
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAbnormal adsorption and desorption behavior of pharmaceutical drugs on polystyrene microspheres†
Teng Wang‡
a,
Yajun Zheng‡a,
Xiaoting Wanga,
Qiang Wang*b,
Congyu Kea,
Daniel E. Austin*c,
Xiaoxiao Hana and
Zhiping Zhang *a
*a
aSchool of Chemistry and Chemical Engineering, Xi'an Shiyou University, Xi'an 710065, China. E-mail: zhangzp0304@gmail.com
bSchool of Chemistry and Molecular Engineering, Nanjing Tech University, Nanjing 211816, China. E-mail: wangqiang@njtech.edu.cn
cDepartment of Chemistry and Biochemistry, Brigham Young University, Provo, Utah 84602, USA. E-mail: austin@chem.byu.edu
First published on 3rd April 2017
Abstract
Herein we report an abnormal adsorption and desorption behavior where a stronger adsorption interaction between polystyrene particles and pharmaceutical drugs results in preferable desorption behavior. This behavior is contrary to the conventional view, in which a weaker adsorption interaction would lead to a more favorable desorption behavior of target molecules at solid surfaces. Different from other materials, numerous experimental results from a combination of mass spectrometry and infrared spectroscopy indicated quantitatively that the adsorption and desorption behavior of pharmaceutical drugs on polystyrene were independent of drug structure and solvent, while the intermolecular hydrogen bond interaction between polystyrene and the drug played a critical role in determining the adsorption and desorption behavior.
Adsorption and desorption are fundamentally related to the practical applications of chromatographic and spectrometric analyses,1–3 catalysis,4 coating,5 suspension,6 flocculation7 and dispersion.8 It is generally accepted that a weaker adsorption interaction (lower interaction energy of the adsorbate on a surface) would lead to a more favorable desorption behavior (faster desorption from a surface) based on numerous theoretical and experimental studies.9–17 By contrast, herein we present one instance of abnormal adsorption and desorption behavior: the stronger the adsorption interactions between polystyrene and pharmaceutical drugs, the more favorable the desorption of those drugs from polystyrene.
Polystyrene (PS) is a synthetic aromatic polymer made from the monomer, styrene. For many years, micro-scale PS particles have found a wide variety of applications in many advanced materials and optical devices because of their homogeneous size, large surface area, negligible solubility in water, and invariant surface characteristics.18–21 PS has also been extensively used as an adsorbent in the treatment of wastewater,22–26 chromatographic separations,27–33 solid-phase extraction,34–40 and adsorption of proteins/peptides.18–21,41–45 Although PS represents a powerful substrate that allows high efficiency treatment of various compounds in complex matrices, a complete analysis of its surface interactions is essential to further understand its performance and to develop improved materials. The characterization of molecular interactions at PS surfaces has involved X-ray photoelectron spectroscopy (XPS),42 Fourier transform infrared spectroscopy (FTIR),46 reflection fluorescence (RF),18 solid-state NMR,47 electrophoresis,48 chromatography,49 laser Doppler velocimetry (LDV),41 enzyme-linked immunosorbent assay (ELISA),50 dynamic light scattering (DLS),43 second harmonic generation (SHG),51 and titration calorimetry.52 However, these techniques are essentially limited to exploring the molecular interaction modes at PS surfaces such as electrostatic interactions43 and hydrophobic interactions,53 and study the desorption behaviors of analytes on PS substrates.30,32,33,49,54–59 For the purpose of fully understanding the interactions between PS and adsorbed molecules, the adsorption and desorption of various compounds to the surfaces of PS particles have been widely analyzed,60–64 but attention has been focused mainly on the reversible and irreversible adsorption mechanisms61–63 or the effect of experimental parameters on adsorption interaction or performance.60,64 There is little available quantitative experimental data on the correlation between adsorption and desorption behaviors of various analytes on PS substrates from different sources, which restricts greatly the understanding and interpretation of different molecular interactions on PS supports even composed of the same matrix.33 In view of this fact, in the current study, we illustrated the adsorption and desorption behaviors of several pharmaceutical drugs due to their significance in clinic applications,65,66 on the surfaces of a series of PS microspheres prepared via dispersion polymerization of styrene67,68 in ethanol/water system. The effects of experimental conditions on the adsorption and desorption capacities of the resulting PS particles have been explored quantitatively using paper spray mass spectrometry owing to its highly simplified procedures and high specificity, sensitivity and precision.69 For example, the desorption experiments were carried out by first depositing 2 μL of sample solution containing studied drugs on a small PS coated paper triangle. After drying, 25 μL methanol and 3.5 kV DC voltage were subsequently applied on the paper substrate for paper spray. The desorption behaviors of studied drugs on PS particles were quantitatively evaluated by the corresponding peak intensity in the obtained mass spectra. The results revealed for the first time that a stronger adsorption interaction between studied drugs and PS surfaces led to a more favorable desorption behavior.
A series of PS microspheres was first prepared according to the reported dispersion polymerization of styrene67,68 in ethanol/water using benzoyl peroxide (BPO) as the initiator and poly(N-vinylpyrrolidone) (PVP) as a steric stabilizer. With variation in the reaction parameters such as the concentrations of styrene, BPO or PVP, PS particles with adjustable diameters ranging from 1.0 to 16 μm can be synthesized. Only our effort on PS prepared from varying the concentration of styrene is reported in detail here, however. Fig. 1a and b demonstrates the typical scanning electron microscope (SEM) images of PS achieved by reaction of 5 mL styrene and 1.0 g BPO in the presence of 1.0 g PVP using 80 mL ethanol as solvent, and uniform microspheres with diameters of around 2.0 μm were obtained. The FT-IR spectrum (Fig. S1†) shows that the sample is a typical PS with characteristics of C–H stretching modes for mono-substituted phenyl rings (bands at 3060 and 3026 cm−1), C–H stretching modes for the CH and CH2 groups on the alkyl chain of the polymer (bands at 2924 and 2849 cm−1), C–C stretching modes for phenyl rings (bands at 1601, 1493 and 1452 cm−1).70,71 To enable on-line desorption experiments of studied drugs at PS surfaces, we coated as-synthesized PS particles onto filter paper (Fig. 1c and d) through a vacuum filtration approach developed in our lab.72,73 Note that the nearly monodisperse PS particles form a dense layer on the filter paper. This paper-based substrate is easy to fabricate, and can then be used for paper spray mass spectrometry (MS)69 experiments to probe the adsorption/desorption behavior. The experimental workflow consists of three steps: (i) fabrication of paper substrate; (ii) deposition of sample solution on PS coated paper; (iii) MS analysis. For example, to evaluate the desorption performance of verapamil from PS coated paper, 2 μL of methanol solution containing 1 μg mL−1 verapamil was spotted on the PS coated paper triangle with around 11 mm height and 9.5 mm base width, and the diameter of the wetted spot was about 7.0 mm (Fig. S2a†). After drying in air for around 4 h, 25 μL of a methanol solution spiked with 10 ng mL−1 D6-verapamil (internal standard) was applied at the paper surface followed immediately by application of the high voltage needed for paper spray. The D6-verapamil did not have enough time to adsorb thoroughly to the PS prior to paper spray analysis due to the floating of applied solvent at the surface of PS coated paper substrate (Fig. S2b†) and immediate spray after applying high voltage. Quantitation of verapamil was achieved using the ratio of the fragment ion (m/z 303) abundance from verapamil to that of the corresponding fragment ion (m/z 309) generated from D6-verapamil (Fig. S3†). Fig. 1e shows the normalized concentration of detected verapamil from PS coated paper substrates, in which PS was prepared in the presence of differing volumes of styrene (5–40 mL) by fixing the amounts of BPO (1.0 g) and PVP (1.0 g) in 80 mL ethanol solution. Notably, the desorption behavior of verapamil demonstrates first an increasing trend followed by a decrease as the styrene volume in PS preparation increases. When the percentage of styrene was 23.4%, namely 25 mL in volume, verapamil illustrated the most favorable desorption performance from PS particles. It should be pointed out that the on-line desorption experiment of verapamil from PS coated paper involved high voltage, paper substrate and an adhesive agent (soluble starch), but extensive investigation, in which the adsorption/desorption experiments were performed by eliminating these factors, demonstrated that they had little impact on the desorption performance (Fig. S4†). These results demonstrate that the on-line desorption experiments are not a result of the paper spray methodology.
 | ||
| Fig. 1 (a) and (b) SEM images of spherical PS obtained by reaction of 5 mL styrene and 1.0 g BPO in the presence of 1.0 g PVP by using 80 mL ethanol as solvent: (a) panoramic morphologies and (b) individual sphere; (c) and (d) SEM images of the surface structure of PS coated paper substrate; (c) as-prepared PS coated paper and (d) close-up image of typical surface structure; (e) desorption behavior of verapamil from PS coated paper substrates and (f) adsorption behavior of verapamil onto PS particles in methanol solution, in which PS was prepared in the presence of various volumes of styrene (5–40 mL) by fixing the amounts of BPO (1.0 g) and PVP (1.0 g) in 80 mL ethanol solution [note: the quantitative analysis was performed with paper spray mass spectrometry, and the desorption and adsorption performance of various PS particles were evaluated by the normalized peak intensity ratios between the characteristic fragment ion m/z 303 from protonated verapamil (m/z 455) and the characteristic fragment ion m/z 309 from the internal standard, protonated verapamil (m/z 461) spiked in the samples; spray solvent or sample, 25 μL methanol or adsorption solution; applied voltage, 3.5 kV]. | ||
The adsorption performance of these PS particles was carried out by mixing 0.2 g PS particles and 10 mL of methanol solution containing 1.0 μg mL−1 verapamil followed by stirring for 30 min. Afterwards, the upper clear solution was collected, separated and diluted with D6-verapamil solution. Fig. 1f exhibits the remaining content of verapamil in the adsorption solution as measured using paper spray mass spectrometry with uncoated filter paper. The error bars represent standard deviations from four replicate experiments. It is notable that with variation of styrene in the PS preparation, the detected amount of verapamil shows the opposite trend as that from the desorption experiments (Fig. 1e). When the volume of styrene was 25 mL in PS preparation, the generated product had the strongest adsorption capacity to verapamil. By comparing the desorption (Fig. 1e) and adsorption (Fig. 1f) behaviors of verapamil on the PS particles from various volumes of styrene, it is interesting that a stronger adsorption performance would lead to a more favorable desorption, which is contrary to the general view.
To demonstrate the generalization of the above phenomenon, a series of experiments with different parameters was carried out, which included various structures of molecules (e.g., amitriptyline, clozapine, quetiapine, risperidone and aripiprazole as shown in Fig. S5†), solvent systems (e.g., ethanol as Fig. S6, propanol as Fig. S7 and isopropanol as Fig. S8†), PS particles prepared through varying the amounts of PVP (Fig. S9a and b†), BPO (Fig. S9c and d†), H2O (Fig. S9e and f†) and styrene/BPO (Fig. S9g and h†) and from commercial sources with different particle sizes (Fig. S10†). The same results were observed for these experiments, indicating that the unique desorption and adsorption behaviors at PS surfaces were independent of molecular structure, solvent system and PS sources, and were a general phenomenon on PS surfaces. In contrast, when other materials, including different metal oxides (e.g., TiO2, Al2O3, ZnO, MgO and ZrO2 as Fig. S11†) and metal–organic frameworks [MOFs, e.g., MIL-53(Al), ZIP-8, and UiO-66(Zr) as Fig. S12†], were employed, the opposite result was obtained. Specifically, a weaker adsorption would lead to a more favorable desorption behavior, in good agreement with the general adsorption–desorption view. These results suggest that the adsorption–desorption behavior of molecules on the PS surfaces is different from that on other materials, which might be attributable to the unique physicochemical properties of PS particles.
To gain an insight into the unique adsorption and desorption behaviors of PS particles, verapamil was used as a model compound and the adsorption kinetic processes were explored. As shown in Tables S1 and S2 and Fig. S13,† the adsorption process of verapamil on PS particles followed a pseudo-second-order kinetic model and the Langmuir monolayer adsorption model. As shown in Fig. 2a, the initial adsorption of verapamil in the PS particles is quite fast followed by continued adsorption at a slower rate. Careful observation found that the adsorption rate exhibited a first increasing followed by decreasing with increase in the volume of styrene in PS preparation, and the PS from 25 mL styrene demonstrated the fastest adsorption rate to verapamil. As the prepared PS particles were coated on paper substrates for on-line desorption experiments, with increase in the elution number for the same paper substrate bearing a single sample spot,74 the desorption behavior of verapamil demonstrated a tendency (Fig. 2b) that is opposite that in the adsorption process (Fig. 2a), and the PS from 25 mL styrene gave the most favorable desorption behavior, in agreement with the desorption behavior of verapamil in conventional solution system (Fig. S14†). However, when the PS coated papers were used as substrates for thin layer chromatography of methylene blue, a reverse pattern was observed. Desorption from particulate surfaces (e.g., thin-layer chromatography experiments as Fig. S15,† and the on-line as Fig. 1a and 2b and the off-line desorption experiments as Fig. S4b†) takes place as adsorbate molecules overcome interaction energy with the surface (minus solvation energy) and are transferred from surface to solvent. Diffusion through the solvent limits the rate of desorption among particle surfaces. Taken these factors into account, it can be concluded that the former is realized through the transfer of solute molecules among PS particles, and the adsorption interactions between solutes and PS particles play a crucial role in determining the elution or desorption efficiency. For the latter, the desorption performance depends on not only the adsorption interactions between PS particles and solutes, but also the diffusion rate of solutes from solid surfaces to the bulk solution.
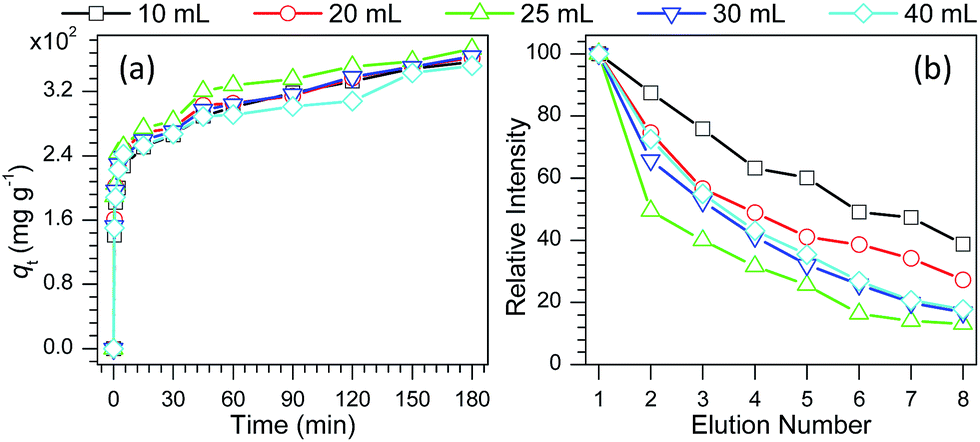 | ||
| Fig. 2 (a) Time profiles of verapamil adsorption on PS particles in methanol solution containing 1.0 μg mL−1 verapamil and (b) elution curves of verapamil from PS coated paper substrates, in which PS was prepared by the reaction between various volumes of styrene (10–40 mL) and 1.0 g BPO and 1.0 g PVP in 80 mL ethanol. | ||
With variation in the preparation parameters, the surface structures (e.g., particle size and texture properties) and degree of polymerization of the PS particles generally have a great effect on their properties.67,68 However, no direct correlation was found between performance of the PS particles in desorption and adsorption of pharmaceutical drugs and their particle sizes, texture properties and molecular weights (Tables S3 and S4†). Infrared spectrometry, as a powerful technique to characterize the surface properties of organic compounds, was also employed to study the surface properties of as-prepared PS particles. Fig. S16a† shows the typical FT-IR spectra of PS particles obtained by reaction of various volumes of styrene (10–40 mL) and 1.0 g BPO in the presence of 1.0 g PVP. Some absorption bands such as 1601 cm−1 and 1028 cm−1 (Fig. S16b†), demonstrated first a decreasing trend followed by increasing as the volume of styrene in preparation of PS particles increased. This trend mirrors the adsorption and desorption performance of the PS particles as discussed above (Fig. 1 and 2). Although the observed change is very small, these results provide a clue as to what causes the variation in desorption/adsorption capabilities of PS particles obtained from different preparation conditions. To confirm these changes, the PS particles, prepared by reaction of 25 mL styrene and 1.0 g BPO in the presence of 1.0 g PVP, were used as a model to adsorb different concentrations of verapamil (1.0–1000 μg mL−1) in methanol solutions for 3 h, respectively. As shown in Fig. S17,† the above IR adsorption bands presented a gradually decreasing trend, suggesting that with increase in the adsorption amount of verapamil at the PS surfaces, it would have a more significant impact on the IR spectra of PS particles. Based on this fact, the IR spectra of the above PS particles obtained from different volumes of styrene (10–40 mL) were examined further after adsorption of verapamil in methanol solution containing 1000 μg mL−1 for 3 h. Compared to the non-adsorbed PS particles (Fig. S16a†), the IR spectra of adsorbed PS samples kept almost constant (Fig. 3a), and no obvious characteristic absorption bands of verapamil were observed (Fig. S18a†) probably due to the small amount of verapamil adsorbed on PS particles (Fig. S18b†). The major difference between those spectra is the intensity of the adsorption bands at 1720, 1601, 1400, 1271, 1028, 906, 756 and 540 cm−1. To demonstrate clearly their differences, transmittance difference (ΔT, Fig. 3b) was used to characterize the effect of styrene amount on the resulting PS particles. As shown in Fig. 3c, with varying the volume of styrene in PS preparation, the ΔT values present first a decreasing trend followed by increasing, in which the PS from 25 mL of styrene in preparation illustrates the lowest value for each case, in good agreement with the adsorption and desorption performances of the particles (Fig. 1 and 2). It should be mentioned here that when the volume of styrene is above than 25 mL in preparation, the surface properties of the obtained PS are also altered rather than maintaining constant. Detailed reasons are not clear and further investigation is still needed. For other PS samples, a similar phenomenon was also observed (Fig. S19–22†). According to density functional theory (DFT) calculations, two types of intermolecular interactions (Fig. S23†), namely the H atom on the phenyl ring of PS and the N atom at the cyano group of verapamil (H–N hydrogen bond) and the H atoms on the phenyl ring of PS and the O atoms at the ether group of verapamil (H–O hydrogen bond), are mainly responsible for the adsorption of verapamil at PS surfaces. As discussed above such as the bands at 1720 cm−1 (C–C stretching vibration for phenyl ring), 1271 cm−1 (C–C stretching vibration for the CH and CH2 groups directly attached to phenyl ring) and 540 cm−1 (C–C skeleton stretching vibration for phenyl ring), the H atoms on the phenyl ring of PS particles played a crucial role in determining the adsorption interactions of verapamil via hydrogen bond interactions and therefore, the characteristic IR bands of C–C skeleton stretching vibration either for phenyl ring or directly attached to phenyl ring in PS samples were greatly affected. With increase in the adsorption interaction of PS particles to verapamil, the IR bands changed significantly. As a result, the pattern of adsorption interaction for the PS particles, obtained by reaction of various volumes of styrene (10–40 mL) and 1.0 g BPO in the presence of 1.0 g PVP in 80 mL ethanol solution (Fig. 1 and 2), was proportional to the change in the band intensity of the above characteristic IR absorption (Fig. 3).
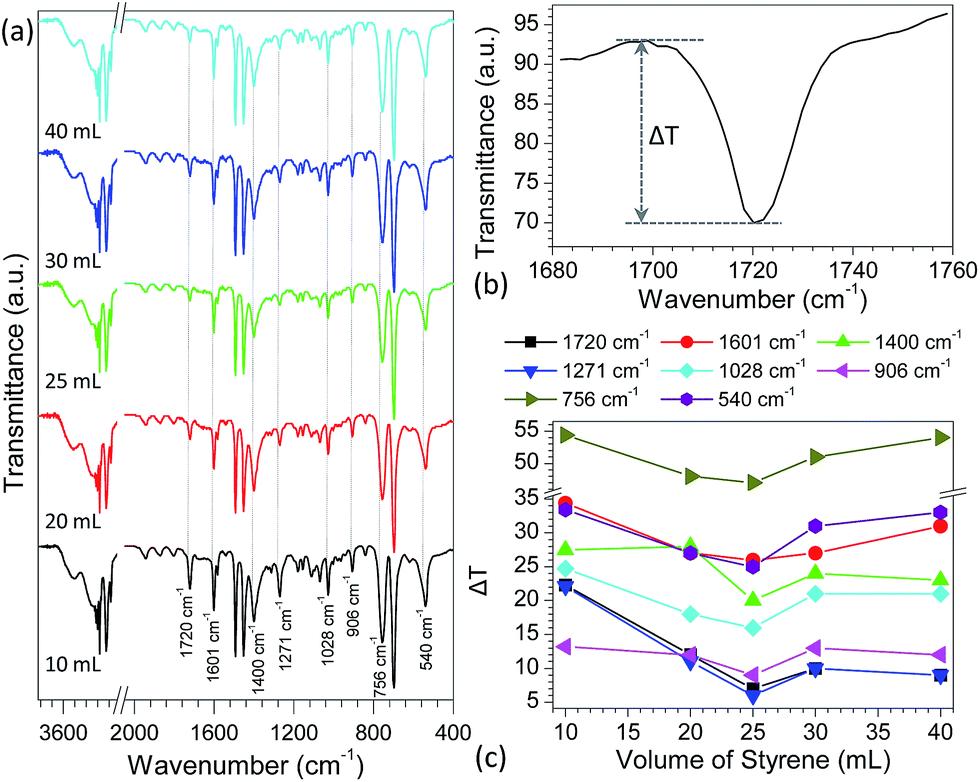 | ||
| Fig. 3 (a) FT-IR spectra of PS particles after adsorption of verapamil in methanol solution containing 1000 μg mL−1 verapamil for 3 h, in which the PS samples were prepared by reaction of various volumes of styrene (10–40 mL) and 1.0 g BPO in the presence of 1.0 g PVP in 80 mL ethanol solution; (b) definition of transmittance difference (ΔT) using the characteristic absorption band at 1720 cm−1 as an example; (c) plots of the transmittance differences of different characteristic absorption bands with varying the volume of styrene in PS preparation. | ||
In conclusion, the combination of mass spectrometry with infrared spectrometry has demonstrated that a stronger adsorption interaction between PS particles and pharmaceutical drugs would lead to a more favorable desorption behavior. This observed phenomenon was independent of the source of PS particles, drug structure and used solvent, which was unique to PS in contrast to other materials such as metal oxides and MOFs. FT-IR results illustrated that the adsorption interaction of PS particles was related closely to the hydrogen bond interaction between studied drug and PS, and a weaker IR absorption band would result in a stronger adsorption interaction at PS surfaces. These results are expected to help us better understand the adsorption and desorption interaction between PS particles and target molecules.
Acknowledgements
We thank Dr Milton Lee for helpful discussion and suggestions on the manuscript. This work was supported by the National Natural Science Foundation of China (21373112 and 21575112) and Shaanxi S&T Research Development Project of China (No. 2014K13-16 and 2016GY-231).Notes and references
- J. Falkenhagen and S. Weidner, Anal. Chem., 2009, 81, 282–287 CrossRef CAS PubMed.
- S. Saha, L. C. Chen, M. K. Mandal and K. Hiraoka, J. Am. Soc. Mass Spectrom., 2013, 24, 341–347 CrossRef CAS PubMed.
- S. Saha, M. K. Mandal, L. C. Chen, S. Ninomiya, Y. Shida and K. Hiraoka, Mass Spectrom., 2013, 2, S0008 Search PubMed.
- K. T. Rim, D. Eom, S.-W. Chan, M. Flytzani-Stephanopoulos, G. W. Flynn, X.-D. Wen and E. R. Batista, J. Am. Chem. Soc., 2012, 134, 18979–18985 CrossRef CAS PubMed.
- L. Qi, J.-P. Chapel, J.-C. Castaing, J. Fresnais and J.-F. Berret, Langmuir, 2007, 23, 11996–11998 CrossRef CAS PubMed.
- S. J. Jonas, A. Z. Stieg, W. Richardson, S. Guo, D. N. Powers, J. Wohlschlegel and B. Dunn, J. Phys. Chem. Lett., 2015, 6, 388–393 CrossRef CAS PubMed.
- O. León, E. Contreras, E. Rogel, G. Dambakli, J. Espidel and S. Acevedo, Energy Fuels, 2001, 15, 1028–1032 CrossRef.
- B. P. Williams, S. Pinge, Y.-K. Kim, J. Kim and Y. L. Joo, Langmuir, 2015, 31, 8989–8997 CrossRef CAS PubMed.
- A. Dąbrowski, Adv. Colloid Interface Sci., 2001, 93, 135–224 CrossRef.
- B. Mu and A. Wang, J. Environ. Chem. Eng., 2016, 4, 1274–1294 CrossRef CAS.
- L. Largitte and R. Pasquier, Chem. Eng. Res. Des., 2016, 109, 495–504 CrossRef CAS.
- K. Karimi and M. J. Taherzadeh, Bioresour. Technol., 2016, 203, 348–356 CrossRef CAS PubMed.
- S. Hokkanen, A. Bhatnagar and M. Sillanpää, Water Res., 2016, 91, 156–173 CrossRef CAS PubMed.
- K. Unger, Angew. Chem., Int. Ed., 1972, 11, 267–278 CrossRef CAS.
- I. Higuchi, T. Ree and H. Eyring, J. Am. Chem. Soc., 1957, 79, 1330–1337 CrossRef CAS.
- B. L. Harris, Ind. Eng. Chem., 1956, 48, 472–481 CrossRef CAS.
- A. B. Ray, Ind. Eng. Chem., 1947, 39, 12–13 CrossRef CAS.
- R. D. Tilton, C. R. Robertson and A. P. Gast, Langmuir, 1991, 7, 2710–2718 CrossRef CAS.
- B. D. Fair and A. M. Jamieson, J. Colloid Interface Sci., 1980, 77, 525–534 CrossRef CAS.
- C. Boisson-Vidal, J. Jozefonvicz and J. L. Brash, J. Biomed. Mater. Res., 1991, 25, 67–84 CrossRef CAS PubMed.
- W. Norde, F. G. Gonzalez and C. A. Haynes, Polym. Adv. Technol., 1995, 6, 518–525 CrossRef CAS.
- K. Jainae, K. Sanuwong, J. Nuangjamnong, N. Sukpirom and F. Unob, Chem. Eng. J., 2010, 160, 586–593 CrossRef CAS.
- J. Kluczka, T. Korolewicz, M. Zołotajkin and J. Adamek, Water Resources and Industry, 2015, 11, 46–57 CrossRef.
- K. Xiao, F. Xu, L. Jiang, Z. Dan and N. Duan, Chemosphere, 2016, 156, 326–333 CrossRef CAS PubMed.
- N. Chaukura, B. B. Mamba and S. B. Mishra, J. Environ. Manage., 2017, 193, 280–289 CrossRef CAS PubMed.
- B. Emo, C. T. Eberlin, K. R. Hixon, E. A. Growney Kalaf, J. M. Laktas and S. A. Sell, J. Environ. Chem. Eng., 2017, 5, 232–239 CrossRef CAS.
- J. C. Masini, J. Sep. Sci., 2016, 39, 1648–1655 CrossRef CAS PubMed.
- J.-B. Qu, G.-S. Huan, Y.-L. Chen, W.-Q. Zhou, J.-G. Liu and F. Huang, ACS Appl. Mater. Interfaces, 2014, 6, 12752–12760 CAS.
- X. Cao, Q. Kong, R. Cai, K. Zhu, X. Ye, J. Chen, W. Mo and J. Wang, J. Sep. Sci., 2014, 37, 3677–3683 CrossRef CAS PubMed.
- X. Huang, S. Zhang, G. A. Schultz and J. Henion, Anal. Chem., 2002, 74, 2336–2344 CrossRef CAS PubMed.
- D. Sacco and E. Dellacherie, Anal. Chem., 1984, 56, 1521–1524 CrossRef CAS.
- Z. Iskandarani and D. J. Pietrzyk, Anal. Chem., 1982, 54, 2427–2431 CrossRef CAS.
- S. Mori, Anal. Chem., 1978, 50, 745–748 CrossRef CAS.
- M. E. León-González and L. V. Pérez-Arribas, J. Chromatogr. A, 2000, 902, 3–16 CrossRef.
- H. W. Kauczor and A. Meyer, Hydrometallurgy, 1978, 3, 65–73 CrossRef CAS.
- V. V. Tolmacheva, V. V. Apyari, A. A. Furletov, S. G. Dmitrienko and Y. A. Zolotov, Talanta, 2016, 152, 203–210 CrossRef CAS PubMed.
- M. Trautmann, S. Lubahn and H.-J. Holdt, React. Funct. Polym., 2014, 83, 84–97 CrossRef CAS.
- W. W. Yang, G. S. Luo and X. C. Gong, Sep. Purif. Technol., 2005, 43, 175–182 CrossRef CAS.
- T. Saitoh, S. Suzuki and M. Hiraide, J. Chromatogr. A, 2005, 1097, 179–182 CrossRef CAS PubMed.
- S. Jara, C. Lysebo, T. Greibrokk and E. Lundanes, Anal. Chim. Acta, 2000, 407, 165–171 CrossRef CAS.
- K. Sofińska, Z. Adamczyk and J. Barbasz, Colloids Surf., B, 2016, 137, 183–190 CrossRef PubMed.
- A. Vesel and K. Elersic, Appl. Surf. Sci., 2012, 258, 5558–5560 CrossRef CAS.
- R. Jain and D. Forciniti, Langmuir, 2012, 28, 15323–15335 CrossRef CAS PubMed.
- M. M. Browne, G. V. Lubarsky, M. R. Davidson and R. H. Bradley, Surf. Sci., 2004, 553, 155–167 CrossRef CAS.
- J. Wang, S. M. Buck, M. A. Even and Z. Chen, J. Am. Chem. Soc., 2002, 124, 13302–13305 CrossRef CAS PubMed.
- J. Wang, X. Chen, M. L. Clarke and Z. Chen, J. Phys. Chem. B, 2006, 110, 5017–5024 CrossRef CAS PubMed.
- J. R. Long, N. Oyler, G. P. Drobny and P. S. Stayton, J. Am. Chem. Soc., 2002, 124, 6297–6303 CrossRef CAS PubMed.
- K. Aoki and Y. Adachi, J. Colloid Interface Sci., 2006, 300, 69–77 CrossRef CAS PubMed.
- M. G. Kontominas, R. Gavara and J. R. Giacin, Eur. Polym. J., 1994, 30, 271–277 CrossRef CAS.
- S. Sakai, J. Kim, B. Hexig, Y. Okahata, C. S. Cho and T. Akaike, Colloids Surf., B, 2012, 94, 192–198 CrossRef CAS PubMed.
- H. M. Eckenrode and H.-L. Dai, Langmuir, 2004, 20, 9202–9209 CrossRef CAS PubMed.
- M. J. Pollitt, G. Buckton, S. Brocchini and H. O. Alpar, Int. J. Pharm., 2005, 298, 333–338 CrossRef CAS PubMed.
- S. F. Turner, S. M. Clarke, A. R. Rennie, P. N. Thirtle, Z. X. Li, R. K. Thomas, S. Langridge and J. Penfold, Langmuir, 2005, 21, 10082–10088 CrossRef CAS PubMed.
- T. Hanai and J. Hubert, Chromatographia, 1983, 17, 633–639 CAS.
- M. Martinez, A. Carrancio, J. L. Casillas and J. Aracil, Ind. Eng. Chem. Res., 1995, 34, 4486–4493 CrossRef CAS.
- R. V. Snyder, R. J. Angelici and R. B. Meck, J. Am. Chem. Soc., 1972, 94, 2660–2663 CrossRef CAS PubMed.
- C. S. Su and D. Patterson, Macromolecules, 1977, 10, 708–710 CrossRef CAS.
- R. H. Schuster, H. Graeter and H. J. Cantow, Macromolecules, 1984, 17, 619–625 CrossRef CAS.
- B. M. Van Liedekerke, H. J. Nelis, W. E. Lambert and A. P. De Leenheer, Anal. Chem., 1989, 61, 728–732 CrossRef CAS.
- Q. B. Meng, G.-S. Yang and Y.-S. Lee, Microporous Mesoporous Mater., 2013, 181, 222–227 CrossRef CAS.
- M. S. Romero-Cano, A. Martín-Rodríguez and F. J. de las Nieves, J. Colloid Interface Sci., 2000, 227, 329–333 CrossRef CAS PubMed.
- M. S. Romero-Cano, A. Martín-Rodríguez and F. J. de las Nieves, J. Colloid Interface Sci., 2000, 227, 322–328 CrossRef CAS PubMed.
- A. Nieto, A. Gayá, C. Moreno, M. Jansá and J. Vives, Ann. Inst. Pasteur/Immunol., 1986, 137, 161–172 CrossRef.
- R. A. Lauten, A.-L. Kjøniksen and B. Nyström, Langmuir, 1984, 16, 4478–4484 CrossRef.
- X. Wang, Y. Zheng, T. Wang, X. Xiong, X. Fang and Z. Zhang, Anal. Methods, 2016, 8, 8004–8014 RSC.
- M. Schulz, S. Iwersen-Bergmann, H. Andresen and A. Schmoldt, Crit. Care, 2012, 16, R136 CrossRef PubMed.
- A. J. Paine, W. Luymes and J. McNulty, Macromolecules, 1990, 23, 3104–3109 CrossRef CAS.
- S. T. Ha, O. O. Park and S. H. Im, Macromol. Res., 2010, 18, 935–943 CrossRef CAS.
- H. Wang, J. Liu, R. G. Cooks and Z. Ouyang, Angew. Chem., Int. Ed., 2010, 49, 877–880 CrossRef CAS PubMed.
- Z. Pei, L. Busenlehner, S. D. Worley, Y. Tang and C. W. Curtis, J. Phys. Chem. B, 1997, 101, 10450–10454 CrossRef CAS.
- F. J. Torres, B. Civalleri, C. Pisani, P. Musto, A. R. Albunia and G. Guerra, J. Phys. Chem. B, 2007, 111, 6327–6335 CrossRef CAS PubMed.
- Y. Zheng, X. Zhang, H. Yang, X. Liu, X. Zhang, Q. Wang and Z. Zhang, Anal. Methods, 2015, 7, 5381–5386 RSC.
- Y. Zheng, Q. Wang, X. Wang, Y. Chen, X. Wang, X. Zhang, Z. Bai, X. Han and Z. Zhang, Anal. Chem., 2016, 88, 7005–7013 CrossRef CAS PubMed.
- Z. Zhang, W. Xu, N. E. Manicke, R. G. Cooks and Z. Ouyang, Anal. Chem., 2012, 84, 931–938 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Mass spectra, text, and figures giving experimental details. See DOI: 10.1039/c7ra01693j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |