
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and anticancer activity of prodigiosenes bearing C-ring esters and amides†
Kate-lyn A. R. Lund,
Carlotta Figliola,
Aleksandra K. Kajetanowicz and
Alison Thompson *
*
Department of Chemistry, Dalhousie University, P.O. Box 15000, Halifax, Nova Scotia B3H 4R2, Canada. E-mail: Alison.Thompson@dal.ca
First published on 27th March 2017
Abstract
Prodigiosenes, a class of compounds characterised by their 4-methoxypyrrolyldipyrrin framework, are known to possess anti-cancer activity. Structural modification of the C-ring of prodigiosenes presented ten new prodigiosenes bearing pendant alkyl esters and amides. These prodigiosenes were evaluated biologically and displayed notable anticancer in vitro activity, particularly when featuring a hexyl chain.
Introduction
Prodigiosin (Fig. 1) is a secondary metabolite isolated from certain Gram-positive and Gram-negative bacteria, including Serratia marcescens.1 This compound is the parent member of the class of red-pigmented prodigiosenes,2 characterised by the 4-methoxypyrrolyldipyrrin framework, and exhibits anticancer,3–6 immunosuppressive,7 antimicrobial4,8 and antimalarial9–11 activities.12–15 Despite longstanding interest in the properties and applications of prodigiosin, there remains a wealth of current research dedicated to this natural product and its analogues. For example, the isolation of prodigiosin from natural sources remains sufficiently challenging that fermentation and the use of extraction/isolation procedures such as functionalised iron-oxide particles are under development.16 As such, the total synthesis of prodigiosin17 has been supplemented by various routes18,19 that are amenable to the preparation of analogues, with the most recent development reported to involve the metal-free coupling of F-BODIPYs and unfunctionalised pyrrole.20 Structurally related hybrubins have also recently emerged, bearing a tetramic acid moiety in place of the C-ring of prodigiosin:21 a total synthesis and biological evaluation has quickly followed.22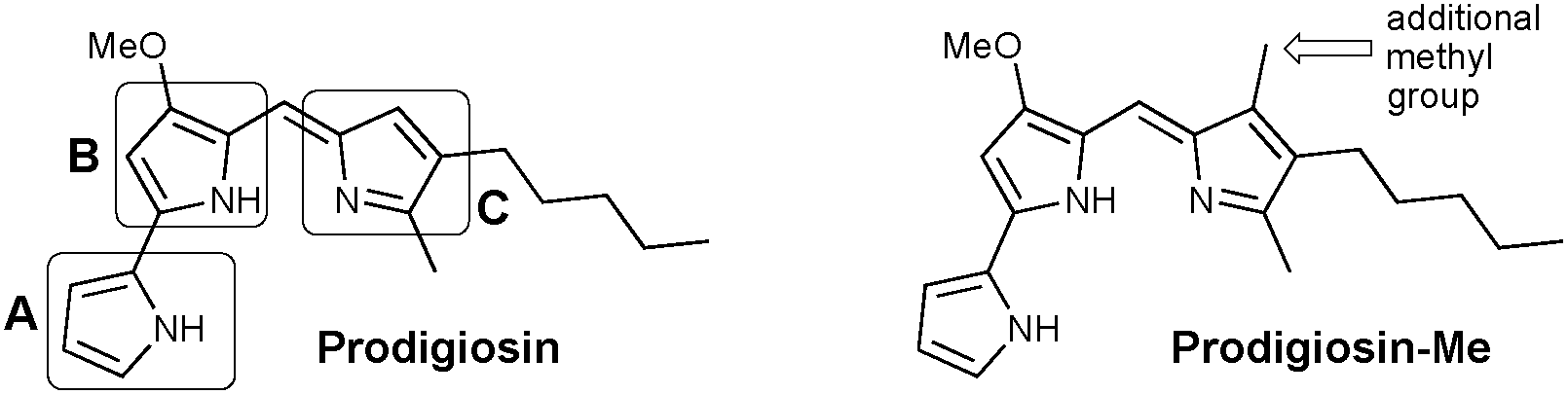 | ||
| Fig. 1 Prodigiosin and the methylated analogue prodigiosin-Me. | ||
Recent reviews have nicely summarised the synthesis, biosynthesis and bioactivity of natural products based on the skeleton of prodigiosin.18,19 Nevertheless, the mechanism(s) via which prodigiosin exerts its bioactivity is still a subject of intense ongoing study, with the most recent developments including: its ability to inhibit Wnt/β-catenin signalling as the basis of clinical potential for treatment of breast cancer;23 its capacity to modify the functions of p53 and p73;24 its ability to disrupt plasma membrane via a chaotropicity-mediated mechanism;25 and the demonstration that it binds G-quadruplex DNA.26 Similarly, the breadth of applications for which prodigiosin itself may be suited has also been recently reviewed,27 with non-drug uses ranging from sunscreens28 to antibacterial dyes for silk29 and cotton.30
Coley's toxin, an early chemotherapeutic, is a mixture of extracts containing prodigiosin which, before being withdrawn by the FDA due to toxicity, was used as a cancer treatment.1 Prodigiosin exhibits systemic toxicity12 at effective cancer doses and as such it is not suitable for clinical development as its therapeutic window is too narrow.31 However, several structurally similar heterocyclic compounds have shown recent clinical potential. For example, Obatoclax (GX15-070) is very similar to prodigiosin, differing by an indole instead of the C-ring pyrrole of prodigiosin, and including a 2,4-dimethyl pyrrole for the C-ring instead of the 2-methyl-3-pentyl C-ring in prodigiosin.32 This drug candidate, courtesy of its ability to inhibit the Bcl-2 family of proteins, has been shepherded through various phase I and II clinical trials including those directed at leukaemia, lymphoma, myelofibrosis and mastocytosis,33 but has not found a route to general clinical use. Very recently, tambjamines and B-ring functionalised prodigiosenes have been shown to be clinically relevant antimalarials with the ability to withstand the development of resistance.34
Against this energetic and multi-faceted backdrop involving analogues of prodigiosin we herein report the synthesis and evaluation of a series of ten prodigiosenes that bear ester and amide substitution about the C-ring of the prodigiosene skeleton.
Previous work in the Thompson group has focused on substituent modifications about the B- and C-rings35,36 of the 4-methoxypyrrolyldipyrrin framework (Fig. 1), with the goals of improving synthetic access37 and evaluating anticancer,38,39 antimicrobial40 and antimalarial41 properties. Key to this endeavour was the discovery that the readily prepared prodigiosin-Me (Fig. 1), containing an extra methyl group on the C-ring, generally maintains the in vitro anticancer activity of prodigiosin and thus serves as a standard in place of the natural product. A series of analogues featuring conjugated ester- and keto-moieties on the C-ring was thus synthesised, and the bioactivity evaluated (Fig. 2, top).36,42 The conjugated carbonyl groups serve to influence the pKa of the target prodigiosenes.36 Furthermore, the ability to prepare C-ring analogues has enabled the conjugation of prodigiosenes to targeting groups such as estrogens.42,43
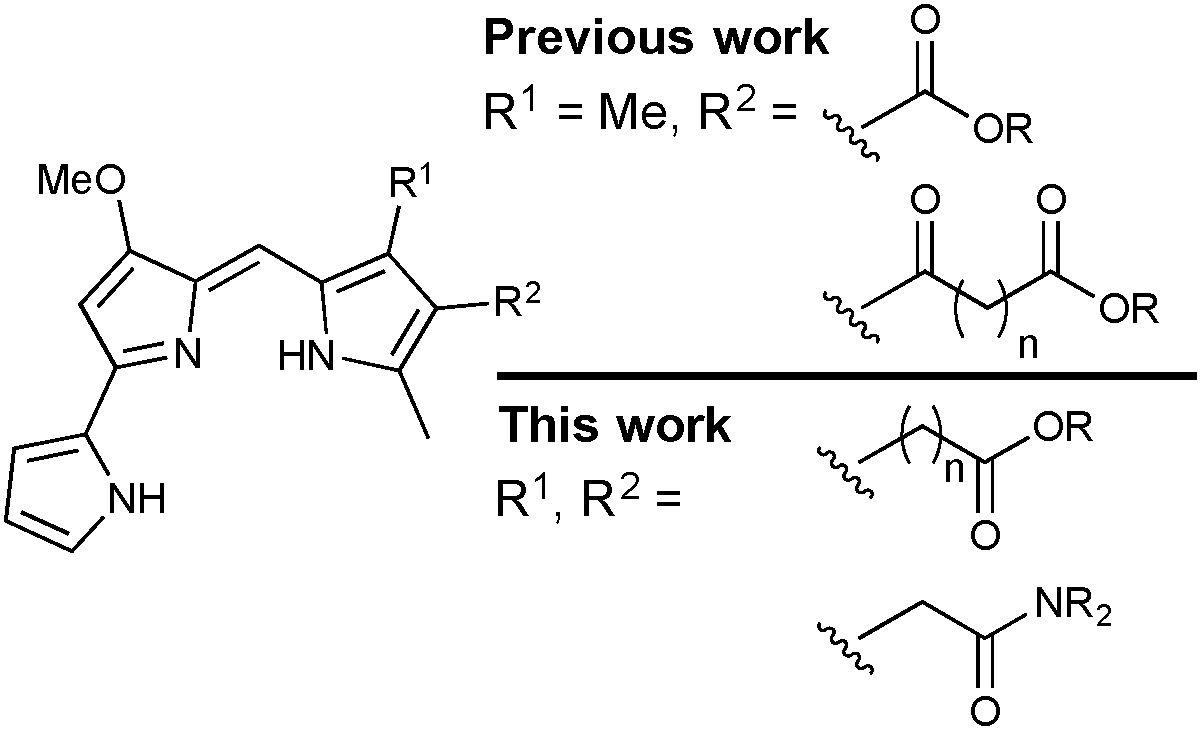 | ||
| Fig. 2 Synthetic prodigiosenes. | ||
The work described herein concerns the synthesis and evaluation of prodigiosenes featuring pendant, non-conjugated alkanoate and amide substitution about the C-ring (Fig. 2, bottom). As such, the pKa of the target prodigiosenes should not be significantly influenced by the nature of ester or amide substituents introduced via modification of the alkanoates. Four compounds of this type have been reported to exhibit interesting selectivity against leukemia cell lines, compared to those representative of eight other human cancers, when evaluated across the National Cancer Institute panel of 60 human cell lines derived from nine different cancer types (NCI-60).44 These were the first prodigiosene analogues to show such obvious selectivity for leukaemia cell lines and they were also the first to bear an alkanoate substitution pattern. In vivo evaluation of these prodigiosenes, using a zebra fish model, revealed that the anti-leukemic properties prevailed in human K562 chronic myelogenous leukaemia cells. Given that the need for new anti-leukaemia agents is pressing,45–47 and that prodigiosenes present different structural features and mechanisms compared to clinically used AraC and anthracyclines, we have prepared ten new prodigiosenes bearing non-conjugated C-ring alkanoate and amide substitution, and evaluated these compounds for their anti-cancer activity, including against six leukaemia cell lines.
Results and discussion
The synthetic route revolved around decorating a Knorr-type pyrrole to render it suitable for condensation to a B-ring surrogate for the prodigiosene skeleton. Activation of the consequent dipyrrolic species, followed by Suzuki coupling with pyrrole boronic acid, would provide the desired prodigiosenes. Given the curious selectivity exhibited by previous C-ring alkanoates,44 we began by preparing derivatives that would enable us to further explore the significance of such substitution. As shown in Fig. 3, the synthesis began with Knorr-type pyrroles 1 and 2. Pyrrole 1 would give rise to the first prodigiosene bearing two propanoates. Pyrrole 2 would enable exploration of the significance of including a directly C-ring conjugated ester alongside an alkanoate. Pyrroles 1 and 2 were prepared in five steps from dibenzyl malonate and benzyl acetoacetate, respectively. Removal of the 2-carboxylate ester substituent from 1 and 2, either through hydrogenolysis (R = Bn) or hydrolysis (R = Et), followed by decarboxylation, enabled the resulting α-free pyrroles to be formylated to produce the 2-formyl pyrroles 3 and 4.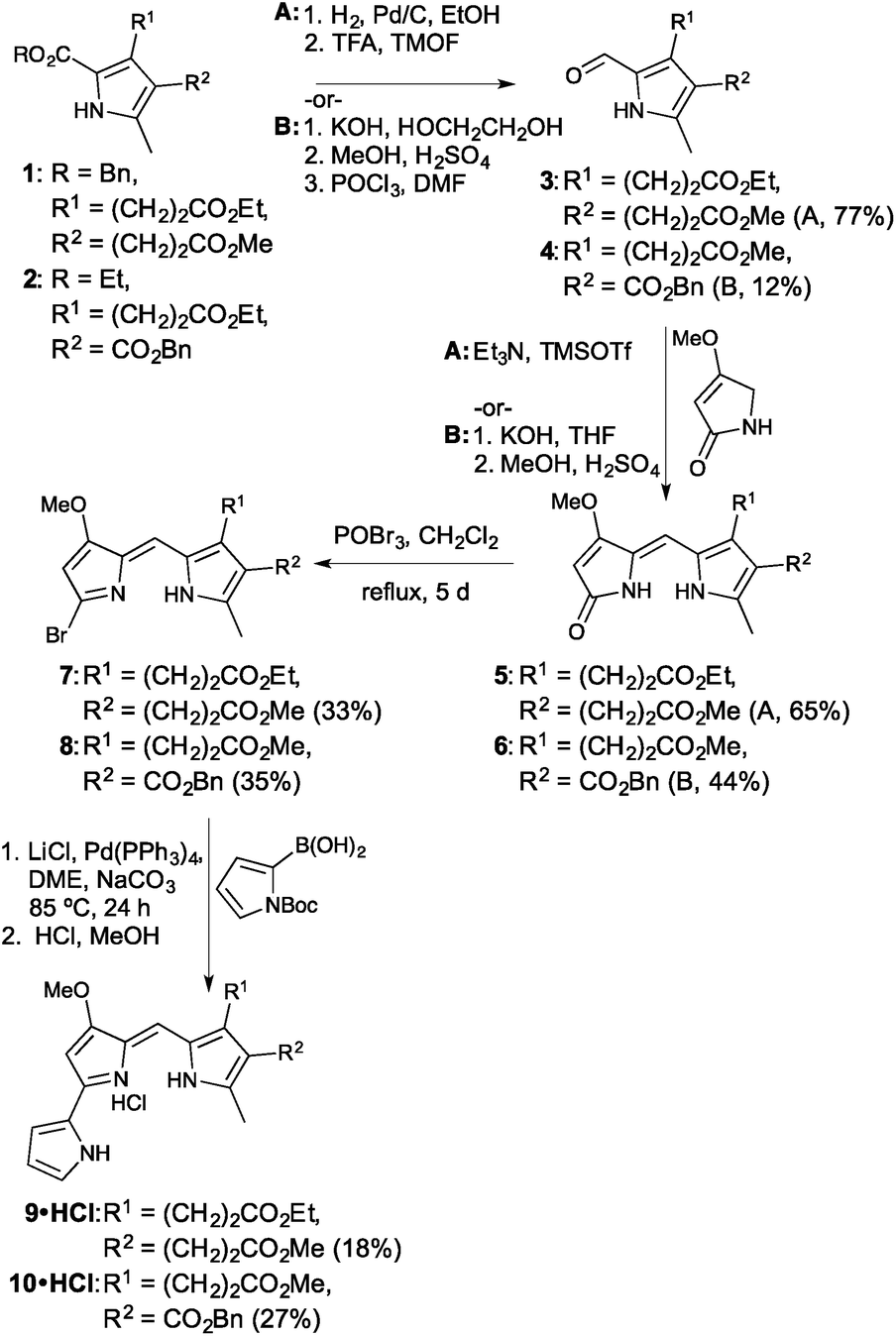 | ||
| Fig. 3 Synthesis of two prodigiosene C-ring alkanoates, 9 and 10. | ||
Condensation of the formyl pyrroles 3 and 4 with 4-methoxy-3-pyrrolin-2-one48 could be achieved under silylative Mukaiyama aldol condensation49 and subsequent elimination of the OTMS functionality,50 or via base-catalysed condensation conditions followed by an acidic quench.39 Although the latter method also serves to saponify any pendant esters, it is useful when the reversibility of the silylative Mukaiyama aldol condensation limits the success of the coupling. In these cases, the use of harsher conditions or extended reaction times serves merely to result in decomposition and so we were forced to use the base-catalysed method, followed by re-esterification, for the fruitful conversion of 4 to 6.
Activation of the dipyrrinones through formation of the bromo-dipyrrins 7 and 8 proceeded smoothly, then Suzuki coupling to N-Boc-pyrrole-2-boronic acid, in a sealed system, resulted in the desired prodigiosenes 9 and 10. Yields were hampered by decomposition of starting material, along with the requirement for two purifications via column chromatography over neutral alumina. The inclusion of alkanoates on the C-ring certainly seems to complicate isolation of the target prodigiosenes.
With two new prodigiosenes bearing C-ring propanoate substitution in hand, we turned our attention to the synthesis of 13 and 14 (Fig. 4). These benzyl esters would complement 9 and 10, as well as enable the comparison of the effect of the benzyl substituent compared to methyl esters.44 Indeed, prodigiosene 13 features a benzyl propanoate and a benzyl ethanoate, whilst 14 features a benzyl propanoate and a methyl substituent at the β-positions of the C-ring. Each was prepared via hydrolysis of the corresponding methyl ester,44 prepared in the same manner as 9 and 10, followed by coupling using benzyl alcohol. The intermediate acids were challenging to manipulate, with success attained via complete removal of the THF and H2O, following completion of the saponification, followed by the addition of 1 M HCl and stirring for 2 h. The acids, as very fine precipitates, could then be isolated via micro-filtration, ready for coupling.
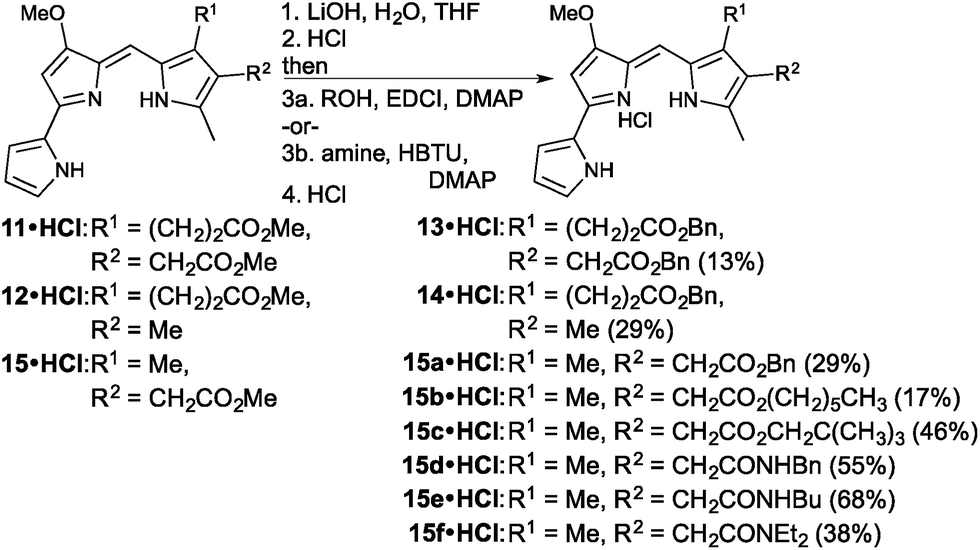 | ||
| Fig. 4 Synthesis of prodigiosenes 13, 14 and 15a–f. | ||
The final set of analogues were derived from prodigiosene 15 which had shown to be most promising in vivo anti-leukaemic activity in a previous study.44 Using a convergent approach, 15 was hydrolysed to give the corresponding ethanoic acid, again isolated as a very fine precipitate. Successful coupling with benzyl, hexyl and neopentyl alcohols led to the isolation of prodigiosenes 15a–c, respectively, to enable evaluation of the significance of the ester type and lipophilicity upon anti-cancer activity. Using a similar approach, and with HBTU as coupling agent, secondary and tertiary amides 15d–f were prepared, cognisant of the fact that amides are less of a metabolic liability than esters.
We were interested to learn how the new alkyl ester and amide prodigiosenes behaved biologically. All ten prodigiosenes (9, 10, 13, 14, 15a–f) were selected for one-dose screening against the NCI panel of 60 human cell lines used in the Development Therapeutics Program (DTP) screening program. The success51 of the NCI in developing new therapeutic agents for the treatment of cancer is evident from a track record of involvement in ≈50% of chemotherapeutic agents currently administered to patients.52 The pattern/fingerprint of cellular sensitivity and resistance of the NCI-60 to a compound correlates well with molecular target expression and identification of desired drugs with mechanisms of action that differ significantly from those of known drugs.53 As such, the rapid go/no go evaluation provided by the NCI serves as an excellent measure of drug potential. In this way, we were able to assess the potential of the compounds against six leukaemia cell lines to determine whether the selectivity shown by the four published alkanoates44 was maintained by the ten variants synthesised herein. Furthermore, evaluation against the NCI-60 would enable us to determine growth inhibition and cytotoxicity effects of our compounds against a further eight cancer types via >50 cell lines representing non-small cell lung, colon, CNS, ovarian, renal, prostate and breast cancers, as well as melanoma.
Fig. 5 plots the growth inhibition of six leukaemia cell lines when treated with 10 μM of prodigiosenes 9, 10, 13, 14 and 15a–f. Fig. 5 reveals that although 9 does little to affect the growth of any of the six cell lines, the other nine prodigiosenes exhibit anticancer activity. Compounds 15b and 15c result in significant death of cells in all six lines, whereas the activity of the other analogues varies between significant growth inhibition and cell death. The HL-60(TB) and RPMI-8226 cell lines are affected differently by esters (10, 13, 14, 15a–c) vs. amides (15d–f). However, in contrast to the bioactivity exhibited by the four previously published alkanoates,44 this one-dose evaluation of C-ring alkanoates or amides revealed that only esters 15a and 15b exhibit significant activity against all the leukaemia cell lines. This contrast demonstrates that substitution of the C-ring results in significant changes in bioactivity. Furthermore, amide derivatives 15d–f showed varying activity against the leukaemia cell lines, again demonstrating the relationship between structure and activity.
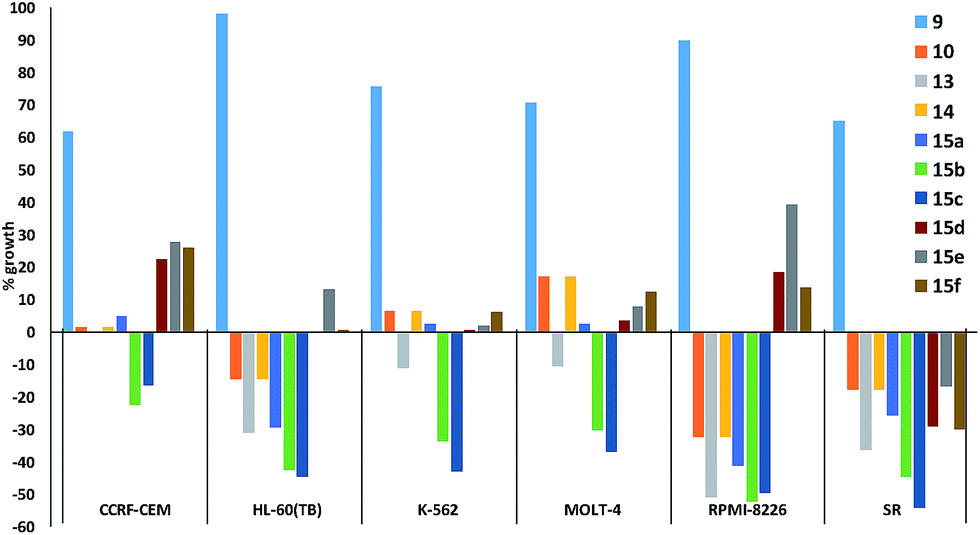 | ||
| Fig. 5 In vitro acidity (% growth) of prodigiosenes 9, 10, 13, 14, 15a–f at 10 μM concentration against six leukaemia cell lines; negative growth represents cell death (http://dtp.cancer.gov). | ||
With the exception of 9 all prodigiosenes showed adequate activity to warrant further five-dose evaluation by the NCI. The average results of the in vitro evaluation, across all lines, is shown in Table 1 (note the negative log scale; activities in terms of drug concentration are reported in the ESI†), alongside those previously reported for prodigiosin and prodigiosin-Me, plus their calculated relative total polar surface area (tPSA).54,55 Three measures of dose–response are included in Table 1: GI50 is the drug concentration that results in a 50% reduction in cell growth; TGI is the drug concentration that results in total growth inhibition; and LC50 is the drug concentration that results in 50% reduction in the measured protein at the end of the drug treatment, i.e. 50% cell loss. As shown in Table 1, most of the evaluated prodigiosenes exhibit modest in vitro activity when compared to the natural product prodigiosin and the model prodigiosin-Me. Intriguingly, the GI50 of 15b reveals that this compound is significantly more active, in terms of growth inhibition of cells, than any of the other esters and amides, potentially pointing to the advantageous role of pentyl or hexyl chains featured in 15b, prodigiosin and prodigiosin-Me. However, the LC50 for 15b is higher than that of prodigiosin and prodigiosin-Me, thus pointing to differentiation between growth inhibition and cell death for this new analogue compared to the natural product and its synthetic mimic. When comparing methyl prodigiosenes 11 and 12 to their corresponding benzyl ester derivatives 13 and 14, respectively, it appears that benzylation results in improved activity in both cases. Furthermore, replacement of the methyl ester of 15 with the more lipophilic hexyl ester (15b) results in significantly enhanced activity. Although increasing the alkyl ester chain length and frequency has little effect upon activity (compare 15 with 10 and 13), tPSA becomes too high making these less likely contenders in terms of drug candidature. Replacement of the ester of 15 with a secondary amide functionality resulted in more than a 15× decrease in activity for 15d–e. However, introduction of a tertiary amide (15f) enabled recovery of GI50 but with losses of effectiveness in terms of TGI and LC50. However, inclusion of the amide serves to stabilise tPSA, despite the introduction of two additional ethyl groups. Based on these results, it appears that having a secondary amide (free NH) on the C-ring is detrimental to the biological activity, but that the inclusion of tertiary amides may be worth exploring further.
| Compound | log10 mean GI50a | log10 mean TGIb | log10 mean LC50c | tPSA (Å2) |
|---|---|---|---|---|
| a GI50 = half maximal growth inhibition concentration.b TGI = total growth inhibition concentration.c LC50 = half maximal lethal concentration. | ||||
| Prodigiosin | −7.85 | −5.68 | −6.65 | 45.65 |
| Prodigiosin-Me | −7.82 | −6.86 | −5.97 | 45.65 |
| 10 | −6.07 | −5.31 | −4.35 | 98.25 |
| 11 | −5.45 | −4.82 | −4.29 | 98.25 |
| 12 | −6.27 | −5.27 | −4.52 | 71.95 |
| 13 | −6.43 | −5.60 | −4.56 | 98.25 |
| 14 | −6.45 | −5.69 | −4.77 | 71.95 |
| 15 | −6.70 | −5.63 | −4.69 | 71.95 |
| 15a | −6.49 | −5.74 | −4.79 | 71.95 |
| 15b | −7.04 | −6.00 | −5.23 | 71.95 |
| 15c | −6.66 | −5.92 | −5.26 | 71.95 |
| 15d | −5.85 | −5.52 | −5.22 | 74.75 |
| 15e | −5.62 | −5.06 | −4.34 | 74.75 |
| 15f | −6.31 | −4.95 | −4.19 | 65.96 |
The five-dose data for the activity of the prodigiosenes against each cell line in the NCI-60 enables selectivity to be appreciated. Select GI50 data is shown in Fig. 6, which plots the activity of methyl ester 15 alongside those of the corresponding benzyl (15a) and hexyl esters (15b), and the corresponding butyl amide (15e) and diethyl amide (15f). The enhanced activity of the hexyl derivative (15b, green in Fig. 6) is evident across most cell lines, and reaches nanomolar effectiveness against some. Although the methyl analogue (15, red) matches 15b in some cases, particularly colon cancer lines, the benzyl analogue (15a, yellow) is generally less effective. As evident from the averaged GI50 data, the secondary amine (15e, brown in Fig. 6) falls comprehensively short of the activity demonstrated by analogous esters, whilst the activity of tertiary amide (15f, olive) recovers such as to parallel that of the corresponding esters in some lines.
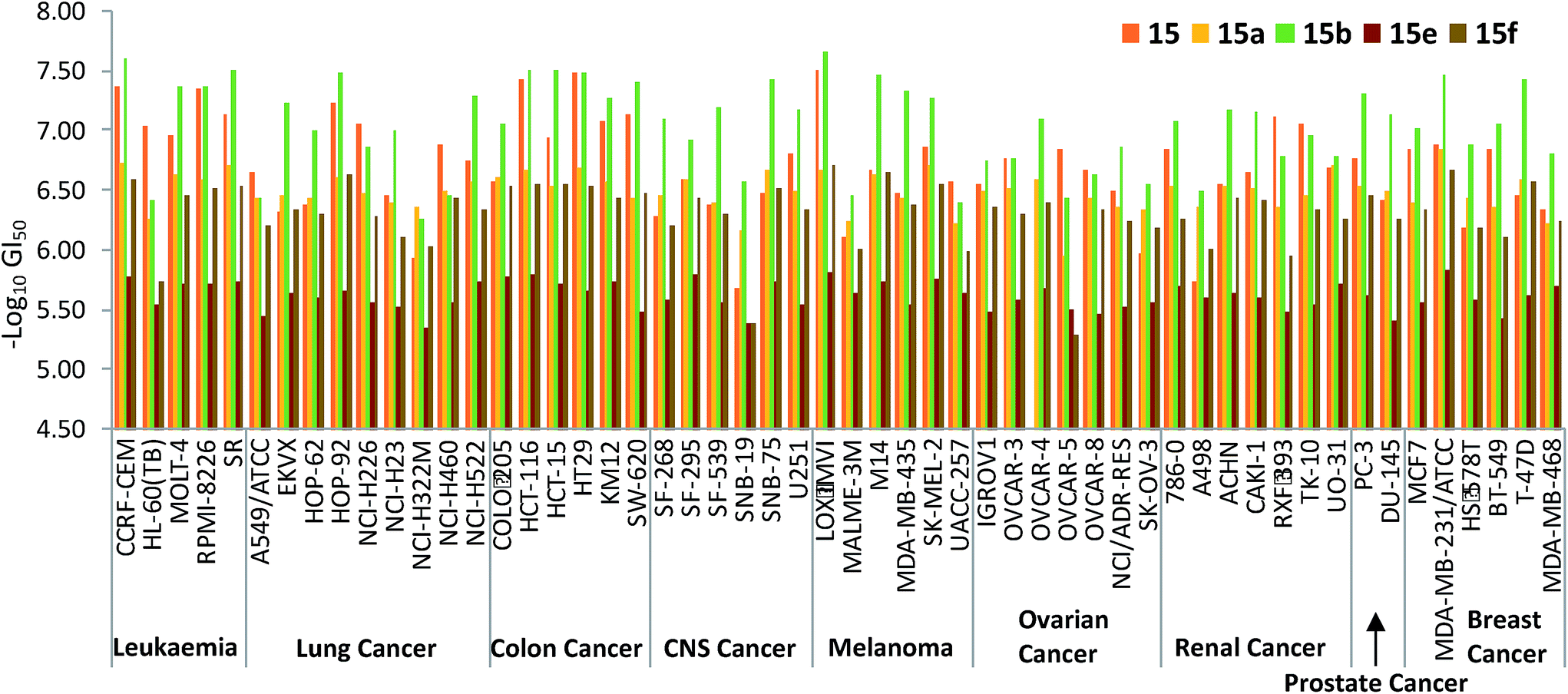 | ||
| Fig. 6 log10 GI50 concentrations (half maximal growth inhibition concentrations) of prodigiosenes 15, 15a, 15b, 15e and 15f against the NCI-60 human cancer cell lines representing 9 different cancer types; see http://http:dtp.cancer.gov. | ||
Fig. 7 plots the GI50 data corresponding to the diesters 10, 11 and 13 alongside those of the monoesters 14 and 15. The benzylated dialkanoate (13, pink in Fig. 7) was significantly more active than the methyl analogue (11, dark green). Furthermore, replacement of a C-ring conjugated benzyl ester (10, light green in Fig. 7) with a methyl substituent (14, yellow) results in a dramatic increase in activity suggesting that pKa,36 or the physical properties incurred through inclusion of conjugated ester functionality at this position, gravely affect the ability of prodigiosenes to interfere with the growth and propagation of the cancer cells against which compounds were evaluated.
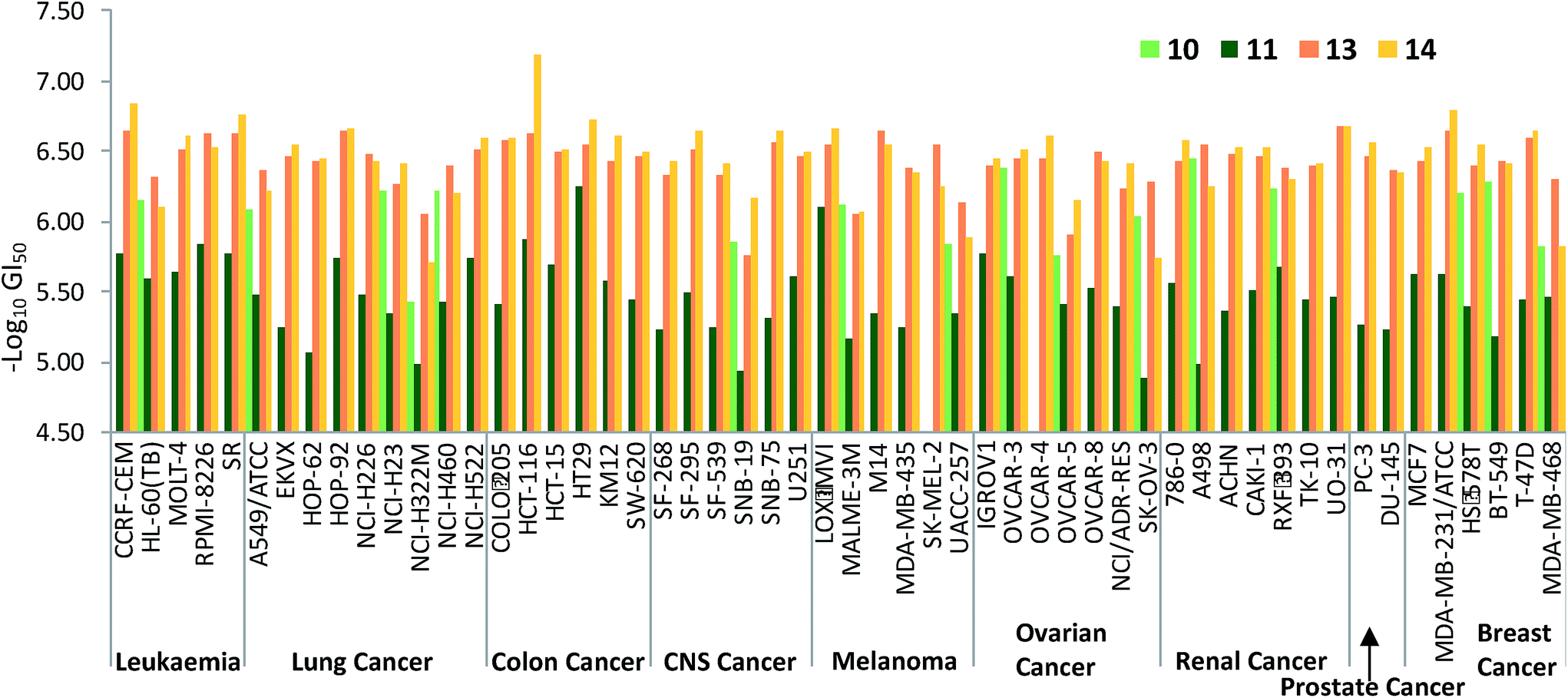 | ||
| Fig. 7 log10 GI50 concentrations (half maximal growth inhibition concentrations) of prodigiosenes 10, 11, 13 and 14 against the NCI-60 human cancer cell lines representing 9 different cancer types; see http://http:dtp.cancer.gov. | ||
Conclusions
Overall, strategies for the synthesis of new prodigiosenes bearing a variety of alkanoate and amides on the C-ring were developed. Ten new prodigiosenes were synthesised and evaluated for their anticancer activity. These compounds showed modest average activity against the NCI standard panel against 60 human cell lines derived from nine cancer types, with increased GI50 values against specific lines for compounds incorporating tertiary amide functionality, as well as lipophilic properties through hexyl substitution. Likewise the presence of an ester moiety directly conjugated to the C-ring resulted in a loss of activity. This small SAR study revealed that, although changing the nature or frequency of C-ring ester and amide substituents did not enhance selectivity, activity can certainly be modified through variation at these positions.Experimental
All chemicals were purchased and used as received unless otherwise indicated. Moisture-sensitive reactions were performed in flame-dried glassware under a positive pressure of nitrogen or argon. Solutions of air- and moisture-sensitive compounds were introduced via syringe or cannula through a septum. Flash chromatography was performed using Silicycle ultra pure silica (230–400 mm) or 150 mesh Brockmann III activated neutral alumina oxide as indicated. The NMR spectra were recorded using a 500 MHz or 300 MHz spectrometer instrument using CDCl3 as solvent and are reported in part per million (δ) using the solvent signals at 7.26 ppm for 1H and at 77.16 ppm for 13C as an internal reference with J values given in hertz. Mass spectra were obtained using TOF and LCQ Duo ion trap instruments operating in ESI+ mode. Melting points were determined using a Fisher-Johns melting point apparatus, and are uncorrected. N-Boc pyrrole-2-boronic acid was purchased from Frontier Scientific. 4-Methoxy-3-pyrrolin-2-one,48 2,56 11,44 12,44 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 44 were prepared according to literature procedures.
44 were prepared according to literature procedures.
Benzyl 3-(3-ethoxy-3-oxopropyl)-4-(3-methoxy-3-oxopropyl)-5-methyl-1H-pyrrole-2-carboxylate (1)
A solution of dibenzyl oximinomalonate57 (6.3 g, 20.0 mmol, 1.6 equiv.) in AcOH (12.4 mL) was added to a stirred solution of 8-ethyl-1-methyl-4-acetyl-5-oxooctanedioate58 (3.4 g, 12.5 mmol, 1 equiv.) in AcOH (15.5 mL) at 70 °C, while a mixture of zinc (4.9 g, 75.0 mmol, 6 equiv.) and NaOAc (2.6 g, 31.3 mmol, 2.5 equiv.) was added portion-wise to the reaction mixture. After the addition was complete, the resulting mixture was heated at reflux temperature for 1 h and then allowed to cool to room temperature. The reaction mixture was then poured into iced water (150 mL) and extracted with Et2O (3 × 100 mL). The combined organic layers were washed with water (200 mL), brine (200 mL), dried over MgSO4 and concentrated under reduced pressure. The crude mixture was purified via column chromatography on silica eluting with 10–30% EtOAc in hexanes to afford the title compound as off-white solid (1.3 g, 27%).1H NMR (300 MHz, CDCl3) δ: 8.58 (br s, 1H), 7.41–7.31 (m, 5H), 5.27 (s, 2H), 4.09 (q, J = 7.1 Hz, 2H), 3.65 (s, 3H), 2.99 (t, J = 7.8 Hz, 2H), 2.72 (t, J = 7.8 Hz, 2H), 2.51 (t, J = 7.8 Hz, 2H), 2.43 (t, J = 7.8 Hz, 2H), 2.20 (s, 3H), 1.22 (t, J = 7.1 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ: 173.4, 173.2, 160.8, 136.4, 130.5 (×2), 128.7, 128.4, 128.3, 120.0, 116.7, 65.9, 60.3, 51.7, 35.5, 20.7, 19.5, 14.3, 11.6 (1 carbon atom unaccounted for); HRMS-ESI (m/z): [M + Na]+ calcd for C22H27NNaO6 424.1731; found, 424.1716.4-[(Methoxycarbonyl)ethyl]-3-[2-(ethoxycarbonyl)ethyl]-5-methyl pyrrole-2-carboxaldehyde (3)
To a solution of 1 (6.0 g, 15.5 mmol, 1 equiv.) in EtOH (400 mL), Et3N (60 drops) was added and the mixture was degassed for 10 minutes. 10% Pd/C (0.6 g, 10% by weight of 1) was added and the reaction mixture was again degassed for 5 minutes before being placed under H2 (balloon) and stirred overnight. The reaction mixture was filtered through Celite, which was then washed with MeOH. The combined fractions were removed in vacuo to give the crude carboxylic acid which was dissolved in dry CH2Cl2 (40 mL). TFA (13.1 mL, 171 mmol, 11 equiv.) was then added drop-wise at 0 °C, under N2. The reaction was monitored using TLC and once there was no starting material left (∼10 min), TMOF (8.5 mL, 78 mmol, 5 equiv.) was added drop-wise and the reaction mixture was warmed to r.t. and monitored until completion using TLC (∼15 min). The reaction mixture was poured onto ice-water and 6 M NaOH (21 mL, 124 mmol, 8 equiv.) was added slowly. This mixture was extracted with CH2Cl2 (2 × 80 mL). The combined organic fractions were washed with saturated NaHCO3 (2 × 80 mL), H2O (80 mL) and brine (80 mL), dried with Na2SO4 and concentrated in vacuo. The crude material was then purified via column chromatography on silica eluting with 30% EtOAc/hexanes to afford the title compound as an off-white solid (3.3 g, 77%): 1H NMR (300 MHz, CDCl3) δ: 10.24 (br s, 1H), 9.45 (s, 1H), 4.11 (q, J = 7.1 Hz, 2H), 3.65 (s, 3H), 3.01 (t, J = 7.8 Hz, 2H), 2.72 (t, J = 7.8 Hz, 2H), 2.54 (t, J = 7.8 Hz, 2H), 2.45 (t, J = 7.8 Hz, 2H), 2.27 (s, 3H), 1.22 (t, J = 7.1 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ: 176.0, 173.2, 172.3, 136.5, 134.7, 127.5, 120.6, 60.6, 51.6, 36.7, 34.9 (×2), 19.2, 14.2, 11.7; HRMS-ESI (m/z): [M + Na]+ calcd for C15H21NNaO5 318.1312; found, 318.1297.4-(Benzylmethanoate)-3[(methoxycarbonyl)ethyl]-5-methyl pyrrole-2-carboxaldehyde (4)
Pyrrole 256 (10 g, 27.9 mmol, 1 equiv.) was dissolved in ethylene glycol (20 mL) with stirring. KOH (2.88 g, 55.8 mmol, 2 equiv.) was added and the reaction mixture was heated to reflux temperature and stirred for 4 h. The reaction mixture was then allowed to cool to r.t., diluted with H2O (50 mL) and extracted with CH2Cl2 (80 mL). The aqueous layer was acidified with 5 M HCl until just acidic, and extracted with CH2Cl2 (3 × 80 mL). The organic combined fractions were dried with Na2SO4 and concentrated in vacuo. The crude material was dissolved in MeOH (500 mL). H2SO4 (27.9 mL, 0.1 M, 2.79 mmol, 0.1 equiv.) was then added and the mixture was heated at reflux temperature for 3 h at which point it was allowed to cool to r.t. and the solvent was removed in vacuo.59 In a separate flask, POCl3 (1.3 mL, 30.7 mmol, 1.1 equiv.) was added drop-wise to DMF (1.1 mL, 30.7 mmol, 1.1 equiv.) in CH2Cl2 (4.5 mL) at 0 °C. This mixture was stirred for 15 min at r.t. and then added drop-wise to a solution of the preceding α-free pyrrole in CH2Cl2 (14 mL) at 0 °C. The reaction mixture was heated to reflux temperature and stirred for 2 h. The reaction mixture was allowed to cool to r.t. and then 1 M NaHCO3 (5 equiv.) was added drop-wise at first, then rapidly. The reaction mixture was returned to reflux temperature for 1 h, before returning to r.t. The organic layer was separated and the aqueous layer was extracted with CH2Cl2. The organic layers were combined, washed with H2O and brine, dried with Na2SO4 and then concentrated in vacuo.37 The crude material was crystallised from 70% EtOAc/hexanes to give the title compound as an off-white solid (1.03 g, 12%): 1H NMR (500 MHz, CDCl3) δ: 9.95 (br s, 1H), 9.65 (s, 1H), 7.43–7.33 (m, 5H), 5.29 (s, 2H), 3.60 (s, 3H), 3.28 (t, J = 7.3 Hz, 2H), 2.63 (t, J = 7.3 Hz, 2H), 2.55 (s, 3H); 13C NMR (125 MHz, CDCl3) δ: 178.3, 173.2, 164.4, 143.8, 137.7, 136.2, 128.8, 128.5, 128.4, 113.1, 66.1, 51.7, 35.4, 20.2, 14.7 (1 carbon atom unaccounted for); HRMS-ESI (m/z): [M + Na]+ calcd for C18H19NNaO5 352.1155; found, 352.1162.
(Z)-Ethyl 3-(4-(3-methoxy-3-oxopropyl)-2-((3-methoxy-5-oxo-1H-pyrrol-2(5H)-ylidene)methyl)-5-methyl-1H-pyrrol-3-yl)propanoate (5)
To a stirred solution of 4-methoxy-3-pyrolin-2-one48 (1.9 g, 17.2 mmol, 2.2 equiv.), Et3N (6.5 mL, 46.8 mmol, 6.0 equiv.) and CH2Cl2 (230 mL) at 0 °C was added TMSOTf (4.2 mL, 23.4 mmol, 3.0 equiv.) and the mixture was stirred for 15 min. 2-Formyl pyrrole 3 (2.3 g, 7.7 mmol, 1.0 equiv.) in CH2Cl2 (90 mL) was added drop-wise and the reaction mixture was stirred for 3 h at 0 °C. The reaction mixture was then poured onto pH 7 phosphate buffer (500 mL). The organic phase was separated, dried with Na2SO4 and concentrated in vacuo. The crude material was diluted with THF (500 mL). HCl (4.3 mL, 12 M, 51.6 mmol, 3 equiv.) was added and the mixture was stirred for 15 min at r.t. before being diluted further with CH2Cl2 (320 mL) and poured onto saturated aq. NaHCO3. The organic layer was separated and the aqueous was extracted with CH2Cl2 (3 × 60 mL). The combined organic layers were dried with Na2SO4, and concentrated in vacuo. The crude material was washed with hexanes and H2O to give the title dipyrrinone as a light brown solid (2.9 g, quant.): 1H NMR (300 MHz, CDCl3) δ: 10.83 (br s, 1H), 10.22 (br s, 1H), 6.32 (s, 1H), 5.09 (s, 1H), 4.13 (q, J = 7.1 Hz, 2H), 3.90 (s, 3H), 3.68 (s, 3H), 2.87 (t, J = 8.0 Hz, 2H), 2.74 (t, J = 8.0 Hz, 2H), 2.49–2.44 (m, 4H), 2.35 (s, 3H), 1.25 (t, J = 7.1 Hz, 3H); 13C NMR (125 MHz, CDCl3); 173.7, 173.3, 173.1, 168.1, 132.5, 128.0, 122.5, 121.7, 118.8, 100.1, 89.9, 60.6, 58.3, 51.7, 36.7, 35.5, 20.0, 19.8, 14.3, 11.6; HRMS-ESI (m/z): [M + Na]+ calcd for C20H26N2NaO6 413.1689; found, 413.1685.(Z)-Benzyl 4-(3-(3-methoxy-3-oxopropyl)-2-((3-methoxy-5-oxo-1H-pyrrole-2(5H)-ylidene)methyl)-5-methyl-1H-pyrrol-3-yl)formate (6)
To a stirred solution of 4-methoxy-3-pyrolin-2-one48 (0.46 g, 4.2 mmol, 1.55 equiv.) in THF (54 mL) was added 4 M KOH (1.4 mL, 5.5 mmol, 2 equiv.), previously bubbled with N2. The resultant mixture was degassed (∼15 minutes) and then placed under a N2 atmosphere. The mixture was then heated to 60 °C with stirring for 1 h, before being cooled slightly. 2-Formyl pyrrole 4 (0.9 g, 2.7 mmol, 1 equiv.) was then added in one portion. The resultant mixture was returned to 60 °C and stirred for 24 h whereupon a yellow precipitate formed. The precipitate (saponified product) was collected via suction filtration, and washed with hexanes. The crude yellow solid was then dissolved in MeOH (500 mL) and conc. H2SO4 (0.5 mL, 5.5 mmol, 2 equiv.) was added drop-wise and the mixture was then stirred at reflux temperature for 3 h, under N2 atmosphere. The reaction mixture was then concentrated in vacuo and the crude product was washed with hexanes and H2O to give the title dipyrrinone as a yellow-brown solid (681 mg, 59%): 1H NMR (500 MHz, DMSO) δ: 10.96 (br s, 1H), 9.66 (br s, 1H), 7.43–7.33 (m, 5H), 6.06 (s, 1H), 5.28 (s, 1H), 5.22 (s, 2H) 3.86 (s, 3H), 3.51 (s, 3H), 2.91 (t, J = 7.6 Hz, 2H), 2.46 (s, 3H), 2.41 (t, J = 7.6 Hz, 2H); 13C NMR (125 MHz, DMSO); 172.5, 170.8, 166.9, 164.1, 139.7, 136.6, 128.4, 128.0, 127.9, 126.5, 126.0, 122.3, 110.3, 94.1, 91.6, 64.9, 58.5, 51.1, 35.0, 20.5, 13.8; HRMS-ESI (m/z): [M + Na]+ calcd for C23H24N2NaO6 447.1527; found, 447.1506.(Z)-Ethyl 3-(2-((5-bromo-3-methoxy-2H-pyrrol-2-ylidene)methyl)-4-(3-methoxy-oxopropyl)-5-methyl-1H-pyrrol-3-yl)propanoate (7)
To a solution of dipyrrinone 5 (1.0 g, 2.7 mmol, 1 equiv.) in CH2Cl2 (100 mL) under N2 was added POBr3 (2.3 g, 5.3 mmol, 2 equiv.). The reaction mixture was heated to reflux temperature and stirred for 24 h. The reaction mixture was cooled slightly, by removing from the heat source, and POBr3 (2.3 g, 5.3 mmol, 2 equiv.) was then added before the mixture was returned to reflux temperature with stirring for 72 h. The reaction mixture was then cooled to r.t. and poured onto saturated NaHCO3 (100 mL) and gently shaken in a separatory funnel. The organic layer was separated and the aqueous layer was extracted with CH2Cl2 (4 × 50 mL). The combined organic layers were dried with Na2SO4 and concentrated in vacuo. The crude material was purified via column chromatography over neutral alumina eluting with CH2Cl2 to afford the title bromodipyrrin as a yellow solid (381 mg, 33%): 1H NMR (500 MHz, CDCl3) δ: 6.86 (s, 1H), 5.57 (s, 1H), 4.12 (q, J = 7.1 Hz, 2H), 3.83 (s, 3H), 3.66 (s, 3H), 2.89 (t, J = 7.9 Hz, 2H), 2.73 (t, J = 7.9 Hz, 2H), 2.51–2.43 (m, 4H), 2.29 (s, 3H), 1.25 (t, J = 7.1 Hz, 3H); 13C NMR (125 MHz, CDCl3); 173.4, 172.7, 167.1, 145.0, 137.6, 137.4, 133.5, 126.0, 121.1, 116.1, 99.5, 60.7, 58.6, 51.8, 36.7, 35.3, 20.0, 19.7, 14.3, 12.5; HRMS-ESI (m/z): [M + H]+ calcd for C20H26BrN2O5 453.1020; found, 453.1024.(Z)-Benzyl 4-(2-((5-bromo-3-methoxy-2H-pyrrol-2-ylidene)methyl)-3-(3-methoxy-3-oxopropyl)-5-methyl-1H-pyrrol-3-yl)formate (8)
To a solution of dipyrrinone 6 (2.0 g, 4.7 mmol, 1 equiv.) in CH2Cl2 (200 mL) under N2 was added POBr3 (2.7 g, 9.4 mmol, 2 equiv.). The reaction mixture was heated to reflux temperature and stirred for 24 h. The reaction mixture was cooled slightly, by removing from the heat source, and POBr3 (2.7 g, 9.4 mmol, 2 equiv.) was then added before the mixture was returned to reflux temperature with stirring for 72 h. The reaction mixture was then cooled to r.t. and poured onto saturated NaHCO3 (100 mL) and gently shaken in a separatory funnel. The organic layer was separated and the aqueous layer was extracted with CH2Cl2 (4 × 50 mL). The combined organic layers were dried with Na2SO4 and concentrated in vacuo. The crude material was purified via column chromatography over neutral alumina eluting with CH2Cl2 to afford the title bromodipyrrin as a yellow solid (816 mg, 35%): 1H NMR (300 MHz, CDCl3) δ: 7.44–7.33 (m, 5H), 6.97 (s, 1H), 5.58 (s, 1H), 5.28 (s, 2H), 3.86 (s, 3H), 3.60 (s, 3H), 3.13 (t, J = 7.6 Hz, 2H), 2.58 (s, 3H), 2.56 (t, J = 7.6 Hz, 2H); 13C NMR (125 MHz, CDCl3) δ: 173.4, 167.8, 164.7, 148.3, 144.3, 140.2, 136.4, 135.7, 128.7, 128.5, 128.3, 126.4, 115.6, 112.9, 100.4, 66.0, 58.8, 51.6, 35.5, 21.2, 15.3; HRMS-ESI (m/z): [M + H]+ calcd for C23H24BrN2O5 487.0863; found, 487.0846.(Z)-Ethyl 3-(2-((4-methoxy-1H-1H′-[2,2′-bipyrrol]-5-yl)methylene)-4-(3-methoxy-3-oxopropyl)-5-methyl-2H-pyrrol-3-yl)propanoate hydrochloride (9·HCl)
To a solution of bromodipyrrin 7 (210 mg, 0.48 mmol, 1 equiv.) in DME (25 mL) was added commercially available N-Boc pyrrole-2-boronic acid (162 mg 0.57 mmol, 1.2 equiv.), LiCl (81 mg, 1.4 mmol, 3 equiv.) and Pd(PPh3)4 (71 mg, 0.05 mmol, 0.1 equiv.), in a Schlenk flask. The reaction mixture was purged with N2 for 15 min and placed under a N2 atmosphere before adding a 2 M aqueous solution of Na2CO3 (1.3 mL, 1.9 mmol, 4 equiv.), previously purged with N2, drop-wise to the reaction mixture. The Schlenk flask was sealed and the reaction mixture was heated to 85 °C and stirred for 24 h. The reaction mixture was cooled to r.t. and H2O (30 mL) was added and the mixture then extracted with EtOAc (4 × 30 mL). The combined organic layers were washed with H2O (60 mL) and brine (60 mL), then dried with Na2SO4 and concentrated in vacuo. The crude product was purified via column chromatography on neutral alumina eluting with 0–30% EtOAc/hexanes. The product was then dissolved in MeOH:H2O (1:1, 10 mL) before 5 M HCl (0.1 mL, 0.48 mmol, 1 equiv.) was added drop-wise and the mixture was stirred for 3 h. The MeOH was partially removed in vacuo and the solid was isolated using Millipore filtration apparatus to give the title prodigiosene HCl salt as a red solid (37 mg, 18%). 1H NMR (500 MHz, CDCl3); 12.75 (br s, 1H), 12.61 (br s, 2H), 7.23 (apparent s, 1H), 7.04 (s, 1H), 6.94–6.93 (m, 1H), 6.36–6.35 (m, 1H), 6.09 (d, J = 1.9 Hz, 1H), 4.13 (q, J = 7.2 Hz, 2H), 4.03 (s, 3H), 3.68 (s, 3H), 2.97 (t, J = 7.9 Hz, 2H), 2.77 (t, J = 7.9 Hz, 2H), 2.56 (s, 3H), 2.51 (t, J = 7.9 Hz, 2H), 2.47 (t, J = 7.9 Hz, 2H), 1.25 (t, J = 7.2 Hz, 3H); 13C NMR; 173.1, 172.4, 166.0, 148.1, 146.4, 139.4, 127.3, 124.0, 123.4, 122.4, 120.9, 117.4, 113.0, 112.0, 93.0, 60.9, 58.9, 51.9, 36.4, 34.7, 20.1, 19.6, 14.3, 12.6; HRMS-ESI (m/z): [M + H]+ calcd for C24H30N3O5 440.2180; found, 440.2162.
(Z)-Benzyl 4-(2-((4-methoxy-1H,1H′-[2,2′-bipyrrol]-5-yl)methylene)-3-(3-methoxy-3-oxopropyl)-5-methyl-2H-pyrrol-3-yl)formate (10·HCl)
To a solution of bromodipyrrin 8 (0.8 g, 1.6 mmol, 1 equiv.) in DME (65 mL) was added commercially available N-Boc pyrrole-2-boronic acid (0.4 mg, 2.0 mmol, 1.2 equiv.), LiCl (0.2 g, 4.9 mmol, 3 equiv.) and Pd(PPh3)4 (179 mg, 0.16 mmol, 0.1 equiv.), in a Schlenk flask. The reaction mixture was purged with N2 for 15 min and placed under a N2 atmosphere before adding a 2 M aqueous solution of Na2CO3 (3.3 mL, 6.6 mmol, 4 equiv.), previously purged with N2, drop-wise to the reaction mixture. The Schlenk flask was sealed and the reaction mixture was heated to 85 °C and stirred for 24 h. The reaction mixture was cooled to r.t. and H2O (70 mL) was added and the mixture then extracted with EtOAc (4 × 50 mL). The combined organic layers were washed with H2O (70 mL) and brine (70 mL), then dried with Na2SO4 and concentrated in vacuo. The crude product was purified via column chromatography on neutral alumina eluting with 0–30% EtOAc/Hexanes. The product was then dissolved in MeOH:H2O (1:1, 30 mL) before 5 M HCl (0.3 mL, 1.6 mmol, 1 equiv.) was added drop-wise and the mixture was stirred for 3 h. The MeOH was partially removed in vacuo and the solid was isolated using Millipore filtration apparatus to give the title prodigiosene HCl salt as a red solid (206 mg, 27%). 1H NMR (500 MHz, CDCl3) δ: 13.04 (br s, 1H), 12.78 (br s, 1H), 12.75 (br s, 1H), 7.43–7.34 (m, 5H), 7.31 (apparent s, 1H), 7.19 (s, 1H), 7.04 (apparent s, 1H), 6.42–6.40 (m, 1H), 6.11 (d, J = 1.4 Hz, 1H), 5.31 (s, 2H), 4.07 (s, 3H), 3.61 (s, 3H), 3.22 (t, J = 7.5 Hz, 2H), 2.81 (s, 3H), 2.60 (t, J = 7.5 Hz, 2H); 13C NMR (125 MHz, CDCl3) δ: 173.2, 167.2, 164.0, 151.0, 150.3, 141.8, 136.2, 129.2, 128.8, 128.5, 128.4, 123.5, 123.2, 122.1, 119.8, 114.7, 112.9, 112.8, 93.6, 66.2, 59.2, 51.8, 35.3, 21.2, 15.1; HRMS-ESI (m/z): [M + H]+ calcd for C27H28N3O5 474.2023; found, 474.2004.
General procedure for hydrolysis of prodigiosene carboxylates (GP1)
LiOH·H2O (2.1 equiv.) was added to a stirred solution of the appropriate prodigiosene (1 equiv.) in THF and H2O (1:1, 0.02 M). The reaction mixture was warmed to 40 °C and then stirred for 1.5 h at which point another portion of LiOH·H2O (0.25 equiv.) was added and stirring was continued overnight. The solvents were removed in vacuo and the resultant solid was dissolved in water (0.06 M). 5 M HCl (4 equiv.) was then added drop-wise and the mixture was stirred for 2 h. The precipitate was collected via suction filtration using a Millipore filter to give the hydrolysed prodigiosene as a dark purple solid.
General procedure for coupling of prodigiosene carboxylic acids with alcohols (GP2)
The prodigiosene carboxylic acid (1 equiv.), prepared via GP1 and used without further purification, was added to a stirred solution of the appropriate alcohol (1 equiv.), EDCI (0.15 equiv.) and DMAP (2.1 equiv.) in dry CH2Cl2 (0.01 M), under N2. The reaction mixture was stirred at r.t. and monitored using TLC for up to 5 days. H2O was then added and the mixture extracted with CH2Cl2 (×3). The combined organic layers were washed with brine, dried with Na2SO4 and concentrated in vacuo. The crude material was purified via column chromatography on neutral alumina eluting with 20% EtOAc/hexanes. The prodigiosene was then dissolved in MeOH:CH2Cl2 (1:1, 0.01 M). Concentrated HCl (1 equiv.) was added drop-wise and the mixture was stirred for ∼30 min. The CH2Cl2 was removed in vacuo and the solid was isolated using Millipore filtration apparatus and washed with MeOH and ether to give the desired ester as its HCl salt.
44 (50 mg, 0.12 mmol, 1 equiv.), and was isolated as a red solid (12 mg, 19%): 1H NMR (300 MHz, CDCl3) δ: 12.79 (br s, 1H), 12.67 (br s, 2H), 7.38–7.23 (m, 10H), 6.85 (s, 1H), 6.68 (br s, 2H), 6.16 (apparent s, 1H), 6.04 (s, 1H), 5.08 (s, 2H), 5.02 (s, 2H), 3.95 (s, 3H), 3.31 (s, 2H), 2.91 (t, J = 7.5 Hz, 2H), 2.54 (t, J = 7.5 Hz, 2H), 1.84 (s, 3H); 13C NMR (125 MHz, CDCl3) δ: 172.8, 171.5, 168.9, 159.8, 139.0, 136.7, 136.1, 136.0, 131.6, 128.7, 128.6 (×2), 128.4, 128.3, 128.2, 127.1, 125.6, 122.7, 114.4, 112.7, 110.3, 95.6, 66.6, 66.4, 58.5, 36.2, 30.4, 20.1, 10.9 (one carbon uncounted for); HRMS-ESI (m/z): [M + H]+ calcd for C34H34N3O5 564.2493; found, 564.2467.44 (50 mg, 0.13 mmol, 1 equiv.), and was isolated as a red solid (28 mg, 44%): 1H NMR (300 MHz, CDCl3) δ: 12.68 (br s, 1H), 12.59 (br s, 2H), 7.37–7.29 (m, 5H), 7.22 (s, 1H), 7.03 (apparent s, 1H), 6.91 (s, 1H), 6.35 (apparent s, 1H), 6.08 (apparent s, 1H), 5.11 (s, 2H), 3.99 (s, 3H), 2.96 (t, J = 7.5 Hz, 2H), 2.57 (t, J = 7.5 Hz, 2H), 2.52 (s, 3H), 1.97 (s, 3H); 13C NMR (125 MHz, CDCl3) δ: 172.3, 165.7, 147.4, 147.2, 139.5, 135.9, 128.7, 128.5, 128.3, 126.8, 123.5, 122.5, 121.5, 120.4, 116.8, 113.0, 111.8, 92.9, 66.6, 58.8, 35.5, 20.3, 12.5, 9.0; HRMS-ESI (m/z): [M + H]+ calcd for C26H28N3O3 430.2125; found, 430.2104.44) (50 mg, 0.14 mmol, 1 equiv.), and was isolated as a red solid (28 mg, 44%): 1H NMR (300 MHz, CDCl3) δ: 12.74 (br s, 1H), 12.65 (br s, 1H), 12.58 (br s, 1H), 7.39–7.30 (m, 5H), 7.22 (apparent s, 1H), 7.04 (s, 1H), 6.91 (apparent s, 1H), 6.35 (apparent s, 1H), 6.08 (s, 1H), 5.12 (s, 2H), 4.01 (s, 3H), 3.46 (s, 2H), 2.54 (s, 3H), 2.22 (s, 3H); 13C NMR (125 MHz, CDCl3) δ: 170.7, 165.8, 147.9, 146.9, 138.2, 135.8, 128.7, 128.5, 128.3, 127.0, 124.1, 122.4, 120.6, 118.5, 117.2, 113.3, 111.9, 93.0, 66.9, 58.9, 30.4, 12.5, 10.3; HRMS-ESI (m/z): [M + H]+ calcd for C25H26N3O3 416.1969; found, 416.1978.44 (60 mg, 0.17 mmol, 1 equiv.), and was isolated as a red solid (17 mg, 23%): 1H NMR (500 MHz, CDCl3) δ: 12.75 (br s, 1H), 12.66 (br s, 1H), 12.58 (br s, 1H), 7.23 (s, 1H), 7.06 (s, 1H), 6.92 (apparent s, 1H), 6.35 (apparent s, 1H), 6.09 (apparent s, 1H), 4.07 (t, J = 7.0 Hz, 2H), 4.02 (s, 3H), 3.41 (s, 2H), 2.57 (s, 3H), 2.26 (s, 3H), 1.63–1.55 (m, 2H), 1.34–1.25 (m, 6H), 0.87 (t, J = 7.0 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ: 170.8, 165.7, 147.7, 147.0, 138.2, 126.9, 124.0, 122.3, 120.4, 118.7, 116.9, 113.3, 111.7, 92.8, 65.2, 58.7, 31.4, 30.3, 28.6, 25.5, 22.5, 14.0, 12.4, 10.2; HRMS-ESI (m/z): [M + H]+ calcd for C24H32N3O3 410.2438; found, 410.2446.44 (60 mg, 0.17 mmol, 1 equiv.), and was isolated as a red solid (78 mg, 46%): 1H NMR (500 MHz, CDCl3) δ: 12.72 (br s, 1H), 12.64 (br s, 1H), 12.56 (br s, 1H), 7.22 (s, 1H), 7.05 (s, 1H), 6.91 (apparent s, 1H), 6.34 (apparent s, 1H), 6.08 (d, J = 3.5 Hz, 1H), 4.01 (s, 3H), 3.77 (s, 2H), 3.43 (s, 2H), 2.58 (s, 3H), 2.27 (s, 3H), 0.89 (s, 9H); 13C NMR (125 MHz, CDCl3) δ: 171.0, 165.8, 147.8, 146.9, 138.2, 127.0, 124.1, 122.4, 120.6, 118.8, 117.1, 113.3, 111.9, 92.9, 74.4, 58.8, 31.5, 30.5, 26.5, 12.5, 10.3; HRMS-ESI (m/z): [M + H]+ calcd for C23H30N3O3 396.2282; found, 396.2270.General procedure for coupling of prodigiosene carboxylic acids with amines (GP3)
To a suspension of the prodigiosene carboxylic acid (1 equiv.), prepared via GP1 and used without further purification, in dry CH2Cl2 (0.02 M) was added the appropriate amine (1 equiv.), DMAP (2 equiv.) and HBTU (2 equiv.) consecutively at 0 °C. The reaction mixture was warmed to room temperature and stirred for 24 h. When the reaction was complete, the reaction mixture was diluted with CH2Cl2 and washed with saturated NaHCO3 (aq.), 2% HCl and brine before being dried with Na2SO4. After filtration and removal of the solvent in vacuo, the crude solid was recrystallised from a CHCl3/hexanes to give the desired amide as its HCl salt.44 (60 mg, 0.17 mmol, 1 equiv.), and was isolated as a red solid (37 mg, 55%): 1H NMR (300 MHz, CDCl3) δ: 12.77 (br s, 1H), 12.68 (br s, 1H), 12.58 (br s, 1H), 7.38–7.18 (m, 5H), 7.02 (s, 1H), 6.99–6.95 (m, 1H), 6.38–6.36 (m, 1H), 6.09 (d, J = 3 Hz, 1H), 5.84–5.80 (m, 1H), 4.41 (d, J = 9.5 Hz, 2H), 4.03 (s, 3H), 3.44 (s, 2H), 2.96 (br s, 1H), 2.53 (s, 3H), 2.19 (s, 3H); 13C NMR (125 MHz, CDCl3) δ: 170.2, 166.2, 148.7, 145.9, 138.1, 137.5, 129.3, 128.9, 127.8, 127.5, 124.2, 122.3, 121.3, 118.5, 118.0, 113.1, 112.2, 93.2, 58.9, 43.8, 31.1, 12.4, 10.3; HRMS-ESI (m/z): [M + H]+ calcd for C25H27N4O2 415.2129; found, 415.2124.44 (60 mg, 0.17 mmol, 1 equiv.), and was isolated as a red solid (19 mg, 68%): 1H NMR (300 MHz, CDCl3) δ: 12.81 (br s, 1H), 12.71 (br s, 1H), 12.59 (br s, 1H), 7.06 (s, 1H), 6.97 (apparent s, 1H), 6.40–6.37 (m, 1H), 6.11 (d, J = 1.5 Hz, 1H), 5.48–5.47 (m, 1H), 4.04 (s, 3H), 3.38 (s, 2H), 3.22 (q, J = 7.0 Hz, 2H), 3.05 (br s, 1H), 2.54 (s, 3H), 2.22 (s, 3H), 1.42 (quin, J = 7.0 Hz, 2H), 1.27 (sex, J = 7.0 Hz, 2H), 0.89 (t, J = 7.0 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ: 170.5, 166.3, 148.8, 145.9, 137.5, 127.8, 124.2, 122.3, 121.4, 118.5, 118.0, 113.0, 112.3, 93.2, 59.0, 39.7, 31.8, 31.7, 20.1, 13.8, 12.4, 10.2; HRMS-ESI (m/z): [M + H]+ calcd for C22H29N4O2 381.2285; found, 381.2269.44 (60 mg, 0.17 mmol, 1 equiv.), and was isolated as a red solid (21 mg, 38%): 1H NMR (500 MHz, CDCl3) δ: 12.71 (br s, 1H), 12.63 (br s, 1H), 12.56 (br s, 1H), 7.23–7.20 (apparent m, 1H), 7.06 (s, 1H), 6.92–6.89 (apparent m, 1H), 6.36–6.33 (apparent m, 1H), 6.09 (s, 1H), 4.02 (s, 3H), 3.46 (s, 2H), 3.37 (br q, J = 7 Hz, 4H), 2.54 (s, 3H), 2.23 (s, 3H), 1.15 (br t, J = 7 Hz, 6H); 13C NMR (125 MHz, CDCl3) δ: 169.1, 165.7, 147.5, 147.3, 138.5, 126.9, 124.3, 122.5, 120.4, 120.1, 116.9, 113.4, 111.8, 92.9, 58.8, 42.4, 40.8, 29.9, 14.4, 13.2, 12.8, 10.5; HRMS-ESI (m/z): [M + H]+ calcd for C22H29N4O2 381.2285; found, 381.2278.Acknowledgements
The authors thank Dr Deborah Smithen for laboratory support. This work was supported by the Canadian Institutes of Health Research (CGS-M to K.-l. A. R. L.; operating grant RNS 89715 to A. T.) and the Nova Scotia Health Research Foundation. A. T. is a Senior Scientist of The Beatrice Hunter Cancer Research Institute. Human cell-line screening and in vivo toxicity evaluations were performed by the Developmental Therapeutics Program, Division of Cancer Treatment and Diagnosis, National Cancer Institute (http://dtp.cancer.gov).References
- G. Rook, Nature, 1992, 357, 545 CrossRef CAS PubMed.
- W. R. Hearn, M. K. Elson, R. H. Williams and J. Medina-Castro, J. Org. Chem., 1970, 35, 142–146 CrossRef CAS PubMed.
- J. W. Bennett and R. Bentley, Adv. Appl. Microbiol., 2000, 47, 1–32 CAS.
- N. N. Gerber, CRC Crit. Rev. Microbiol., 1975, 3, 469–485 CrossRef CAS PubMed.
- N. R. Williamson, P. C. Fineran, T. Gristwood, S. R. Chawrai, F. J. Leeper and G. P. C. Salmond, Future Microbiol., 2007, 2, 605–618 CrossRef CAS PubMed.
- N. R. Williamson, P. C. Fineran, F. J. Leeper and G. P. C. Salmond, Nat. Rev. Microbiol., 2006, 4, 887–899 CrossRef CAS PubMed.
- R. D'Alessio, A. Bargiotti, O. Carlini, F. Colotta, M. Ferrari, P. Gnocchi, A. Isetta, N. Mongelli, P. Motta, A. Rossi, M. Rossi, M. Tibolla and E. Vanotti, J. Med. Chem., 2000, 43, 2557–2565 CrossRef.
- E. Marchal, M. I. Uddin, D. A. Smithen, C. L. A. Hawco, M. Lanteigne, D. P. Overy, R. G. Kerr and A. Thompson, RSC Adv., 2013, 3, 22967–22971 RSC.
- A. J. Castro, Nature, 1967, 213, 903–904 CrossRef CAS PubMed.
- K. Papireddy, M. Smilkstein, J. X. Kelly, S. M. Salem, M. Alhamadsheh, S. W. Haynes, G. L. Challis and K. A. Reynolds, J. Med. Chem., 2011, 54, 5296–5306 CrossRef CAS PubMed.
- E. Marchal, D. A. Smithen, M. I. Uddin, A. W. Robertson, D. L. Jakeman, V. Mollard, C. D. Goodman, K. S. MacDougall, S. A. McFarland, G. I. McFadden and A. Thompson, Org. Biomol. Chem., 2014, 12, 4132–4142 CAS.
- A. Fürstner, Angew. Chem., Int. Ed., 2003, 42, 3582–3603 CrossRef PubMed.
- M. S. Melvin, K. E. Wooton, C. C. Rich, G. R. Saluta, G. L. Kucera, N. Lindquist and R. A. Manderville, J. Inorg. Biochem., 2001, 87, 129–135 CrossRef CAS PubMed.
- G. Park, J. T. Tomlinson, M. S. Melvin, M. W. Wright, C. S. Day and R. A. Manderville, Org. Lett., 2003, 5, 113–116 CrossRef CAS PubMed.
- R. Péréz-Tomas, B. Montaner, E. Llagostera and V. Soto-Cerrato, Biochem. Pharmacol., 2003, 66, 1447–1452 CrossRef.
- K. V. Arivizhivendhan, M. Mahesh, R. Boopathy, K. Patchaimurugan, P. Maharaja, S. Swarnalatha, R. R. Mary and G. Sekaran, J. Phys. Chem. B, 2016, 120, 9685–9696 CrossRef CAS PubMed.
- D. L. Boger and M. Patel, J. Org. Chem., 1988, 53, 1405–1415 CrossRef CAS.
- D. X. Hu, D. M. Withall, G. L. Challis and R. J. Thomson, Chem. Rev., 2016, 116, 7818–7853 CrossRef CAS PubMed.
- Nisha, K. Kumara and V. Kumar, RSC Adv., 2015, 5, 10899–10920 CAS.
- J. Li, Q. Zhang, J. Yin, C. Yu, K. Cheng, Y. Wei, E. Hao and L. Jiao, Org. Lett., 2016, 18, 5696–5699 CrossRef CAS PubMed.
- Z. Zhao, T. Shi, M. Xu, N. L. Brock, Y.-L. Zhao, Y. Wang, Z. Deng, X. Pang and M. Tao, Org. Lett., 2016, 18, 572–575 CrossRef CAS PubMed.
- D. E. Jeffries and C. W. Lindsley, J. Org. Chem., 2017, 82, 431–437 CrossRef CAS PubMed.
- Z. Wang, B. Li, L. Zhou, S. Yu, Z. Su, J. Song, Q. Sun, O. Sha, X. Wang, W. Jiang, K. Willert, L. Wei, D. A. Carson and D. Lu, Proc. Natl. Acad. Sci., 2016, 113, 13150–13155 CrossRef CAS PubMed.
- V. V. Prabhu, B. Hong, J. E. Allen, S. Zhang, A. R. Lulla, D. T. Dicker and W. S. El-Deiry, Cancer Res., 2016, 76, 1989–1999 CrossRef CAS PubMed.
- R. K. Suryawanshi, C. D. Patil, S. H. Koli, J. E. Hallsworth and S. V. Patil, Nat. Prod. Res., 2016, 31, 572–577 CrossRef PubMed.
- S. Rastogi, D. Zhang and J. T. Davis, Supramol. Chem., 2016, 28, 18–28 CrossRef CAS.
- N. Darshan and H. K. Manonmani, Food Sci. Technol., 2015, 52, 5393–5407 CrossRef CAS PubMed.
- R. K. Suryawanshi, C. D. Patil, H. P. Borase, C. P. Narkhede, A. Stevenson, J. E. Hallsworth and S. V. Patil, Int. J. Cosmet. Sci., 2015, 37, 98–107 CrossRef CAS PubMed.
- Y. Kim and J. Choi, Fibers Polym., 2015, 16, 1981–1987 CrossRef CAS.
- Y. Ren, J. Gong, R. Fu, Z. Li, Q. Li, J. Zhang, Z. Yu and X. Cheng, Dyes Pigm., 2017, 138, 147–153 CrossRef CAS.
- A. Fuerstner, Angew. Chem., Int. Ed., 2003, 42, 3582–3603 CrossRef CAS PubMed.
- K. Daïri, Y. Yao, M. Faley, S. Tripathy, E. Rioux, X. Billot, D. Rabouin, G. Gonzalez, J. Lavallée and G. Attardo, Org. Process Res. Dev., 2007, 11, 1051–1054 CrossRef.
- C. A. Goard and A. D. Schimmer, Core Evidence, 2013, 8, 15–26 CrossRef CAS PubMed.
- P. Kancharla, J. X. Kelly and K. A. Reynolds, J. Med. Chem., 2015, 58, 7286–7309 CrossRef CAS PubMed.
- E. Marchal, S. Rastogi, A. Thompson and J. T. Davis, Org. Biomol. Chem., 2014, 12, 7515–7522 CAS.
- S. Rastogi, E. Marchal, I. Uddin, B. Groves, J. Colpitts, S. A. McFarland, J. T. Davis and A. Thompson, Org. Biomol. Chem., 2013, 11, 3834–3845 CAS.
- M. I. Uddin, S. Thirumalairajan, S. M. Crawford, T. S. Cameron and A. Thompson, Synlett, 2010, 2561–2564 CAS.
- R. I. Sáez Díaz, J. Regourd, P. V. Santacroce, J. T. Davis, D. L. Jakeman and A. Thompson, Chem. Commun., 2007, 2701–2703 RSC.
- J. Regourd, A. Al-Sheikh Ali and A. Thompson, J. Med. Chem., 2007, 50, 1528–1536 CrossRef CAS PubMed.
- E. Marchal, M. I. Uddin, D. A. Smithen, L. A. Hawco, M. Lanteigne, D. P. Overy, R. G. Kerr and A. Thompson, RSC Adv., 2013, 3, 22967–22971 RSC.
- E. Marchal, D. A. Smithen, M. I. Uddin, A. W. Robertson, D. L. Jakeman, V. Mollard, C. D. Goodman, K. S. MacDougall, S. A. McFarland, G. I. McFadden and A. Thompson, Org. Biomol. Chem., 2014, 12, 4132–4142 CAS.
- C. L. A. Hawco, E. Marchal, M. I. Uddin, A. E. G. Baker, D. P. Corkery, G. Dellaire and A. Thompson, Bioorg. Med. Chem., 2013, 21, 5995–6002 CrossRef CAS PubMed.
- E. Marchal, M. I. Uddin, C. L. A. Hawco and A. Thompson, Can. J. Chem., 2015, 93, 526–535 CrossRef CAS.
- D. A. Smithen, A. M. Forrester, D. P. Corkery, G. Dellaire, J. Colpitts, S. A. McFarland, J. N. Berman and A. Thompson, Org. Biomol. Chem., 2013, 11, 62–68 CAS.
- J. H. Lee, Y. D. Joo, H. Kim, S. H. Bae, M. K. Kim, D. Y. Zang, J. L. Lee, G. W. Lee, J. H. Lee, J. H. Park, D. Y. Kim, W. S. Lee, H. M. Ryoo, M. S. Hyun, H. J. Kim, Y. J. Min, Y. E. Jang and J. H. Lee, Blood, 2011, 118, 3832–3841 CrossRef CAS PubMed.
- B. Löwenberg, G. J. Ossenkoppele, W. van Putten, H. C. Schouten, C. Graux, A. Ferrant, P. Sonneveld, J. Maertens, M. Jongen-Lavrencic, M. von Lilienfeld-Toal, B. J. Biemond, E. Vellenga, M. van Marwijk Kooy, L. F. Verdonck, J. Beck, H. Döhner, A. Gratwohl, T. Pabst and G. Verhoef, N. Engl. J. Med., 2009, 361, 1235–1248 CrossRef PubMed.
- J. P. Patel and R. L. Levine, Hematology Am. Soc. Hematol. Educ. Program, 2012, 2012, 28–34 Search PubMed.
- R. C. F. Jones and A. D. Bates, Tetrahedron Lett., 1986, 27, 5285–5288 CrossRef CAS.
- C. Curti, A. Sartori, L. Battistini, G. Rassu, P. Burreddu, F. Zanardi and G. Casiraghi, J. Org. Chem., 2008, 73, 5446–5451 CrossRef CAS PubMed.
- M. C. Clift and R. J. Thomson, J. Am. Chem. Soc., 2009, 131, 14579–14583 CrossRef CAS PubMed.
- M. Monga and E. A. Sausville, Leukemia, 2012, 16, 520–526 CrossRef PubMed.
- D. Yamamoto, Y. Uemura, K. Tanaka, K. Nakai, C. Yamamoto, H. Takemoto, K. Kamata, H. Hirata and K. Hioki, Int. J. Cancer, 2000, 88, 121–128 CrossRef CAS PubMed.
- L. Yang, Y. Zhang, G. Yang, Q. Chen and J. S. Ma, Dyes Pigm., 2004, 62, 27–33 CrossRef CAS.
- P. Ertl, B. Rohde and P. Selzer, J. Med. Chem., 2000, 43, 3714–3717 CrossRef CAS PubMed.
- A. K. Ghose and G. M. Crippen, J. Chem. Inf. Comput. Sci., 1987, 27, 21–35 CrossRef CAS PubMed.
- M. F. Chen and S. F. MacDonald, Can. J. Chem., 1974, 52, 1760–1761 CrossRef CAS.
- T. D. Lash, U. N. Mani, E. A. Lyons, P. Thientanavanich and M. A. Jones, J. Org. Chem., 1999, 64, 478–487 CrossRef CAS.
- J. B. Paine III and D. Dolphin, J. Org. Chem., 1985, 50, 5598–5604 CrossRef.
- D. A. Smithen, T. S. Cameron and A. Thompson, Org. Lett., 2011, 13, 5846–5849 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Spectral NMR characterisation data for all new compounds; NCI data, in terms of drug concentration, for compounds 10–15. See DOI: 10.1039/c7ra01628j |
| This journal is © The Royal Society of Chemistry 2017 |