
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhragmalin-type limonoids from the roots of Trichilia sinensis†
Shou-Bai Liu‡
,
Hui-Qin Chen‡,
Zhi-Kai Guo,
Wen-Hua Dong,
Jun Wang,
Wen-Li Mei* and
Hao-Fu Dai *
*
Laboratory of Biology and Genetic Resources of Tropical Crops, Ministry of Agriculture, Institute of Tropical Bioscience and Biotechnology, Chinese Academy of Tropical Agricultural Sciences, Haikou 571101, P. R. China. E-mail: meiwenli@itbb.org.cn; daihaofu@itbb.org.cn; Fax: +86-898-66961869; Tel: +86-898-66961869
First published on 2nd June 2017
Abstract
Twelve new phragmalin-type limonoids, including trichisintons A–D (1–4), which contain an unusual bicyclo[5.2.14,10]decane motif in the tetranortriterpenoid core, and trisinenmalins A–C and E–I (5–7 and 10–14), together with the known compounds 15-acetyltrichagmalin C (8) and trisinenmalin D (9), were isolated from the roots of Trichilia sinensis. Their structures were determined on the basis of the analysis of MS and NMR spectroscopic data, as well as by comparison with the literature. Compounds 5–7 and 10–14 displayed weak inhibitory activity against selected tumor cell lines. Compounds 2–4, 6–9 and 12–14 exhibited weak acetylcholinesterase-inhibiting activity in vitro.
Introduction
Natural products play an important role in the discovery of new drugs, because the great structural diversity of natural products with their various significant biological activities has always been an important source of inspiration for medicinal chemists in their search for new drug leads with pharmacological activities.1–4 Limonoids, which are a class of tetranortriterpenoids with a β-substituted furan ring, are secondary metabolites that are mainly found in the Rutaceae and Meliaceae families5 and have attracted continuous attention owing to their diverse structures and various pharmacological activities, including antitumor, antimicrobial, antiprotozoal6,7 and neuroprotective effects.8Rearranged limonoids with three uncommon carbon frameworks (mexicanolide-type, phragmalin-type, and trijugin-type) have been found in the Trichilia genus,7 among which the phragmalin-type and modified phragmalin-type limonoids possess characteristic tricyclo[3.3.12,10.11,4]decane and bicyclo[5.2.14,10]decane rings.7 Trichilia sinensis Bentv., which is a member of the Trichilia genus (Meliaceae) known to be rich in limonoids,9–15 triterpenoids,16,17 diterpenoids18,19 and steroids,20,21 is a shrub that is mainly distributed in China and Vietnam22 of which the fruits, leaves and roots have historically been used as folk medicines to treat abdominal pain caused by Ascaris lumbricoides, chronic osteomyelitis, scabies, eczema,23 rheumatism and traumatic injury.24 A previous investigation of the chemical constituents of T. sinensis revealed a series of mexicanolide-type limonoids,25 whereas phragmalin-type limonoids have never been reported from this species.
To investigate more structurally interesting and bioactive limonoids, 12 new phragmalin-type limonoids, namely, trichisintons A–D (1–4) and trisinenmalins A–C and E–I (5–7 and 10–14), together with the two known analogs 15-acetyltrichagmalin C (8) and trisinenmalin D (9) (Fig. 1), were isolated from the roots of T. sinensis. Trichisintons A–D (1–4) possess an unusual bicyclo[5.2.14,10]decane motif in the tetranortriterpenoid core. Structurally, the 1,2-dihydroxyphragmalin-type limonoids (6–8, 12, and 14) are the key precursors in the synthesis of the trichiliton-type limonoids (1–4) in a hypothetical biosynthetic pathway.26 Here, the isolation process, structural determination, and bioactivity, including cytotoxicity and acetylcholinesterase-inhibiting activity, of these compounds are described.
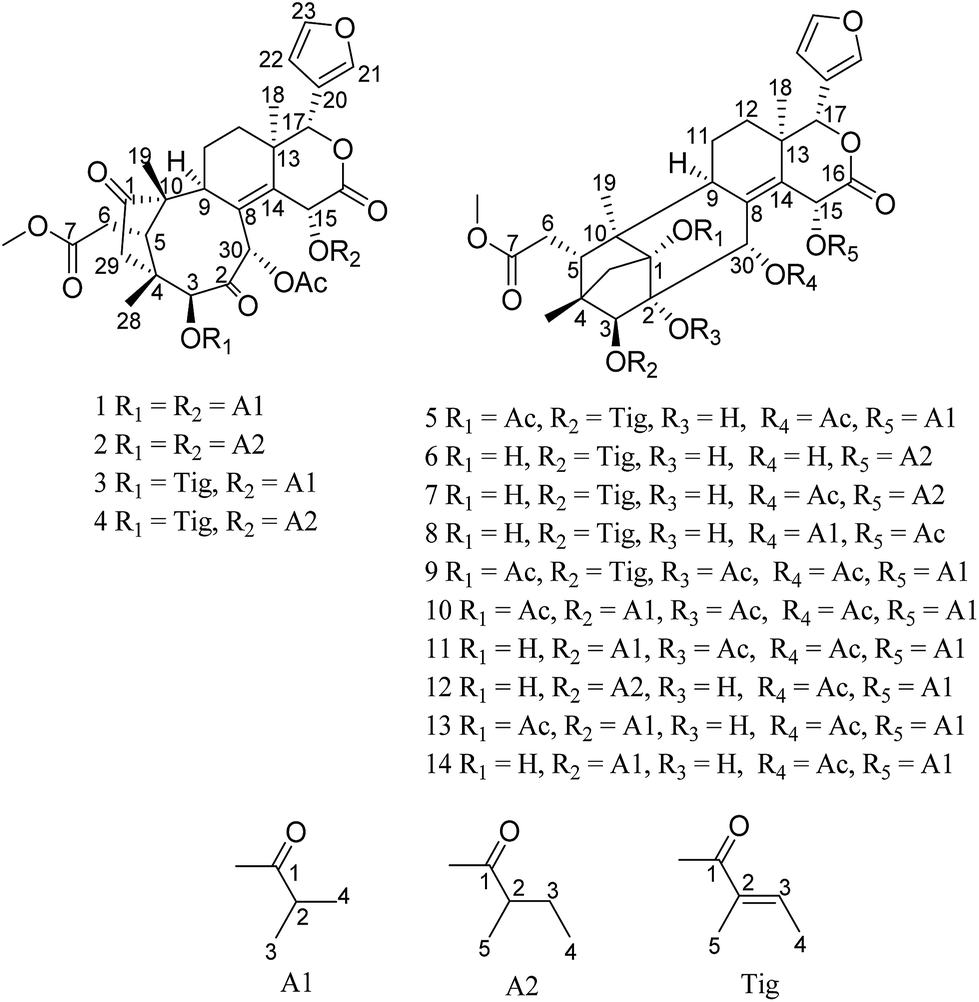 | ||
| Fig. 1 Structures of compounds 1–14. | ||
Results and discussion
Structural determination of new compounds 1–4, 5–7 and 10–14
Trichisinton A (1) was isolated as a white, amorphous powder, and its molecular formula C37H46O13, which was established by HREIMS (m/z 698.2935 [M]+, calcd as 698.2938), requires 15 degrees of unsaturation. The IR absorption at 1750 cm−1 showed the presence of a carbonyl group. The 13C NMR data (Table 2) displayed 37 carbon resonances, which were classified as nine methyl (one O-methyl), four methylene, eleven methine (three olefinic and four oxygenated), and 13 quaternary (three olefinic and seven carbonyl) carbons, which was supported by the DEPT and HSQC spectra. A typical β-substituted furan ring (δH 6.40, 7.41, and 7.43; δC 109.8, 120.3, 142.0 and 143.5), two keto carbonyls (δC 219.2, 199.9), and five ester carbonyls (δC 167.6, 168.7, 173.9, 175.3, and 176.1) were evident from the NMR data (Tables 1 and 2). Furthermore, one acetoxy and two isobutyryloxy groups were inferred according to the 1H–1H COSY and HMBC correlations (Fig. 2). The above observations accounted for 11 of the 15 degrees of unsaturation and suggested that 1 is a limonoid analogue with four additional rings. The two geminal methylene protons [δH 2.54 (d, 16.8 Hz), 2.38 (d, 16.8 Hz); δC 44.4] at C-29 were assigned by the HMBC correlations with the ketone carbonyl C-1 (δC 219.2), C-3 (δC 83.8), C-4 (δC 45.7), C-5 (δC 40.9), C-10 (δC 54.1) and C-28 (δC 21.9). The key HMBC correlations from H-5 (δH 3.62) to C-1, C-3, C-4, C-6 (δC 33.1), C-9 and C-10, as well as from the singlet methyl protons at δH 0.95 (H3-19) to C-1, C-5 and C-10, suggested that a cyclopentanone ring was constructed by C-1, C-29, C-4, C-5 and C-10, which was characteristic of trichiliton-type limonoids with an unusual bicyclo[5.2.14,10]decane motif in the tetranortriterpenoid core. A comparison of the NMR data revealed great similarity to those of trichiliton A.26 Detailed analysis of the 1D (1H NMR, 13C NMR and DEPT) and 2D NMR data (1H–1H COSY, HSQC and HMBC) indicated that both compounds shared the same skeleton, and the major differences were the substituents at C-3, C-15 and C-30, as well as the carbonyl carbon at C-2 instead of the double bond between C-2 and C-3 in the known compound trichiliton A, which was supported by the downfield chemical shift of C-2 (δC 199.9) and the HMBC correlations from H-3 (δH 5.33) and H-30 (δH 5.77) to C-2. In addition, the two isobutyryloxy groups at C-3 and C-15, respectively, and the acetoxy substituent at C-30 were assigned by the relevant HMBC correlations, as shown in Fig. 2. Thus, the planar structure of 1 was established, as shown in Fig. 1.| Position | 1 | 2 | 3 | 4 |
|---|---|---|---|---|
| 3 | 5.33 s | 5.36 s | 5.00 s | 5.00 s |
| 5 | 3.62 br d (12.4) | 3.60 br d (12.0) | 3.70 br d (12.3) | 3.71 br d (12.3) |
| 6a | 2.46 m | 2.46 dd (12.3, 16.3) | 2.52 m | 2.51 d (17.1) |
| 6b | 2.37 dd (16.3, 1.8) | 2.36 d (16.0) | 2.39 m | 2.42 d (17.1) |
| 9 | 3.02 d (3.0) | 3.01 br s | 3.01 d (3.2) | 3.01 d (2.8) |
| 11α | 1.65 m | 1.66 m | 1.61 m | 1.63 m |
| 11β | 1.96 d (14.8) | 1.96 d (14.7) | 1.94 d (14.8) | 1.94 d (14.8) |
| 12α | 1.04 m | 1.06 m | 1.05 m | 1.05 m |
| 12β | 1.23 overlapped | 1.18 m | 1.20 overlapped | 1.18 m |
| 15 | 6.64 d (0.7) | 6.62 br s | 6.41 s | 6.41 s |
| 17 | 5.88 s | 5.90 s | 5.82 s | 5.82 s |
| 18 | 1.23 s | 1.22 s | 1.20 s | 1.20 s |
| 19 | 0.95 s | 0.95 s | 0.95 s | 0.96 s |
| 21 | 7.41 s | 7.43 s | 7.45 s | 7.45 s |
| 22 | 6.40 br s | 6.41 s | 6.41 s | 6.41 s |
| 23 | 7.43 t (1.6) | 7.43 s | 7.43 t (1.9) | 7.44 s |
| 28 | 1.07 s | 1.09 s | 1.15 s | 1.15 s |
| 29α | 2.38 d (16.8) | 2.38 d (17.3) | 2.39 d (17.2) | 2.39 d (17.1) |
| 29β | 2.54 d (16.8) | 2.55 d (17.3) | 2.55 d (17.2) | 2.56 d (17.1) |
| 30 | 5.77 s | 5.79 s | 5.67 s | 5.67 s |
| MeO-7 | 3.71 s | 3.70 s | 3.76 s | 3.76 s |
| R1-2′ | 2.84 m | 2.67 m | ||
| 3′ | 1.29 d (6.9) | 1.81 m 1.55 m | 7.35 dq (7.0, 1.0) | 7.35 q (6.9) |
| 4′ | 1.28 d (6.9) | 0.91 t (7.5) | 1.82 d (7.0) | 1.82 d (6.9) |
| 5′ | 1.28 d (6.8) | 1.98 s | 1.99 s | |
| R2-2′′ | 2.47 m | 2.28 m | 2.42 m | 2.24 m |
| 3′′ | 1.17 d (7.0) | 1.81 m 1.55 m | 1.14 d (7.0) | 1.76 m 1.49 m |
| 4′′ | 1.23 d (7.0) | 0.97 t (7.5) | 1.18 d (7.0) | 0.93 t (7.4) |
| 5′′ | 1.14 d (6.9) | 1.11 d (6.9) | ||
| OAc-30 | 2.11 s | 2.11 s | 2.09 s | 2.09 s |
| Position | 1 | 2 | 3 | 4 |
|---|---|---|---|---|
| 1 | 219.2 | 219.2 | 219.2 | 219.2 |
| 2 | 199.9 | 200.0 | 200.6 | 200.5 |
| 3 | 83.8 | 83.8 | 84.8 | 84.8 |
| 4 | 45.7 | 45.6 | 45.0 | 45.0 |
| 5 | 40.9 | 41.0 | 41.1 | 41.1 |
| 6 | 33.1 | 33.2 | 33.3 | 33.4 |
| 7 | 173.9 | 173.8 | 174.2 | 174.1 |
| 8 | 134.5 | 134.4 | 134.3 | 134.4 |
| 9 | 39.2 | 39.3 | 39.3 | 39.3 |
| 10 | 54.1 | 54.1 | 54.2 | 54.2 |
| 11 | 21.4 | 21.4 | 21.4 | 21.4 |
| 12 | 28.5 | 28.5 | 28.5 | 28.6 |
| 13 | 40.4 | 40.5 | 40.4 | 40.4 |
| 14 | 141.6 | 141.8 | 141.8 | 141.9 |
| 15 | 64.5 | 64.4 | 64.1 | 64.0 |
| 16 | 167.6 | 167.6 | 167.6 | 167.6 |
| 17 | 79.8 | 79.8 | 79.6 | 79.7 |
| 18 | 17.9 | 17.4 | 17.3 | 17.4 |
| 19 | 23.9 | 24.0 | 24.0 | 24.0 |
| 20 | 120.3 | 120.4 | 120.5 | 120.5 |
| 21 | 142.0 | 142.0 | 142.0 | 142.0 |
| 22 | 109.8 | 109.8 | 109.9 | 109.9 |
| 23 | 143.5 | 143.4 | 143.5 | 143.5 |
| 28 | 21.9 | 22.0 | 21.9 | 22.0 |
| 29 | 44.4 | 44.4 | 44.0 | 44.0 |
| 30 | 74.7 | 74.8 | 74.5 | 74.6 |
| MeO-7 | 52.5 | 52.5 | 52.5 | 52.6 |
| R1-1′ | 176.1 | 175.5 | 166.7 | 166.8 |
| 2′ | 34.0 | 41.0 | 129.4 | 129.4 |
| 3′ | 20.7 | 26.9 | 140.1 | 140.1 |
| 4′ | 18.6 | 11.4 | 14.7 | 14.7 |
| 5′ | 15.4 | 12.5 | 12.5 | |
| R2-1′′ | 175.3 | 174.7 | 174.5 | 174.1 |
| 2′′ | 34.5 | 40.3 | 33.8 | 40.3 |
| 3′′ | 17.3 | 26.6 | 19.1 | 26.5 |
| 4′′ | 19.2 | 11.3 | 17.3 | 11.4 |
| 5′′ | 14.8 | 14.9 | ||
| OAc-30 | 168.7 | 168.7 | 168.3 | 168.3 |
| 19.9 | 20.7 | 20.7 | 20.7 |
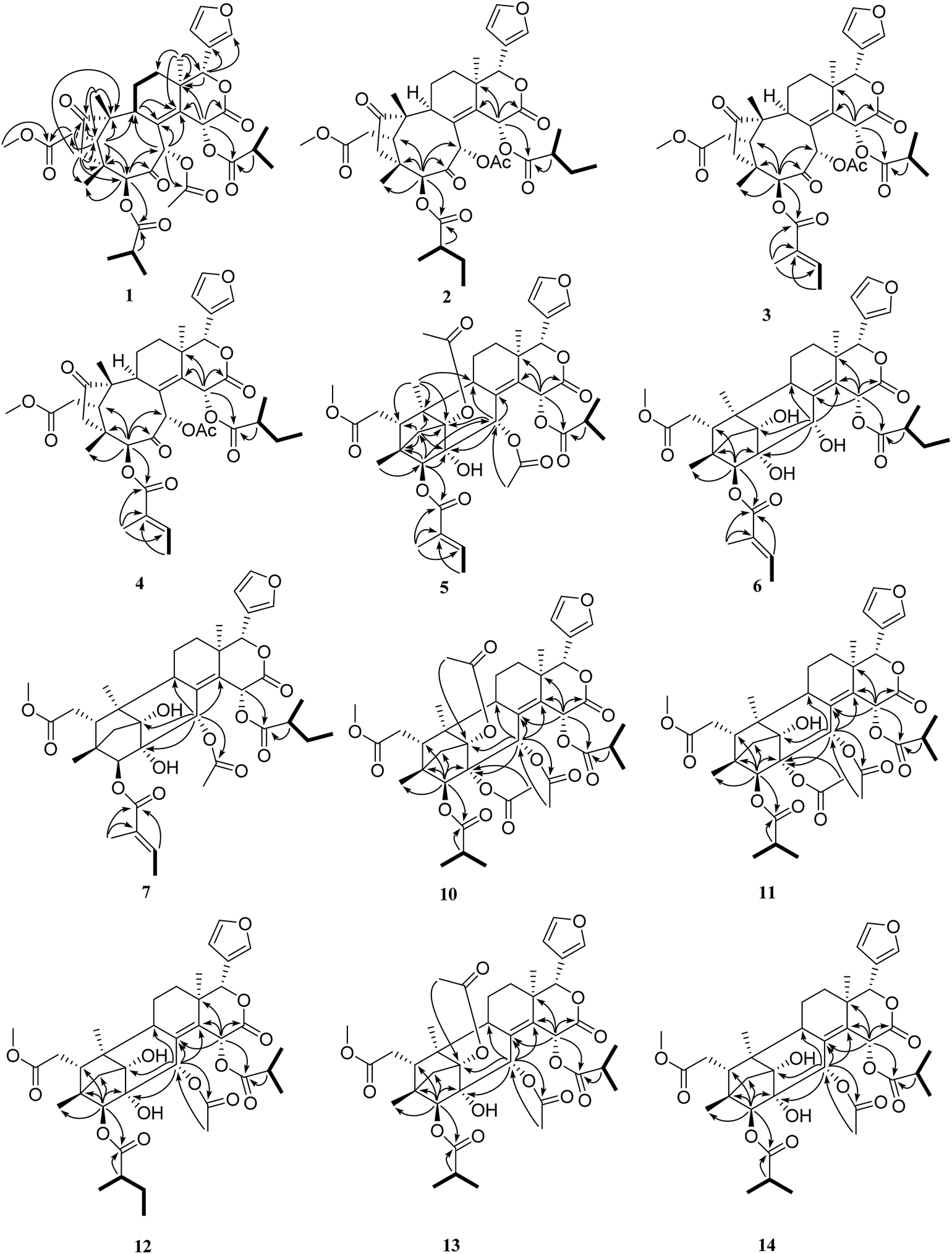 | ||
| Fig. 2 Key 1H–1H COSY (—) and HMBC (H → C) correlations of 1–7 and 10–14. | ||
The relative configuration of 1 (ESI Fig. S92†) was determined by the ROESY spectrum. The ROESY correlations of H-5/H-17, H-5/H3-28, H-5/H-30, H-30/H-15, H-5/H-15, H-15/H-2′, and H-11β/H-19 indicated that these protons and the isobutyryl group at C-3 were all β-oriented, whereas the ROESY correlations of H-22/H3-18, H3-18/H-11α, H-9/H-29α, and H-29α/H-3 revealed the α-orientation of the corresponding protons. Therefore, the structure of compound 1 was finally established.
Trichisinton B (2) was assigned a molecular formula of C39H50O13, as established on the basis of the prominent HRESIMS ion peak at m/z 749.3158 ([M + Na]+, calcd for C39H50O13Na 749.3149). The NMR spectroscopic data indicated that 2 had a similar skeleton to that of 1, except for the different substitution patterns at C-3 and C-15. Instead of isobutyryloxy groups, compound 2 possessed two 2-methylbutanoate groups at C-3 (δC 83.8) and C-15 (δC 64.4), respectively, on the basis of the corresponding 1H–1H COSY correlations of H-2′ (H-2′′)/H-3′ (H-3′′), H-3′ (H-3′′)/H-4′ (H-4′′) and H-2′ (H-2′′)/H-5′ (H-5′′), as well as the HMBC correlations from H-2′ (δH 2.67; H-2′′, δH 2.28) to C-1′ (δC 175.5; C-1′′, δC 174.7). The attachment of these two moieties to C-3 and C-15 was deduced from the HMBC correlations from H-3 (δH 5.36) to C-1′ (δC 175.5) and from H-15 (δH 6.62) to C-1′′ (δC 174.7), respectively (Fig. 2). The relative configuration was found to be the same as that of 1 by an ROESY experiment (ESI Fig. S92†).
Trichisinton C (3), which was a white, amorphous powder, had a molecular formula of C38H46O13 on the basis of its prominent HRESIMS ion peak at m/z 749.2578 ([M + K]+, C38H46O13K calcd as 749.2570). A comparison between the NMR data (Tables 1 and 2) of 1 and 3 showed that the tiglyloxy group at C-3 in 3 was not present in 1, which was confirmed by the HMBC correlations from H-5′ (δH 1.98) to C-1′ (δC 166.7), C-2′ (δC 129.4) and C-3′ (δC 140.1), and from H-4′ (δH 1.82) to C-3′. The attachment of this moiety to C-3 was determined by the cross-peak of H-3 (δH 5.00)/C-1′ in the HMBC spectrum. Similarly, instead of a 2-methylbutanoate group, an isobutyryloxy moiety was found to be connected to C-15 as a result of the HMBC correlations from H-2′′ (δH 2.42) to C-1′′ (δC 174.7) (Fig. 2). Therefore, the structure of trichisinton C (3) was determined, as shown in Fig. 1.
Trichisinton D (4), which was a white, amorphous powder, displayed a peak for [M + K]+ at m/z 763.2726 (calcd for C39H48O13K 763.2726) in the HRESIMS spectrum, which was 14 mass units greater than that of 3. Detailed analysis of the 1H and 13C NMR data of 4 revealed a structure that was closely related to that of 3, and the major difference was the replacement of the isobutyryloxy group at C-15 by a 2-methylbutanoate group in 4. The 2-methylbutanoate group was confirmed by the 1H–1H COSY correlations of H-2′′/H-3′′, H-3′′/H-4′′ and H-2′′/H-5′′, as well as the HMBC correlations from H-2′′ (δH 2.24) to C-1′′ (δC 174.1). The connection with C-15 was confirmed as a result of the HMBC correlations from H-15 (δH 6.41) to C-13 (δC 40.4), C-14 (δC 141.9), C-16 (δC 167.6) and C-1′′ (δC 174.1) (Fig. 2). Therefore, the structure of 4 was established, as shown in Fig. 1, with the same relative configuration as that of 1 according to the ROESY spectrum.
Trisinenmalin A (5) was isolated as a white, amorphous powder, and its molecular formula of C40H50O14, which was established by the HRESIMS spectrum (m/z 777.3099 [M + Na]+, calcd as 777.3098), requires 16 degrees of unsaturation. Its IR absorption bands showed the presence of hydroxy (3444 cm−1) and carbonyl groups (1729 cm−1). The 13C NMR and DEPT spectroscopic data of 5 displayed 40 carbon signals, which was in agreement with the molecular formula, including ten methyl (one methoxy group), four methylene, eleven methine (four oxygenated and four olefinic), and fifteen quaternary carbons (six carbonyl and four olefinic). Furthermore, one tiglyloxy, one isobutyryloxy, and two acetoxy groups were inferred according to the HMBC correlations and 1H–1H COSY correlations (Fig. 2). In addition, the typical β-furan moiety (δH 6.46, 7.39 and 7.57; δC 110.0, 120.6, 142.1 and 143.1) was deduced. The above fragments, together with the remaining 23 carbons, revealed that 5 was a limonoid with six rings, which was consistent with the degree of unsaturation. In the HMBC spectrum, the key correlations from H-30 (δH 5.39) to C-1 (δC 89.1), C-8 (δC 134.2) and C-9 (δC 36.4), from H-3 (δH 4.70) to C-2 (δC 77.8), C-5 (δC 36.1) and C-29 (δC 38.4), and from H-29 (δH 2.40, 2.51) to C-1, C-2, C-4 (δC 44.3), C-5 and C-10 (δC 49.4), as well as from H-19 (δH 1.06) to C-1, C-5, C-9 and C-10, confirmed the presence of a tricyclo[3.3.12,10.11,4]decane moiety, which was characteristic of phragmalin-type limonoids.27 By comparison of the 1H and 13C NMR data of 5 with those of 1,30-diacetyltrichagmalin F,11 it was found that both compounds shared the same skeleton, which was further confirmed by detailed analysis of the 2D NMR data (1H–1H COSY, HSQC and HMBC). The major difference between 5 and the known compound 1,30-diacetyltrichagmalin F was the replacement of the hydroxyisobutyryl group at C-15 by an isobutyryloxy group in 5, which was supported by the HMBC correlations (Fig. 2) from H-15 (δH 6.19) to C-1′′ (δC 174.9), C-16 (δC 167.8), C-14 (δC 136.4) and C-13 (δC 39.0). Therefore, the planar structure of compound 5 was established.
The relative configuration of 5 was deduced by analysis of its ROESY spectrum. As shown in ESI Fig. S92,† the observed ROESY correlations of H3-28/H-5, H-5/H-17, H-17/H-15, H-17/H-3′, and H-15/H-30 indicated that these protons and the tiglyl group at C-3 were all β-oriented, whereas the correlations of H-22/H3-18, H3-18/H-11α, and H-9/H3-19 revealed the α-orientation of the corresponding protons. Therefore, the structure of compound 5 was finally established, as shown in Fig. 1.
Trisinenmalin B (6) was isolated as a white, amorphous powder. Its molecular formula of C37H48O12 was established by HRESIMS (m/z 707.3038, calcd for [M + Na]+ 707.3038). The NMR data of 6 were similar to those of trichagmalin F,11 except for the presence of a 2-methylbutanoate group in 6 instead of the hydroxyisobutyryl group in trichagmalin F. The 2-methylbutanoate group was confirmed by the 1H–1H COSY correlations of H-2′′/H-3′′, H-3′′/H-4′′ and H-2′′/H-5′′, as well as the HMBC cross-peak of H-2′′ (δH 2.44)/C-1′′ (δC 175.3). The connection with C-15 was confirmed by the HMBC cross-peaks from H-15 (δH 6.18) to C-8 (δC 138.3), C-13 (δC 38.6), C-14 (δC 135.4), C-16 (δC 167.6) and C-1′′ (δC 175.3).
The relative configuration of 6 was assigned by an ROESY experiment (ESI Fig. S92†) and was found to be the same as that of 5. Therefore, the structure of 6 was confirmed, as shown in Fig. 1.
Trisinenmalin C (7), which was obtained as a white, amorphous powder, displayed a molecular formula of C39H50O13, as determined by the prominent HRESIMS ion peak at m/z 765.2888 ([M + K]+, calcd as 765.2883), which was 42 mass units more than that of 6. MS and NMR (Tables 3 and 4) analysis indicated that 7 was an acetylated derivative of 6. On the basis of the coherent HMBC correlation from H-30 (δH 5.42) to the carbon resonance at δC 171.1, the acetoxy group was determined to be located at C-30. Thus, 7 was demonstrated to be the 30-acetyl derivative of 6, and its relative configuration was found to be the same as that of 6 by an ROESY experiment. Therefore, the structure of 7 was established, as shown in Fig. 1.
| Position | 5 | 6 | 7 | 9 | 10 | 11 | 12 | 13 | 14 |
|---|---|---|---|---|---|---|---|---|---|
| 3 | 4.70 s | 4.72 s | 4.71 s | 5.42 s | 5.31 s | 5.31 s | 5.07 s | 5.04 s | 5.02 s |
| 5 | 2.81 t (7.0) | 2.85 t (7.0) | 2.86 m | 2.82 m | 2.69 m | 2.75 m | 2.70 m | 2.67 t (7.2) | 2.73 m |
| 6 | 2.31 m | 2.31 d (7.2) | 2.32 m | 2.29 d (7.3) | 2.27 m | 2.28 m | 2.26 m | 2.27 d (7.3) | 2.28 m |
| 9 | 2.70 d (7.0) | 2.61 d (6.8) | 2.70 d (6.8) | 2.82 m | 2.80 d (7.4) | 2.72 m | 2.70 m | 2.71 d (6.7) | 2.70 m |
| 11α | 1.70 m | 1.70 m | 1.72 m | 1.77 m | 1.76 m | 1.88 m | 1.73 m | 1.74 m | 1.73 m |
| 11β | 1.85 m | 1.85 m | 1.88 m | 1.85 m | 1.76 m | 1.88 m | 1.90 m | 1.86 m | 1.89 m |
| 12α | 1.03 m | 1.51 m | 1.04 dt (13.6, 3.0) | 1.08 m | 1.17 m | 1.08 m | 1.06 dt (13.6, 3.2) | 1.08 m | 1.07 d (13.3) |
| 12β | 1.38 m | 1.78 m | 1.39 m | 1.45 dt (13.9, 2.2) | 1.47 m | 1.48 m | 1.46 m | 1.42 m | 1.43 t (13.3) |
| 15 | 6.19 d (2.0) | 6.18 s | 6.19 d (1.8) | 6.62 d (2.0) | 6.73 d (2.1) | 6.72 s | 6.56 d (2.0) | 6.58 d (2.0) | 6.55 s |
| 17 | 5.31 s | 5.27 s | 5.32 s | 5.44 s | 5.48 s | 5.48 s | 5.47 s | 5.48 s | 5.48 s |
| 18 | 1.11 s | 1.13 s | 1.12 s | 1.14 s | 1.17 s | 1.16 s | 1.17 s | 1.16 s | 1.16 s |
| 19 | 1.06 s | 1.11 s | 1.13 s | 1.08 s | 1.07 s | 1.14 s | 1.13 s | 1.06 s | 1.12 s |
| 21 | 7.57 s | 7.57 s | 7.58 s | 7.57 s | 7.57 s | 7.58 s | 7.61 s | 7.56 s | 7.57 s |
| 22 | 6.46 d (1.6) | 6.46 s | 6.46 d (1.0) | 6.47 br s | 6.48 d (1.7) | 6.48 s | 6.50 d (1.6) | 6.46 d (1.1) | 6.47 s |
| 23 | 7.39 t (1.6) | 7.40 s | 7.40 t (1.6) | 7.40 br s | 7.40 t (1.7) | 7.39 s | 7.40 t (1.6) | 7.40 t (1.6) | 7.40 s |
| 28 | 0.85 s | 0.84 s | 0.82 s | 0.81 s | 0.81 s | 0.77 s | 0.79 s | 0.81 s | 0.77 s |
| 29a | 2.40 m | 1.61 d (10.4) | 1.56 m | 2.39 d (12.0) | 2.37 d (11.7) | 1.49 m | 1.60 d (10.6) | 2.35 m | 1.58 d (10.9) |
| 29b | 2.51 d (11.3) | 1.86 d (10.4) | 1.83 m | 2.47 m | 2.46 d (11.7) | 1.49 m | 1.81 d (10.6) | 2.48 m | 1.81 d (10.9) |
| 30 | 5.39 s | 4.13 s | 5.42 s | 6.13 s | 6.14 s | 6.21 s | 5.51 s | 5.50 s | 5.49 s |
| MeO-7 | 3.71 s | 3.71 s | 3.72 s | 3.68 s | 3.66 s | 3.65 s | 3.66 s | 3.66 s | 3.66 s |
| R2-2′ | 2.62 m | 2.60 m | 2.37 m | 2.57 m | 2.57 m | ||||
| 3′ | 7.10 dq (7.0, 1.2) | 7.07 dq (6.9, 0.9) | 7.11 dq (7.0, 1.1) | 6.58 q (6.8) | 1.23 d (6.9) | 1.31 d (6.8) | 1.76 m, 1.46 m | 1.22 d (7.0) | 1.26 d (6.8) |
| 4′ | 1.74 d (7.0) | 1.74 d (6.9) | 1.75 d (7.0) | 1.69 d (6.8) | 1.31 d (6.9) | 1.22 d (6.8) | 0.86 t (7.5) | 1.22 d (7.0) | 1.26 d (6.8) |
| 5′ | 1.99 s | 1.92 s | 1.99 s | 2.02 s | 1.27 d (7.3) | ||||
| R5-2′′ | 2.40 m | 2.44 m | 2.20 m | 2.46 m | 2.48 m | 2.46 m | 2.44 m | 2.47 m | 2.44 m |
| 3′′ | 1.21 d (7.0) | 1.51 m, 1.44 m | 1.79 m, 1.53 m | 1.20 d (7.0) | 1.22 d (6.8) | 1.23 d (7.4) | 1.27 d (6.8) | 1.26 d (7.0) | 1.22 d (6.6) |
| 4′′ | 1.19 d (7.0) | 0.96 t (7.4) | 0.96 t (7.4) | 1.21 d (7.0) | 1.22 d (6.8) | 1.21 d (7.4) | 1.22 d (6.8) | 1.26 d (7.0) | 1.22 d (6.6) |
| 5′′ | 1.17 d (6.9) | 1.17 d (6.9) | |||||||
| OAc-1 | 2.05 s | 2.07 s | 2.07 s | 2.06 s | |||||
| OAc-2 | 1.88 s | 1.87 s | 1.96 s | ||||||
| OAc-30 | 2.02 s | 2.05 s | 2.02 s | 2.02 s | 2.04 s | 2.07 s | 2.04 s | 2.07 s |
| Position | 5 | 6 | 7 | 9 | 10 | 11 | 12 | 13 | 14 |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 89.1 | 84.3 | 83.8 | 88.8 | 88.6 | 84.1 | 83.6 | 89.3 | 83.6 |
| 2 | 77.8 | 76.6 | 77.0 | 83.5 | 83.7 | 83.9 | 77.3 | 78.0 | 77.2 |
| 3 | 87.9 | 87.4 | 88.3 | 79.6 | 82.3 | 82.9 | 87.0 | 86.4 | 87.0 |
| 4 | 44.3 | 43.3 | 43.2 | 45.9 | 46.1 | 45.0 | 44.3 | 45.4 | 44.2 |
| 5 | 36.1 | 37.7 | 37.7 | 35.9 | 36.0 | 37.3 | 37.8 | 36.1 | 37.6 |
| 6 | 33.7 | 33.9 | 33.8 | 33.8 | 33.7 | 33.8 | 33.7 | 33.7 | 33.7 |
| 7 | 174.1 | 174.2 | 174.3 | 174.0 | 173.8 | 173.9 | 174.1 | 173.9 | 174.1 |
| 8 | 134.2 | 138.3 | 134.7 | 133.0 | 133.7 | 134.0 | 134.7 | 134.4 | 134.8 |
| 9 | 36.4 | 34.9 | 36.1 | 35.9 | 35.9 | 35.8 | 36.0 | 36.4 | 36.0 |
| 10 | 49.4 | 47.8 | 47.5 | 50.0 | 50.0 | 48.7 | 47.6 | 49.3 | 47.5 |
| 11 | 18.3 | 18.4 | 18.4 | 18.3 | 18.3 | 18.4 | 18.4 | 18.4 | 18.4 |
| 12 | 28.5 | 29.0 | 28.6 | 28.6 | 28.6 | 28.6 | 28.5 | 28.5 | 28.5 |
| 13 | 39.0 | 38.6 | 39.0 | 39.0 | 39.2 | 39.2 | 39.3 | 39.2 | 39.3 |
| 14 | 136.4 | 135.4 | 136.3 | 136.6 | 136.6 | 136.6 | 136.1 | 136.2 | 136.0 |
| 15 | 63.9 | 63.6 | 63.9 | 64.7 | 64.5 | 64.5 | 64.5 | 64.4 | 64.4 |
| 16 | 167.8 | 167.6 | 167.8 | 168.2 | 168.0 | 168.1 | 167.9 | 167.9 | 167.9 |
| 17 | 80.4 | 81.2 | 80.5 | 79.6 | 80.6 | 80.6 | 80.9 | 80.7 | 80.8 |
| 18 | 16.8 | 17.0 | 16.9 | 17.2 | 17.0 | 17.0 | 16.9 | 17.0 | 16.9 |
| 19 | 19.3 | 17.7 | 17.5 | 18.1 | 18.1 | 17.7 | 17.6 | 17.9 | 17.5 |
| 20 | 120.6 | 120.7 | 120.7 | 120.6 | 120.5 | 120.5 | 120.5 | 120.5 | 120.5 |
| 21 | 142.1 | 142.0 | 142.2 | 142.2 | 142.3 | 142.3 | 142.4 | 142.3 | 142.3 |
| 22 | 110.0 | 110.0 | 110.0 | 110.1 | 110.0 | 110.0 | 110.0 | 110.0 | 110.0 |
| 23 | 143.1 | 143.1 | 143.1 | 143.2 | 143.1 | 143.1 | 143.1 | 143.1 | 143.1 |
| 28 | 14.9 | 15.0 | 14.6 | 14.6 | 15.3 | 15.2 | 15.3 | 15.2 | 15.1 |
| 29 | 38.4 | 39.7 | 39.9 | 38.9 | 39.0 | 40.2 | 40.0 | 38.5 | 40.0 |
| 30 | 69.7 | 68.9 | 70.3 | 67.6 | 67.5 | 68.6 | 70.6 | 69.8 | 70.5 |
| MeO-7 | 52.2 | 52.1 | 52.1 | 52.2 | 52.2 | 52.1 | 52.0 | 52.2 | 52.1 |
| R2-1′ | 168.4 | 168.4 | 168.6 | 168.2 | 176.3 | 176.4 | 176.8 | 177.1 | 177.3 |
| 2′ | 130.0 | 129.7 | 130.0 | 132.1 | 34.6 | 34.6 | 41.7 | 34.9 | 34.9 |
| 3′ | 139.1 | 139.2 | 139.3 | 132.4 | 18.8 | 18.2 | 27.5 | 18.5 | 18.5 |
| 4′ | 14.6 | 14.6 | 15.0 | 13.7 | 18.2 | 18.8 | 11.5 | 18.2 | 17.8 |
| 5′ | 12.4 | 12.3 | 12.4 | 13.3 | 15.6 | ||||
| R5-1′′ | 174.9 | 175.3 | 174.5 | 175.0 | 175.0 | 175.0 | 175.7 | 175.6 | 175.7 |
| 2′′ | 34.1 | 40.9 | 40.4 | 34.1 | 34.2 | 34.1 | 34.2 | 34.2 | 34.2 |
| 3′′ | 18.1 | 26.9 | 26.6 | 19.4 | 20.2 | 20.2 | 19.5 | 19.5 | 19.5 |
| 4′′ | 18.0 | 11.6 | 11.4 | 18.2 | 19.4 | 19.4 | 17.8 | 20.3 | 20.3 |
| 5′′ | 16.3 | 14.6 | |||||||
| OAc-1 | 170.2 | 169.5 | 169.5 | 170.5 | |||||
| 22.0 | 22.0 | 22.0 | 21.3 | ||||||
| OAc-2 | 167.8 | 168.4 | 168.1 | ||||||
| 21.2 | 21.1 | 21.6 | |||||||
| OAc-30 | 170.2 | 171.1 | 168.4 | 168.4 | 168.4 | 171.6 | 170.2 | 171.6 | |
| 21.2 | 21.3 | 21.4 | 21.5 | 21.2 | 21.4 | 22.0 | 21.4 |
Trisinenmalin E (10) was obtained as a white, amorphous powder with a molecular formula of C41H52O15 (prominent HRESIMS ion peak at m/z 807.3208 [M + Na]+, calcd as 807.3204). The 1H and 13C NMR data of 10 indicated that its structure was closely related to that of trisinenmalin D (9), and the only difference lay in the presence of an isobutyryloxy group in 10 instead of the tiglyloxy moiety in 9, which was confirmed by the 1H–1H COSY correlations of H-2′/H-3′ and H-2′/H-4′ and the HMBC correlation from H-2′ (δH 2.62) to C-1′ (δC 176.3). The attachment of the isobutyryloxy group to C-3 was deduced from the HMBC correlation from H-3 (δH 5.31) to the carbon resonance at δC 176.3. Accordingly, the structure of trisinenmalin E (10) was established, as shown in Fig. 1, and the relative configuration was found to be the same as that of 9 according to the respective ROESY correlations.
Trisinenmalin F (11), which was a white amorphous powder, had the molecular formula of C39H50O14, as determined by the prominent HRESIMS ion peak at m/z 765.3109 ([M + Na]+, calcd as 765.3098). Analysis of the MS and NMR data of 11 indicated that its structure was closely related to that of trisinenmalin E (10), except for the absence of signals for acetoxy groups and the presence of a hydroxyl group in 11, which was consistent with the reduction of 42 amu in molecular weight, as was observed. Moreover, the chemical shift of C-1 (δC 84.1) in 11 was shifted upfield (ΔδC 4.5) in comparison with that in trisinenmalin E (10), which further confirmed that C-1 was substituted by a hydroxy group instead of an acetoxy moiety. The relative configuration was found to be the same as that of 10 by an ROESY experiment (ESI Fig. S92†). Accordingly, the structure of trisinenmalin F (11) was determined, as shown in Fig. 1.
Trisinenmalin G (12) was afforded as a white, amorphous powder having a molecular formula of C38H50O13, as established on the basis of the prominent HRESIMS ion peak at m/z 753.2890 ([M + K]+, calcd for C38H50O13K 753.2883). The 1H and 13C NMR data revealed that 12 was a structural congener of 11. Detailed analysis of the 1D and 2D NMR data indicated that the major differences between these two compounds were the substituent groups at C-2 and C-3. The hydroxy group at C-2 and the 2-methylbutanoate group at C-3 were inferred from the corresponding HMBC correlations, respectively. On the basis of extensive analysis of all the data, compound 12, which was given the trivial name of trisinenmalin G, was established, as shown in Fig. 1, and assigned the same relative configuration as that of 11 by analysis of the ROESY spectrum.
Trisinenmalin H (13) was isolated as a white, amorphous powder. The molecular formula of C39H50O14 was deduced from the positive prominent HRESIMS ion peak at m/z 781.2833 ([M + K]+, calcd for C39H50O14K 781.2832). By comparison, the 1H and 13C NMR data (Tables 3 and 4) of 13 were similar to those of trisinenmalin E (10), except for the absence of signals for acetoxy groups and the presence of a hydroxy group in 13. This inference was confirmed by the HMBC spectrum, in which the correlations from H-3 (δH 5.04) to C-2 (δC 78.0) and from H-30 (δH 5.50) to C-1 (δC 89.3) and C-2 were observed; moreover, this conclusion was consistent with the reduction of 42 amu in the molecular weight in comparison with that of 10. The ROESY spectrum displayed signals for the same relative configuration as that of 10. Thus, the structure of 13 was identified, as shown in Fig. 1.
Trisinenmalin I (14) had an adduct ion peak at m/z 739.2727 [M + K]+ in the HRESIMS spectrum, which was 42 amu less than that of 13, which indicated that 14 was a deacetylated derivative of 13. Moreover, the chemical shift of C-1 (δC 83.6) in 14 was shifted upfield (ΔδC 5.7) in comparison with that of C-1 (δC 89.3) in trisinenmalin H (13), which indicated that C-1 was substituted by a hydroxy group. By extensive analysis of the 1D and 2D NMR data, the structure of 14 was established, as shown in Fig. 1.
Two known phragmalin-type limonoids were also isolated from the same extract. Compound 8 was identified as 15-acetyltrichagmalin C11 on the basis of a comparison of its NMR and ESI-MS data with those of the reported compound, and compound 9 was confirmed to be trisinenmalin D28 by spectroscopic data (UV, IR, 1D and 2D NMR) and HRESIMS analysis, as well as by comparison with the literature.
Trichisintons A–D were assigned to the class of limonoids that contain the unusual bicyclo[5.2.14,10]decane ring system. So far, compounds with this type of skeleton have only been found in four species, namely, Trichilia connaroides,13,26 Xylocarpus granatum, X. moluccensis29 and Chisocheton ceramicus.30 Fang et al.26 proposed a biosynthetic pathway of this type of compound, of which the direct precursors were 1,2-dihydroxyphragmalin-type limonoids. However, they did not obtain any phragmalin-type limonoids during their isolation experiments. In this study, five 1,2-dihydroxyphragmalin-type limonoids (6–8, 12, and 14) were identified, which provided a crucial factor to prove the abovementioned hypothesis of the biosynthesis of these types of compound. Accordingly, 7 and 14 were the direct precursors of 4 and 1, respectively, and were processed by cleavage of the carbon bond in the 1,2-diol.
In summary, 12 new and 2 known phragmalin-type limonoids were isolated from the roots of T. sinensis for the first time.
Bioactivity evaluation
All the isolated compounds (1–14) were tested for cytotoxicity (Table 5) and AChE inhibitory activity (Table 6). Compounds 5–7 and 10–14 displayed weak inhibitory activity against selected tumor cell lines. Compounds 2–4, 6–9 and 12–14 exhibited weak AChE inhibitory activity, with inhibition percentages ranging from 16.4% to 34.9%.| Compound | Cell line, IC50 (μM) | ||
|---|---|---|---|
| K562 | SGC-7901 | BEL-7402 | |
| a Positive control.b Only IC50 values of <40 μM are reported. | |||
| 1 | >40b | >40 | >40 |
| 2 | >40 | >40 | >40 |
| 3 | >40 | >40 | >40 |
| 4 | >40 | >40 | >40 |
| 5 | 15.75 ± 0.23 | 15.54 ± 0.27 | 10.63 ± 0.71 |
| 6 | >40 | >40 | 38.57 ± 0.86 |
| 7 | 24.81 ± 1.12 | 14.56 ± 0.46 | 11.87 ± 0.62 |
| 8 | >40 | >40 | >40 |
| 9 | >40 | >40 | >40 |
| 10 | >40 | 27.99 ± 0.90 | 36.11 ± 0.84 |
| 11 | >40 | >40 | 37.30 ± 0.40 |
| 12 | 26.77 ± 0.52 | 15.22 ± 0.78 | 11.72 ± 0.53 |
| 13 | >40 | >40 | 27.14 ± 0.77 |
| 14 | 27.65 ± 0.46 | 17.15 ± 0.29 | 19.15 ± 0.31 |
| Taxola | 2.07 ± 0.17 | 4.26 ± 0.30 | 6.69 ± 0.10 |
| Compound | Percentage inhibition (%) | Compound | Percentage inhibition (%) | Compound | Percentage inhibition (%) |
|---|---|---|---|---|---|
| a Positive control (0.08 μg mL−1). | |||||
| 1 | <10 | 6 | 24.4 ± 0.9 | 11 | <10 |
| 2 | 10.6 ± 0.6 | 7 | 19.7 ± 0.8 | 12 | 17.2 ± 1.0 |
| 3 | 34.9 ± 2.0 | 8 | 21.8 ± 1.2 | 13 | 19.4 ± 1.0 |
| 4 | 19.5 ± 1.0 | 9 | 16.4 ± 1.0 | 14 | 16.9 ± 1.2 |
| 5 | <10 | 10 | <10 | Tacrinea | 68.6 ± 0.7 |
Experimental section
General
Optical rotations were measured in CHCl3 with a Rudolph Autopol III polarimeter (Rudolph Research Analytical, USA) at 25 °C. UV spectra were recorded using a Shimadzu UV-2550 spectrometer (Beckman, USA) and a DU800 spectrophotometer (Beckman, Brea, CA, USA). IR absorption was measured with a Nicolet 380 FT-IR instrument (Thermo, USA) using KBr pellets. 1D and 2D NMR spectra were recorded on Bruker Avance 500 NMR spectrometers (Bruker, Germany) using TMS as an internal standard. HREIMS spectra were recorded with an AutoSpecPremier spectrometer (Waters, Milford, MA, USA). HRESIMS spectra were recorded with an API QSTAR Pulsar mass spectrometer (Bruker, Germany). Column chromatography was performed with silica gel (60–80, 200–300 mesh, Qingdao Haiyang Chemical Co. Ltd., China), ODS gel (20–45 μm, Fuji Silysia Chemical Co. Ltd., USA) and Sephadex LH-20 (Merck, Germany). TLC was carried out on precoated silica gel G plates (Qingdao Haiyang Chemical Co. Ltd.), and spots were detected by spraying with 5% H2SO4 in EtOH followed by heating.Plant material
Roots of Trichilia sinensis were collected from Wanning, Hainan province, People's Republic of China in November 2011. The plant was identified by Prof. Zhengfu Dai of the Institute of Tropical Bioscience and Biotechnology, Chinese Academy of Tropical Agricultural Sciences. A voucher specimen (no. 20111120) was deposited at the Institute of Tropical Bioscience and Biotechnology, Chinese Academy of Tropical Agricultural Sciences.Extraction and isolation
Air-dried and powdered roots of Trichilia sinensis (13.2 kg) were extracted three times with 95% EtOH at room temperature to afford a crude extract (450.0 g). The extract was then dissolved in 3.0 L water and subsequently partitioned with petroleum ether (PE) (3.0 L × 3), EtOAc (3.0 L × 3) and n-BuOH (3.0 L × 3) to give three portions. The EtOAc portion (212.0 g) was subjected to passage over a silica gel vacuum liquid chromatograph (VLC) and eluted with gradients of PE–EtOAc (from 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to 1:1) and CHCl3–MeOH (from 25:1 to 0:1) to yield ten major fractions (Fr.1–Fr.10). Fr.3 (35.0 g) was applied to a silica gel VLC and eluted with a gradient of CHCl3–MeOH (from 1:0 to 20:1) to give four portions (Fr.3A–Fr.3D). Fr.3A (8.0 g) was applied to silica gel with CHCl3–EtOAc (20:1 to 1:1) as the eluent to give four fractions (Fr.3A1–Fr.3A4). Fr.3A4 (1.9 g) was separated on a column of Sephadex LH-20 by elution with CHCl3–MeOH (1:1) to obtain Fr.3A4A (950.0 mg), which was then chromatographed on a silica gel column and eluted with CHCl3–EtOAc (4:1) to yield 8 (12.0 mg) and 10 (9.8 mg). By the same purification procedures, Fr.3B (9.3 g) afforded 1 (16.0 mg) and 2 (8.0 mg). Fr.4 (17.0 g) was chromatographed on an ODS gel column and eluted with MeOH–H2O (gradient from 40:60 to 100:0) to give seven fractions (Fr.4A–Fr.4G). Fr.4C (1.5 g) was chromatographed on a Sephadex LH-20 column and eluted with MeOH to give two fractions (Fr.4C1 and Fr.4C2), and then Fr.4C1 (600.0 mg) was separated on a silica gel column by elution with CHCl3–EtOAc (15:1) to obtain 3 (12.0 mg); Fr.4C2 yielded 14 (12.0 mg). Fr.4E (700.0 mg) was applied to a silica gel column (PE–CHCl3–isopropyl alcohol, 7:3:0.1) to obtain two subfractions (Fr.4E1 and Fr.4E2), and then Fr.4E2 (42.5 mg) was applied to a silica gel column (CHCl3–MeOH, 150:1) to yield 4 (6.0 mg). By the same purification procedures, Fr.4F afforded 6 (6.5 mg), 7 (5.0 mg), 9 (6.0 mg) and 12 (7.0 mg). Fr.6 (30.5 g) was applied to an ODS gel column and eluted with MeOH–H2O (gradient from 40:60 to 100:0) to give seven fractions (Fr.6A–Fr.6G). Fr.6E–Fr.6G were purified on a Sephadex LH-20 column by elution with CHCl3–MeOH (1:1) and then further separated by repeated silica gel column chromatography to afford 11 (14.0 mg), 13 (10.0 mg) and 5 (40.0 mg), respectively.
ε) 239 (2.90) nm; IR (KBr) νmax 3452, 2322, 1750, 1640, 1381, 1219, 1028 cm−1; 1H and 13C NMR data: Tables 1 and 2; HREIMS m/z 698.2935 [M]+ (calcd for C37H46O13, 698.2938).ε) 238 (2.85), 229 (1.18) nm; IR (KBr) νmax 3460, 2925, 1748, 1632, 1383, 1032 cm−1; 1H and 13C NMR data: Tables 1 and 2; HRESIMS m/z 749.3158 [M + Na]+ (calcd for C39H50O13Na, 749.3149).ε) 272 (2.50), 256 (2.43) nm; IR (KBr) νmax 3452, 2925, 1742, 1638, 1385 cm−1; 1H and 13C NMR data: Tables 1 and 2; HRESIMS m/z 749.2578 [M + K]+ (calcd for C38H46O13K, 749.2570).ε) 272 (2.75), 258 (2.73) nm; IR (KBr) νmax 3449, 2924, 1748, 1647, 1378, 1031 cm−1; 1H and 13C NMR data: Tables 1 and 2; HRESIMS m/z 763.2726 [M + K]+ (calcd for C39H48O13K, 763.2726).ε) 225 (3.82) nm; IR (KBr) νmax 3444, 2927, 1729, 1634, 1382, 1232, 1029 cm−1; 1H and 13C NMR data: Tables 3 and 4; HRESIMS m/z 777.3099 [M + Na]+ (calcd for C40H50O14Na, 777.3098).ε) 272 (2.94), 256 (2.88) nm; IR (KBr) νmax 3450, 2925, 1719, 1637, 1384 cm−1; 1H and 13C NMR data: Tables 3 and 4; HRESIMS m/z 707.3038 [M + Na]+ (calcd for C37H48O12Na, 707.3038).ε) 272 (2.92), 254 (2.84) nm; IR (KBr) νmax 3451, 2924, 1726, 1636, 1462, 1382, 1228, 1024 cm−1; 1H and 13C NMR data: Tables 3 and 4; HRESIMS m/z 765.2888 [M + K]+ (calcd for C39H50O13K, 765.2883).ε) 272 (2.85), 256 (2.81) nm; IR (KBr) νmax 3442, 2954, 1744, 1635, 1371, 1247, 1038 cm−1; 1H and 13C NMR data: Tables 3 and 4; HRESIMS m/z 819.3202 [M + Na]+ (calcd for C42H52O15Na, 819.3198).ε) 229 (3.71), 218 (3.90) nm; IR (KBr) νmax 3460, 2928, 1748, 1627, 1250, 1033 cm−1; 1H and 13C NMR data: Tables 3 and 4; HRESIMS m/z 807.3208 [M + Na]+ (calcd for C41H52O15Na, 807.3204).ε) 230 (3.84) nm; IR (KBr) νmax 3453, 2969, 1747, 1636, 1383, 1235, 1027 cm−1; 1H and 13C NMR data: Tables 3 and 4; HRESIMS m/z 765.3109 [M + Na]+ (calcd for C39H50O14Na, 765.3098).ε) 272 (2.96), 252 (2.93) nm; IR (KBr) νmax 3450, 2924, 1731, 1636, 1460, 1382, 1228, 1022 cm−1; 1H and 13C NMR data: Tables 3 and 4; HRESIMS m/z 753.2890 [M + K]+ (calcd for C38H50O13K, 753.2883).ε) 272 (2.69), 252 (2.61) nm; IR (KBr) νmax 3458, 2924, 1745, 1644, 1461, 1380, 1228, 1024 cm−1; 1H and 13C NMR data: Tables 3 and 4; HRESIMS m/z 781.2833 [M + K]+ (calcd for C39H50O14K, 781.2832).ε) 272 (2.92), 256 (2.90) nm; IR (KBr) νmax 3456, 2924, 1743, 1641, 1460, 1379, 1227, 1135, 1023 cm−1; 1H and 13C NMR data: Tables 3 and 4; HRESIMS m/z 739.2727 [M + K]+ (calcd for C37H48O13K, 739.2726).
:1 to 1:1) and CHCl3–MeOH (from 25:1 to 0:1) to yield ten major fractions (Fr.1–Fr.10). Fr.3 (35.0 g) was applied to a silica gel VLC and eluted with a gradient of CHCl3–MeOH (from 1:0 to 20:1) to give four portions (Fr.3A–Fr.3D). Fr.3A (8.0 g) was applied to silica gel with CHCl3–EtOAc (20:1 to 1:1) as the eluent to give four fractions (Fr.3A1–Fr.3A4). Fr.3A4 (1.9 g) was separated on a column of Sephadex LH-20 by elution with CHCl3–MeOH (1:1) to obtain Fr.3A4A (950.0 mg), which was then chromatographed on a silica gel column and eluted with CHCl3–EtOAc (4:1) to yield 8 (12.0 mg) and 10 (9.8 mg). By the same purification procedures, Fr.3B (9.3 g) afforded 1 (16.0 mg) and 2 (8.0 mg). Fr.4 (17.0 g) was chromatographed on an ODS gel column and eluted with MeOH–H2O (gradient from 40:60 to 100:0) to give seven fractions (Fr.4A–Fr.4G). Fr.4C (1.5 g) was chromatographed on a Sephadex LH-20 column and eluted with MeOH to give two fractions (Fr.4C1 and Fr.4C2), and then Fr.4C1 (600.0 mg) was separated on a silica gel column by elution with CHCl3–EtOAc (15:1) to obtain 3 (12.0 mg); Fr.4C2 yielded 14 (12.0 mg). Fr.4E (700.0 mg) was applied to a silica gel column (PE–CHCl3–isopropyl alcohol, 7:3:0.1) to obtain two subfractions (Fr.4E1 and Fr.4E2), and then Fr.4E2 (42.5 mg) was applied to a silica gel column (CHCl3–MeOH, 150:1) to yield 4 (6.0 mg). By the same purification procedures, Fr.4F afforded 6 (6.5 mg), 7 (5.0 mg), 9 (6.0 mg) and 12 (7.0 mg). Fr.6 (30.5 g) was applied to an ODS gel column and eluted with MeOH–H2O (gradient from 40:60 to 100:0) to give seven fractions (Fr.6A–Fr.6G). Fr.6E–Fr.6G were purified on a Sephadex LH-20 column by elution with CHCl3–MeOH (1:1) and then further separated by repeated silica gel column chromatography to afford 11 (14.0 mg), 13 (10.0 mg) and 5 (40.0 mg), respectively.
ε) 239 (2.90) nm; IR (KBr) νmax 3452, 2322, 1750, 1640, 1381, 1219, 1028 cm−1; 1H and 13C NMR data: Tables 1 and 2; HREIMS m/z 698.2935 [M]+ (calcd for C37H46O13, 698.2938).ε) 238 (2.85), 229 (1.18) nm; IR (KBr) νmax 3460, 2925, 1748, 1632, 1383, 1032 cm−1; 1H and 13C NMR data: Tables 1 and 2; HRESIMS m/z 749.3158 [M + Na]+ (calcd for C39H50O13Na, 749.3149).ε) 272 (2.50), 256 (2.43) nm; IR (KBr) νmax 3452, 2925, 1742, 1638, 1385 cm−1; 1H and 13C NMR data: Tables 1 and 2; HRESIMS m/z 749.2578 [M + K]+ (calcd for C38H46O13K, 749.2570).ε) 272 (2.75), 258 (2.73) nm; IR (KBr) νmax 3449, 2924, 1748, 1647, 1378, 1031 cm−1; 1H and 13C NMR data: Tables 1 and 2; HRESIMS m/z 763.2726 [M + K]+ (calcd for C39H48O13K, 763.2726).ε) 225 (3.82) nm; IR (KBr) νmax 3444, 2927, 1729, 1634, 1382, 1232, 1029 cm−1; 1H and 13C NMR data: Tables 3 and 4; HRESIMS m/z 777.3099 [M + Na]+ (calcd for C40H50O14Na, 777.3098).ε) 272 (2.94), 256 (2.88) nm; IR (KBr) νmax 3450, 2925, 1719, 1637, 1384 cm−1; 1H and 13C NMR data: Tables 3 and 4; HRESIMS m/z 707.3038 [M + Na]+ (calcd for C37H48O12Na, 707.3038).ε) 272 (2.92), 254 (2.84) nm; IR (KBr) νmax 3451, 2924, 1726, 1636, 1462, 1382, 1228, 1024 cm−1; 1H and 13C NMR data: Tables 3 and 4; HRESIMS m/z 765.2888 [M + K]+ (calcd for C39H50O13K, 765.2883).ε) 272 (2.85), 256 (2.81) nm; IR (KBr) νmax 3442, 2954, 1744, 1635, 1371, 1247, 1038 cm−1; 1H and 13C NMR data: Tables 3 and 4; HRESIMS m/z 819.3202 [M + Na]+ (calcd for C42H52O15Na, 819.3198).ε) 229 (3.71), 218 (3.90) nm; IR (KBr) νmax 3460, 2928, 1748, 1627, 1250, 1033 cm−1; 1H and 13C NMR data: Tables 3 and 4; HRESIMS m/z 807.3208 [M + Na]+ (calcd for C41H52O15Na, 807.3204).ε) 230 (3.84) nm; IR (KBr) νmax 3453, 2969, 1747, 1636, 1383, 1235, 1027 cm−1; 1H and 13C NMR data: Tables 3 and 4; HRESIMS m/z 765.3109 [M + Na]+ (calcd for C39H50O14Na, 765.3098).ε) 272 (2.96), 252 (2.93) nm; IR (KBr) νmax 3450, 2924, 1731, 1636, 1460, 1382, 1228, 1022 cm−1; 1H and 13C NMR data: Tables 3 and 4; HRESIMS m/z 753.2890 [M + K]+ (calcd for C38H50O13K, 753.2883).ε) 272 (2.69), 252 (2.61) nm; IR (KBr) νmax 3458, 2924, 1745, 1644, 1461, 1380, 1228, 1024 cm−1; 1H and 13C NMR data: Tables 3 and 4; HRESIMS m/z 781.2833 [M + K]+ (calcd for C39H50O14K, 781.2832).ε) 272 (2.92), 256 (2.90) nm; IR (KBr) νmax 3456, 2924, 1743, 1641, 1460, 1379, 1227, 1135, 1023 cm−1; 1H and 13C NMR data: Tables 3 and 4; HRESIMS m/z 739.2727 [M + K]+ (calcd for C37H48O13K, 739.2726).Cytotoxicity assay
A human chronic myelogenous leukemia cell line (K562), a human gastric carcinoma cell line (SGC-7901), and a human hepatocellular carcinoma cell line (BEL-7402) were bought from the Cell Bank of the Type Culture Collection of the Chinese Academy of Sciences, Shanghai Institute of Cell Biology. An MTT assay was used to determine inhibition of the growth of K562, SGC-7901, and BEL-7402 cells in vitro, and taxol was used as a positive control. Cells were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum, 100 IU mL−1 penicillin, and 100 mg mL−1 streptomycin at 37 °C under a 5% CO2 atmosphere with 90% humidity. Cells in the logarithmic phase were used for experiments. The specific experimental procedures were the same as those described previously.31Bioassay of AChE inhibitory activity
The AChE inhibitory activity of these compounds was assayed by the spectrophotometric method developed by Ellman32 with slight modifications. S-Acetylthiocholine iodide, 5,5′-dithio-bis(2-nitrobenzoic acid) (DNTB, Ellman's reagent) and AChE were purchased from Sigma Chemicals. The compounds were dissolved in DMSO. The reaction mixture (total 200 μL) containing phosphate buffer (pH 8.0), the test compound (50 μg mL−1), and acetylcholinesterase (0.02 U mL−1) was incubated for 20 min (30 °C). The reaction was initiated by the addition of 20 μL DNTB (2.48 mg mL−1) and 20 μL acetylthiocholine iodide (1.81 mg mL−1) for the assay of AChE inhibitory activity. The hydrolysis of acetylthiocholine was monitored at 405 nm for one hour. Tacrine (Sigma-Aldrich, 99%) was used as a positive control with a final concentration of 0.08 μg mL−1, and DMSO was used as a negative control with a final concentration of 0.1%. All the reactions were performed in triplicate. The percentage inhibition was calculated as follows: % inhibition = (E − S)/E × 100 (where E is the absorbance at 405 nm of the solution without the test compound and S is the absorbance at 405 nm of the solution with the test compound). The values are expressed as the mean ± SD of triplicate experiments.Acknowledgements
This research was financially supported by the Special Fund for Agro-scientific Research in the Public Interest (201303117), Special Grant for Modernization of Traditional Chinese Medicine of Hainan Province (2015ZY04), and Key Research and Development Scheme of Hainan Province (ZDYF2016140).Notes and references
- D. J. Newman, J. Med. Chem., 2008, 51, 2589–2599 CrossRef CAS PubMed.
- K. H. Lee, J. Nat. Prod., 2010, 73, 500–516 CrossRef CAS PubMed.
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2012, 75, 311–335 CrossRef CAS PubMed.
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2016, 79(3), 629–661 CrossRef CAS PubMed.
- G. Jayaprakasha, R. Singh, J. Pereira and K. Sakariah, Phytochemistry, 1997, 44, 843–846 CrossRef CAS PubMed.
- X. Fang, Y. T. Di and X. J. Hao, Curr. Org. Chem., 2011, 15, 1363–1391 CrossRef CAS.
- Q. G. Tan and X. D. Luo, Chem. Rev., 2011, 111, 7437–7522 CrossRef CAS PubMed.
- J. B. Sun, N. Jiang, M. Y. Lv, P. Wang, F. G. Xu, J. Y. Liang and W. Qu, RSC Adv., 2015, 5, 24750–24757 RSC.
- H. P. Zhang, S. H. Wu, Y. M. Shen, Y. B. Ma, D. G. Wu, S. H. Qi and X. D. Luo, Can. J. Chem., 2003, 81, 253–257 CrossRef CAS.
- X. N. Wang, C. Q. Fan, S. Yin, L. S. Gan and J. M. Yue, Phytochemistry, 2008, 69, 1319–1327 CrossRef CAS PubMed.
- Q. Zhang, Y. T. Di, H. P. He, X. Fang, D. L. Chen, X. H. Yan, F. Zhu, T. Q. Yang, L. L. Liu and X. J. Hao, J. Nat. Prod., 2011, 74, 152–157 CrossRef CAS PubMed.
- H. Y. Wang, J. S. Wang, S. M. Shan, X. B. Wang, J. Luo, M. H. Yang and L. Y. Kong, Planta Med., 2013, 79, 1767–1774 CrossRef CAS PubMed.
- H. Y. Wang, J. S. Wang, Y. Zhang, J. Luo, M. H. Yang, X. B. Wang and L. Y. Kong, Chem. Pharm. Bull., 2013, 61, 1075–1080 CrossRef CAS PubMed.
- C. P. Liu, J. B. Xu, Y. S. Han, M. A. Wainberg and J. M. Yue, Org. Lett., 2014, 16, 5478–5481 CrossRef CAS PubMed.
- F. L. An, J. Luo, X. B. Wang, M. H. Yang and L. Y. Kong, Org. Biomol. Chem., 2016, 4, 1231–1235 Search PubMed.
- T. A. Olugbade and S. A. Adesanya, Phytochemistry, 2000, 54, 867–870 CrossRef CAS PubMed.
- I. J. C. Vieira, O. de Aquino Azevedo, J. J. de Souza, R. Braz-Filho, M. dos Santos Gonçalves and M. F. de Araújo, Molecules, 2013, 18, 2589–2597 CrossRef CAS PubMed.
- A. J. Aladesanmi and S. A. Odediran, Fitoterapia, 2000, 71, 179–182 CrossRef CAS PubMed.
- M. del Carmen Ramírez, R. A. Toscano, J. Arnason, S. Omar, C. M. Cerda-García-Rojas and R. Mata, Tetrahedron, 2000, 56, 5085–5091 CrossRef.
- W. W. Harding, H. Jacobs, P. A. Lewis, S. McLean and W. F. Reynolds, Nat. Prod. Lett., 2001, 15, 253–260 CrossRef CAS PubMed.
- Q. Zhang, Y. Zhang, H. P. He, Y. T. Di and X. J. Hao, Nat. Prod. Commun., 2012, 7, 1267–1268 CAS.
- S. K. Chen, B. Y. Chen and H. Li, in Flora Republicae Popularis Sinicae (Zhongguo Zhiwu Zhi), Science Press, Beijing, 1997, vol. 34, p. 68 Search PubMed.
- Editorial Committee of Chinese Materia Medica; the Administration Bureau of Traditional Chinese Medicine, Chinese Materia Medica (Zhonghua Bencao), Shanghai Science & Technology Press, Shanghai, 2000, vol. 13, pp. 3880–3881 Search PubMed.
- H. F. Dai, X. L. Zheng, F. W. Xing and W. L. Mei, Records of Li Folk Medicine, China Science & Technology Press, Beijing, 2014, vol. 3, pp. 135–136 Search PubMed.
- J. B. Xu, Y. Lin, S. H. Dong, F. Wang and J. M. Yue, J. Nat. Prod., 2013, 76, 1872–1880 CrossRef CAS PubMed.
- X. Fang, Y. T. Di, Z. L. Geng, C. J. Tan, J. Guo, J. Ning and X. J. Hao, Eur. J. Org. Chem., 2010, 2010, 1381–1387 CrossRef.
- D. A. Mulholland, B. Parel and P. H. Coombes, Curr. Org. Chem., 2000, 4, 1011–1054 CrossRef CAS.
- W. Yang, D. M. Fang, H. P. He, X. J. Hao, Z. J. Wu and G. L. Zhang, Rapid Commun. Mass Spectrom., 2013, 27, 1203–1212 CrossRef CAS PubMed.
- J. Li, M. Y. Li, T. Bruhn, D. C. Götz, Q. Xiao, T. Satyanandamurty, J. Wu and G. Bringmann, Chem.–Eur. J., 2012, 18, 14342–14351 CrossRef CAS PubMed.
- I. A. Najmuldeen, A. H. A. Hadi, K. Awang, K. Mohamad, K. A. Ketuly, M. R. Mukhtar, S.-L. Chong, G. Chan, M. A. Nafiah, N. S. Weng, O. Shirota, T. Hosoya, A. E. Nugroho and O. Shirota, J. Nat. Prod., 2011, 74, 1313–1317 CrossRef CAS PubMed.
- Y. X. Li, W. L. Mei, W. J. Zuo, Y. X. Zhao, W. H. Dong and H. F. Dai, Phytochem. Lett., 2012, 5, 41–44 CrossRef CAS.
- G. L. Ellman, K. D. Courtney, A. J. Valentino and R. M. Featherstone, Biochem. Pharmacol., 1961, 7, 88–95 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: 1D and 2D NMR spectra and mass spectra of 1–7 and 9–14. See DOI: 10.1039/c7ra01785e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |