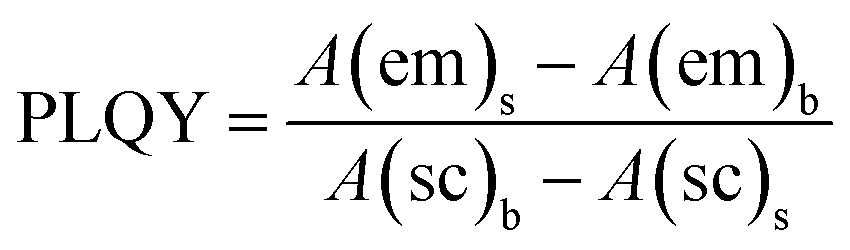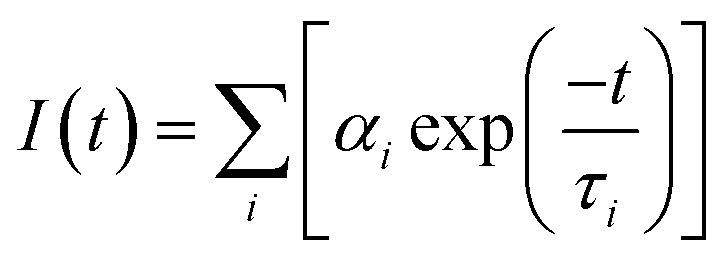DOI:
10.1039/C7RA01804E
(Paper)
RSC Adv., 2017,
7, 16637-16643
The effect of oxygen on the microwave-assisted synthesis of carbon quantum dots from polyethylene glycol†
Received
13th February 2017
, Accepted 4th March 2017
First published on 15th March 2017
Abstract
Fluorescent carbon quantum dots (CQDs) were synthesized using polyethylene glycol (PEG) as both the carbon source and solvent in various atmospheres including air, nitrogen, carbon dioxide and oxygen by following a microwave irradiation method. The CQDs were characterized by performing ultraviolet-visible absorption spectroscopy, Fourier transform infrared spectroscopy, transmission electron microscopy, and fluorescence spectroscopy. We found that different reaction atmospheres yielded significantly different reaction rates and properties of the CQDs. In particular, including oxygen in the synthesis of the CQDs clearly shortened the reaction time, significantly enhanced the photoluminescence (PL) intensity and quantum yield (PLQY) of the CQDs, and extended their average lifetime. The PLQY and fluorescence lifetime of the CQDs were improved from 5.78% and 2.4 ns, respectively, when synthesized in a medium open to air to 7.84% and 3.0 ns, respectively, when synthesized in an O2 atmosphere. The CQDs synthesized using microwave irradiation showed strong blue fluorescence under ultraviolet light.
Introduction
Carbon quantum dots (CQDs) constitute a new carbon nanomaterial with dimensions below 10 nm and have become a group of newly emerged fluorescent nanomaterials. Since CQDs were found during the purification of single-walled carbon nanotubes (CNTs) in 2004,1 they have been increasingly garnering the interest of researchers because of their advantages over the quantum dots of heavy metal oxides, with these advantages including their low toxicity, biocompatibility, low cost and chemical inertness. As such, CQDs are expected to be widely used in various applications including as chemical molecular and ion probes2–5 and in bioimaging,5,6 drug delivery7 and photocatalysis.8
At present, the methods used to synthesize CQDs are mainly classified into top-down and bottom-up methods. The top-down processes including laser ablation,9–11 electrochemical oxidation,12 chemical oxidation using nitric acid,13–15 and confined combustion16 are currently regarded as conventional methods to synthesize CQDs from the exfoliation of graphite oxide and activated carbon, the cleavage of graphene, or the unzipping of CNTs. However, these processes are time- and energy-consuming, and require expensive reagents or equipment. Bottom-up approaches have received more attention for preparing CQDs by dehydration, carbonization and aromatization of carbonaceous precursors (L-ascorbic acid,17 glucose18 and citric acids5). However, these CQDs typically require further surface oxidation and/or passivation to render or enhance their fluorescence and simultaneously the hydrophilicity of their surfaces.19 Thus, it is still a challenge to develop a simple method for synthesizing self-passivated CQDs.
Polyethylene glycol (PEG) is a polyhydroxy compound and has been used as a surface passivating agent in CQD syntheses. Recently, PEG has attracted much interest as a carbonaceous precursor in the synthesis of CQDs due to its non-toxicity, non-immunogenicity and reactivity with other biomolecules.20–22 So far, a few reports on the synthesis of fluorescent CQDs from PEG have appeared. PEG, as a water-soluble oligomer, shows excellent potential in improving the solubility and biocompatibility of nanoparticles.23 In 2013, Rui-Jun Fan et al.24 obtained CQDs derived from an aqueous solution of PEG 400 by carrying out a hydrothermal treatment of this solution at 120 °C. However, the photoluminescence quantum yield (PLQY) was very low, only 3.5%, and such a route involving heat operation and intricate post-processes is time-consuming. In 2014, Chen et al.25 reported a simple one-pot thermal treatment to fabricate water-soluble fluorescent CQDs from PEG at a high temperature of 160 °C. Recently, Kumar et al.26 reported that blue fluorescent CQDs could be directly prepared from PEG 400 by carrying out ultrasonic treatments at various temperatures. Although the CQDs prepared using this method showed a high PLQY, this approach requires a long ultrasonic treatment time of 2.5 h.
Microwave irradiation, by interacting with the matter in a solution, can elevate the temperature of the solution up to 1000 °C within only one minute,27 and use of such irradiation is thus energy efficient. Some efforts have made to synthesize CQDs by microwave heating of aqueous solutions of PEG or PEG and sugar in various conditions.28,29 Nevertheless, little attention was paid to the effect of the atmosphere on the formation of CQDs from PEG in these liquid reaction processes. In our current work, we discovered that oxygen could be used to accelerate the synthesis of CQDs from PEG and remarkably influenced the properties of the CQDs using PEG as a single-component precursor without subsequent passivation.
Experimental
Synthesis of CQDs
PEG with a molecular weight of 400 Da (PEG 400) was purchased from Chengdu Kelong Chemical Reagent Factory (Cheng Du, China), and was used as both a carbon source and solvent. For synthesizing CQDs, a volume of 40 mL of PEG 400 was transferred into a dried 100 mL single-necked flask and then heated in a 500 W microwave reactor (MAS-I, Sineo Microwave Chemistry Technology Co., Ltd, Shanghai, China) to 100 °C with a stirring speed of 500 rpm by using a magnetic agitator. During the synthesis, a gas (O2, N2 or CO2) was bubbled into the flask, or the flask was left open to air, in order to investigate the effects of these atmospheres on the synthesized CQDs. Finally, in each case, a golden yellow solution containing CQDs was obtained and named “CQDs-a-b, Q(A) = c mL min−1”, where a, b, A and c indicate the reaction time, the reaction temperature, the name of atmosphere involving the PEG, and the flow rate of the corresponding atmosphere, respectively. For example, “CQDs-60-100, Q(O2) = 100 mL min−1” stands for CQDs prepared from 40 mL PEG 400 by carrying out a 500 W microwave heating at 100 °C for 60 min and involving 100 mL min−1 oxygen in the reaction. When CQDs were synthesized under an open-air atmosphere, Q(A) was omitted.
Characterizations
The fluorescence of the CQDs was measured by using an LS 55 fluorescence spectrophotometer (PE Corporation, USA). The CQD dimensions were determined by using a high-resolution transmission electron microscope (HRTEM, JEOL, Japan) operating at an accelerating voltage of 200 kV. The samples for the TEM were prepared by drying 5 μL of an isopropanol solution of PEG containing CQDs on a carbon-coated copper grid (thinner pure carbon support films of 7–10 nm) in a vacuum drying oven at 80 °C for more than three days. Ultraviolet-visible (UV-vis) absorption spectra were obtained using a UV-2450 UV-vis spectrophotometer (Shimadzu Corporation, Japan). Fourier transform infrared (FTIR) spectra were acquired from a 360 FTIR spectrometer (Thermo Nicolet Corporation, USA) with wavenumbers ranging from 500 to 4000 cm−1. Time-resolved fluorescence intensity decay of the CQDs was recorded using a FM-4P-TCSPC spectrofluorometer (Horiba Jobin Yvon, France) with a laser excitation wavelength of 375 nm. 1H NMR and 13C NMR spectra of the CQDs and raw PEG were recorded in deuterated chloroform on an AVANCE III HD (Bruker Biospin, Switzerland) 600 MHz spectrometer.| |
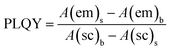 | (1-1) |
Quantum yield and fluorescence lifetime measurements
In this work, an FM-4P-TCSPC transient fluorescence spectro-meter was used to directly measure the absolute PLQY and fluorescence lifetime of the CQDs. The absolute PLQY of the CQDs was calculated using the formula. where A(em) and A(sc) are the integrated fluorescence emission intensity and integrated Rayleigh scattering intensity, respectively, and the subscripts s and b refer to sample (CQDs) and blank (solvent), respectively.
Fluorescence lifetime, defined as the characteristic period that the CQDs remained in their excited states prior to returning to their ground states, was determined by using a transient fluorescence spectrometer with a 370 nm-wavelength laser. The decay trace of the PL intensity of the CQDs was fitted using multi-exponential functions I(t) based on a non-linear least squares analysis using the equation:
| |
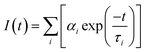 | (1-2) |
where
αi are the fractional contributions of the time-resolved decay lifetime of
τi.
Results and discussion
Effect of atmosphere on CQD synthesis
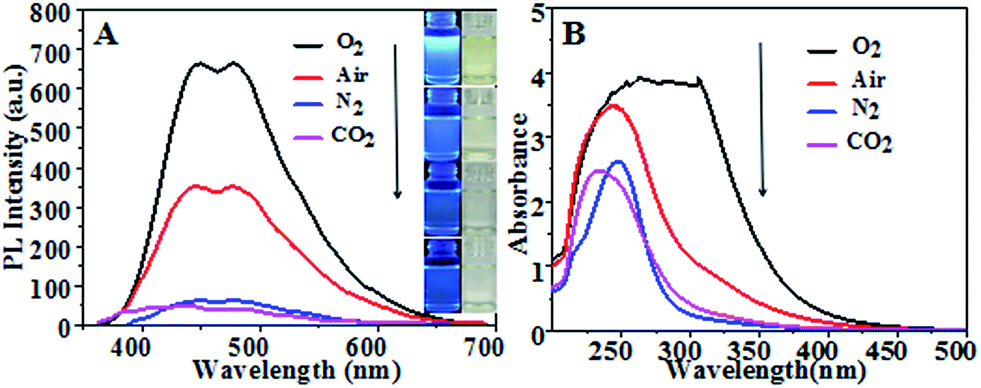
Fig. 1(A) shows fluorescence emission spectra and corresponding optical images (inset) of CQDs synthesized in different atmospheres. They consistently showed CQDs synthesized in pure oxygen and in air to have much stronger PL intensity levels than those in N2 and in CO2, with the strongest PL intensity for pure oxygen. Fig. 1(B) shows UV-vis absorption spectra of the CQDs. The CQDs synthesized in pure oxygen clearly yielded the broadest and strongest absorption band in the UV-visible region with wavelengths from 225 nm to 325 nm, indicating these CQDs to have the greatest relative number of aromatic sp2 centers.30–32 The CQDs synthesized in the inert N2 and CO2 atmospheres exhibited relatively low PL intensities and weak UV adsorption.
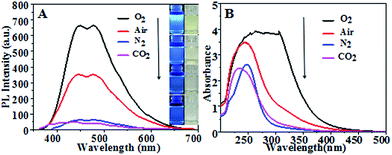 |
| | Fig. 1 (A) Fluorescence emission spectra of the various CQDs. (Insets are optical images of CQDs synthesized in various gas atmospheres under 365 nm-wavelength UV light (left) and sunlight (right)). (B) UV-vis absorption spectra of CQDs synthesized at 100 °C for 60 min in different gas atmospheres with a flow rate of 100 mL min−1. | |
Fig. 2 shows TEM images of the synthesized CQDs and their corresponding particle size distributions. The CQDs in Fig. 2(A), (C) and (D) were observed to be very well dispersed and to have a narrow size distribution, while those in Fig. 2(B), i.e., “CQDs-60-100, in air”, were poorly dispersed with a relatively broad size distribution. In addition, the “CQDs-60-100, in air” sample was observed to have a rougher surface and larger mean particle size than the other three samples, possibly resulting in the low PLQYs of these CQDs, which are shown in Table 1. To test the effect of the CQDs synthesis conditions on the CQDs fluorescence, the CQDs solutions, all at the same concentration, were excited with the same number of photons. More photons were observed to be emitted from the fluorescent CQDs having the uniform particle size distribution and good dispersibility than from the other CQDs. Moreover, the fluorescence emission of the CQDs having the broad particle size distribution with poor dispersibility was more self absorbed than the fluorescence emission of the other CQDs since the former CQDs cast a greater “shade”.
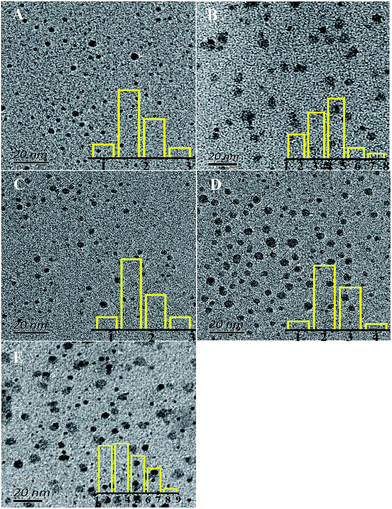 |
| | Fig. 2 TEM images of the synthesized CQDs with different reaction atmospheres: (A) CQDs-60-100, Q(O2) = 100 mL min−1, 1.8 nm; (B) CQDs-60-100, air, 3.8 nm; (C) CQDs-60-100, Q(N2) = 100 mL min−1 1.7 nm; (D) CQDs-60-100, Q(CO2) = 100 mL min−1, 2.4 nm. (E) CQDs-60-120, Q(O2) = 100 mL min−1, with the overlays being the corresponding particle size distribution histogram. | |
Table 1 The PLQY and average lifetime (t) values of the CQDs
| CQDs samples |
PLQY, % |
τ, ns |
Size, nm |
| CQDs-60-100, in air |
4.05 |
0.33 |
3.8 |
| CQDs-60-100, Q(O2) = 100 mL min−1 |
5.82 |
0.43 |
1.8 |
| CQDs-60-100, Q(N2) = 100 mL min−1 |
4.45 |
0.28 |
1.7 |
| CQDs-60-100, Q(CO2) = 100 mL min−1 |
8.83 |
<0.1 |
2.4 |
Table 1 lists the PLQY and average lifetime values of the synthesized CQDs. The inclusion of an oxygen atmosphere during the synthesis was found to increase both the PL intensity and the PLQY of the CQDs. The enhanced PLQY of the CQDs in the O2 atmosphere was inferred, based on eqn (1-1) in section 2.2, to be due to the improvement of their PL intensity. The significantly enhanced PLQY in CO2 may have been caused by an increased A(sc) (see eqn (1-1)). Using eqn (1-2) and the resulting equation τ = ∑αiτi, the average lifetimes of the CQDs were calculated from the fluorescence decay curves presented in Fig. S1.† The results showed that O2 involved in the reaction can also extend the average lifetime of CQDs from 0.33 ns to 0.43 ns (Table 1). Both the N2 and CO2 atmospheres shortened the lifetime of the synthesized CQDs. The average lifetime of “CQDs-60-100, Q(CO2) = 100 mL min−1” was in fact so short that it could not be determined using our instrument because its fluorescence decay curve (red curve) coincided with that of the silicone background (blue curve) with hardly any differences (in Fig. S1(D)†).
Effect of reaction conditions in oxygen atmosphere
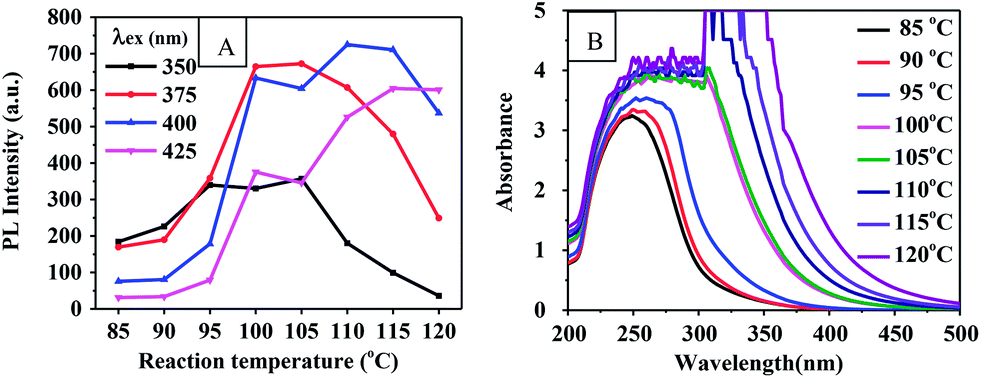
Reaction temperature. In order to study the influence of the reaction temperature on the properties of the CQDs, PEG 400 was heated using 500 W microwave irradiation to temperatures of 90 °C, 100 °C, 110 °C, or 120 °C for 60 min. Fig. 3(A) shows the effect of the reaction temperature on the PL intensities of the CQDs. The PL intensities were generally observed to increase and then decrease as the reaction temperature was increased in this range. Note that the excitation wavelength used increased with temperature. Fig. 3(B) shows the UV-vis absorption spectra at the indicated reaction temperatures. The absorption bands became broader and stronger when the reaction temperature was increased from 85 °C to 120 °C. A very strong absorption appeared at 320 nm when the temperature was increased to above 110 °C. However, the absorbance and PL intensity of the CQDs were observed to sharply increase with an increase in the reaction temperature from 95 to 100 °C and then to slowly increase from 100 to 105 °C. This result may be attributed to the rapid evaporation of the water produced from the reaction at 100 °C having shifted the reaction equilibrium advantageously through a continuous removal of the product.
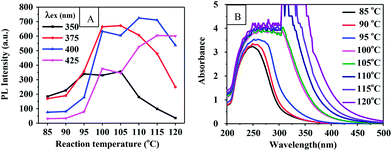 |
| | Fig. 3 (A) PL intensity-reaction temperature curve of CQDs prepared for 60 min. (B) UV-vis absorption spectrum of CQDs at corresponding reaction temperatures. | |
Optical images of these CQD solutions are shown in Fig. S2.† These solutions, when obtained at temperatures above 100 °C, were observed to be dark. Although using a higher temperature can shorten the reaction time, it may lead to an uneven size distribution of the CQD particles or excessive carbonization. This supposition was supported by the TEM image of the CQDs synthesized at 120 °C for 1 h as shown in Fig. 2(E). The reactions at temperatures below 100 °C needed to be carried out for a long time and produced CQDs with weak PL intensities. Fig. S3† shows the fluorescence emission spectra of CQDs obtained at various reaction temperatures for 60 min. The PL intensities of the synthesized CQDs increased first and then slightly decreased between excitation wavelengths of 350 and 425 nm as the reaction temperature was increased from 90 °C to 120 °C. The PL intensities of the CQDs peaked at an excitation wavelength of 375 nm when the reaction temperature was 100 °C, which indicated the optimal reaction temperature to be 100 °C. We also found that the CQDs exhibited an excitation-dependent PL behavior (Fig. S3†), as reported previously.24,29,31 With an increase in the excitation wavelength from 300 to 500 nm, the emission from the CQDs gradually shifted to a longer wavelength. Similarly, a red shift for the CQD emission occurred as the reaction temperature was increased. This red shift may have been due to the enhanced aromatization at increased reaction temperatures.
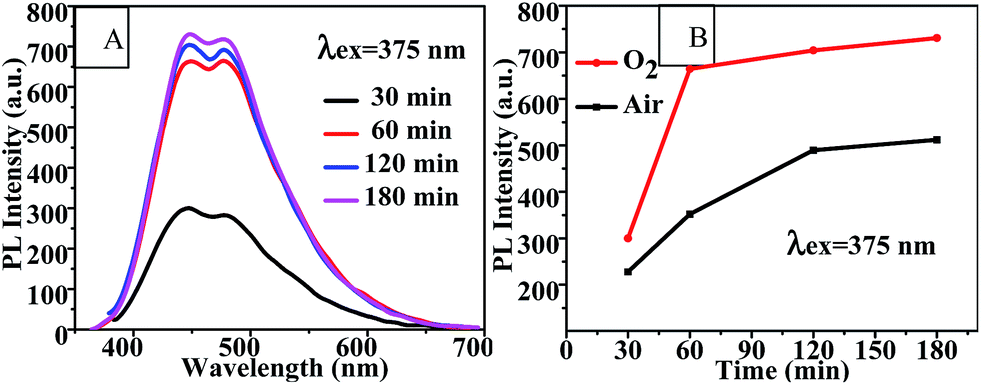
Reaction time. As shown in Fig. 4(A) and (B), the PL intensity of CQDs synthesized in an O2 flow increased as the reaction time was extended from 30 min to 60 min and then leveled off. This increased PL intensity was more significant for CQDs synthesized in the oxygen atmosphere than for that in air. This observation suggested that oxygen was involved in the CQD formation reaction. Therefore, using an oxygen atmosphere could significantly reduce the reaction time for synthesizing the CQDs with a high PL intensity. Additionally, the PL intensities of the CQDs obtained using the various reaction times peaked at an excitation wavelength of 375 nm, and also exhibited an excitation-dependent PL behavior (Fig. S4†).
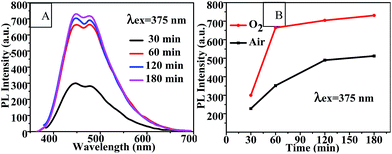 |
| | Fig. 4 (A) Fluorescence emission spectra and (B) PL intensity-reaction time curve of CQDs prepared with various different reaction times at 100 °C. | |
The PLQY and lifetime values of the CQDs synthesized in air and in oxygen for 120 min are shown in Table 2. The lifetimes of these CQDs increased from 0.33 ns and 0.43 ns, respectively, to 2.4 ns and 3.0 ns, respectively, (Table 2) as the reaction time was increased from 60 min (Table 1) to 120 min. And the PLQY increased, simultaneously with PL intensity, from 4.05% and 5.82% to 5.78% and 7.84% as the reaction time was increased from 60 to 120 min. Moreover, the PLQY and lifetime of the CQDs increased as pure oxygen was bubbled into the reaction solution.
Table 2 The PLQY and average lifetime (t) values of CQDs
| CQDs samples |
λex |
PLQY |
τ |
| CQDs-120-100, in air |
375 nm |
5.78% |
2.4 ns |
| CQDs-120-100, Q(O2) = 100 mL min−1 |
375 nm |
7.84% |
3.0 ns |
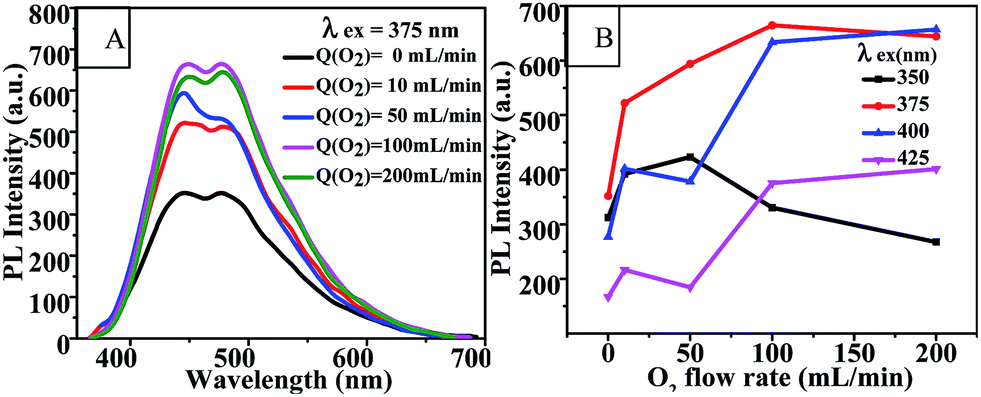
Oxygen flow rate. Fig. 5(A) and (B) show the effects of oxygen flow rate on the fluorescence emission of CQD solutions at an excitation wavelength of 375 nm and on the PL intensity of the CQDs at various excitation wavelengths. It is clear that the PL intensity of the CQDs could be enhanced by increasing the oxygen flow rate up to 100 mL min−1, and peaked at an excitation wavelength of 375 nm. This result further indicated that oxygen was involved in the synthesis reaction of CQDs. The fluorescence emission spectra of CQDs synthesized with various oxygen flow rates and with a series of excitation wavelengths are shown in Fig. S5.† Similarly, these CQDs also exhibited excitation-dependent PL behavior.
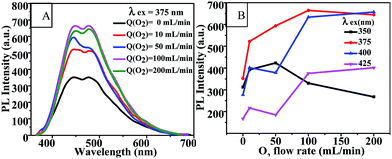 |
| | Fig. 5 (A) Fluorescence emission spectra and (B) PL intensity-O2 flow rate curves of CQDs prepared under various oxygen flow rates at 100 °C for 60 min. Q(O2) = 0 mL min−1 indicates the CQDs were synthesized in a medium open to air. | |
Chemical structure of CQDs
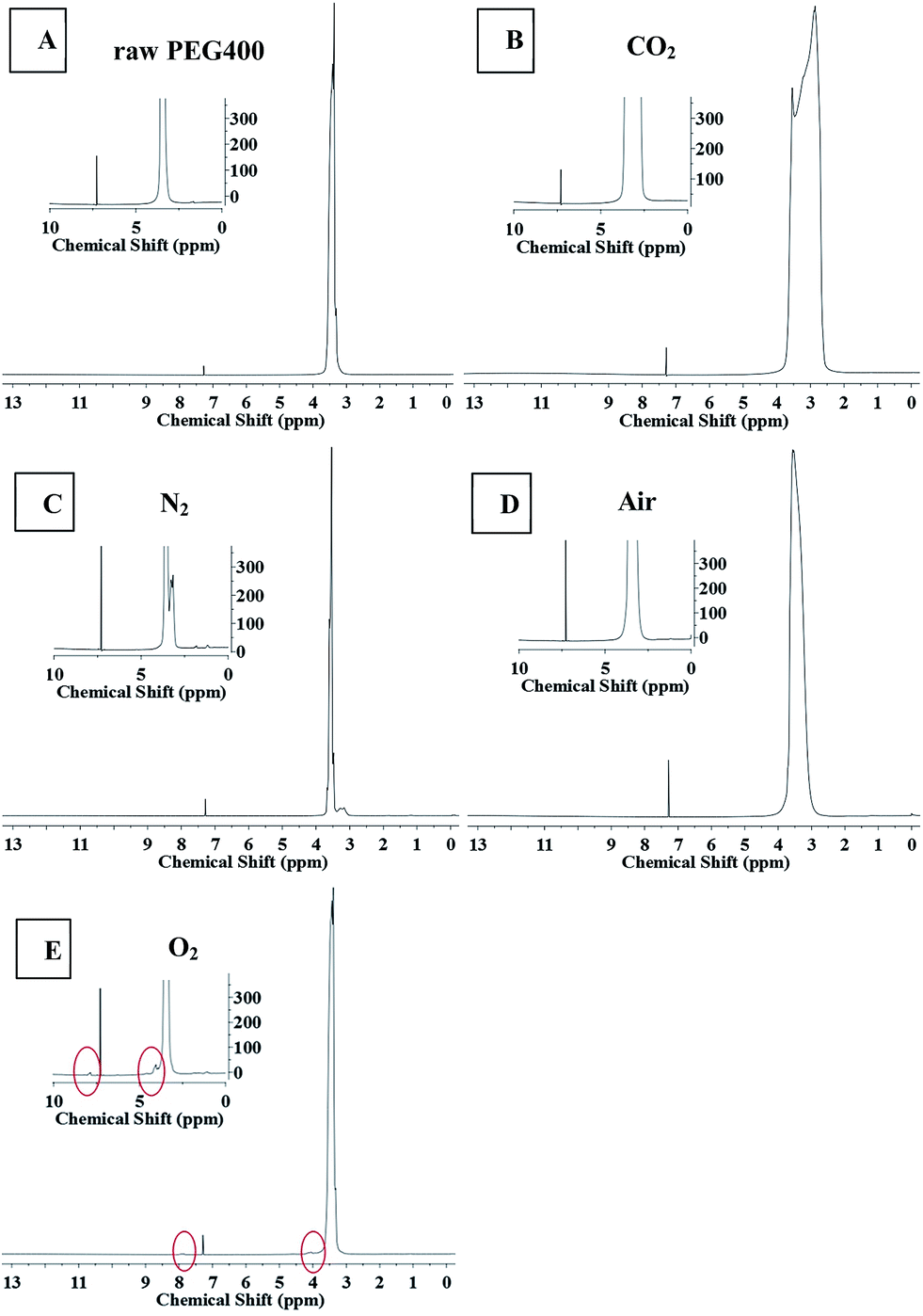
NMR spectroscopy and FTIR experiments were carried out to determine the chemical structures of the CQDs synthesized under different atmospheres, as shown in Fig. 6 and 7. Slight differences between the 1H NMR spectrum of raw PEG (Fig. 6(A)) and that of the “CQDs-60-100, Q(O2) = 100 mL min−1” sample (Fig. 6(E)) were found only in two domains, with chemical shifts of 8–7.5 ppm (assigned to hydrogen of the aromatic ring or sp2 carbon) and 4.5–4.0 ppm (assigned to the protons attached to hydroxyl, ether or carbonyl groups), respectively.33 And even less of a difference was found between the 1H NMR spectra of the CQDs synthesized in the other atmospheres. In the 1H NMR spectra of PEG and its derived CQDs, the predominant peak at the chemical shift of 3.5 ppm was attributed to –CH2–O (from PEG 400), and a weak peak at about 3 ppm (Fig. 6(B) and (C)) may be assigned to PEG fragments of various molecular weights.
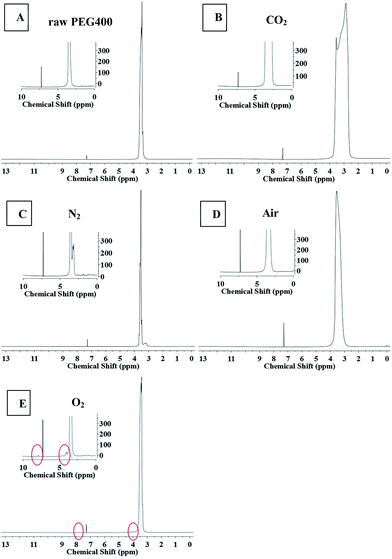 |
| | Fig. 6 The 1H NMR spectra of raw PEG and CQDs synthesized in the different atmospheres. (The illustration is a partial enlarged view.) A raw PEG 400, B CO2, C N2, D air, E O2. | |
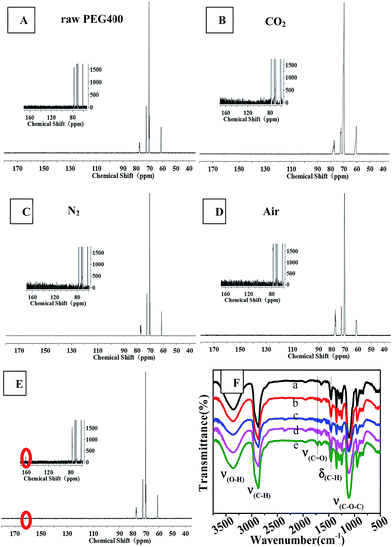 |
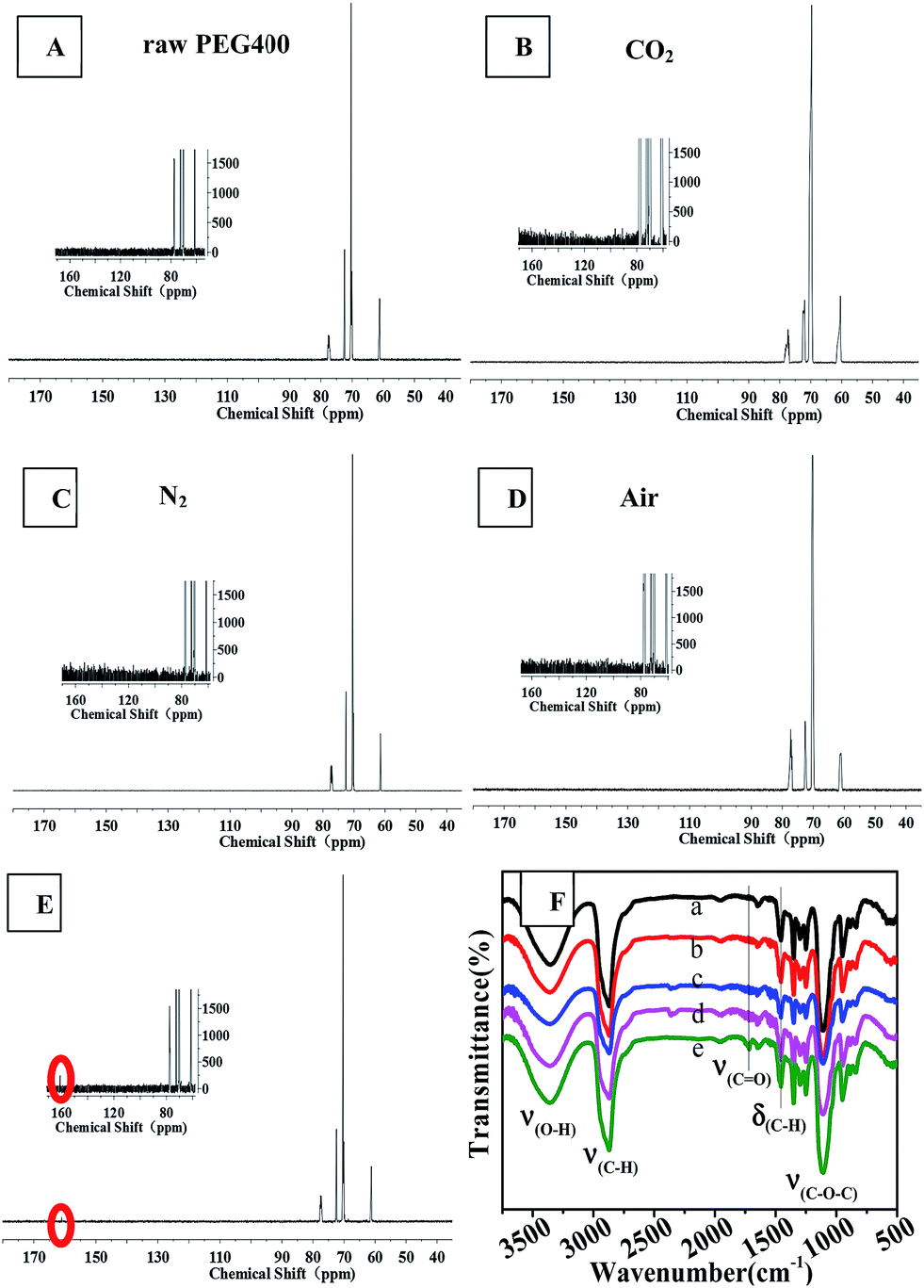
| | Fig. 7 13C NMR (from A to E, the illustration is a partial enlarged view) and FTIR (F) spectra of raw PEG and CQDs prepared in different atmospheres: F (a) raw PEG400, (b) CO2, (c) N2, (d) air, (e) O2. | |
Fig. 7 shows 13C NMR spectra and FTIR spectra. The 13C NMR spectra of the synthesized CQDs were similar and contained four main peaks at chemical shifts of 61.2 ppm, 70.2 ppm and 72 ppm, and 77.2 ppm, which were assigned to carbon atoms in the linkages of CH2–O, CH2–CH2 (from PEG 400), and the deuterated reagent CDCl3, respectively. An obvious difference between the 13C NMR spectra was observed: the CQDs synthesized in an O2 atmosphere yielded a new peak at 160 ppm, which was assigned to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C of an aromatic ether or aromatic sp2 carbons. This peak was not observed in the spectra of the CQDs synthesized in the other atmospheres. This result revealed that heating PEG in the oxygen atmosphere produced a higher degree of aromatization than did heating it in inert atmospheres such as nitrogen and carbon dioxide.
C of an aromatic ether or aromatic sp2 carbons. This peak was not observed in the spectra of the CQDs synthesized in the other atmospheres. This result revealed that heating PEG in the oxygen atmosphere produced a higher degree of aromatization than did heating it in inert atmospheres such as nitrogen and carbon dioxide.
The results obtained from FTIR observations (Fig. 7(F)) supported the above-mentioned inference that oxygen promoted the aromatization of PEG during the synthesis of CQDs. In the FTIR spectra, the bands at 3358 cm−1, 2870 cm−1 and 1400–1000 cm−1 (ref. 25) were assigned to the stretching vibrations of C–OH, C–H and C–O–C linkages, which are characteristic of PEG 400. The synthesized CQDs exhibited many weak peaks in the range of 1500–2000 cm−1, indicative of the formation of aromatic rings due to dehydration. The IR spectrum of “CQDs-60-100, Q(O2) = 100 mL min−1” showed a new, albeit weak peak at 1716 cm−1, which corresponded to the stretching vibrations of CO in the carboxylic moiety,25 and relatively sharp peaks at 1456 cm−1 and 800–900 cm−1, which corresponded to the bending vibration bands of C–H and C–H bond vibrations in aromatics.11,34,35
Mechanism of formation of the CQDs
Based on the above results, the fluorescence of the CQDs arose from the delocalized π-electron transition (π–π* transition) of the CQDs that absorbed UV-vis light with a wavelength of about 250 nm. In the microwave-assisted formation of CQDs, PEG underwent successive reactions, including dehydrogenation, polymerization, cyclization and aromatization. The general reaction pathway is shown in Scheme 1. The formed CQDs contained oxygen functional groups such as phenolic and alcoholic hydroxyl groups, cyclic ether bonds, and carboxyl groups.25,36–38 In proposed specific pathways, ether bonds of PEG first decomposed into free radicals that subsequently isomerized into alcohol hydroxyl groups,25,38 and these alcohol hydroxyls formed double bonds as a result of an elimination reaction. Here, three conjugated double bonds converted into a six-membered ring by way of the Diels–Alder reaction.37 At this time, according to this pathway, oxygen could interact with the α-C of the ether bond to form two free radicals,36 which would have facilitated the aromatization of carbon atoms.
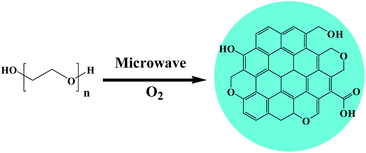 |
| | Scheme 1 Microwave-assisted synthesis of CQDs from polyethylene glycol. | |
Conclusions
Using an oxygen atmosphere in the synthesis of CQDs from PEG 400 can dramatically enhance both the PL intensity and PLQY of the CQDs, and prolong their average lifetime at the same time. The mechanism of formation of these CQDs was indicated to involve PEG successively undergoing dehydration, dehydrogenation, polymerization and aromatization reactions, leading to the formation of the CQDs. Here, including oxygen in the dehydrogenation reaction can increase the degree of conjugation of the formed CQDs and increase the strength of the PL intensity.
Acknowledgements
We are grateful for the financial support of the National Natural Science Foundation of China (Grant No. 31470600) and Top-notch Academic Programs Project of Jiangsu Higher Education Institutions (PPZY2015C221).
Notes and references
- X. Xu, R. Ray, Y. Gu, H. J. Ploehn, L. Gearheart, K. Raker and W. A. Scrivens, J. Am. Chem. Soc., 2004, 126, 12736–12737 CrossRef CAS PubMed.
- A. Cayuela, M. L. Soriano and M. Valcárcel, Anal. Chim. Acta, 2013, 804C, 246–251 CrossRef PubMed.
- F. Yan, Y. Zou, M. Wang, X. Mu, N. Yang and L. Chen, Sens. Actuators, B, 2014, 192, 488–495 CrossRef CAS.
- L. Q. Liu, Y. F. Li, L. Zhan, Y. Liu and C. Z. Huang, Sci. China: Chem., 2011, 54, 1342–1347 CrossRef CAS.
- M. Zhou, Z. Zhou, A. Gong, Y. Zhang and Q. Li, Talanta, 2015, 143, 107–113 CrossRef CAS PubMed.
- S. Chandra, P. Das, S. Bag, D. Laha and P. Pramanik, Nanoscale, 2011, 3, 1533–1540 RSC.
- W. Zonghua, X. Jianfei, Z. Chengfeng, V. Brian, X. Yanzhi, Z. Feifei, L. Yanhui, X. Linhua and T. Jie, Colloids Surf., B, 2013, 112C, 192–196 Search PubMed.
- S. Zhuo, M. Shao and S. T. Lee, ACS Nano, 2016, 6, 1059–1064 CrossRef PubMed.
- Y. P. Sun, B. Zhou, Y. Lin, W. Wang, K. A. S. Fernando, P. Pathak, M. J. Meziani, B. A. Harruff, X. Wang and H. Wang, J. Am. Chem. Soc., 2006, 128, 7756–7757 CrossRef CAS PubMed.
- S. L. Hu, K. Y. Niu, J. Sun, J. Yang, N. Q. Zhao and X. W. Du, J. Mater. Chem., 2009, 19, 484–488 RSC.
- J. Wang, C. F. Wang and S. Chen, Angew. Chem., 2012, 51, 9297–9301 CrossRef CAS PubMed.
- Q. L. Zhao, Z. L. Zhang, B. H. Huang, J. Peng, M. Zhang and D. W. Pang, Chem. Commun., 2008, 41, 5116–5118 RSC.
- H. Liu, T. Ye and C. Mao, Angew. Chem., Int. Ed., 2007, 46, 6473–6475 CrossRef CAS PubMed.
- L. Tian, D. Ghosh, W. Chen, S. Pradhan, X. Chang and S. Chen, Chem. Mater., 2009, 21, 2803–2809 CrossRef CAS.
- M. J. Krysmann, A. Kelarakis and E. P. Giannelis, Green Chem., 2012, 14, 3141–3145 RSC.
- A. Rahy, C. Zhou, J. Zheng, S. Y. Park, M. J. Kim, I. Jang, S. J. Cho and D. J. Yang, Carbon, 2012, 50, 1298–1302 CrossRef CAS.
- B. Zhang, C. Y. Liu and Y. Liu, Ber. Dtsch. Chem. Ges., 2010, 2010, 4411–4414 Search PubMed.
- Z. Ma, H. Ming, H. Huang, Y. Liu and Z. Kang, New J. Chem., 2012, 36, 861–864 RSC.
- C. Fowley, B. Mccaughan, A. Devlin, I. Yildiz, F. M. Raymo and J. F. Callan, Chem. Commun., 2012, 48, 9361–9363 RSC.
- F. M. Veronese and G. Pasut, Drug Discovery Today, 2005, 10, 1451–1458 CrossRef CAS PubMed.
- A. S. Karakoti, S. Das, S. Thevuthasan and S. Seal, Angew. Chem., 2011, 50, 1980–1994 CrossRef CAS PubMed.
- V. Milosavljevic, H. V. Nguyen, P. Michalek, A. Moulick, P. Kopel, R. Kizek and V. Adam, Chem. Pap., 2014, 69, 192–201 Search PubMed.
- R. Liu, D. Wu, S. Liu, K. Koynov, W. Knoll and Q. Li, Angew. Chem., Int. Ed., 2009, 48, 4598–4601 CrossRef CAS PubMed.
- R. J. Fan, Q. Sun, L. Zhang, Y. Zhang and A. H. Lu, Carbon, 2014, 71, 87–93 CrossRef CAS.
- M. Chen, W. Wang and X. Wu, J. Mater. Chem. B, 2014, 2, 3937–3945 RSC.
- V. B. Kumar, Z. Porat and A. Gedanken, Ultrason. Sonochem., 2016, 28, 367–375 CrossRef CAS PubMed.
- K. Mackenzie, O. Dunens and A. T. Harris, Sep. Purif. Technol., 2009, 66, 209–222 CrossRef CAS.
- H. Zhu, X. Wang, Y. Li, Z. Wang, F. Yang and X. Yang, Chem. Commun., 2009, 34, 5118–5120 RSC.
- A. Jaiswal, S. S. Ghosh and A. Chattopadhyay, Chem. Commun., 2012, 48, 407–409 RSC.
- X. Sui and Z. Wang, Polym. Adv. Technol., 2013, 24, 593–599 CrossRef CAS.
- S. Yang, J. Sun, C. Zhu, P. He, Z. Peng and G. Ding, Analyst, 2015, 141, 1052–1059 RSC.
- D. Pan, J. Zhang, Z. Li, C. Wu, X. Yan and M. Wu, Chem. Commun., 2010, 46, 3681–3683 RSC.
- B. De and N. Karak, RSC Adv., 2013, 3, 8286–8290 RSC.
- S. Zhu, J. Zhang, L. Wang, Y. Song, G. Zhang, H. Wang and B. Yang, Chem. Commun., 2012, 48, 10889–10891 RSC.
- Z. C. Yang, X. Li and J. Wang, Carbon, 2011, 49, 5207–5212 CrossRef CAS.
- Z. Diao, B. Li, B. Yu, A. Liu and L. He, J. Energy Chem., 2013, 22, 363–367 CrossRef CAS.
- B. M. Jacobson, G. M. Arvanitis, C. A. Eliasen and R. Mitelman, J. Org. Chem., 1985, 50, 194–201 CrossRef CAS.
- J. N. Hemenway, T. C. Carvalho, V. M. Rao, Y. Wu, J. K. Levons, A. S. Narang, S. R. Paruchuri, H. J. Stamato and S. A. Varia, J. Pharm. Sci., 2012, 101, 3305–3318 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra01804e |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Songlin Zuo
,
Songlin Zuo