DOI:
10.1039/C7RA01649B
(Paper)
RSC Adv., 2017,
7, 14816-14823
Selective hydroformylation of alkyl acrylates using [2,2′-bis(dipyrrolylphosphinooxy)-1,1′-(±)-binaphthyl]/Rh catalyst: reversal of regioselectivity†
Received
9th February 2017
, Accepted 23rd February 2017
First published on 6th March 2017
Abstract
The rhodium-catalyzed hydroformylation of alkyl acrylates with different P–N diphosphine ligands is investigated here. Under mild conditions (syngas pressure: 2 MPa, 20 °C), 2,2′-bis(dipyrrolylphosphinooxy)-1,1′-(±)-binaphthyl (L1) rhodium catalyst could give good conversion of ethyl acrylate (82.9%) in 12 h and exclusive branched aldehyde selectivity of >99.0%. More importantly, on elevating the temperature to 90 °C, this Rh system could preferentially afford the linear aldehyde with 96.1% regioselectivity, and the TOF could reach up to 9000 h−1. Deuterioformylation was conducted to explore the mechanism of regioselectivity reversal, and the results established that the reversible rhodium hydride addition to form the Rh-alkyl species might play a vital role in this reversal. The β-hydride elimination of branched Rh-alkyl species was comparatively stronger than that of linear ones under increased temperature, probably because L1 could cause comparatively larger steric repulsion in branched Rh-alkyl species under high temperature, due to its bulky and rigid binaphthyl backbone characteristics. In turn, the linear Rh-alkyl species progress to linear aldehyde was facilitated.
Introduction
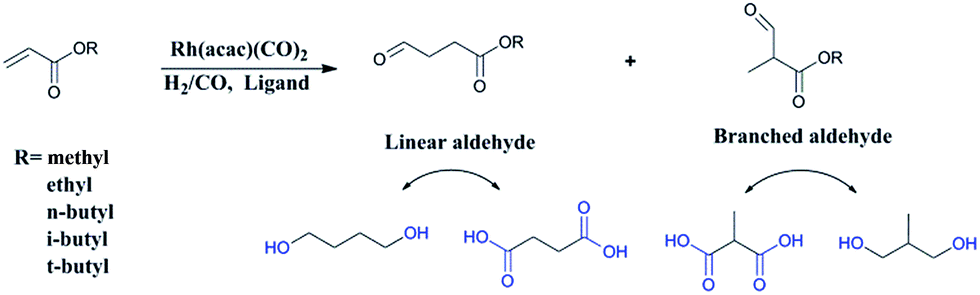
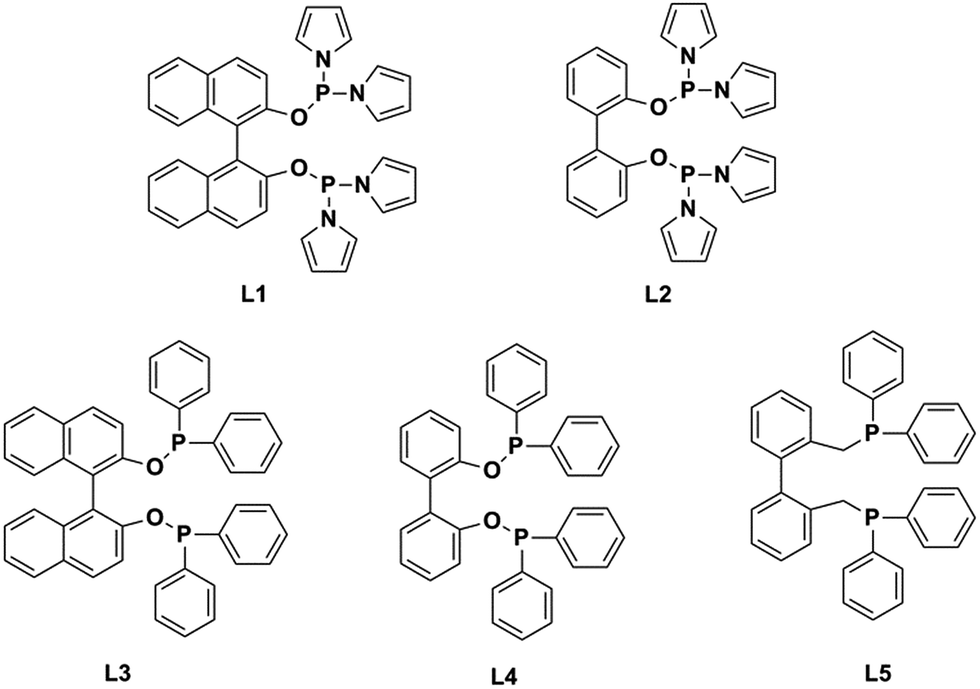
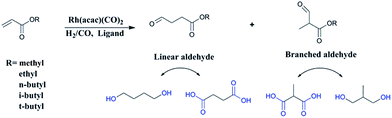
Recently, researchers have developed a consuming passion for the hydroformylation of functionalized olefins, like styrene,1,2 dicyclopentadiene,3–5 vinylacetate,6 allyl alcohol,7,8 alkyl acrylate9–12 and so on. Hydroformylation of alkyl acrylate could provide 1,3- and 1,4-bifunctional carbonyl compounds (corresponding to branched and linear aldehydes), and these compounds could transform to malonic acids, 1,4-dicarboxylic acid, lactones, 2-methyl-1,3-propanediol and 1,4-butanediol13,14 (shown in Scheme 1), which are useful intermediates for the production of fine chemicals, pharmaceuticals, resin, adhesives, oil paints, plasticizers and so on. Nevertheless, acrylate was regarded as a kind of less reactive substrate compared with general olefins,15–17 and the problem might lie in the formation of thermodynamically stable five- or six-membered rings through the coordination of the carbonyl group to the Rh center,18 hence high temperature or high pressure was required.9,12 A number of approaches have been adopted to improve the activity, like using supported aqueous- or ionic-phase catalyst,19 or conducting the hydroformylation in supercritical CO2.10,11 In addition, the hydroformylation generally gave the branched aldehyde as the major product. For instance, a branched aldehyde selectivity of 93% (b/l = 20) was observed in the hydroformylation of ethyl acrylate by using Rh/PPh3-supported ionic liquid-phase catalyst under the condition of 4 MPa and 60 °C (S/C was around 436).19 Xiao et al. found that with the electron withdrawing CF3-substituted dppb ligand rhodium catalyst could afford a conversion of 95% and exclusive branched aldehyde selectivity of 95% in hydroformylation of butyl acrylate, which was conducted at 80 °C and 2 MPa (S/C: 1314).10,11,20 Exclusively, Zhang and co-workers realized high selectivity towards linear aldehyde by using an Rh complex with a pyrrolyl-substituted tetraphosphorus ligand under 1 MPa and 100 °C,9 however, the synthesis of the tetraphosphorus ligand seems to be a little complicated and be of high cost. And the mechanism of high linear regioselectivity was unclear. The above catalytic system either produced mainly branched or linear aldehyde. Due to the wide application of 1,3- and 1,4-bifunctional compounds,21–23 it is highly desirable to seek an accessible and controllable ligand/Rh system that could efficiently and selectively afford branched or linear aldehyde as the major product. It is suggested that phosphorus ligands possessing strong π-acceptor were effective in terms of activity and regioselectivity, for example, the good performance made with the above CF3- or pyrrolyl-substituted phosphorus ligands could be ascribed to that the electron-withdrawing character improved the π-acceptor ability of phosphorus.9,20,24 In this study, five accessible ligands L1–L5 (Fig. 1) were employed to Rh-catalyzed hydroformylation of alkyl acrylate.25–29 Intriguingly, with the presence of L1 linear aldehyde was exclusively obtained as the predominant product under mild conditions; moreover, high selectivity to branched aldehyde could also be realized just by tuning the reaction conditions. Deuterium tests combining with the 1H and 2H NMR measurements were performed to probe the mechanism.
 |
| | Scheme 1 Hydroformylation of alkyl acrylate. | |
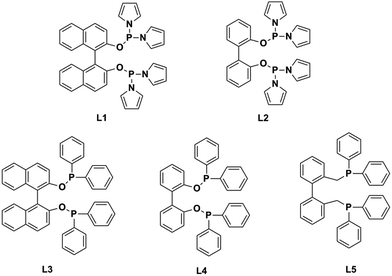 |
| | Fig. 1 Structure of the ligands L1–L5. | |
Experimental
Synthetic procedures
All reactions were carried out under an argon atmosphere with standard Schlenk techniques. All glasswares were oven-dried before use. Toluene was distilled over sodium under argon atmosphere prior to use. Ligands (L1–L5) were prepared based on references and confirmed by 1H and 31P NMR measurements.25–29
General procedures for hydroformylation of ethyl acrylate in toluene or in d6-benzene
The rhodium catalyst was prepared in situ by mixing the ligand L1 with Rh(acac)(CO)2 (acac = acetylacetonate) in toluene. The reaction proceeded in toluene and was analyzed by gas chromatography (GC). The substrate/catalyst ratio was 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 and the Rh(acac)(CO)2 concentration was 1.7 × 10−4 mol L−1. The hydroformylation reaction was performed in a 60 mL steel autoclave equipped with a pressure gauge, a magnetic stirrer and a thermocouple. The substrate alkyl acrylate (1.0 mL) was mixed with a toluene solution, which containing Rh(acac)(CO)2 (1 μmol, 260 μL) and ligand (5 μmol). Then, the autoclave was sealed and purged with syngas (CO/H2) over three times and subsequently pressurized to a preset value and started to stir. When the reactor was heated to the preset temperature, the reaction time was accounted. After the reaction was finished, the products were analyzed by GC.
000 and the Rh(acac)(CO)2 concentration was 1.7 × 10−4 mol L−1. The hydroformylation reaction was performed in a 60 mL steel autoclave equipped with a pressure gauge, a magnetic stirrer and a thermocouple. The substrate alkyl acrylate (1.0 mL) was mixed with a toluene solution, which containing Rh(acac)(CO)2 (1 μmol, 260 μL) and ligand (5 μmol). Then, the autoclave was sealed and purged with syngas (CO/H2) over three times and subsequently pressurized to a preset value and started to stir. When the reactor was heated to the preset temperature, the reaction time was accounted. After the reaction was finished, the products were analyzed by GC.
d6-Benzene was used as the solvent for convenient 1H NMR analysis of hydroformylation products.
General procedures for deuterioformylation in d6-benzene
The deuterioformylation reaction was performed in a 60 mL steel autoclave equipped with a pressure gauge, a magnetic stirrer and a thermocouple. The substrate ethyl acrylate (0.005 mol, 0.5 mL) was mixed with benzene-d6, which containing Rh(acac)(CO)2 (0.5 μmol, 130 μL) and ligand (2.5 μmol). Then, the autoclave was sealed and purged with syngas (CO/D2) over three times and subsequently pressurized to 0.5 MPa 1:1 (CO/D2) and started to stir. When the reactor was heated to the preset temperature, the reaction time was accounted. After the reaction was finished, the products were analyzed by 1H and 2H NMR and GC.
Results and discussion
Hydroformylation of ethyl acrylate using ligand L1
L1–L5 were prepared and used as the ligands in Rh-catalyzed hydroformylation of alkyl acrylates. The hydroformylation of ethyl acrylate with L1 was investigated as the model reaction. As the hydroformylation reaction highly relies on the reaction condition, the influence of P/Rh ratio, temperature, and pressure on regioselectivity was evaluated. The rhodium catalyst was prepared in situ by mixing the ligand L1 with Rh(acac)(CO)2 (acac = acetylacetonate) in toluene. The reaction proceeded in toluene and was analyzed by gas chromatography (GC). The substrate/catalyst ratio was 10000 and the Rh(acac)(CO)2 concentration was 1.7 × 10−4 mol L−1. As shown in Table 1, the ligand/metal ratio had a dramatic effect on the hydroformylation. The reaction hardly occurred without ligand (entry 1), yet a big jump in conversion and regioselectivity was observed after small amount of ligand was introduced (entry 2). The linear/branched (l/b) ratio was improved when the ligand/metal ratio increased from 1:1 to 10:1 (Table 1, entries 2–6), further increasing the ligand/metal ratio resulted in a little drop of l/b ratio (Table 1, entries 7–8). An optimal ligand/metal ratio of 10 is preferred to achieve high conversion and high selectivity to linear aldehyde. The syngas (CO/H2 1/1) pressure also influenced the reaction. Apparently, low pressure benefited the formation of linear aldehyde, whereas impeded the conversion of the substrate a little (Table 1, entries 10–13). The temperature had a prominent impact on the regioselectivity (Table 1, entries 14–19). High temperature was in favor of linear aldehyde and hydrogenation. Increasing the temperature higher than 90 °C caused the l/b ratio improved a little but the selectivity to aldehyde declined, meanwhile, the conversion maintained around 99.0% (entries 17–19). When the hydroformylation was performed under 0.5 MPa and 90 °C, a l/b ratio of 24.8 (equiv. to 96.1% linear aldehyde selectivity) was obtained, the TOF could reach 9000 h−1. The performance was comparable with that achieved by the tetraphosphorus/Rh system, which afforded approximately 99.9% selectivity to linear aldehyde in the hydroformylation of alkyl acrylate under 100 °C and 1 MPa, and a conversion of 64% after 1 h (TOF: 5400 h−1).9
Table 1 Hydroformylation of ethyl acrylate using ligand L1a
| Entry |
P/Rh |
T (°C) |
P (MPa) |
t (h) |
Conv.b (%) |
Hydro.c (%) |
Aldehyded (%) |
l/be |
TOFf (h−1) |
| Conditions: S/C = 10000, [Rh] = 1.7 × 10−4 mol L−1, reaction time = 2 h. Conversion of the substrate based on GC. Selectivity for hydrogenation product based on GC. Selectivity for aldehyde based on GC. The molar ratio of linear to branched aldehyde. The turnover frequency of ethyl acrylate. Reaction time = 1 h. |
| 1 |
0 |
100 |
1 |
2 |
Trace |
Trace |
n.d. |
— |
— |
| 2 |
1 |
100 |
1 |
2 |
67.3 |
14.9 |
85.1 |
6.7 |
4255 |
| 3 |
2 |
100 |
1 |
2 |
86.1 |
14.1 |
85.9 |
8.6 |
4295 |
| 4 |
4 |
100 |
1 |
2 |
89.6 |
17.2 |
82.8 |
9.1 |
4140 |
| 5 |
8 |
100 |
1 |
2 |
95.9 |
15.0 |
85.0 |
9.5 |
4250 |
| 6 |
10 |
100 |
1 |
2 |
98.9 |
15.1 |
84.9 |
11.1 |
4245 |
| 7 |
15 |
100 |
1 |
2 |
98.7 |
15.0 |
85.0 |
10.1 |
4250 |
| 8 |
20 |
100 |
1 |
2 |
99.6 |
12.3 |
87.7 |
10.1 |
4385 |
| 9g |
10 |
90 |
0.5 |
1 |
95.0 |
10.2 |
89.8 |
24.8 |
9000 |
| 10 |
10 |
90 |
0.5 |
2 |
97.2 |
11.8 |
88.2 |
26.6 |
4410 |
| 11 |
10 |
90 |
1 |
2 |
98.7 |
14.6 |
86.4 |
12.9 |
4320 |
| 12 |
10 |
90 |
1.5 |
2 |
99.8 |
10.0 |
90.0 |
6.0 |
4500 |
| 13 |
10 |
90 |
2 |
2 |
99.9 |
14.4 |
85.6 |
2.0 |
4280 |
| 14 |
10 |
40 |
1 |
2 |
86.2 |
6.3 |
93.7 |
0.2 |
4685 |
| 15 |
10 |
50 |
1 |
2 |
96.3 |
9.9 |
90.1 |
1.0 |
4505 |
| 16 |
10 |
70 |
1 |
2 |
98.9 |
9.3 |
90.7 |
6.3 |
4535 |
| 17 |
10 |
90 |
1 |
2 |
98.7 |
14.6 |
86.4 |
12.9 |
4320 |
| 18 |
10 |
100 |
1 |
2 |
98.9 |
15.1 |
84.9 |
11.1 |
4245 |
| 19 |
10 |
120 |
1 |
2 |
99.4 |
23.6 |
76.4 |
13.3 |
3820 |
From entries 10–13 and entries 14–19 in Table 1, it seemed that the generation of linear aldehyde could be impeded at lower temperature or higher pressure, which implied that the formation of the branched aldehyde, in turn, might be favorable under these conditions. To verify it, the hydroformylation of ethyl acrylate using ligand L1 under low temperatures (20–35 °C) was also performed and the results were shown in Table 2. Similar to the manner under 100 °C, the reaction did not take place without ligand (entry 1). Increasing the P/Rh ratio from 1 to 5 accelerated the transformation of the substrate, but did not contribute too much to the hydrogenation and the selectivity to aldehyde (entries 2–4), further increasing the P/Rh ratio led to a little drop of conversion, meanwhile, the hydrogenation and the selectivity to aldehyde maintained around 4.1% and 95.9% (entries 5–7), respectively. Compare Table 1 with 2, it is not difficult to make these findings: (a) the branched aldehyde predominated at lower temperature as expected; (b) the optimal conversion for the low temperature tests was maintained at lower P/Rh range than that for high temperature; (c) apparently, b/l increased along with the pressure rising at low temperature (Table 2, entries 8–12), for the situation of high temperature, the l/b increased with the pressure decreased (Table 1, entries 9–13). The possible reason is explained later. Noting that further lowering the temperature led to higher b/l ratio (Table 2, entries 13–15), i.e., heating at 20 °C resulted in branched aldehyde selectivity higher than 99.0% (2 MPa, S/C: 10000, b/l ratio > 99). For the point view of selective formation of branched aldehyde, the result achieved under more mild conditions was comparable with those reported by Xiao (S/C: 1314, 80 °C, 2 MPa, branched aldehyde selectivity of 95%)20 and Clarke (S/C: 10000, 75 °C, 7.5 MPa, branched aldehyde yield of 85%).12 However, low temperature also hindered the conversion of the substrate, and longer reaction time (i.e., 12 h) could guarantee the conversion (entry 13).
Table 2 Hydroformylation of ethyl acrylate using ligand L1 under low temperaturesa
| Entry |
P/Rh |
T (°C) |
P (MPa) |
t (h) |
Conv.b (%) |
Hydro.c (%) |
Aldehyded (%) |
b/le |
| Conditions: S/C = 10000, [Rh] = 1.7 × 10−4 mol L−1. See Table 1. See Table 1. See Table 1. The molar ratio of branched to linear aldehyde. |
| 1 |
0 |
35 |
2 |
2 |
n.d. |
n.d. |
n.d. |
n.d. |
| 2 |
1 |
35 |
2 |
2 |
55.3 |
4.3 |
95.7 |
43.1 |
| 3 |
2 |
35 |
2 |
2 |
64.6 |
4.0 |
96.0 |
40.3 |
| 4 |
5 |
35 |
2 |
2 |
75.6 |
3.8 |
96.2 |
41.8 |
| 5 |
8 |
35 |
2 |
2 |
70.4 |
4.5 |
95.5 |
43.8 |
| 6 |
10 |
35 |
2 |
2 |
66.3 |
4.1 |
95.9 |
44.4 |
| 7 |
15 |
35 |
2 |
2 |
66.4 |
3.6 |
96.4 |
44.7 |
| 8 |
5 |
35 |
1 |
2 |
61.8 |
5.0 |
95.0 |
14.4 |
| 9 |
5 |
35 |
2 |
2 |
75.6 |
3.8 |
96.2 |
41.8 |
| 10 |
5 |
35 |
3 |
2 |
68.0 |
2.6 |
95.6 |
54.2 |
| 11 |
5 |
35 |
4 |
2 |
67.2 |
2.1 |
96.3 |
64.7 |
| 12 |
5 |
35 |
5 |
2 |
66.4 |
1.7 |
98.3 |
71.6 |
| 13 |
5 |
35 |
2 |
12 |
99.9 |
3.5 |
96.5 |
40.9 |
| 14 |
5 |
26 |
2 |
12 |
98.7 |
2.5 |
97.5 |
33.4 |
| 15 |
5 |
20 |
2 |
12 |
82.9 |
1.9 |
98.1 |
>99 |
| 16 |
5 |
20 |
2 |
4 |
42.4 |
1.9 |
98.1 |
>99 |
| 17 |
5 |
20 |
2 |
8 |
77.7 |
3.0 |
97.0 |
>99 |
| 18 |
5 |
20 |
2 |
12 |
82.9 |
1.9 |
98.1 |
>99 |
Moreover, except for aldehyde and hydrogenation products, no polymetric product was detected by GC analysis, whatever under high or low temperatures. Considering that GC was unable to determine the polymetric product, the residue after hydroformylation under 90 °C and 20 °C was collected for 1H NMR analysis, the characteristic shift of the terminal vinylidene (ca. 5.5 and 6.2 ppm) was not observed (see Fig. S11 and S12 in the ESI†),30 which confirmed that no polymetric product was generated in L1/Rh catalyzed hydroformylation of ethyl acrylate. 1H NMR also proved that branched product was dominant at 20 °C, since the characteristic chemical shifts of branched aldehyde (9.45 ppm, CHO, 1H), its keto–enol isomer (11.75 ppm, OH, 1.48H) and trace amount of hydrogenation product (2.18 ppm, –CH2CO–, 0.14H) were observed, which hinted that the molar ratio of enol to branched aldehyde was around 1.48 (Fig. S11†). As for the reaction products under 90 °C, linear aldehyde (9.30 ppm, CHO, 4.4H) indeed predominant accompanied by little hydrogenation product (2.18 ppm, –CH2CO–, 3.23H), substrate (6.26 ppm, 0.98H; 5.96 ppm, 0.96H; 5.35 ppm, 1H, CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCO–) and trace amount of branched product and its enol isomer (Fig. S12†).
CHCO–) and trace amount of branched product and its enol isomer (Fig. S12†).
Hydroformylation of ethyl acrylate using ligand L2–L5
With the optimal reaction parameters for high- or low-temperature tests in hand, ligands L2–L5 were also employed in the hydroformylation of ethyl acrylate for two purposes: (a) obtaining some information that how the steric and the electronic properties of ligands influenced the activity and regioselectivity; (b) looking for an optimal ligand with which high regioselectivity could be realized. As shown in Tables 3 and 4, the tendency at low temperature is similar to that at high temperature. L1/Rh and L2/Rh exerted much better performance towards the conversion and the regioselectivity than other L/Rh system, and L1/Rh displayed higher selectivity to linear aldehyde than L2/Rh at high temperature. L3–L5 exerted inferior activity and regioselectivity. L3/Rh only took effect under high temperature accompanied with lower conversion and l/b (entry 3). At 90 °C, L4/Rh and L5/Rh showed almost no reaction activity. At 20 °C, only the latter one exerted a low reaction activity but high branched aldehyde selectivity. The accessible ligand L1 could be the optimal ligand in terms of high activity and high regioselectivity.
Table 3 Hydroformylation of ethyl acrylate with various ligands at 90 °Ca
| Entry |
Ligand |
Conv.b (%) |
Hydro.c (%) |
Aldehyded (%) |
l/be |
| Conditions: S/C = 10000, [Rh] = 1.7 × 10−4 mol L−1, P/Rh = 10/1, temperature = 90 °C, syngas = 0.5 MPa, reaction time = 2 h. See Table 1. See Table 1. See Table 1. See Table 1. |
| 1 |
L1 |
97.2 |
11.8 |
88.2 |
26.6 |
| 2 |
L2 |
71.1 |
12.5 |
87.5 |
18.4 |
| 3 |
L3 |
31.7 |
24.3 |
75.7 |
0.25 |
| 4 |
L4 |
1.38 |
1.38 |
n.d. |
n.d. |
| 5 |
L5 |
0.95 |
0.95 |
n.d. |
n.d. |
Table 4 Hydroformylation of ethyl acrylate with various ligands at 20 °Ca
| Entry |
Ligand |
Conv.b (%) |
Hydro.c (%) |
Aldehyded (%) |
b/le |
| Conditions: S/C = 10000, [Rh] = 1.7 × 10−4 mol L−1, P/Rh = 10/1, temperature = 20 °C, syngas = 2 MPa, reaction time = 12 h. See Table 1. See Table 1. See Table 1. The molar ratio of branched to linear aldehyde. |
| 1 |
L1 |
82.9 |
1.9 |
98.1 |
>99 |
| 2 |
L2 |
64.2 |
0.9 |
99.1 |
>99 |
| 3 |
L3 |
Trace |
n.d. |
Trace |
— |
| 4 |
L4 |
n.d. |
n.d. |
n.d. |
n.d. |
| 5 |
L5 |
15.1 |
1.3 |
98.7 |
>99 |
According to the reported paper, a dynamic equilibrium between ee and ea complex isomers was exist for rhodium complex with diphosphine ligands.31,32 In a certain range, catalysts with larger cone angle were in favor of forming ee isomers which will lead to higher l/b ratio. For L1 and L5, the cone angle is 99° and 113° respectively,2,33 and their bidentate ee-coordination in trigonal bipyramidal [HRhL(1 or 5)(CO)2] seems to be the preferred isomer. And there is no accurate result about the cone angle for L2, L3, L4. But van leeuwen group27 found that [HRhL(2 or 4)(CO)2] most probably adopted the ee isomers through the characteristic of IR and NMR. This indicates that all these ligands are likely to produce high l/b ratio potentially. Actually, our experimental data revealed that the cone angle may not exert significant influence on selectivity or activity of the catalyst, as that L4 and L5 showed poor selectivity and activity at 90 °C. At 20 °C, L3 and L4 also presented poor selectivity and activity. The unique electronic effect of ligand L1 accompanying with the temperature and pressure may have more impact on the selectivity and activity.
Hydroformylation of other alkyl acrylates with L1
As L1 exhibited a superior performance to other ligands in the hydroformylation of ethyl acrylate, and most importantly, the regioselectivity could be reversed through the control of reaction condition. To expand the substrate, other various unsaturated acrylates were also hydroformylated using L1 under 90 or 20 °C, respectively. As summarized in Tables 5 and 6, L1/Rh system had benign substrate compatibility. High selectivity to linear or branched aldehyde was observed at 90 °C or 20 °C, respectively. In particular, ethyl acrylate exerted best reactivity and regioselectivity at high temperature, and methyl acrylate owned better reactivity at 20 °C.
Table 5 Hydroformylation of alkyl acrylates with L1 under 90 °Ca
| Entry |
Substrate |
Conv.b (%) |
Hydro.c (%) |
Aldehyded (%) |
l/be |
| See Table 3. See Table 3. See Table 3. See Table 3. See Table 3. |
| 1 |
Methyl acrylate |
84.8 |
11.7 |
88.3 |
23.2 |
| 2 |
Ethyl acrylate |
97.2 |
11.8 |
88.2 |
26.6 |
| 3 |
Butyl acrylate |
43.1 |
14.4 |
85.4 |
16.5 |
| 4 |
i-Bu acrylate |
66.6 |
8.7 |
91.3 |
10.9 |
| 5 |
t-Bu acrylate |
44.3 |
15.3 |
84.5 |
12.9 |
Table 6 Hydroformylation of alkyl acrylates with L1 under 20 °Ca
| Entry |
Substrate |
Conv.b (%) |
Hydro.c (%) |
Aldehyded (%) |
b/le |
| See Table 4. See Table 4. See Table 4. See Table 4. See Table 4. |
| 1 |
Methyl acrylate |
95.6 |
2.0 |
98.0 |
84.2 |
| 2 |
Ethyl acrylate |
82.9 |
1.9 |
98.1 |
>99 |
| 3 |
Butyl acrylate |
69.7 |
2.2 |
97.8 |
67.2 |
| 4 |
i-Bu acrylate |
90.9 |
2.5 |
97.5 |
62.3 |
| 5 |
t-Bu acrylate |
50.1 |
1.6 |
98.4 |
81.2 |
Proposal mechanism for the hydroformylation of alkyl acrylate
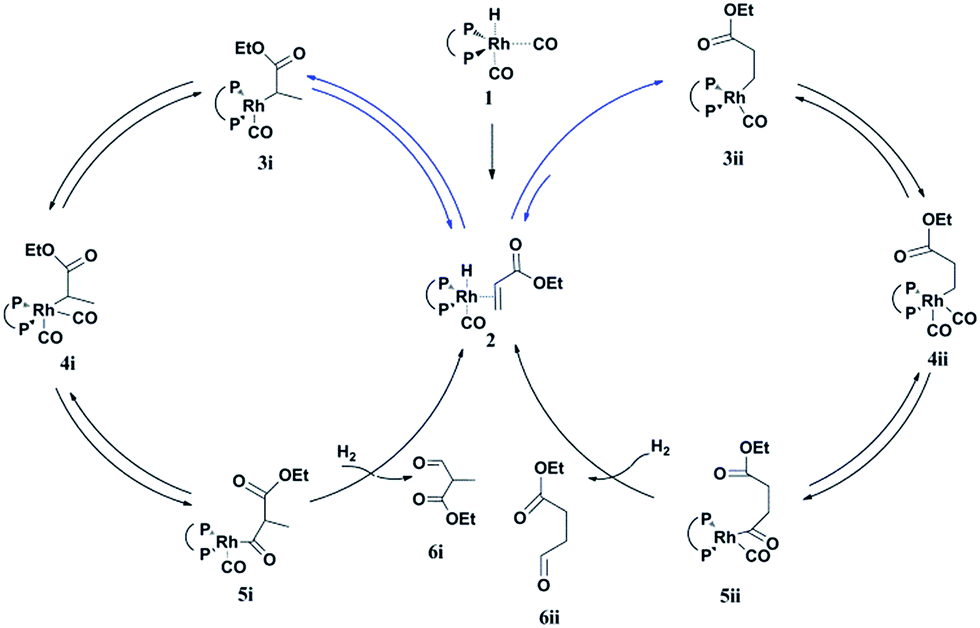
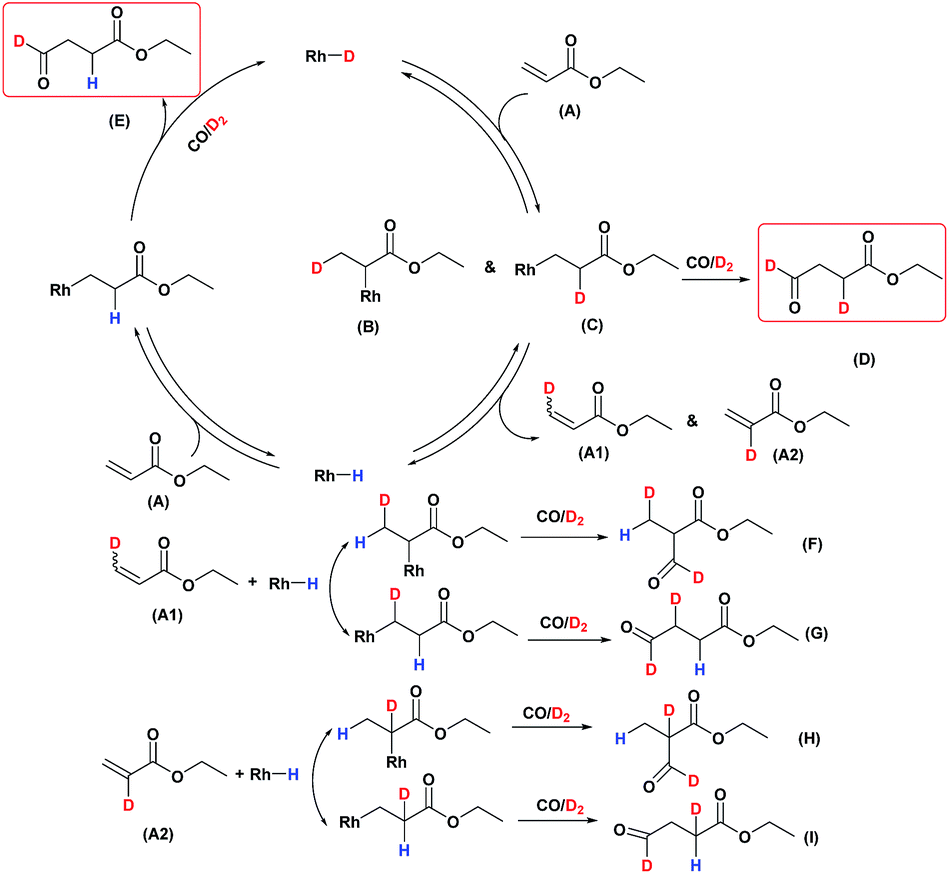
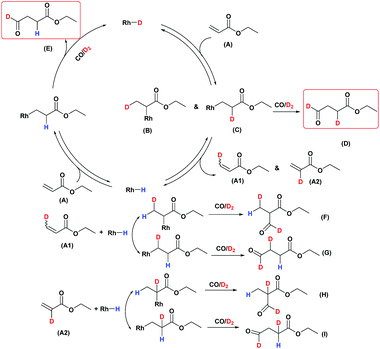
A possible mechanism for the hydroformylation of ethyl acrylate was drawn in Scheme 2. Owing to the stronger electronegativity of oxygen atom than carbon, the carbonyl is a strongly electron withdrawing group which could render hydride transfer to the β carbon easier than to the α carbon when the rhodium hydride addition to the coordinated ethyl acrylate, thus the transformation of 2 to 3i (branched Rh-alkyl, Scheme 2) might be easier than 2 to 3ii (linear Rh-alkyl), which might be one of the driving force for the branched aldehyde as the major product in general. L1/Rh and L2/Rh exerted much better performance towards the conversion and the regioselectivity than other L/Rh system, probably as a result of the easy carbon monoxide dissociation owing to bearing the electron withdrawing N-pyrrolyl, and in turn facilitated the alkene coordination to metal.34,35 In a word, the transformation of 1 to 2 was accelerated by the withdrawing N-pyrrolyl moiety. L1/Rh resulted in higher selectivity to linear aldehyde than L2/Rh at high temperature. L1 owned a bulky binaphthyl backbone and was more rigid than L2, we were wondering that if this larger steric hindrance around Rh center could influence the comparative stability of 3i and in turn favor 3ii dominant at high temperature and consequently reverse the regioselectivity. The Rh-alkyl complexes 3i and 3ii are 16 VE unsaturated, and could become saturated by reversing back to 2 via β-H elimination or by further coordinating with CO to form 4i or 4ii. Lazzaroni et al. found that the formation of the linear and the branched Rh-alkyl species in hydroformylation of styrene are not reversible at low temperature (equals to complexes 3i and 3ii in our case) but reversible at high temperature.36 In order to probe the reversibility of rhodium hydride addition to coordinated ethyl acrylate, H2 was replaced by D2 for deuterioformylation. If hydride addition is irreversible, no deuterium could be found at the α- or β-vinyl position of the recovered ethyl acrylate. Appearance of deuterium in the α- or β-vinyl position requires the linear or branched Rh-alkyl species occurring β-hydride elimination, respectively.
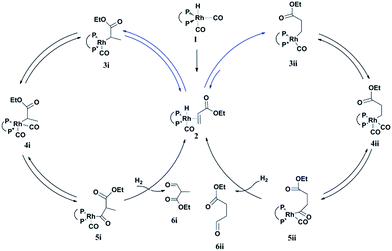 |
| | Scheme 2 Proposed mechanism for the hydroformylation of alkyl acrylate. | |
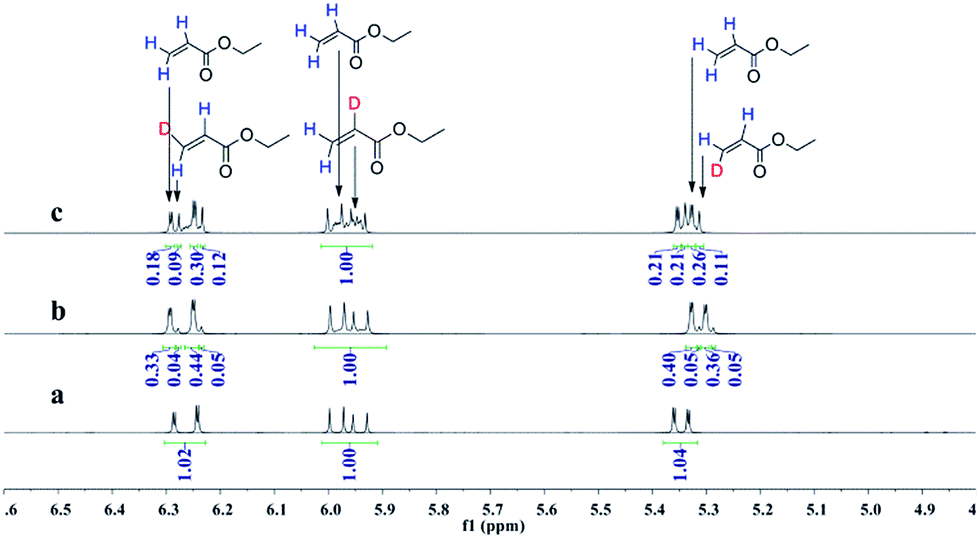
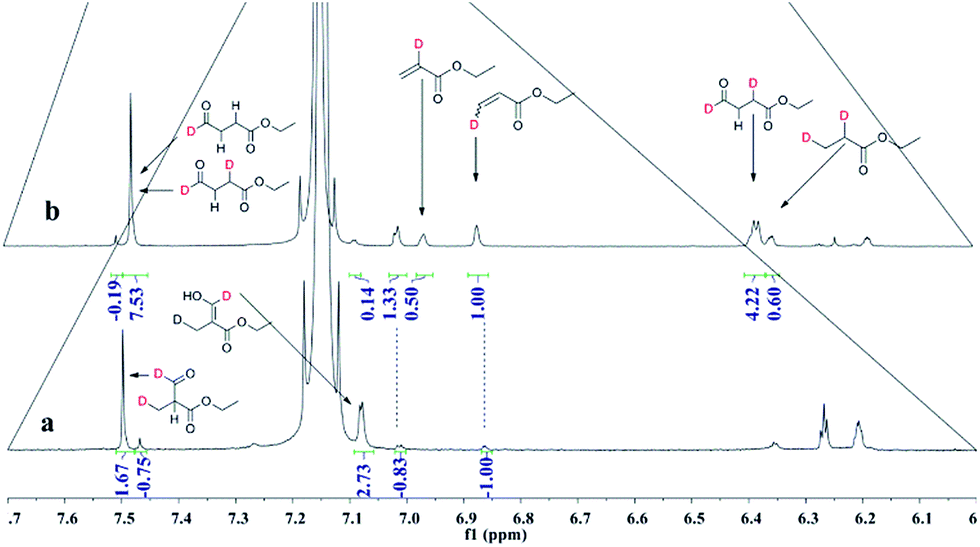
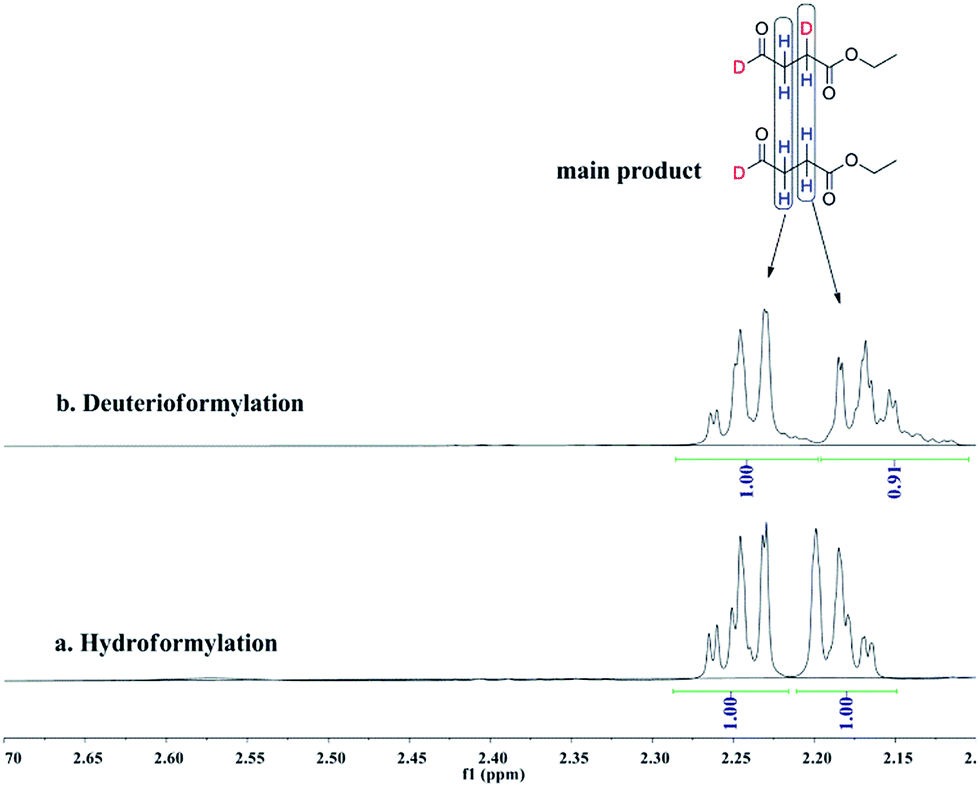
Deuterioformylation of ethyl acrylate catalyzed by Rh(acac)(CO)2/L1 was conducted in C6D6 at 90 °C and 20 °C, respectively. The syngas pressure was 0.5 MPa 1:1 CO/D2. After the two reactions approximately approached to proximate conversion, the reaction residue was analyzed by 1H and 2H NMR measurements. As demonstrated in Fig. 2, after magnified the region from 5.0 ppm to 6.5 ppm in 1H NMR which were the resonance of recovered ethyl acrylate, some interesting phenomena were observed when compared with the 1H NMR of hydroformylation at 90 °C (Fig. 2a). Two couple of new shoulders at around 6.25 ppm and 5.30 ppm which could be attributed to the β-d1-ethyl acrylate came into appearance at 20 °C (Fig. 2b), which indicated that the branched Rh-alkyl species encountered obvious β-hydride elimination. In contrast, the α-d1-ethyl acrylate was scarcely detected at 20 °C (Fig. 2b), probably because that the linear Rh-alkyl species was formed less than 3i as mentioned above or difficult to occur β-hydride elimination. At 90 °C, the two couple of new shoulders at around 6.25 ppm and 5.30 ppm was enhanced (Fig. 2c), which might prove that the β-H elimination of branched Rh-alkyl was effectively promoted. A few ‘noise’ appeared around 5.96 ppm just in Fig. 2c, which likely stemmed from small amount of α-d1-ethyl acrylate. 2H NMR (Fig. 3) showed significantly less β- and α-d1-ethyl acrylate at 20 °C than that at 90 °C, and the intensity of β-d1-ethyl acrylate was higher than that for α-d1-ethyl acrylate at 90 °C, which was in agreement with the 1H NMR results. Apparently, more deuterium incorporated into the β- than into the α-vinyl position, which indicated that the branched Rh-alkyl species might go through stronger β-H elimination than linear Rh-alkyl species at 90 °C. The reversible hydride addition could yield incorporation of deuterium in the aldehyde products and in the recovered ethyl acrylate. A possible pathway for the formation of linear aldehyde at 90 °C was proposed and drawn in Scheme 3. 2H NMR (Fig. 3a) showed the integration of linear d1-formyl group was higher than that of the adjacent two methylenes. Its 1H NMR (Fig. 4b) displayed the integrations of the two methylenes were 1.0 and 0.91, thus, the linear aldehyde was possibly made up by D and E. Since at 90 °C mainly the branched d1-Rh-alkyl species (B) β-hydride eliminates to give β-d1-ethyl acrylate (A1) and Rh-H species which could rapidly incorporate into the unreacted ethyl acrylate by a fast hydride addition and later generate E (Scheme 3). And the linear Rh-D addition to ethyl acrylate could give linear Rh-alkyl species (C) and subsequently form D.
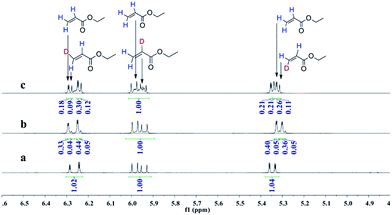 |
| | Fig. 2 1H NMR spectra of double bond region for recovered ethyl acrylate collected after (a) hydroformylation at 90 °C, (b) deuterioformylation at 20 °C, (c) deuterioformylation at 90 °C. | |
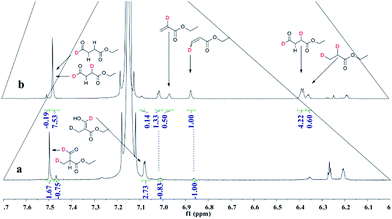 |
| | Fig. 3 2H NMR spectra of products after deuterioformylation at (a) 20 °C and (b) 90 °C in C6D6. | |
 |
| | Scheme 3 Possible pathway for deuterioformylation of ethyl acrylate with L1 at 90 °C. | |
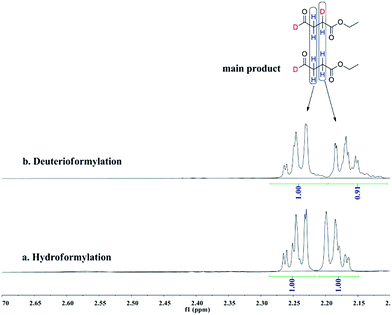 |
| | Fig. 4 1H NMR spectra of the two methylenes on linear aldehyde after (a) hydroformylation of ethyl acrylate at 90 °C, (b) deuterioformylation of ethyl acrylate at 90 °C in C6D6. | |
Based on the above results, the branched Rh-alkyl species (3i) was easier formed with respect to the linear Rh-alkyl (3ii). In addition, both 1H and 2H NMR confirmed that the β-H elimination of 3i was quite lower at 20 °C and under this condition 3i was difficult to form or β-H eliminate to 2, as a consequence, the 16 VE unsaturated branched Rh-alkyl species (3i) seems to be readily progress CO coordination to yield branched aldehyde (6i) without extensive competitive reversion to ethyl acrylate (left cycle, Scheme 2). Further increasing the partial pressure of CO at 20 °C (Table S4, ESI†), the b/l ratio could be further enhanced as the CO insertion step was facilitated. The inherent characteristics of L1, the bulky and rigid binaphthyl backbone might cause large steric hindrance around Rh center and previous report also revealed [RhHL1(CO)2] was in an ee coordination fashion,31 which might cause higher steric repulsion to 3i and encourage 3i β-hydride eliminate to 2 under increased temperature (i.e., 90 °C). The transformation of 2 to 3ii or 2 to 3i could be enhanced by ascending the temperature. 3ii with less steric repulsion might suffer less β-hydride elimination and appears much more likely to undergo CO insertion to yield linear aldehyde 6ii at 90 °C (right cycle, Scheme 2). Meanwhile, the in situ generated Rh-H species from the β-hydride elimination of 3i would go through fast hydride addition and later yield 6ii. Our experiment data displayed the optimal conversion for 20 °C was maintained at lower P/Rh range than that for 90 °C (entries 1–8 in Table 1 vs. entries 1–7 in Table 2), this might be an indirect proof of more crowded 3i and less crowded 3ii dominant at 20 °C and 90 °C, respectively. Further increasing the partial pressure of CO at 90 °C, the l/b ratio could be further increased a little which suggested that partial pressure of CO might have less impact on the regioselectivity. It is likely that the reversible rhodium hydride addition to form the Rh-alkyl species might play a vital role on the regioselectivity in hydroformylation of alkyl acrylate. The synergic effect of the larger steric hindrance of L1 and elevating the temperature could help β-H elimination of branched Rh-alkyl species. L2 with less steri hindrance could afford inferior regioselectivity reversal. Therefore, highly selectivity to both branched and linear aldehyde became feasible in L1/Rh system, just reasonably lowering or elevating the temperatures.
Conclusions
Highly active and selective hydroformylation of alkyl acrylates in terms of linear or branched aldehyde was realized with a rhodium catalyst modified by 2,2′-bis(dipyrrolylphosphinooxy)-1,1′-binaphthyl ligand (L1), just by tuning the reaction conditions. At low temperature, more branched Rh-alkyl species predominantly progress to branched aldehyde due to easier formation than linear Rh-alkyl species and less β-hydride elimination than that at higher temperature. At high temperature, more linear Rh-alkyl species progress to linear aldehyde probably due to its increased formation and less β-hydride elimination than branched Rh-alkyl species, since the large steric hindrance around Rh center may cause higher repulsion to branched Rh-alkyl species than to the linear one due to the bulky and rigid binaphthyl backbone characteristics of L1, hence, the branched Rh-alkyl is rather more readily to experience β-H elimination, especially at high temperature. Consequently, the reversal of the regioselectivity was observed at the Rh/L1 system. These results enable the Rh/L1 to be a potential candidate for controllable hydroformylation of alkyl acrylates or other functionalized olefin.
Acknowledgements
The authors thank the financial support from the National Natural Science Foundation of China (No. 201202108), from the Opening Project of Key laboratory of green chemistry of Sichuan institutes of higher education (LZJ1042), and from the Sichuan university outstanding scholar research found (No. 2015SCU04A05). We are grateful to Thank Chengdu Institute of Biology, Chinese Academy of Sciences for the 2H NMR measurements.
Notes and references
- C. Y. Zheng, M. Mo, H. R. Liang, X. L. Zheng, H. Y. Fu, M. L. Yuan, R. X. Li and H. Chen, Appl. Organomet. Chem., 2013, 27, 474–478 CrossRef CAS.
- E. Boymans, M. Janssen, C. Müller, M. Lutz and D. Vogt, Dalton Trans., 2013, 42, 137–142 RSC.
- Y. Fan, S. Ren, B. Xu, T. Shi and Y. Chen, C. N. Patent, 101053843 A, 2007.
- H. K. Reinius and A. O. I. Krause, Catal. Lett., 2000, 70, 149–154 CrossRef CAS.
- R. Luo, H. R. Liang, X. L. Zheng, H. Y. Fu, M. L. Yuan, R. X. Li and H. Chen, Catal. Commun., 2014, 50, 29–33 CrossRef CAS.
- S. Breeden, D. J. Cole-Hamilton, D. F. Foster, G. J. Schwartz and M. Wills, Angew. Chem., Int. Ed., 2000, 39, 4106 CrossRef CAS PubMed.
- E. Drent and W. W. Jager, Shell, GB Pat., 2282137, 1995.
- K. N. Bhatt and S. B. Halligudi, J. Mol. Catal., 1994, 91, 187 CrossRef CAS.
- S. C. Yu, Y. M. Chie and X. M. Zhang, Adv. Synth. Catal., 2009, 351, 537–540 CrossRef CAS.
- Y. L. Hu, W. P. Chen, A. M. B. Osuna, A. M. Stuart, E. G. Hope and J. Xiao, Chem. Commun., 2001, 725–726 RSC.
- Y. L. Hu, W. P. Chen, A. M. B. Osuna, J. A. Iggo and J. Xiao, Chem. Commun., 2002, 7, 788–789 RSC.
- M. L. Clarke and G. J. Roff, Green Chem., 2007, 9, 792–796 RSC.
- K. Nakamura, T. Miyai, K. Ushio, S. Oka and A. Ohno, Bull. Chem. Soc. Jpn., 1988, 61, 2089–2093 CrossRef CAS.
- Y. Kawai, K. Hida, D. H. Dao and A. Ohno, Tetrahedron Lett., 1998, 39, 9219–9222 CrossRef CAS.
- C. K. Brown and G. Wilkinson, J. Chem. Soc. A, 1970, 17, 2753 RSC.
- G. Fremy, Y. Castanet, J.-F. Carpentier, E. Monflier and A. Mortreux, Angew. Chem., Int. Ed. Engl., 1995, 34, 1474 CrossRef CAS.
- G. Fremy, Y. Castanet, R. Grzybek, E. Monflier, A. Mortreux, A. M. Trzeciak and J. J. Ziolowski, J. Organomet. Chem., 1995, 505, 11 CrossRef CAS.
- G. Fremy, E. Monflier, J. F. Carpentier, Y. Castanet and A. Mortreux, J. Mol. Catal. A: Chem., 1998, 129, 35–40 CrossRef CAS.
- A. G. Panda, M. D. Bhor, S. R. Jagtap and B. M. Bhanage, Appl. Catal., A, 2008, 347, 142–147 CrossRef CAS.
- O. Saidi, S. F. Liu and J. L. Xiao, J. Mol. Catal. A: Chem., 2009, 305, 130–134 CrossRef CAS.
- A. Gambacorta, M. E. Farah and D. Tofani, Tetrahedron, 1999, 55, 12615–12628 CrossRef CAS.
- C. Botteghi, R. Ganzerla, M. Lenarda and G. Moretti, J. Mol. Catal., 1987, 40, 129–182 CrossRef CAS.
- R. L. Pruett, Adv. Organomet. Chem., 1979, 17, 1–60 CrossRef CAS.
- K. G. Moloy and J. L. Petersen, J. Am. Chem. Soc., 1995, 117, 7696–7710 CrossRef CAS.
- K. Ding and B. Zhao, C. N. Patent, 1857776 A, 2006.
- W. Liu, M. Yuan, H. Fu, H. Chen, R. Li and X. Li, Chem. Lett., 2009, 38, 596–597 CrossRef CAS.
- S. C. van der Slot, J. Duran, J. Luten, P. C. J. Kamer and P. W. N. M. van Leeuwen, Organometallics, 2002, 21, 3873 CrossRef CAS.
- Y. G. Zhou, W. Tang, W. B. Wang, W. Li and X. Zhang, J. Am. Chem. Soc., 2002, 124, 2 Search PubMed.
- T. A. Puckette, T. J. Devou, G. W. Phillips and J. L. Stavinoha, US Pat., 4879416, 1989.
- C. L. Quan and M. Soroush, Macromolecules, 2005, 38, 7619–7628 CrossRef CAS.
- C. P. Casey, E. L. Paulsen, E. W. Beuttenmueller, B. R. Proft, L. M. Petrovich, B. A. Matter and D. R. Powell, J. Am. Chem. Soc., 1997, 119, 11817 CrossRef CAS.
- L. A. van der Veen, M. D. K. Boele, F. R. Bregman, P. C. J. Kamer, P. W. N. M. van Leeuwen, K. Goubitz, J. Fraanje, H. Schenk and C. Bo, J. Am. Chem. Soc., 1998, 120, 11616 CrossRef CAS.
- C. P. Casey, G. T. Whiteker, M. G. Melville, L. M. Petrovich, J. Gamey and D. R. Powell, J. Am. Chem. Soc., 1992, 114, 5535–5543 CrossRef CAS.
- R. Jackstell, H. Klein, M. Beller, K. D. Wiese and D. Rottger, Eur. J. Org. Chem., 2001, 3871–3877 CrossRef CAS.
- E. Rafter, D. G. Gilheany, J. N. H. Reek and P. W. N. M. van Leeuwen, ChemCatChem, 2010, 2, 387–391 CrossRef CAS.
- R. Lazzaroni, A. Raffaelli, R. Settambolo, S. Bertozzi and G. Vitulli, J. Mol. Catal., 1989, 50, 1 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra01649b |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *a,
Haiyan Fua,
Bin Xub,
Ruixiang Lia,
Chunchun Zhangc and
Hua Chen
*a,
Haiyan Fua,
Bin Xub,
Ruixiang Lia,
Chunchun Zhangc and
Hua Chen