
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Regioselective chitosan end-group activation: the triskelion approach†
V. D. Pickenhahn,
M. Grange,
G. De Crescenzo,
M. Lavertu* and
M. D. Buschmann*
Dept. Chemical Engineering and Inst. Biomedical Engineering, Ecole Polytechnique, Montreal, QC, Canada. E-mail: marc.lavertu@polymtl.ca; michael.buschmann@polymtl.ca
First published on 27th March 2017
Abstract
Chitosan (CS) end-group conjugation methods are rarely reported in the literature, mainly since the CS terminal aldehyde moiety produced by nitrous acid depolymerization is only present in trace amounts in its reactive form. In a previous study, our group proposed an intermolecular thioacetylation process that allowed terminal conjugation of thiol-reactive species to chitosan with 50% efficiency. However, this reaction is incompatible with acid-labile substituents and the conversion efficiency of CS end-groups could be limited by the size of the thiol-reactive species engaged in the reaction, mainly by steric hindrance since two substituents are required to obtain the stabilized thioacetal derivative. In the present study, we developed a novel CS end-group thioacetylation approach relying on a new regioselective linker that bears three thiol moieties. This trivalent linker, referred to as triskelion here, was specifically designed for activation of the CS 2,5-anhydro-D-mannose (M-Unit) end-group and consists of a thiol-hook for efficient aldehyde conjugation through an intramolecular reaction and a thiol-tail that remains available for subsequent end-group functionalization with any thiol-reactive species. The chemical synthesis of this linker provided the desired material with high yields over three steps. The in situ intramolecular thioacetylation process between the triskelion linker and 2,5-anhydro-D-mannose (M-Unit, monomeric) was assessed by semi-quantitative LC-MS studies, revealing that the corresponding intramolecular thioacetal largely predominated (∼90%). This regioselective derivatization was also performed onto M-Unit CS aldehydes and the desired CS-b-triskelion conjugates were obtained with functionalization degrees over 85%, as confirmed by NMR spectroscopy (1H and DOSY). As a final assessment of the CS-b-triskelion thiol-tail reactivity, these conjugates were successfully engaged with thiol-reactive magnetic beads into disulfide bond displacement with 50% efficiency. The proposed CS terminal activation with the triskelion linker opens new perspectives for biomedical applications, especially brush-like surface modifications and other copolymer formation through disulfide linkages or Michael-type additions.
Introduction
Chitosan (CS) and its derivatives hold great interest for biomedical applications in regenerative medicine,1 tissue engineering2–4 and drug/gene delivery5–7 fields. CS graft-modifications located on the C-2 amines and/or the O-6 hydroxyls along the CS backbone, such as in polymer conjugation,8,9 ligands/fluorescent dye addition10–13 have been proposed in order to provide the biopolymer with new/enhanced properties, for various drug/gene delivery,14–16 imaging17 and tissue engineering18,19 applications. However, such lateral grafting may negatively impact on the ability of CS to bind anionic components such as nucleic acid, either by reducing its positive charge (N-grafts) or by steric hindrance (for N/O-grafts copolymers).20 Moreover, given that many reactive moieties are available along the CS backbone, obtaining graft-conjugates that are structurally well-defined may prove challenging.20,21 Block-copolymerization is a promising approach when compared to lateral grafting methods, not only because it circumvents the aforementioned issues, but also because it allows for the synthesis of chitosan derivatives with distinct and unique properties and applications (e.g. polymeric micelles formation, brush-like surface modification).The production of chitosan-based copolymers through end-group modifications such as fluorophore conjugation and PEGylation relying on oxime-click chemistry has been reported.22–24 As an alternative strategy for CS-based block copolymers synthesis, we recently developed an aqueous thiol-based chemistry approach, namely thioacetylation (Fig. 1 – part A).25 The aldehyde functions present on CS termini following nitrous acid (HONO) depolymerisation26 were regioselectively modified in acidic aqueous conditions with linear thiol bearing molecules, without any interference from C2-amines and C6-hydroxyl CS moieties. Even though the CS end-group aldehyde reactive form is only present in trace amounts (the non-reactive gem-diol form largely predominates in aqueous conditions), thiol bearing polymers such as PEG–thiols were conjugated to the CS end group with an efficiency close to 50%. The CS end-group thioacetylation presents significant advantages compared to the alternative oxime-click pathway: (1) CS derivatization is effective in acidic aqueous conditions so that any interference with CS amine groups that are fully protonated and thus unreactive27 is prevented and (2) there is no need for an external chemical treatment to stabilize the final structure.28 The aqueous thioacetylation process occurs with a first thiol attack on the CS aldehyde end-group that forms a hemithioacetal intermediate (Fig. 1 – structure 1) that must be subsequently stabilized in situ by a second thiol nucleophilic attack, the latter being facilitated by concentrating the reaction medium through a freeze-drying (FD) process (Fig. 1 – structure 2). Indeed, FD was shown to substantially increase the conversion degree of this transformation, especially for cases where thiol-molecules were used in slight to moderate excess vs. chitosan end-groups. It is worth mentioning that the intermolecular stabilization of the hemithioacetal intermediate may be sterically hindered by the presence of the first reacted group for large thiol-bearing substituents, potentially limiting the conjugation efficiency. Additionally, although the need to use strong acidic conditions for this thioacetylation process to be effective also ensures the regioselectivity of the reaction by protonating all CS amines, it may nevertheless compromise the integrity of moieties to be conjugated.
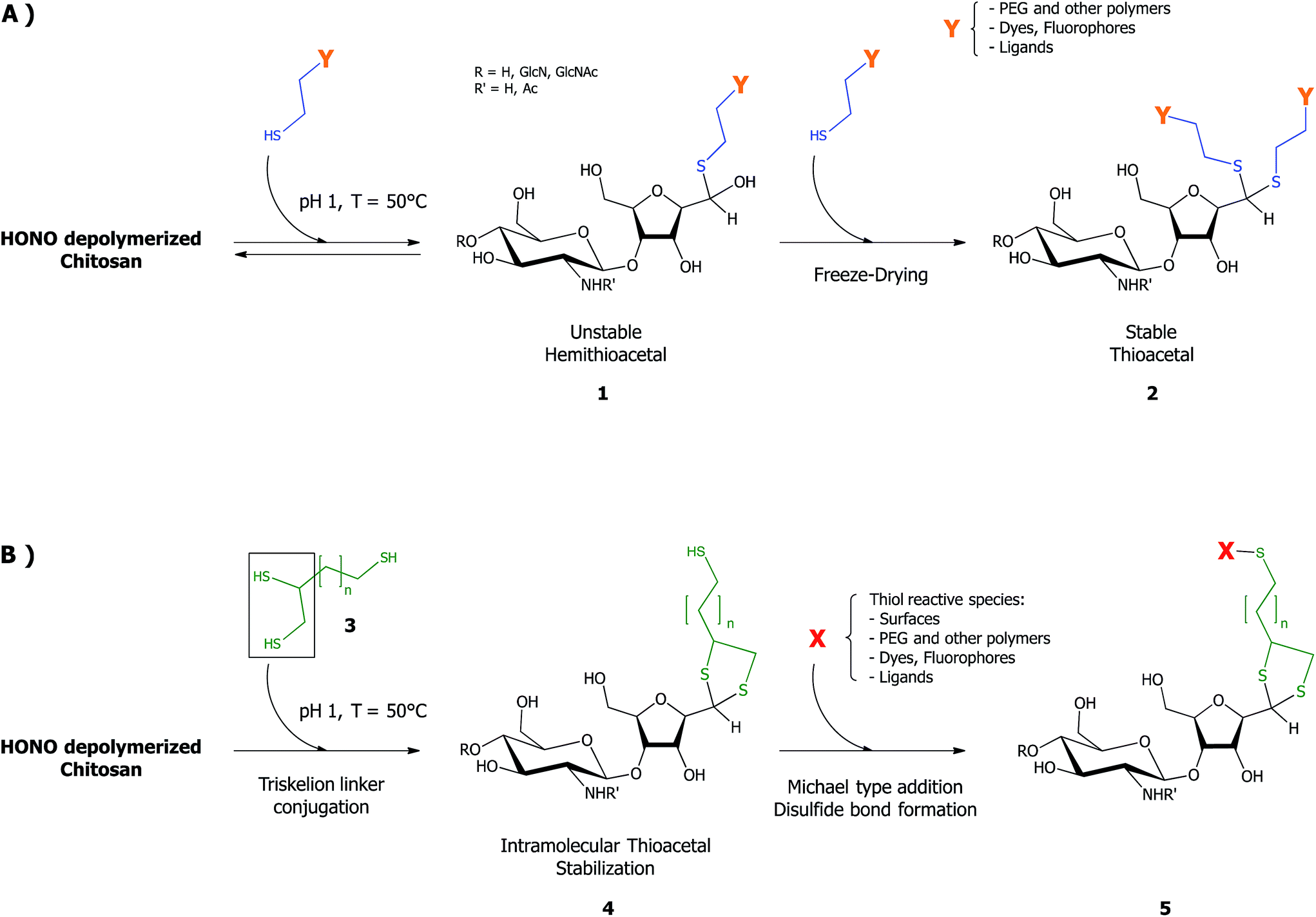 | ||
| Fig. 1 Schematic representation of the CS end-group thioacetylation processes developed by our group: (A) summary of the previously described chitosan end-group thioacetylation process.25 Briefly, CS nitrous acid (HONO) depolymerization induces the formation of 2,5-anhydro-D-mannose (M-Unit) that carries an aldehyde moiety at the end of the cleaved polymer. Despite the fact that this aldehyde is present almost completely in its hydrated and non-reactive form (gem-diol), thiolated macromolecules react with it in strong acidic conditions (pH 1) through a pH dependent equilibrium, forming the corresponding hemithioacetal intermediate (structure 1). The stabilization of the latter into its thioacetal form (structure 2) occurs with a second thiolated macromolecule attack on the same location, an attack that may be sterically hindered by the presence of the first macromolecule. (B) The novel HONO-depolymerized CS end-group thioacetylation process. The trivalent linker (structure 3), referred to as triskelion, comprises a thiol-hook (framed) and a thiol-tail. The derivatization of CS end-groups with triskelion should predominantly lead to the formation of structure 4 by promoting the thiol-hook attack on the CS aldehyde through a facilitated and more efficient intramolecular thioacetylation process vs. the previously described intermolecular strategy.25 The terminally-activated CS (i.e. the CS-b-triskelion adduct) can freely react with any thiol-reactive species or structure through its remaining thiol-tail, as represented in structure 5. | ||
The main objective of this study, depicted in Fig. 1 – part B, was to develop an innovative and improved CS end-group thioacetylation process that would address the shortcomings mentioned above and allow for the effective terminal activation of the polymer and its subsequent conjugation with any thiol reactive structure. In order to achieve this goal, we designed and synthesized a trivalent linker, referred to in this manuscript as “Triskelion”, which bears three thiol moieties (Fig. 1 – structure 3). We hypothesized that this specific trivalent structure would provide the linker with the following characteristics: (1) on one side, two thiol moieties forming a thiol-hook should allow for the two thiol attacks onto the CS reducing-end required for stabilization to occur almost simultaneously (intramolecular vs. intermolecular thioacetylation; Fig. 1 – structure 4) and therefore significantly improve the conjugation efficiency, while (2) the other extremity of the linker carrying the third thiol moiety, referred to as the thiol-tail, should allow for subsequent functionalization in mild conditions with any structure bearing a thiol-reactive moiety (Fig. 1 – structure 5).
The effectiveness and the regioselectivity of the condensation reaction between the triskelion linker and 2,5-anhydro-D-mannose (M-Unit), the CS terminal monomer unit bearing an aldehyde function following HONO depolymerisation, were examined by Liquid Chromatography-Mass Spectrometry (LCMS). For comparison purpose, a small molecule bearing a similar thiol-hook, namely ethanedithiol (EDT), was also reacted with the M-Unit. These mechanistic studies were followed by quantitative experiments where HONO depolymerized CS bearing an M-Unit end was conjugated with the triskelion linker and EDT. The conjugation efficiency was determined by 1H NMR spectroscopy. Diffusion Ordered Spectroscopy (DOSY) experiments were also performed to assess the covalent conjugation in order to prevent physical mixture of reagents. Finally, reactions between triskelion-modified CS and microparticles coated with pyridyl disulfide moieties were performed as a direct application of this novel conjugation strategy.
Materials and methods
Each chemical reaction was performed at least three times independently (N ≥ 3), using fresh reactants in Ar degassed solvents to minimize disulfide bond formation.Reagents, materials
Chitosan with a degree of deacetylation (% DDA) of 91.7%, Mn = 193 kg mol−1 (PDI = 1.256) was provided by Marinard Biotech Inc. deuterium oxide (Cat #151882), deuterium chloride 35 wt% in deuterium oxide (Cat #543047), chloroform-d (Cat #151858), sodium nitrite (Cat #431605), hydrochloric acid standard solution – 1.0 N in H2O (Cat #31894-9), hydrochloric acid 37% (Cat #320331), sodium hydroxide solution 1.0 M (Cat #319511), sodium acetate (Cat #241245), GlcNH2 D-(+)-glucosamine hydrochloride 99% (Cat #C-1276), sodium acetate trihydrate BioXtra (Cat #S7670), Dowex® 50WX8-100 [H+] (Cat #217506), Dowex® 1X8-50 [Cl−] (Cat #217417), EDT (1,2-ethanedithiol) ≥98.0% (Cat #02390), 2-propanol anhydrous 99.5% (Cat #278475), 1,2,6-hexanetriol 96% (Cat #T66206), methanesulfonyl chloride ≥99.7% (Cat #471259), potassium thioacetate 98% (Cat #241776), dichloromethane anhydrous ≥99.8% (Cat #270997), tetrahydrofurane anhydrous (Cat #186562), triethylamine BioUltra ≥99.5% (Cat #90335), sodium carbonate BioXtra ≥99.0% (Cat #S7795), sodium chloride BioXtra ≥99.5% (Cat #S7653), magnesium sulfate anhydrous ReagentPlus® ≥99.5% (Cat #M7506), ethyl acetate CHROMASOLV® for HPLC ≥99.8% (Cat #439169), cyclohexane for HPLC ≥99.9% (Cat #650455), N,N-dimethylformamide biotech. grade, ≥99.9% (Cat #494488), sodium methoxide 0.5 M in methanol (Cat #71751), methanol for HPLC, ≥99.9% (Cat #34860), tris(2-carboxyethyl)phosphine hydrochloride BioUltra, ≥98% (Cat #75259) were purchased from Sigma-Aldrich. Macrosep Advance Centrifugal Devices with 1 kDa MWCO Omega Membrane (Cat # MAP001C37), fluorescein-5-maleimide (Cat #16383) and thiol-activated MagBeads (Cat #50605) were purchased from Pall Corporation, Cayman Chemical and PureCube, respectively.Trivalent linker – triskelion synthesis
As starting material, the commercially available 1,2,6-hexanetriol (Fig. 2 – structure 1) has been chosen because of its potential to form a stable 5-membered thioacetal ring once conjugated to the aldehyde carried by the CS terminal 2,5-anhydro-D-mannose (M-Unit)29,30 (Fig. 1 – structure 4). The triol starting material also comprises a “thiol-tail”, a spacer composed of 6 bonds that is likely to provide sufficient flexibility and mobility to the third remaining thiol group in order to minimize steric hindrance effects and facilitate subsequent CS–triskelion conjugation reactions.31,32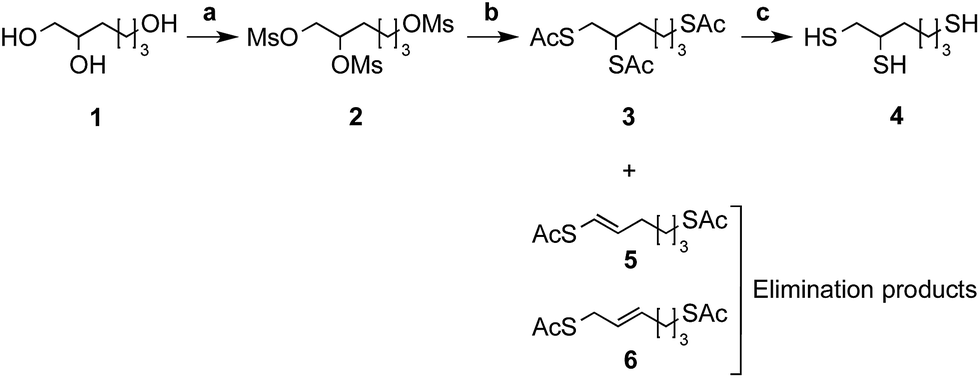 | ||
Fig. 2 Schematic representation of the triskelion synthesis pathway. The triol starting material, namely 1,2,6-hexanetriol (1), was activated with methanesulfonyl chloride (MsCl) (2) in an anhydrous 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of dichloromethane (DCM) and tetrahydrofurane (THF) (a). Mesylate leaving groups (–OMs) were displaced by potassium thioacetate (AcSK) in anhydrous dimethylformamide (DMF) (b) to give the acetyl-protected triskelion linker (3) as major product. The cleavage of acetyl groups was performed using sodium methoxide (MeONa) (c) in order to obtain the desired triskelion linker (4) in high yields (66% over 3 steps). Compounds (5) and (6) correspond to the elimination products obtained after mesylate group displacement. The detailed synthesis protocols are available in ESI; Section 1.† Summary of conditions: (a) MsCl in anhydrous [2:1] DCM/THF at room temperature for 3 h; (b) AcSK in anhydrous DMF at 0–5 °C overnight; (c) MeONa in MeOH at room temperature for 5–10 min. :1 mixture of dichloromethane (DCM) and tetrahydrofurane (THF) (a). Mesylate leaving groups (–OMs) were displaced by potassium thioacetate (AcSK) in anhydrous dimethylformamide (DMF) (b) to give the acetyl-protected triskelion linker (3) as major product. The cleavage of acetyl groups was performed using sodium methoxide (MeONa) (c) in order to obtain the desired triskelion linker (4) in high yields (66% over 3 steps). Compounds (5) and (6) correspond to the elimination products obtained after mesylate group displacement. The detailed synthesis protocols are available in ESI; Section 1.† Summary of conditions: (a) MsCl in anhydrous [2:1] DCM/THF at room temperature for 3 h; (b) AcSK in anhydrous DMF at 0–5 °C overnight; (c) MeONa in MeOH at room temperature for 5–10 min. | ||
As depicted in Fig. 2, the triol starting material (Fig. 2 – structure 1) was engaged in a 3-step synthesis inspired from van der Vlies et al.33 where hydroxyls were transformed into mesylate (–OMs) leaving groups (Fig. 2 – structure 2). The mesylates were then displaced by sodium thioacetate (–SAc) and the acetate protected triskelion linker (Fig. 2 – structure 3) was deprotected with sodium methoxide (MeONa) to obtain the desired trithiol linker (Fig. 2 – structure 4), namely the triskelion. The detailed protocols of the triskelion synthesis and characterization (1H, 13C NMR spectroscopy and mass spectrometry) are available in ESI – Section 1.†
Intramolecular thioacetylation of CS end-group studies involving molecules bearing a thiol-hook
With the triskelion linker synthesized, the conjugation process with 2,5-anhydro-D-mannose (M-Unit) and CS M-Unit aldehyde was assessed semi-quantitatively and then quantitatively, using Liquid Chromatography-Mass Spectrometry (LC-MS) and Nuclear Magnetic Resonance (1H and DOSY NMR) experiments, respectively. The experimental flowchart of these experiments is presented in Fig. 3.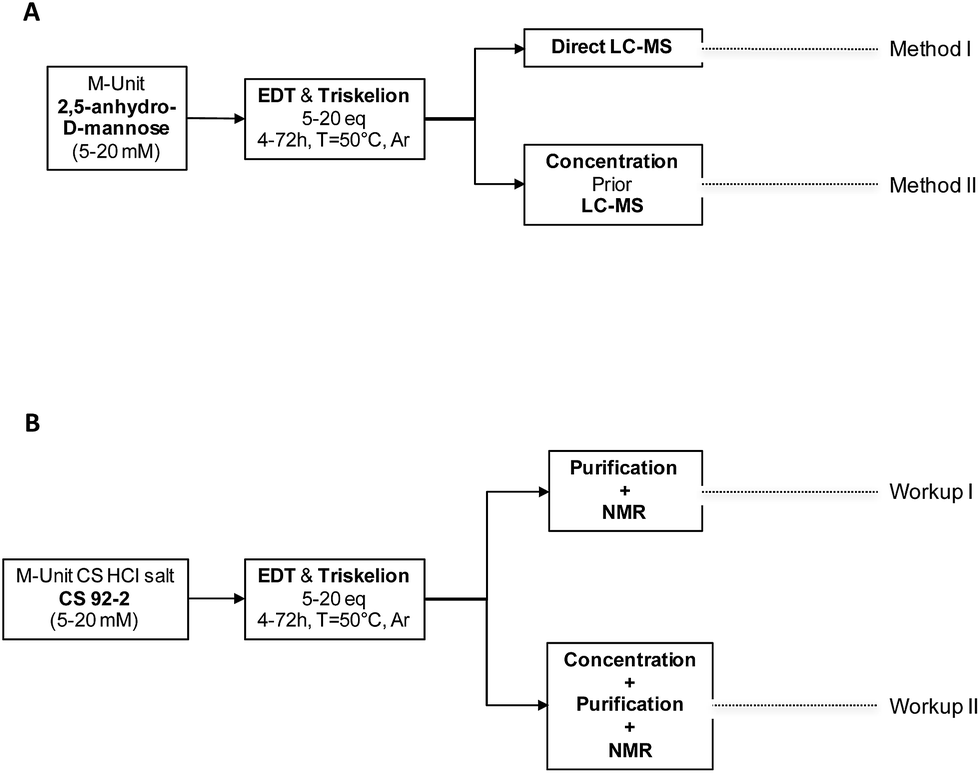 | ||
| Fig. 3 Experimental design flowchart. (A) Mechanistic studies. M-Unit (2,5-anhydro-D-mannose) obtained by HONO treatment of glucosamine, was reacted with molecules bearing a thiol-hook (ethanedithiol-EDT and triskelion linker). The products of these reactions were treated using either Method I: direct LC-MS of the reaction mixture to determine to which extent intramolecular thioacetal formation occurs in situ or Method II: concentration prior to perform LC-MS analysis to determine the influence of such dehydration/concentration step on the final products proportion and also to detect some by-products formation upon concentration. (B) M-Unit CS HCl salt reactivity. M-Unit CS HCl salt was also reacted with ethanedithiol – EDT- and triskelion linkers. After reaction completion, reaction media were treated using either: Workup I: unreacted thiol-hook molecules removal by reprecipitation + NaOH treatment + reprecipitation in order to determine the in situ conjugation process efficiency or Workup II: concentration by FD or rota-evaporation + Workup I in order to quantify a potential increase in conjugation efficiency upon concentration. Conjugation process efficiencies were determined by 1H NMR using both eqn (1) & (2) as detailed in the section below. | ||
Qualitative evaluation of the 2,5-anhydro-D-mannose – molecules bearing a thiol-hook conjugation by LCMS (Fig. 3 – part A)
The expected derivatized M-Unit products were analyzed by comparing the chromatogram integration peaks of determined m/z values corresponding to both proton and sodium adducts within the same run. Such an approximation was deemed appropriate given that potential products (Fig. 7) have similar structures and are therefore expected to have nearly equal sensitivity to ionization.
Quantitative evaluation of the M-Unit CS aldehyde – thiol-hook molecules conjugation by NMR (Fig. 3 – part B)
Quantification of CS derivatization efficiency: functionalization degree calculation of non-purified modified CSs
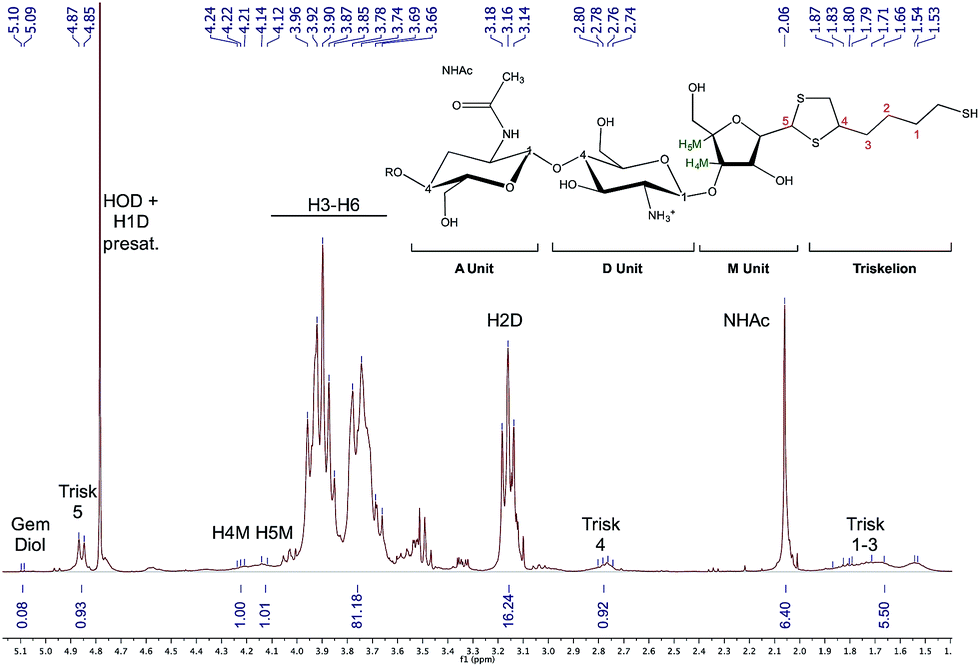
Characterization of chitosan starting materials (DDA and Mn) as well as conjugation efficiencies were performed by 1H NMR spectroscopy using a Bruker Avance III 400 spectrometer equipped with a Bruker CryoProbe Prodigy™. For optimal quantification accuracy, a pulse angle of 30° coupled to an acquisition time of 2 s and repetition times of 5–10 s were used, depending on the sample analyzed. Diffusion ordered spectroscopy experiments (DOSY) were used to assess the covalent conjugation and were performed on the same instrument, using 16 gradients (2.5–50.0 gauss per cm) with a gradient pulse (d) of 3 ms and a diffusion time (D) of 150 ms.The regioselectivity as well as the stoichiometry of the thioacetylation process in acidic aqueous medium has been described by our group in a previous study.25 Briefly, in the case of independent linear molecules bearing a single thiol moiety, two successive thiol attacks on the CS end-group aldehyde are required to stabilize the hemithioacetal intermediate into the thioacetal final product. In the case where molecules bearing a thiol-hook are reacted with M-Unit CS aldehyde in similar conditions, both thiol attacks are expected to occur intramolecularly and almost simultaneously, leading to the formation of a stable 5-membered ring thioacetal (Fig. 1 – structure 4). Consequently, we used the previously described equations by considering one thiol-hook molecule per aldehyde for the following calculations:
The functionalization degree (F) of each regioselective conjugation was assessed by 1H NMR and calculated per the following equations:
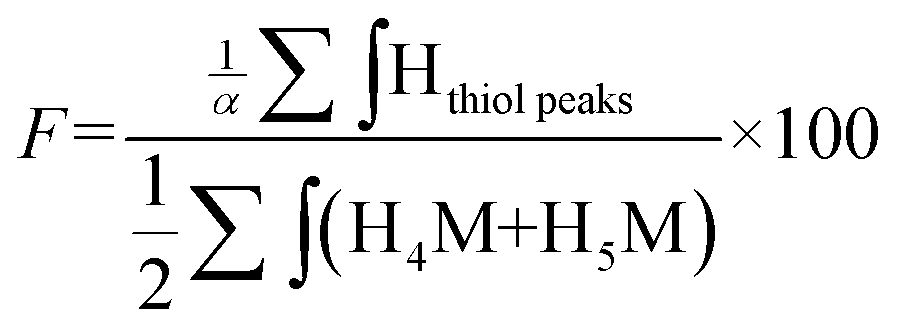 | (1) |
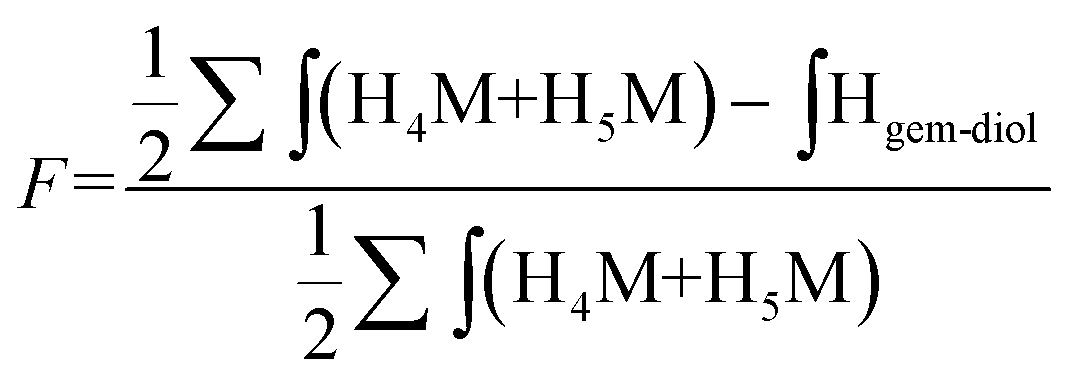 | (2) |
Since M-Unit corresponds to the CS chain extremity, both equations were normalized using its characteristic peaks, namely H4M & H5M (Fig. 4). Eqn (1) involves the integration of the conjugated linker characteristic peaks only (Hthiol peaks), whereas eqn (2) is based on the diminution of the gem-diol peak intensity (Hgem-diol); Hgem-diol representing the proportion of unmodified CS end-groups.36 Both integrations in eqn (1) and (2) are normalized to the number of protons used for the calculation, namely α = 4 for EDT derivatized products and α = 6 for triskelion's α = 1 and 2 for the gem-diol and M-Unit (H4M & H5M), respectively (Fig. 4). It is worth mentioning that the stoichiometry (and the regioselectivity) of the transformation can be validated by comparing the results of these two equations. Indeed, given that eqn (2) is independent from reaction stoichiometry, it will be consistent with eqn (1) result only if the stoichiometry chosen for calculation in eqn (1) is correct, namely one thiol-hook per CS terminal unit.
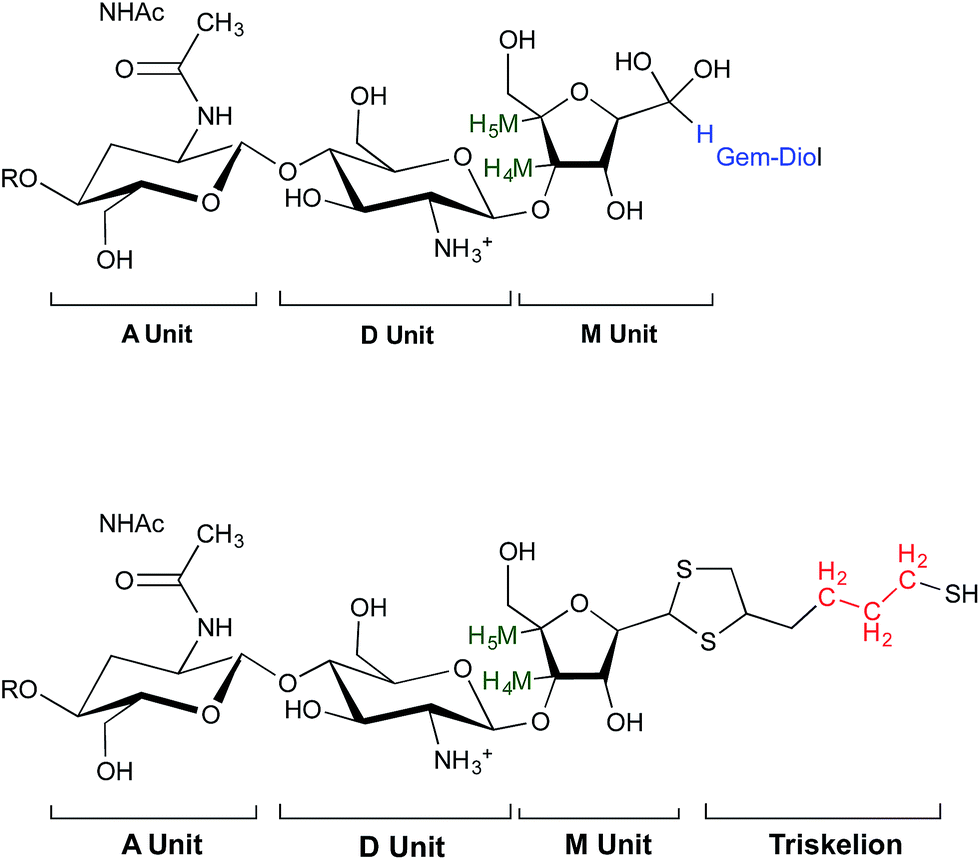 | ||
| Fig. 4 Structure of unmodified CS (top) and CS-b-triskelion adduct (bottom). The protons corresponding to the 1H NMR peaks used for the calculations of the functionalization degree in eqn (1) and (2) are shown in green, blue and red. | ||
CS-b-triskelion conjugation to magnetic microparticles
CS-b-triskelion conjugates synthesized as described above were engaged with pyridyl disulfide-activated magnetic beads in order to assess the thiol-tail reactivity of such conjugates towards thiol-reactive species through disulfide bond displacement (Fig. 5). Briefly, a reduced CS-b-triskelion mixture in 50 mM acetate buffer pH 4 + 200 mM NaCl (m = 10 mg, F = 70%, nSH = 3 μmol, c = 10 mg mL−1) was incubated with thiol-activated magnetic beads (v = 1 mL, 1.5 μmol mL−1 functional group density) for 24 h under inert atmosphere at room temperature. The efficiency of the conjugation process was assessed by measuring the absorbance of the 2-thiopyridone group released upon disulfide linkage displacement (λ = 343 nm, ε = 7175 M−1 cm−1 in the conjugation buffer). The selectivity of the approach was examined using a negative control by engaging non-modified CS with freshly activated beads. The concentration of non-modified CS within the supernatant of the conjugation medium was measured using the spectrophotometric Orange II assay.37 The selectivity of the conjugation reaction was also assessed by 1H NMR analyses and by comparing the functionalization degree (F) of the CS that was released from the beads surface after careful removal of unreacted CS to that of the original mixture engaged in the conjugation process. The detailed protocol of this transformation and associated analyses are provided in ESI – Section 4.†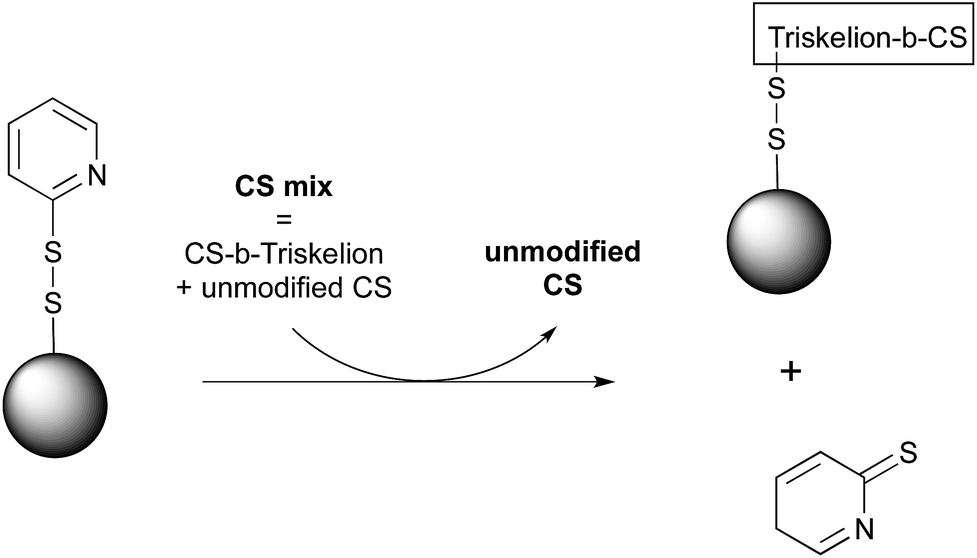 | ||
| Fig. 5 Schematic representation of thiol-activated magnetic beads surface modification. CS-b-triskelion was attached to thiol-activated magnetic microparticles through pyridyl disulfide bond displacement with the release of the pyridinethione leaving group. | ||
Results and discussion
The pure trivalent linker was efficiently synthesized in 3 steps
The complete synthesis pathway depicted in Fig. 2 representing the 3-step synthesis of the trivalent linker, namely triskelion, is discussed in the following section.The first step of the synthesis dealt with the functionalization of the triol hydroxyl group into leaving groups, namely mesylate groups. Methanesulfonyl chloride (MsCl) was preferred to its p-toluenesulfonyl chloride counterpart because it is less bulky and thus favors the transformation of the sterically hindered secondary alcohol while ensuring higher stability of the –OMs intermediates.38 Since the triol starting material is not soluble in dichloromethane (DCM), it was dissolved in a [2:1] anhydrous mixture of dichloromethane/tetrahydrofurane (THF) to ensure complete solubility of the species engaged, namely 1,2,6-haxanetriol, methanesulfonyl chloride and triethylamine. Three equivalents of MsCl per hydroxyl moiety were used to avoid side reactions (–OMs group displacement by unreacted hydroxyls) and were also preferred to the 5 equivalents initially tested, as the amount of salts was too important with the latter and lowered the reaction medium stirring efficiency. The reaction was monitored by thin layer chromatography (TLC) and 1H NMR (data not shown) and was complete in 3 h at room temperature. Acidic workup was performed twice to get rid of salts that formed during the process. The tri-OMs intermediate (Fig. 6 – step 1) was pure enough to be engaged in the next step without further purification (yield ≥ 93%, F = 100% for all experiments; N = 4).
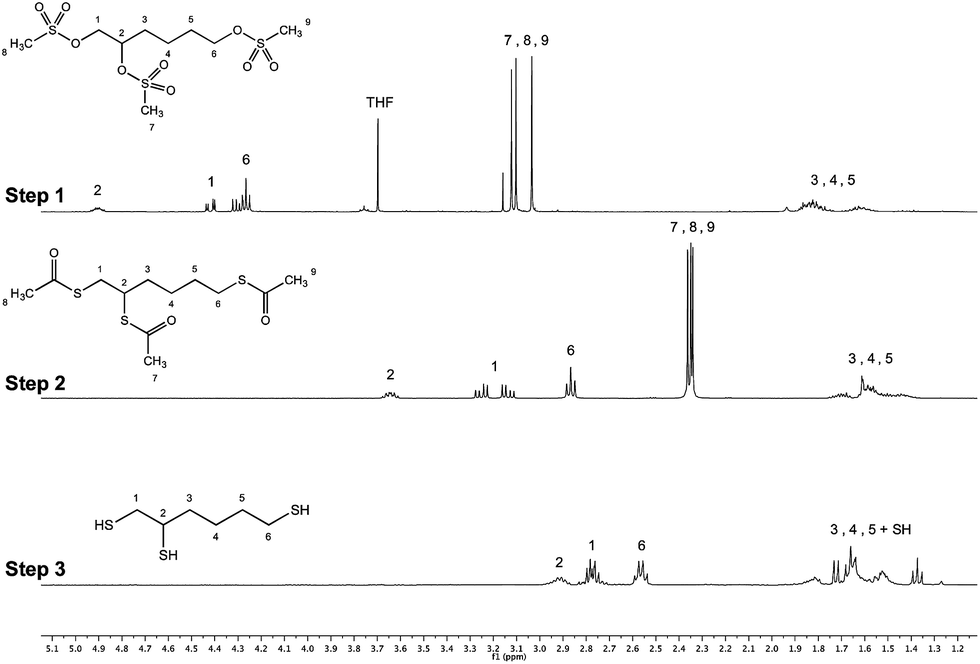 | ||
| Fig. 6 Stacked 1H NMR spectra (400 MHz, CDCl3, ns = 32 scans, acquisition time = 2 s, d1 = 2 s) of the trivalent triskelion linker 3-step synthesis. Step 1 (conversion of tri-OH into tri-OMs) was obtained with 93% yield, step 2 (conversion of tri-OMs into tri-SAc) led to the desired tri-thioacetate with 75% yield and step 3 (conversion of tri-SAc into tri-SH) gave the deprotected triskelion linker with 95% yield. The products of the synthesis were also characterized by 13C NMR spectroscopy and mass spectrometry (ESI – Section 1†). | ||
The second step of the synthesis is the displacement of the mesylate leaving groups by sodium thioacetate (AcSK; 5 equivalents per –OMs), a reaction favored in anhydrous dimethylformamide (DMF), a polar aprotic solvent.33 Initial attempts performed at room temperature revealed a [1:1] mixture of both substitution and elimination products, SN2 (Fig. 2 – structure 3) and E2 products respectively (Fig. 2 – structures 5 & 6). Since a decrease in temperature of the reaction medium may favour the substitution mechanism vs. elimination,39 further experiments were performed in an ice bath (T = 0–5 °C overnight). The amount of E2 products was diminished by 50% in this case, leading to 75% formation of the product of interest after flash chromatography. The synthetic pathway described in Fig. 2 has the advantage to give the triskelion linker under its acetyl-protected form with more than 70% yield over 2 steps (Fig. 6 – step 2), avoiding any intra- and inter-molecular disulfide linkage formation and allowing for long time storage stability (up to 8 months at −20 °C, data not shown).
The last step of the synthesis proposed herein consists in the acetyl deprotection by methanolysis using sodium methoxide, namely the Zemplén transesterification, engaging slightly more than a stoichiometric amount of sodium methoxide per S-acetyl moiety (1.5 equivalent). Sodium methoxide was preferred to other bases like sodium hydroxide because it proceeds quickly without generating β-elimination and other racemization.40,41 Complete acetyl deprotection was observed after 5–10 min (monitored by TLC and 1H NMR analysis; data not shown), maintaining the triskelion linker in its acetyl-deprotected form with 95% yield (Fig. 6 – step 3). However, as reported by Wilson et al.,42 the yield of the reaction was limited by concomitant decomposition of the final product when longer reaction durations were performed (stacked 1H NMR spectra are available in ESI – Fig. S4†). Once deprotected, the triskelion linker was ready to be directly engaged in the M-Unit aldehyde thioacetylation process, without further purification.
The 2,5-anhydro-D-mannose aldehyde derivatization in MeOH with molecules bearing a thiol-hook leads to the intramolecular thioacetal as major product
In a preliminary study, ethanedithiol (EDT) was engaged in the intramolecular thioacetylation process with the M-Unit aldehyde as described in the “Materials and methods” section above; except that a proportion of 30% isopropanol in 0.1 M HCl was added to ensure complete solubilization of the species. The results of this semi-quantitative study indicated that the intramolecular thioacetal (5-membered ring thioacetal derivatized M-Units) is the only structure detected, independently from the reaction duration and the post-reaction method used (data not shown). The fact that no hemithioacetal intermediate was detected confirms that the in situ intramolecular thioacetylation process predominates when molecules bearing a thiol-hook are engaged with the M-Unit aldehyde.The triskelion linker was reacted with the 2,5-anhydro-D-mannose under the same conditions as for EDT, except that the co-solvent was changed to 30% THF or 90% MeOH in 0.1 M HCl. These co-solvents were selected to ensure complete solubility of the triskelion, 2,5-anhydro-D-mannose and CS, thus allowing direct comparison of the reactions performed with the former and the latter. Since both the thiol-hook and the thiol-tail of the triskelion linker may react with the M-Unit aldehyde, three different conjugates can form, as depicted in Fig. 7: the intramolecular thioacetal A (through thiol-hook attack), the hemithioacetal B (after a first thiol-tail attack) and its stabilized linear thioacetal form C (through two successive thiol-tail attacks). LC-MS analyses were used to assess the influence of the reaction conditions (co-solvent and method post-reaction) on the nature and the proportion of each possible final product.
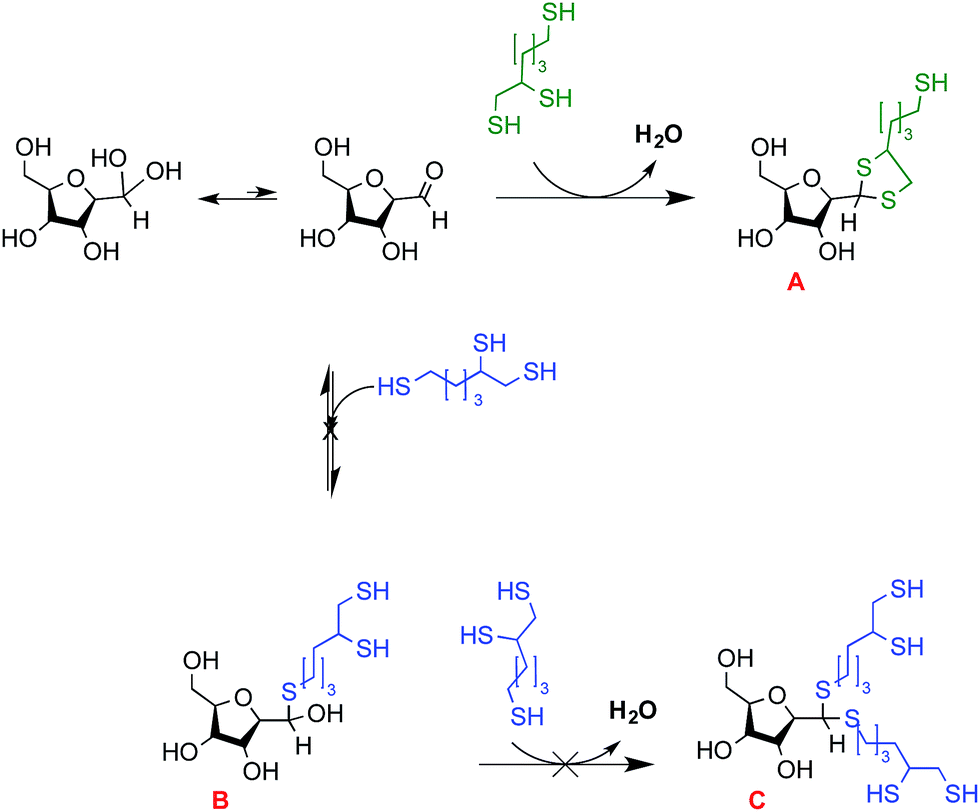 | ||
| Fig. 7 Schematic representation of potential triskelion-derivatized 2,5-anhydro-D-mannose (M-Unit) products. Since the triskelion linker carries three reactive thiol moieties able to engage into the thioacetylation process (“thiol-hook” vs. “thiol-tail”), there are three potential observable structures: structure A which corresponds to the expected intramolecular thioacetal obtained from thiol-hook reaction with the M-Unit aldehyde; structure B represents the hemithioacetal intermediate that is formed upon reaction of the thiol-tail with the M-Unit aldehyde. This structure is unstable and it can be stabilized by a second triskelion attack to form the structure C, the intermolecular thioacetal. All the potential disulfide related products such as A-ss-A, B and C with a disulfide closed hook were considered within the calculations without being represented in this figure. The LC-MS results indicate that the intramolecular thioacetal (structure A) corresponds to the major derivatization product observed by LC-MS when MeOH is used as co-solvent, whereas the intermolecular thioacetal (structure C) is favored when THF is used as co-solvent. | ||
The results of the reactions performed between the triskelion linker and 2,5-anhydro-D-mannose are reported in Table 1 and Fig. 8. When MeOH was used as co-solvent, the results indicated that the triskelion thiol-hook attack on the aldehyde of 2,5-anhydro-D-mannose lead predominantly to the formation of the intramolecular thioacetal (A). This finding is in agreement with what was observed with EDT, where results indicated that the stabilization of hemithioacetals occurred in situ through a 5-membered ring cyclization process. On the other hand, the use of THF as co-solvent appeared to favor the intermolecular thioacetal formation (C) at the expense of its intramolecular counterpart (A). This difference in reactivity could possibly be explained by the presence of peroxides that would have formed upon THF air oxidation.43,44 Indeed, THF peroxide traces may have deactivated the thiol-hook by oxidation while leaving the remaining thiol-tails free to react with the M-Unit aldehyde. In order to favor the intramolecular thioacetal formation (structure A), THF was not used as a co-solvent for the triskelion linker conjugation to chitosans.
| Final products (see Fig. 7) | Chemical formula | Expected m/z [M + Na]+ | Observed m/z [M + Na]+ |
|---|---|---|---|
| Intramolecular thioacetal (A) | C12H22O4S3 | 349.0572 | 349.0562 |
| C24H42O8S6 | 673.1096 | 673.1092 | |
| Hemithioacetal (B) | C12H22O5S3 | 365.0522 | 365.0500 |
| Intermolecular thioacetal (C) | C18H36O4S6 | 531.0830 | 531.0849 |
| C18H34O4S6 | 529.0674 | 529.0662 | |
| C18H32O4S6 | 527.0517 | 527.0525 |
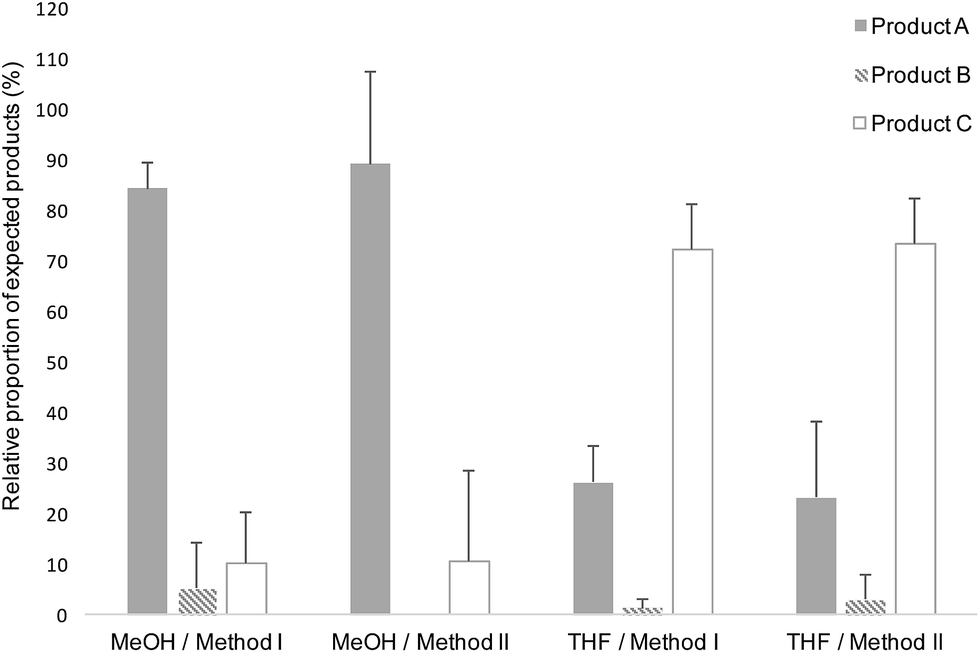 | ||
| Fig. 8 Relative proportion of the M-Unit/triskelion conjugation products observed by LC-MS (see Fig. 7 and Table 1). Products A, B and C represent the intramolecular thioacetal, the hemithioacetal intermediate and the intermolecular or linear thioacetal, respectively. Each conjugation reaction (10–20 mM aldehyde, pH 1, T = 50 °C, t = 72 h, 30% THF or 90% MeOH) was performed in triplicates (N = 3) and the reaction media were treated as follows: Method I refers to a direct LC-MS analysis of the reaction medium whereas Method II stands for a concentration to dryness step prior to performing LC-MS. All the potential disulfide related products such as A-ss-A, B and C with a disulfide closed hook were considered within the calculations (Table 1). The results of this semi-quantitative study suggest that the intramolecular thioacetylation (A) is the favored mechanism of M-Unit/triskelion conjugation when MeOH is used as co-solvent. On the other hand, the intermolecular thioacetal (C) is predominantly formed when THF is used as co-solvent, possibly because of some thiol-hook deactivation occurring due to the presence of peroxides within THF. | ||
CS end-groups were successfully activated with the triskelion linker through an efficient intramolecular thioacetylation process
M-Unit CS 92-2 HCl salt were derivatized with molecules bearing a thiol-hook (EDT and triskelion linker) and the purified conjugates were characterized by NMR spectroscopy, as described in Fig. 3 – part B. EDT experiments where performed in 30% iPrOH, whereas triskelion conjugations were conducted in 90% MeOH. The functionalization degrees F of such derivatizations were calculated from the 1H NMR spectrum of each purified conjugate using both eqn (1) and (2). The results obtained with eqn (2) are shown in Fig. 9.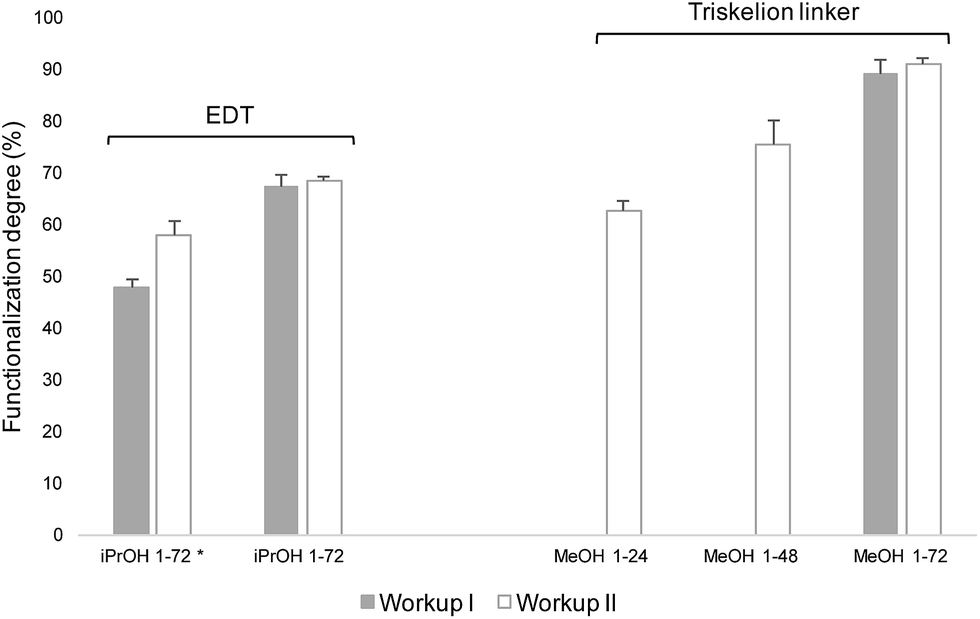 | ||
| Fig. 9 Conjugation efficiency of the M-Unit CS HCl salt reacted with 20 equivalents of molecules bearing a thiol-hook (EDT and the triskelion linker) per CS end-group for 24–72 h at pH 1 and 50 °C. 30% isopropanol (iPrOH) and 90% methanol (MeOH) were used as co-solvent for EDT and triskelion linker solubilization, respectively. The nomenclature used in the graphical representation herein is: “Solvent pH-duration” (i.e., iPrOH 1–72 stands for 30% isopropanol at pH 1 for 72 h). Reaction media were treated according to Workup I (alkali treatment + precipitation in THF) and Workup II (concentration to dryness + alkali treatment + precipitation in THF). Functionalization degrees (F) were calculated using eqn (2) (decrease in gem-diol peak intensity) and confirmed with eqn (1) considering 1 molecule bearing a thiol-hook per purified CS adduct. Functionalization degrees calculated with eqn (1) are not shown here since they were with 2% of those obtained with eqn (2). (* corresponds to the results of the conjugations obtained with 5 equivalents of thiol-hook per M-Unit aldehyde instead of 20). | ||
In a preliminary study, 5–20 equivalents of EDT were conjugated per M-Unit CS aldehyde in order to study the influence of increasing thiol-hook equivalents on the derivatization efficiency. As observed in our previous study,25 a concentration step post-reaction enhances the intermolecular thioacetylation process when a small amount of thiol-bearing molecules are engaged by favoring both the formation of the hemithioacetal intermediate and the second thiol molecule attack to form the final stable thioacetal. As shown in Fig. 9, the in situ intramolecular thioacetal formation efficiency when 5 equivalents of EDT per CS end-group were used (iPrOH 1–72* Workup I) was enhanced with a concentration step post-reaction (iPrOH 1-72* Workup II), therefore leading to better conversion degrees (from 48 ± 1 to 58 ± 3%). On the other hand, when 20 equivalents of EDT were added, such concentration step post-reaction did not affect the conjugation efficiency since similar functionalization degrees were calculated independently from the Workup implemented (68 ± 2 and 69 ± 1% for iPrOH 1–72 Workups I and II, respectively).
Similar experiments as presented above with EDT were performed with 20 equivalents of triskelion linker, except that 90% MeOH was used as co-solvent. After 72 h reaction (Fig. 9; MeOH 1–72), the drying step post-reaction does not have any significant impact on the conjugation efficiency since comparable functionalization degrees were obtained regardless the Workup performed (89 ± 3% and 91 ± 1% (Fig. 10) using Workup I and II, respectively). As observed in our previous study,25 our results reveal that a significant in situ thioacetal formation can be achieved when a large excess of thiol-bearing molecules is used.
 | ||
| Fig. 10 1H NMR, 400 MHz, D2O, T = 25 °C, ns = 64 scans, acquisition time = 2 s, d1 = 10 s. The sample herein corresponds to the CS-b-triskelion conjugate obtained after 72 h reaction in 90% MeOH (MeOH 1–72) and processed as per Workup II. Functionalization degrees F of 91% and 92% were obtained using eqn (1) and (2), respectively. | ||
A time-point experiment was also performed in order to determine if the reaction duration may be lowered to 48 h and then to 24 h while giving such high functionalization degrees by concentrating the reaction media (Workup II). Both eqn (1) and (2) indicate 63 ± 2% efficiency after 24 h, whereas 78 ± 1% of CS end-group aldehydes were converted after 48 h reaction (Fig. 9; MeOH 1–24 & MeOH 1–48).
These results indicate that the equilibrium between the M-Unit aldehyde and the hemithioacetal formed upon thiol-hook molecule addition (Fig. 11) can be displaced towards the latter by increasing both the equivalents of thiol-bearing molecules and the reaction duration.
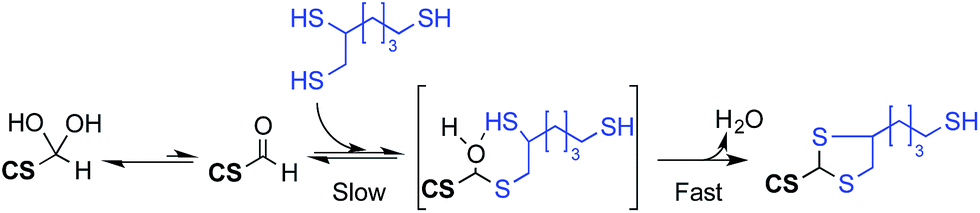 | ||
| Fig. 11 Proposed mechanism of the triskelion linker conjugation to M-Unit CS under acidic aqueous conditions. Although the rate limiting step of this conjugation process is the transformation of the gem-diol into its corresponding active aldehyde, the latter is engaged in an equilibrium with the hemithioacetal intermediate (in brackets) upon thiol-hook addition (triskelion linker shown herein). As suggested by LCMS studies, the hemithioacetal intermediate is readily transformed into its stable thioacetal counterpart with the release of a water molecule. The equilibrium between the M-Unit aldehyde and the hemithioacetal can be displaced towards the latter by increasing both the amount of thiol-hook engaged in the reaction and the reaction duration. | ||
The effectiveness of such transformation was assessed by DOSY NMR experiments (e.g., Fig. 12), which provided evidence that the trivalent linker was covalently attached to the CS chain. Indeed, both the triskelion linker and CS chains had the same translational diffusion coefficient in D2O at 25 °C, despite of their significant molecular weight difference (182.03 g mol−1 vs. 3200 g mol−1, for the trivalent linker and CS chain, respectively).
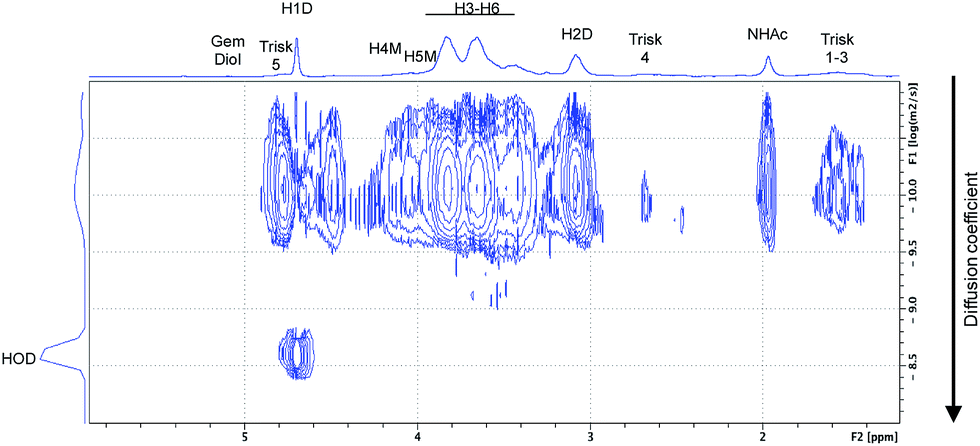 | ||
| Fig. 12 DOSY spectrum of the CS-b-triskelion conjugate (MeOH 1–72, Workup II). 16 gradients between 2.5 and 50.0 gauss per cm with a gradient pulse (δ) of 3 ms, a diffusion time (Δ) of 150 ms. Both CS and triskelion have the same translational diffusion coefficient at 25 °C in D2O, indicating that they are joined together by a covalent bond. | ||
LC-MS and NMR reveal the regioselectivity of the CS-end-group aldehyde intramolecular thioacetylation process
The regioselectivity and the stoichiometry of this transformation has been already assessed by LC-MS, where one molecule bearing a thiol-hook was covalently conjugated per 2,5-anhydro-D-mannose aldehyde via a 5-membered ring formation. Both the regioselectivity and the stoichiometry of the CS end-group aldehyde derivatization with thiol-hook molecules were also confirmed by calculating and comparing the functionalization degrees of these transformations with eqn (1) (based on thiol-hook representative peaks integration values and where 1 thiol-hook per CS end-group was considered) and eqn (2) (based on the concomitant diminution of the gem-diol peak intensity and which is independent from the number of conjugated molecules bearing a thiol-hook). Thus, for each conjugation reported in Fig. 9, similar functionalization degrees (F ± 2%) were calculated using both equations (data not shown), suggesting that (1) the thioacetylation occurred on the CS extremity, namely the M-Unit aldehyde, and (2) that the intramolecular thioacetylation process dominates with respect to the intermolecular process.The terminally activated CS efficiently and selectively react with thiol reactive surface
Reduced CS-b-triskelion mixture (MeOH 1–48 processed with Workup II, F = 70%) was incubated with thiol-activated magnetic beads for 24 h under inert atmosphere at room temperature. The agarose-coated PureCube thiol-activated magnetic beads carrying ethylenediamine spacers were chosen to minimize non-specific binding with CS. Undesirable reactions were also minimized by solubilizing CS mixtures in acetate buffer pH 4 + 200 mM NaCl as conjugation buffer, ensuring CS amines complete protonation while screening electrostatic repulsions between CS chains that could potentially reduce grafting efficiency as the process evolves.A 2-fold excess of activated CS (nSH = 3 μmol vs. 1.5 μmol pyridyl disulfide moieties) was reacted with the beads to ensure maximum coating efficiency onto bead surface. Both the bead functional group density and the reaction efficiency were determined by absorbance measurements of the pyridinethione released group (λ = 343 nm, ε = 7175 L M−1 cm−1 in 50 mM acetate buffer pH 4 + 200 mM NaCl; Data not shown).
After unreacted CS removal, CS-b-triskelion was released by tris(2-carboxyethyl)phosphine (TCEP) reduction, giving the desired CS-b-triskelion conjugate with 51% yield. Considering the functionalization degree of the initial mixture (F = 70%) engaged in this coating process, the maximum amount of CS-b-triskelion expected to be selectively conjugated to the beads would be 3.5 mg (2:1 CS-b-triskelion/pyridyl disulfide functional group stoichiometry) and m = 1.8 mg CS-b-triskelion was recovered. TCEP was preferred to other reductants (such as dithiothreitol or β-mercaptoethanol) because of its effectiveness in acidic conditions.45 Nonetheless, since TCEP bears 3 carboxylic acid moieties that have low pKa as 4.3, 3.6 and 2.9,46 electrostatic interactions with CS amines were avoided by rinsing the conjugates with 0.1 M HCl solution in order to neutralize the carboxyl moieties.
To assess the selectivity of the reaction, the conjugation process described above was also performed with non-modified CS. After 24 h incubation with freshly-activated beads, CS quantification by the Orange II assay revealed that more than 95% of the non-modified polymer remained within the supernatant, whereas less than 5% of CS was recovered after the entire process (data not shown). Given the precision of the assay, these results indicate that non-modified CS did not adsorb to or react with the surface during the process and that only thiol-activated CS bind to the bead surface through disulfide linkage.
The selectivity of the reaction was further assessed by recovering and analyzing the conjugated CS from the beads using TCEP, after careful removal of any non-reacted CS from the conjugation mixture (removal of supernatant). In order be able to confirm the selectivity of the conjugation reaction, a CS with F = 70% was specifically chosen for this experiment. Indeed, if only CS-b-triskelion adducts reacted with the beads, the functionalization degree of the recovered CS population should be higher than that of starting material, given that unreacted/non-modified CS were discarded within the supernatant. 1H NMR spectra were used to calculate the functionalization degree of the CS mixture engaged in the process (Fig. 13A) and that of the recovered CS (Fig. 13B). For comparison purpose, both spectra were normalized based on the CS acetyl peaks (NHAc; δ = 2.06 ppm) since the latter remains unchanged upon this treatment. Both eqn (1) & (2) indicated 70% functionalization in starting CS vs. 95% in recovered CS, indicating that only activated CS reacted with the surface functional groups, demonstrating the selectivity of the reaction. This result also indicated that such chemistry could be used in HPLC columns to obtain highly purified activated triskelion CS.
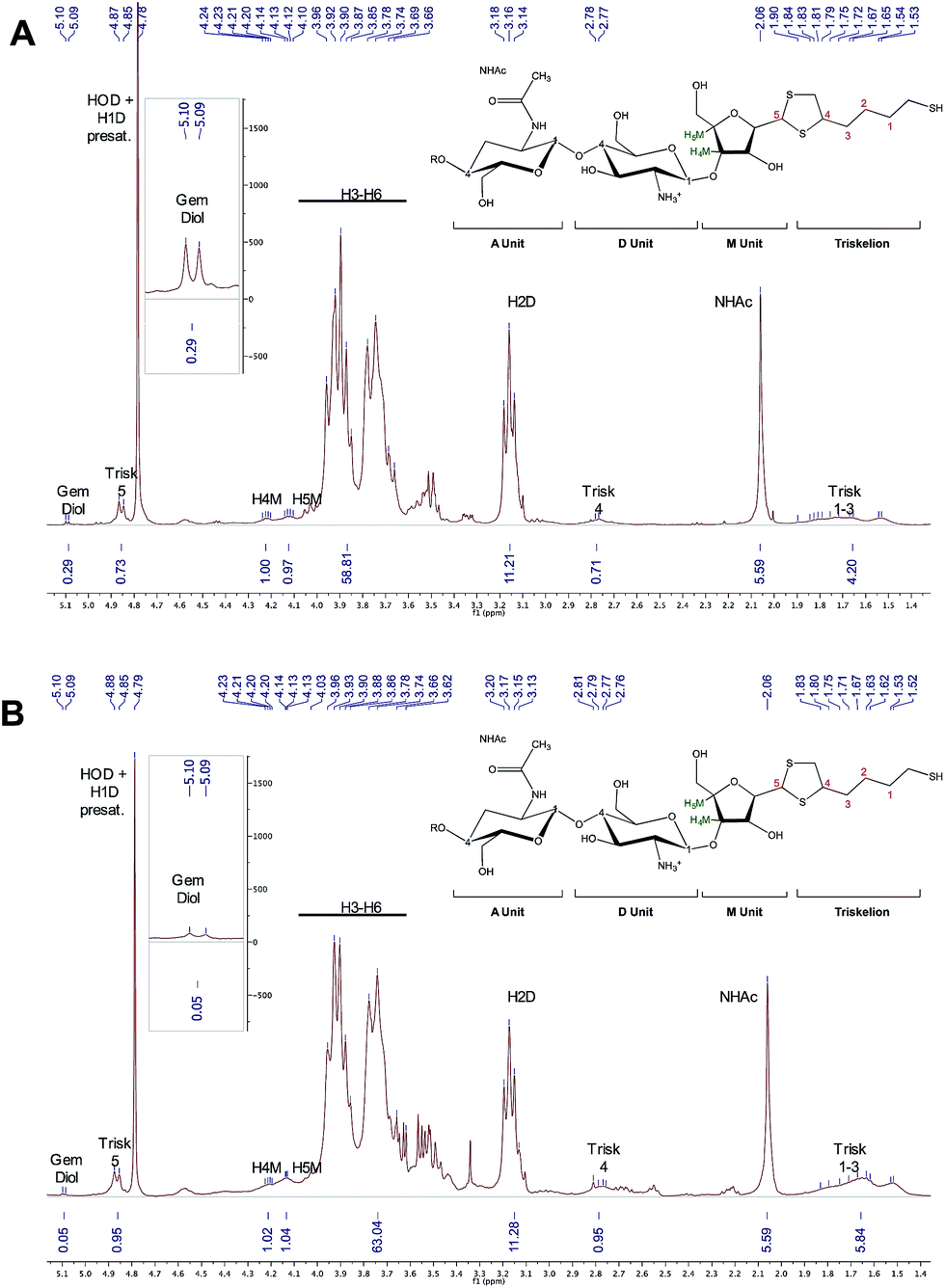 | ||
| Fig. 13 1H NMR spectra (400 MHz, D2O, T = 25 °C, ns = 256 scans, acquisition time = 2 s, d1 = 10 s) representing (A) the initial CS + CS-b-triskelion mixture that was engaged with the thiol-activated magnetic beads and (B) the released CS-b-triskelion post treatment. Both spectra were normalized using the unmodified CS acetyl peak (δ = 2.06 ppm, 5.59 H) for comparison purpose. Both eqn (1) & (2) were used to calculate functionalization degree of CS mixture and released CS calculations, giving 70% (±2) and 95% (±2), respectively. This result indicates that only CS-b-triskelion adducts bind to the thiol-activated beads through disulfide linkage displacement. | ||
Conclusions
This study was dedicated to the development of a novel and improved thioacetylation process to terminally activate chitosan. This process relies on the use of a specifically designed and synthesized trivalent linker (the triskelion) that bears a thiol-hook and a thiol-tail, allowing for chitosan terminal activation through an efficient intramolecular conjugation mechanism and subsequent functionalization with any thiol-reactive structure, respectively. A combination of mass spectrometry and NMR spectroscopy was used to assess the efficiency and confirm the regioselectivity of the reaction. The function of such terminally activated chitosan was validated in a direct application where CS-b-triskelion conjugates were used to modify the surface of magnetic beads through labile disulfide linkages formation. Compared to our previously proposed thioacetylation process, where two successive thiol attacks on the CS end-group aldehyde are necessary to stabilize the hemithioacetal intermediates into their final thioacetal structures,25 this novel approach allows for a more efficient one-step CS end-group activation by thioacetylation. Importantly, the use of the triskelion linker directly overcomes the two main shortcomings of our previously proposed approach, namely, the potential limited efficiency due to steric hindrance of large thiol-bearing substituents and the incompatibility of the process with acid-labile moieties to be conjugated.Ongoing studies are focused on the purification of CS-b-triskelion conjugates by chromatography, using pyridyl disulfide activated resin; and on the development of novel conjugation strategies producing Michael type addition and disulfide linked adducts.
The herein described CS-b-triskelion synthesis offers new perspectives for CS biomedical applications such as CS-block modifications and brush-like surface coatings through labile/non-labile linkages.
Acknowledgements
This work was supported by the Natural Sciences and Engineering Research Council of Canada (NSERC) and ANRis Pharmaceuticals. The authors would like to acknowledge Alexandra Furtos (Mass Spectrometry, Université de Montréal), Cédric Malveau and Sylvie Bilodeau (NMR spectroscopy, Université de Montréal) for their help in characterization.References
- C. Shi, Y. Zhu, X. Ran, M. Wang, Y. Su and T. Cheng, J. Surg. Res., 2006, 133, 185–192 CrossRef CAS PubMed.
- S. K. L. Levengood and M. Zhang, J. Mater. Chem. B, 2014, 2, 3161–3184 RSC.
- F. Croisier and C. Jérôme, Eur. Polym. J., 2013, 49, 780–792 CrossRef CAS.
- P. K. Dutta, K. Rinki and J. Dutta, in Chitosan for Biomaterials II, eds. R. Jayakumar, M. Prabaharan and A. R. A. Muzzarelli, Springer Berlin Heidelberg, Berlin, Heidelberg, 2011, pp. 45–79 Search PubMed.
- L. Hu, Y. Sun and Y. Wu, Nanoscale, 2013, 5, 3103–3111 RSC.
- M. Dash, F. Chiellini, R. M. Ottenbrite and E. Chiellini, Prog. Polym. Sci., 2011, 36, 981–1014 CrossRef CAS.
- M. D. Buschmann, A. Merzouki, M. Lavertu, M. Thibault, M. Jean and V. Darras, Adv. Drug Delivery Rev., 2013, 65, 1234–1270 CrossRef CAS PubMed.
- I. K. Park, T. H. Kim, Y. H. Park, B. A. Shin, E. S. Choi, E. H. Chowdhury, T. Akaike and C. S. Cho, J. Controlled Release, 2001, 76, 349–362 CrossRef CAS PubMed.
- O. Germershaus, S. Mao, J. Sitterberg, U. Bakowsky and T. Kissel, J. Controlled Release, 2008, 125, 145–154 CrossRef CAS PubMed.
- J. You, F. Q. Hu, Y. Z. Du and H. Yuan, Biomacromolecules, 2007, 8, 2450–2456 CrossRef CAS PubMed.
- J. H. Na, H. Koo, S. Lee, K. H. Min, K. Park, H. Yoo, S. H. Lee, J. H. Park, I. C. Kwon, S. Y. Jeong and K. Kim, Biomaterials, 2011, 32, 5252–5261 CrossRef CAS PubMed.
- H. Prichystalova, N. Almonasy, A. M. Abdel-Mohsen, R. M. Abdel-Rahman, M. M. Fouda, L. Vojtova, L. Kobera, Z. Spotz, L. Burgert and J. Jancar, Int. J. Biol. Macromol., 2014, 65, 234–240 CrossRef CAS PubMed.
- L. Upadhyaya, J. Singh, V. Agarwal and R. P. Tewari, J. Controlled Release, 2014, 186, 54–87 CrossRef CAS PubMed.
- Z. Liu, Z. Zhang, C. Zhou and Y. Jiao, Prog. Polym. Sci., 2010, 35, 1144–1162 CrossRef CAS.
- L. Casettari and L. Illum, J. Controlled Release, 2014, 190, 189–200 CrossRef CAS PubMed.
- J. Sarvaiya and Y. K. Agrawal, Int. J. Biol. Macromol., 2015, 72, 454–465 CrossRef CAS PubMed.
- X. Fei and Y. Gu, Prog. Nat. Sci., 2009, 19, 1–7 CrossRef CAS.
- R. LogithKumar, A. KeshavNarayan, S. Dhivya, A. Chawla, S. Saravanan and N. Selvamurugan, Carbohydr. Polym., 2016, 151, 172–188 CrossRef CAS PubMed.
- M. Larsson, W.-C. Huang, M.-H. Hsiao, Y.-J. Wang, M. Nydén, S.-H. Chiou and D.-M. Liu, Prog. Polym. Sci., 2013, 38, 1307–1328 CrossRef CAS.
- L. Casettari, D. Vllasaliu, E. Castagnino, S. Stolnik, S. Howdle and L. Illum, Prog. Polym. Sci., 2012, 37, 659–685 CrossRef CAS.
- S. P. McManus, A. Kozlowski, and P. D. Youso, NEKTAR Therapeutics, US Pat., US 2012/0238735, 2012.
- B. E. Benediktsdottir, K. K. Sorensen, M. B. Thygesen, K. J. Jensen, T. Gudjonsson, O. Baldursson and M. Masson, Carbohydr. Polym., 2012, 90, 1273–1280 CrossRef CAS PubMed.
- R. Novoa-Carballal and A. H. E. Muller, Chem. Commun., 2012, 48, 3781–3783 RSC.
- R. Novoa-Carballal, C. Silva, S. Moller, M. Schnabelrauch, R. L. Reis and I. Pashkuleva, J. Mater. Chem. B, 2014, 2, 4177–4184 RSC.
- V. D. Pickenhahn, V. Darras, F. Dziopa, K. Biniecki, G. De Crescenzo, M. Lavertu and M. D. Buschmann, Chem. Sci., 2015, 6, 4650–4664 RSC.
- G. G. Allan and M. Peyron, Carbohydr. Res., 1995, 277, 257–272 CrossRef CAS PubMed.
- D. Filion, M. Lavertu and M. D. Buschmann, Biomacromolecules, 2007, 8, 3224–3234 CrossRef CAS PubMed.
- T. S. Zatsepin, D. A. Stetsenko, A. A. Arzumanov, E. A. Romanova, M. J. Gait and T. S. Oretskaya, Bioconjugate Chem., 2002, 13, 822–830 CrossRef CAS PubMed.
- D. P. N. Satchell and R. S. Satchell, Chem. Soc. Rev., 1990, 19, 55–81 RSC.
- K. Pihlaja and P. Pasanen, in Ethers, Crown Ethers, Hydroxyl Groups and their Sulphur Analogues, John Wiley & Sons, Ltd, 1981, pp. 821–858 Search PubMed.
- G. Delaittre, A. M. Greiner, T. Pauloehrl, M. Bastmeyer and C. Barner-Kowollik, Soft Matter, 2012, 8, 7323–7347 RSC.
- J. M. Goddard and J. H. Hotchkiss, Prog. Polym. Sci., 2007, 32, 698–725 CrossRef CAS.
- A. J. van der Vlies, U. Hasegawa and J. A. Hubbell, Mol. Pharm., 2012, 9, 2812–2818 CrossRef CAS PubMed.
- S. Claustre, F. Bringaud, L. Azéma, R. Baron, J. Périé and M. Willson, Carbohydr. Res., 1999, 315, 339–344 CrossRef CAS PubMed.
- R. E. Barnett and W. P. Jencks, J. Am. Chem. Soc., 1967, 89, 5963–5964 CrossRef CAS.
- K. Tømmeraas, K. M. Vårum, B. E. Christensen and O. Smidsrød, Carbohydr. Res., 2001, 333, 137–144 CrossRef.
- P. L. Ma, M. D. Buschmann and F. M. Winnik, Anal. Chem., 2010, 82, 9636–9643 CrossRef CAS PubMed.
- R. K. Crossland and K. L. Servis, J. Org. Chem., 1970, 35, 3195–3196 CrossRef CAS.
- C. R. Noller, C. A. Luchetti, E. M. Acton and R. A. Bernhard, J. Am. Chem. Soc., 1953, 75, 3851–3852 CrossRef CAS.
- L. Zervas, I. Photaki and N. Ghelis, J. Am. Chem. Soc., 1963, 85, 1337–1341 CrossRef CAS.
- B. Ren, M. Wang, J. Liu, J. Ge, X. Zhang and H. Dong, Green Chem., 2015, 17, 1390–1394 RSC.
- H. Wilson, S. Byrne and K. M. Mullen, Chem.–Asian J., 2015, 10, 715–721 CrossRef CAS PubMed.
- D. R. Burfield, J. Org. Chem., 1982, 47, 3821–3824 CrossRef CAS.
- B. Zhang, M. Cho, J. D. Fortner, J. Lee, C.-H. Huang, J. B. Hughes and J.-H. Kim, Environ. Sci. Technol., 2009, 43, 108–113 CrossRef CAS PubMed.
- E. B. Getz, M. Xiao, T. Chakrabarty, R. Cooke and P. R. Selvin, Anal. Biochem., 1999, 273, 73–80 CrossRef CAS PubMed.
- A. Krȩżel, R. Latajka, G. D. Bujacz and W. Bal, Inorg. Chem., 2003, 42, 1994–2003 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra01348e |
| This journal is © The Royal Society of Chemistry 2017 |