT motif. These results will be highly useful for both theoretical work for further information on noncovalent aromatic clusters, which are vital for the structural stabilization and function of biomolecules, and for the design and synthesis of aromatic clusters using various biomolecules.
Acknowledgements
S. D. S. thanks the UGC-NRC, C. K. K. thanks the UGC-DSKPDF, and UPE-II for providing funding.References
- (a) T. P. Sauer and C. D. Sherrill, J. Phys. Chem. A, 2005, 109, 10475 CrossRef PubMed; (b) E. Lanzarotti, R. R. Biekofsky, D. A. Estrin, M. A. Marti and A. G. Turjanski, J. Chem. Inf. Model., 2011, 51, 1623 CrossRef CAS PubMed and references therein; (c) S. R. Gadre, S. D. Yeole and N. Sahu, Chem. Rev., 2014, 114, 12132 CrossRef CAS PubMed; (d) A. S. Mahadevi and G. N. Sastry, Chem. Rev., 2016, 116, 2775 CrossRef CAS PubMed; (e) C. Janiak, J. Chem. Soc., Dalton Trans., 2000, 3885 RSC; (f) M. Nishio, Phys. Chem. Chem. Phys., 2011, 13, 13873 RSC; (g) O. Takahashi, Y. Kohno and M. Nishio, Chem. Rev., 2010, 110, 6049 CrossRef CAS PubMed.
- (a) B. W. Van De Waal, Chem. Phys. Lett., 1986, 123, 69 CrossRef CAS; (b) A. De Meijere and F. Huisken, J. Chem. Phys., 1990, 92, 5826 CrossRef CAS; (c) H. Krause, B. Ernstberger and H. J. Neusser, Chem. Phys. Lett., 1991, 184, 411 CrossRef CAS; (d) C. A. Hunter, J. Singh and J. M. Thornton, J. Mol. Biol., 1991, 218, 837 CrossRef CAS PubMed; (e) W. J. Pullan, J. Chem. Inf. Model., 1997, 37, 1189 CrossRef CAS; (f) O. Engkvist, P. Hobza, H. L. Selzle and E. W. Schlag, J. Chem. Phys., 1999, 110, 5758 CrossRef CAS; (g) N. Kannan and S. Vishveshwara, Protein Eng., 2000, 13, 753 CrossRef CAS PubMed; (h) C. Gonzalez and E. C. Lim, J. Phys. Chem. A, 2001, 105, 1904 CrossRef CAS; (i) C. Gonzalez and E. C. Lim, J. Phys. Chem. A, 2001, 105, 10583 CrossRef CAS.
- (a) T. Iimori, Y. Aoki and Y. Ohshima, J. Chem. Phys., 2002, 117, 3675 CrossRef CAS; (b) X. Ye, Z. Li, W. Wang, K. Fan, W. Xu and Z. Hua, Chem. Phys. Lett., 2004, 397, 56 CrossRef CAS; (c) D. C. Easter, D. A. Terrell and J. A. Roof, J. Phys. Chem. A, 2005, 109, 673 CrossRef CAS PubMed; (d) M. Swart, T. van der Wijst, C. F. Guerra and F. M. Bickelhaupt, J. Mol. Model., 2007, 13, 1245 CrossRef CAS PubMed; (e) H. Takeuchi, J. Chem. Inf. Model., 2007, 47, 104 CrossRef CAS PubMed; (f) A. S. Mahadevi, A. Rahalkar, S. R. Gadre and G. N. Sastry, J. Chem. Phys., 2010, 133, 164308 CrossRef PubMed; (g) M. Chourasia, G. M. Sastry and G. N. Sastry, Int. J. Biol. Macromol., 2011, 48, 540 CrossRef CAS PubMed; (h) J. Yang and M. P. Waller, J. Phys. Chem. A, 2013, 117, 174 CrossRef CAS PubMed.
- S. Kumar and A. Das, J. Chem. Phys., 2012, 136, 174302 CrossRef PubMed.
- (a) T. Morimoto, H. Uno and H. Furuta, Angew. Chem., Int. Ed. Engl., 2007, 46, 3672 CrossRef CAS PubMed; (b) M. Sathiyendiran, J. Y. Wu, M. Velayudham, G. H. Lee, S. M. Peng and K. L. Lu, Chem. Commun., 2009, 3795 RSC; (c) S. H. Lin, C. I. Yang, T. S. Kuo, M. H. Chiang, K. C. Hsu and K. L. Lu, Dalton Trans., 2012, 41, 1448 RSC; (d) J. Zaho, K. Peng, Y. Guo, J. Zhang, S. Chen and J. Hu, New J. Chem., 2015, 39, 6016 RSC; (e) I. Kato, T. Nakagaki, T. Shimasaki and T. Shinmyozu, CrystEngComm, 2008, 10, 483 RSC; (f) N. Koch and M. Mazik, Synthesis, 2013, 45, 3341 CrossRef CAS; (g) B. E. Conn, A. Atnagulov, B. Yoon, R. N. Barnett, U. Landman and T. P. Bigioni, Sci. Adv., 2016, 2, e1601609 Search PubMed.
- (a) P. Elumalai, P. Rajakannu, F. Hussain and M. Sathiyendiran, RSC Adv., 2013, 3, 2171 RSC; (b) S. D. Sathiyashivan, B. Shankar, P. Rajakannu, P. Vishnoi, D. T. Masram and M. Sathiyendiran, RSC Adv., 2015, 5, 74705 RSC.
- (a) C. Y. Su, Y. P. Cai, C. L. Chen, F. Issner, B. S. Kang and W. Kaim, Angew. Chem., Int. Ed., 2002, 41, 3371 CrossRef CAS; (b) S. Janich, R. Frohlich, A. Wakamiya, S. Yamaguchi and E. U. Wurthwein, Chem.–Eur. J., 2009, 15, 10457 CrossRef CAS PubMed; (c) D. Y. Lee, N. Singh, M. J. Kim and D. O. Jang, Org. Lett., 2011, 13, 3024 CrossRef CAS PubMed; (d) B. Shankar, F. Hussain and M. Sathiyendiran, J. Organomet. Chem., 2012, 719, 26 CrossRef CAS; (e) P. Rajakannu, P. Elumalai, F. Hussain and M. Sathiyendiran, J. Organomet. Chem., 2013, 725, 1 CrossRef CAS.
- (a) C. Y. Su, Y. P. Cai, C. L. Chen, M. D. Smith, W. Kaim and H. C. zur Loye, J. Am. Chem. Soc., 2003, 125, 8595 CrossRef CAS PubMed; (b) V. Amendola, G. Bergamaschi, M. Boiocchi, L. Fabbrizzi and N. Fuscoa, Dalton Trans., 2011, 40, 8367 RSC; (c) C. Y. Su, M. D. Smith and H. C. zur Loye, Angew. Chem., Int. Ed., 2003, 42, 4085 CrossRef CAS PubMed; (d) S. Samai and K. Biradha, Cryst. Growth Des., 2011, 11, 5723 CrossRef CAS; (e) F. Jin, X. F. Yang, S. L. Li, Z. Zheng, Z. P. Yu, L. Kong, F. Y. Hao, J. X. Yang, J. Y. Wu, Y. P. Tian and H. P. Zhou, CrystEngComm, 2012, 14, 8409 RSC; (f) J. D. Crowley and P. H. Bandeen, Dalton Trans., 2010, 39, 612 RSC; (g) S. Bhattacharya and B. K. Saha, J. Chem. Sci., 2016, 128, 207 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: X-ray crystallographic file in CIF format for 3 and 6. CCDC 1531052 and 1531800. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra01682d |
| This journal is © The Royal Society of Chemistry 2017 |