
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProgrammed cell death 4 inhibits proliferation and differentiation and induces apoptosis of human mesenchymal stem cells through suppressing the Wnt/β-catenin pathway
Yang Liu†
a,
Dongyue Su†a and
Tao Song *b
*b
aDepartment of Endocrinology, Huaihe Hospital of Henan University, Kaifeng, 475000, China
bDepartment of Orthopaedics, The People's Liberation Army 155 Hospital, No.3 Prior Street of Hospital, Kaifeng, 475000, China. E-mail: doysong@163.com; Tel: +86-0371-22258520
First published on 17th May 2017
Abstract
Background: Programmed cell death 4 (PDCD4) has been shown to act as a tumor suppressor involved in several cancers. However, the function and possible molecular mechanisms of PDCD4 in human mesenchymal stem cells (hMSCs) are still unclear. Methods: The expression of PDCD4 mRNA were detected by quantitative real-time PCR (qRT-PCR) in N-hMSCs and OP-hMSCs. Western blot assays were used to examine the protein levels of PDCD4, osteogenic markers Runx2 and Osterix, and Wnt/β-catenin pathway related proteins β-catenin, C-myc, CyclinD1 and Wnt1, Lgr5 and Axin2. CCK-8 assay and flow cytometry analysis were performed to determine the proliferation and apoptosis of N-hMSCs and OP-hMSCs, respectively. Results: PDCD4 was expressed more highly in OP-hMSCs than N-hMSCs. The proliferation was decreased, and apoptosis was increased in OP-hMSCs compared with N-hMSCs. PDCD4 knockdown promoted proliferation and differentiation and suppressed apoptosis of OP-hMSCs, while PDCD4 overexpression in N-hMSCs showed the reverse effect. Moreover, suppression of PDCD4 activated the Wnt/β-catenin pathway in OP-hMSCs, whereas overexpression of PDCD4 blocked the Wnt/β-catenin pathway in N-hMSCs. Furthermore, Wnt/β-catenin pathway inhibitor, XAV939, inhibited proliferation and differentiation and induced apoptosis of OP-hMSCs and N-hMSCs. Finally, XAV939 reversed the enhanced proliferation and differentiation and suppressed apoptosis of OP-hMSCs mediated by PDCD4 knockdown. Conclusion: PDCD4 inhibits proliferation and differentiation and induces apoptosis of hMSCs by repressing the Wnt/β-catenin pathway. The present study elucidates a novel PDCD4-Wnt/β-catenin regulatory pathway in osteoporosis and provides a promising therapeutic target for osteoporosis.
1. Introduction
Osteoporosis, a metabolic skeletal disorder, is characterized by low bone mass and deteriorated bone structure and is a common health problem affecting the aging population.1 The imbalance between bone resorption and bone formation is generally considered to cause osteoporosis.2 Additionally, dysfunction of bone marrow-derived mesenchymal stem cells (BM-MSCs) in mediating bone formation has been regarded as a major contributor to the progression of osteoporosis.3 Therefore, the molecular mechanism underlying MSC differentiation may provide more effective therapeutic targets for osteoporosis.MSCs are pluripotent stem cells, which are characterized by being able to differentiate into chondrocytes, adipocytes and osteocytes as well as other mesodermal and non-mesodermal cell types such as endoderm-like and neural cells.4,5 MSCs possess the capacity of releasing immunosuppressive cytokines,6 directed migration7 and self-renew.8 Due to their multipotency and paracrine effect,9,10 MSCs have been found to be a useful cell-based therapeutic schedule for regenerating tissue in some pathological conditions including neurodegenerative disorders,11 myocardial infarction12 and bone loss.13 BM-MSCs can eventually generate osteocytes and cartilage cells, crucial for maintenance of bone metabolism balance under physiological and pathological conditions.14
Wnt family is comprised of at least 19 conventional secreted glycoproteins and plays important roles during development and throughout life.15 The typical Wnt/β-catenin signaling pathway is initiated through Wnt binding to frizzled receptor to recruit LRP5/6, leading to the release of cytoplasmic β-catenin from a protein complex comprising casein kinase 1 (CK1), APC, Axin1/2, and glycogen synthase kinase 3b (GSK3b).16 Then, β-catenin translocates into the nucleus, interacts with transcription factor 4 (TCF-4) or lymphoid enhancer-binding factor 1 (LEF-1) and activates the expression of target genes essential for cell proliferation and differentiation.17,18 Wnt signaling pathway is primary associated with the osteogenesis. Wnt3a and 10a could activate classic Wnt signaling pathway while Wnt4a and 5a activate non-classic Wnt signaling pathway, both of which may promote the differentiation of BM-MSCs and the bone formation.19 Moreover, a previous study suggest that flavonoids might induce osteogenic differentiation of BM-MSCs through the activation of Wnt/β-catenin signaling.20 Although numerous mechanistic studies have reported that many signaling pathways take part in the regulation of osteogenesis, such as the MAPK, BMP, Wnt, and NF-κB signaling,21 the precise mechanisms involved in the proliferation and differentiation of BM-MSCs into osteoblasts are largely unknown.
The programmed cell death 4 (PDCD4), containing nuclear localization signals and two MA-3 domains associated with protein–protein interactions in eIF4GI and eIF4GII with the ATP-dependent RNA helicase eIF4A proposing the repression of translation initiation, is identified as a novel tumor suppressor.22–25 The aberrant expressions of PDCD4 were observed in several cancers, such as pregnancy-associated breast cancer,26 pancreatic cancer,27 and primary lung cancer.28 In addition, PDCD4 plays significant roles in tumor promotion, progression, differentiation and metastasis.29–31 However, the function of PDCD4 in MSCs differentiation is still unclear.
In the present study, we aimed to investigate the function of PDCD4 in MSCs proliferation and differentiation and its underlying molecular mechanisms.
2. Materials and methods
2.1. Cell culture
The normal human marrow mesenchymal stem cells (N-hMSCs) and osteoporosis-derived human marrow mesenchymal stem cells (OP-hMSCs) were obtained from Cambrex (Walkersville, MD, USA). All cells were cultured in Dulbecco's modified Eagle's medium (DMEM; Gibco, Grand Island, NY, USA) supplemented with 10% fetal bovine serum (Gibco), 4 mM L-glutamine and 1% penicillin/streptomycin (Invitrogen, Carlsbad, CA, USA) at 37 °C in an atmosphere of 95% air and 5% CO2.2.2. Quantitative real-time PCR (qRT-PCR)
Total RNA was extracted by Tirol reagent (Invitrogen). The High Capacity cDNA Archive Kit (Applied Biosystems, Foster City, CA, USA) was used for mRNA reverse transcription. qRT-PCR was conducted using SYBR Premix kit (Takara Bio, Madison, WI, USA) with an ABI7300 real-time PCR machine (Life Technologies, Gaithersburg, MD, USA). β-Actin was used as endogenous controls. The relative expression of PDCD4 mRNA was calculated by normalizing to β-actin mRNA expression using the ΔΔCt method.2.3. Western blot analysis
Western blot analysis was performed as previously described.32 Briefly, cells were cultured for 48 hours and total proteins were isolated using RIPA buffer. Extracted proteins were separated by SDS-PAGE, transferred to nitrocellulose membranes (Bio-Rad, Hercules, CA, USA), and incubated with first antibodies overnight. After washing with TBST, the membranes were incubated with secondary antibodies for 2 h. Antibodies against PDCD4, Runx2, Osterix, β-catenin, C-myc, CyclinD1, Wnt1, Lgr5 and Axin2 and β-actin were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). β-Actin was used as an endogenous control. The proteins were visualized using the Image Quant software (Molecular Dynamics, Sunnyvale, CA, USA).2.4. Cell transfection
PDCD4 cDNAs were amplified and inserted into pcDNA3.1 vectors to construct pcDNA-PDCD4. Si-control and si-PDCD4 were obtained from Dharmacon (Lafayette, CO, USA). Cell transfection was performed by using Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol.2.5. Cell proliferation assay
N-hMSCs and OP-hMSCs were seeded into the 96-well plate and cultured for 24 h, 48 h and 72 h, respectively. Cell Counting Kit-8 (Beyotime, Jiangsu, China) was used to detect the cell viability according to the manufacturer's instructions. The absorbance at 450 nm was then tested by a microtiter plate reader (Molecular Devices, Sunnyvale, CA, USA).2.6. Cell apoptosis assay
To detect cell apoptosis, the N-hMSCs and OP-shMSCs were harvested by trypsinization 48 h post transfection and resuspended in PBS containing 4% fetal bovine serum. After washing, cells were stained using V-FITC/PI kit (BD PharMingen, San Diego, CA, USA) according to the manufacturer's instructions. Cells were subsequently analyzed using flow cytometry (BD Biosciences, San Jose, CA, USA). The data were analyzed by the BD FACS Diva software V6.1.3 (BD Biosciences).2.7. Statistical analysis
Statistical analyses were performed by SPSS 19.0 software (SPSS, Chicago, IL, USA). The data were presented as means ± SD. Student's t-test or one-way ANOVA was performed to evaluate the statistical significance. P < 0.05 was considered statistically significant.3. Results
3.1. PDCD4 expression, cell proliferation and apoptosis are different in OP-hMSCs and N-hMSCs
In order to observe the function of PDCD4 in osteoporosis, mRNA and protein expression levels of PDCD4 in OP-hMSCs and N-hMSCs were measured by qRT-PCR and western blot, respectively. Compared with N-hMSCs, the levels of PDCD4 mRNA (Fig. 1A) and protein (Fig. 1B) were significantly increased in OP-hMSCs. Then we performed proliferation and apoptosis assay in OP-hMSCs and N-hMSCs. As expected, CCK-8 assay showed that cell viability of OP-hMSCs was significantly reduced compared with N-hMSCs (Fig. 1C). Further, flow cytometry analysis demonstrated that apoptosis of OP-hMSCs was significantly increased compared with N-hMSCs (Fig. 1D). Thus, we speculated that the aberrant expression of PDCD4 may be closely related to the proliferation and the apoptosis of OP-hMSCs in osteoporosis.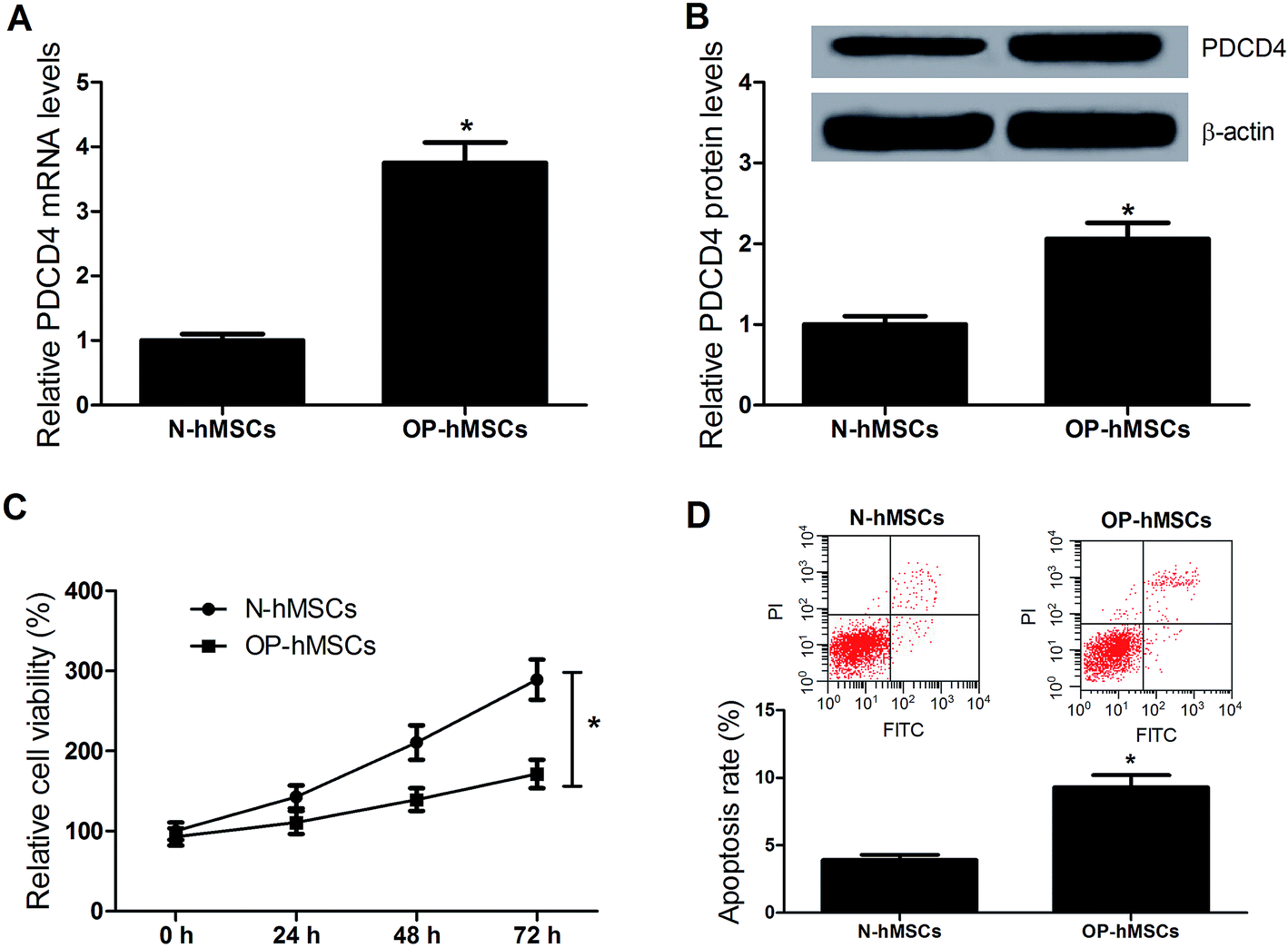 | ||
| Fig. 1 Expression of PDCD4 and cell proliferation and apoptosis in N-hMSCs and OP-hMSCs. (A) The expression levels of PDCD4 were detected by qRT-PCR analysis in N-hMSCs and OP-hMSCs. (B) The protein level of PDCD4 was examined by western blot in N-hMSCs and OP-hMSCs. (C) CCK-8 assay was performed to detect the cell viability of N-hMSCs and OP-hMSCs. (D) Cell apoptosis was detected and calculated by flow cytometry analysis after N-hMSCs and OP-hMSCs were cultured for 48 h. *P < 0.05 vs. N-hMSCs. | ||
3.2. PDCD4 inhibits proliferation and differentiation and induced apoptosis of OP-hMSCs and N-hMSCs
In consideration of the high expression of PDCD4 in OP-hMSCs, pcDNA-PDCD4 was transfected into N-hMSCs and si-PDCD4 was transfected into OP-hMSCs, respectively. As shown in Fig. 2A, the protein level of PDCD4 was successfully overexpressed in N-hMSCs and knocked down in OP-hMSCs. Then, CCK-8 assay was performed to detect the cell viability of different hMSCs at indicated times. The results showed that the cell viability of PDCD4 overexpressing N-hMSCs was reduced 1.44 times at 72 h (Fig. 2B), while the cell viability of OP-hMSCs with PDCD4 knockdown was increased 1.65 times (Fig. 2C) compared with the controls. Moreover, to examine the effect of PDCD4 on the apoptosis of hMSCs, flow cytometry analysis was carried out in N-hMSCs transfected with pcDNA-PDCD4 or OP-hMSCs transfected with si-PDCD4 48 h post transfection. Compared with the control groups, apoptosis rate of N-hMSCs was significantly elevated in pcDNA-PDCD4 group (Fig. 2D), conversely, apoptosis rate of OP-hMSCs was notably decreased in si-PDCD4 group (Fig. 2D). Further, osteogenic markers Runx2 and Osterix was examined by western blot at 48 h after N-hMSCs were transfected with pcDNA-PDCD4 or OP-hMSCs were transfected with si-PDCD4. Results showed that PDCD4 overexpression in N-hMSCs downregulated Runx2 and Osterix (Fig. 2E and F), while si-PDCD4 mediated knockdown in OP-hMSCs upregulated Runx2 and Osterix (Fig. 2G and H). Taken together, these results suggested that PDCD4 hindered proliferation and differentiation and induced apoptosis of hMSCs.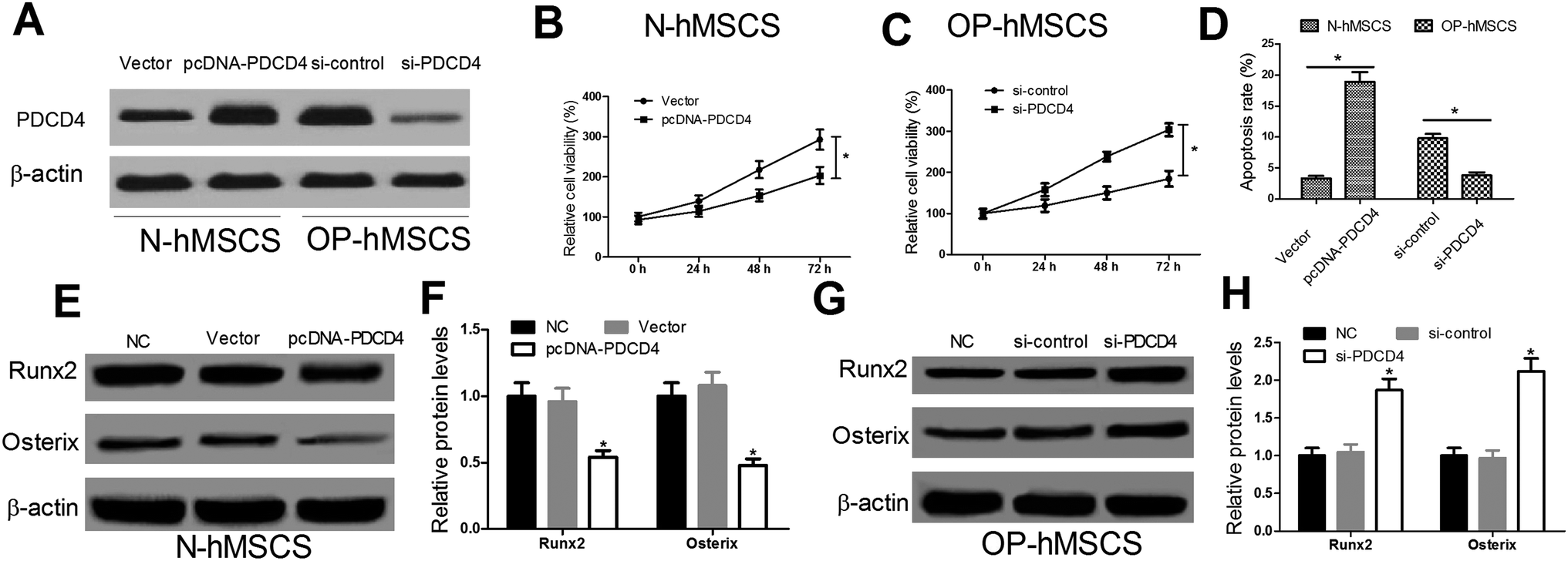 | ||
| Fig. 2 Effect of PDCD4 overexpression in N-hMSCs or PDCD4 knockdown in OP-hMSCs on the cell proliferation, differentiation and apoptosis. (A) Western blot analysis was conducted to measure the protein level of PDCD4 after N-hMSCs were transfected with pcDNA-PDCD4 or OP-hMSCs were transfected with si-PDCD4 for 24 h. (B and C) CCK-8 assay was performed to detect cell viability after N-hMSCs were transfected with pcDNA-PDCD4 or OP-hMSCs were transfected with si-PDCD4 for 24 h, 48 h and 72 h. (D) Cell apoptosis was determined by flow cytometry analysis in N-hMSCs transfected with pcDNA-PDCD4 or OP-hMSCs transfected with si-PDCD4 at 48 h. The osteogenic markers Runx2 and Osterix were examined by western blot after N-hMSCs were transfected with pcDNA-PDCD4 (E and F) or OP-hMSCs were transfected with si-PDCD4 (G and H) for 48 h. *P < 0.05 vs. controls. | ||
3.3. PDCD4 inactivated the Wnt/β-catenin pathway in hMSCs
To explore the effect of PDCD4 on the Wnt/β-catenin pathway in hMSCs, western blot was performed to examine the expressions of Wnt/β-catenin pathway related proteins β-catenin, C-myc and CyclinD1 at 48 h in pcDNA-PDCD4 transfected N-hMSCs or si-PDCD4 transfected OP-hMSCs. Western blot showed that PDCD4 upregulation in N-hMSCs evidently reduced the β-catenin, C-myc and CyclinD1 levels (Fig. 3A and C), while PDCD4 suppression in OP-hMSCs distinctly enhanced their expression (Fig. 3D and F). Wnt target genes such as Wnt1, Lgr5 and Axin2 have provided evidence for Wnts as critical stem cell signals. Thus, the expression of Wnt1, Lgr5 and Axin2 was further detected by western blot analysis. As expected, the protein levels of Wnt1 and Lgr5 were decreased and Axin2 was elevated in pcDNA-PDCD4 transfected N-hMSCs (Fig. 3B and C). On the contrary, si-PDCD4 inhibited Wnt1 and Lgr5 expression and promoted Axin2 expression in OP-hMSCs (Fig. 3E and F). These results revealed that PDCD4 repressed the Wnt/β-catenin pathway in hMSCs.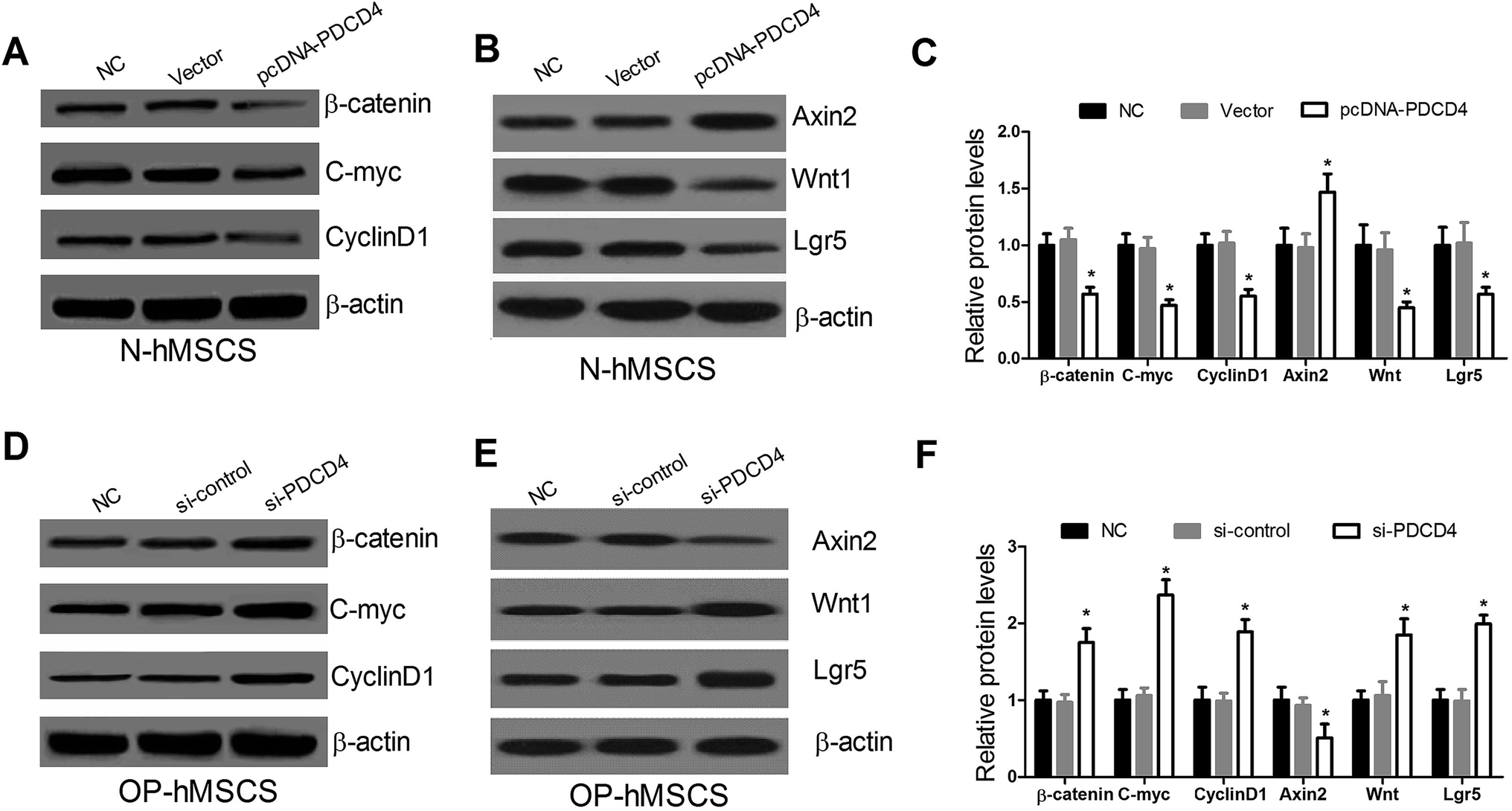 | ||
| Fig. 3 PDCD4 inhibits the Wnt/β-catenin pathway in N-hMSCs and OP-hMSCs. Western blot was performed to detect the expression of the β-catenin, C-myc, CyclinD1, Wnt1, Lgr5 and Axin2 after N-hMSCs were transfected with pcDNA-PDCD4 (A–C) or OP-hMSCs were transfected with si-PDCD4 (D–F) for 48 hours. *P < 0.05 vs. controls. | ||
3.4. Inactivation of Wnt/β-catenin pathway suppresses proliferation and differentiation and induces the apoptosis of hMSCs
To explore the effect of Wnt/β-catenin pathway on the proliferation, differentiation and apoptosis of hMSCs, CCK-8 assay, western blot and flow cytometry analysis were performed after N-hMSCs and OP-hMSCs were treated with 10 μM or 20 μM XAV939, an inhibitor of Wnt/β-catenin pathway. CCK-8 assay showed that XAV939 dramatically impaired cell growth of N-hMSCs and OP-hMSCs (Fig. 4A and B). Similarly, XAV939 notably blocked the expression of osteogenic markers Runx2 and Osterix in N-hMSCs (Fig. 4C and D) and OP-hMSCs (Fig. 4E and F). Moreover, XAV939 drastically induced apoptosis of N-hMSCs and OP-hMSCs (Fig. 4G and H). Taken together, these results demonstrated that XAV939 impeded the proliferation and differentiation and induced the apoptosis of hMSCs.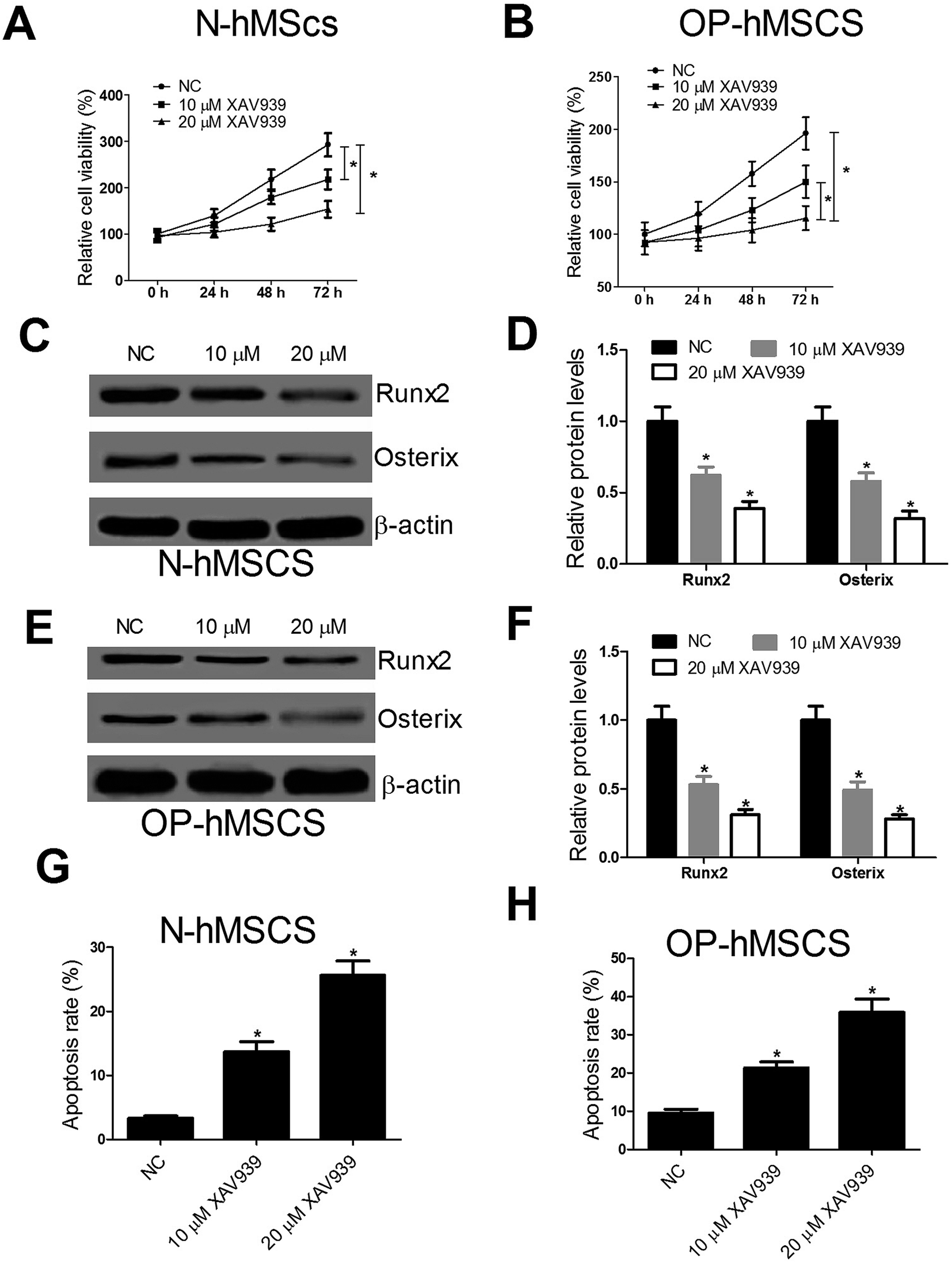 | ||
| Fig. 4 XAV939 suppresses proliferation and differentiation and promotes apoptosis of N-hMSCs and OP-hMSCs. (A and B) CCK-8 assay was performed to detect cell viability after N-hMSCs and OP-hMSCs were treated with 10 μM or 20 μM XAV939. The Runx2 and Osterix expressions were examined by western blot after N-hMSCs (C and D) and OP-hMSCs (E and F) were treated with 10 μM or 20 μM XAV939 for 48 h. (G and H) Flow cytometry analysis was used to determine cell apoptosis after N-hMSCs and OP-hMSCs were treated with 10 μM or 20 μM XAV939 for 48 h. *P < 0.05 vs. controls. | ||
3.5. XAV939 reverses the effect of PDCD4 knockdown on proliferation, differentiation and apoptosis of OP-hMSCs
To further confirm whether PDCD4 knockdown promoted the proliferation and differentiation and suppressed the apoptosis of OP-hMSCs through Wnt/β-catenin pathway, CCK-8 assay, western blot and flow cytometry analysis were performed after OP-hMSCs were treated with si-PDCD4 or si-PDCD4 + XAV939 (10 μM). The results indicated that XAV939 reversed the increased cell viability (Fig. 5A), reduced apoptosis rate (Fig. 5B), the elevated expression of Runx2 and Osterix and the reduced Axin2 level (Fig. 5C and D) in OP-hMSCs triggered by PDCD4 knockdown. All these data illuminated that PDCD4 suppressed proliferation and differentiation and induced apoptosis of OP-hMSCs through inhibiting Wnt/β-catenin pathway.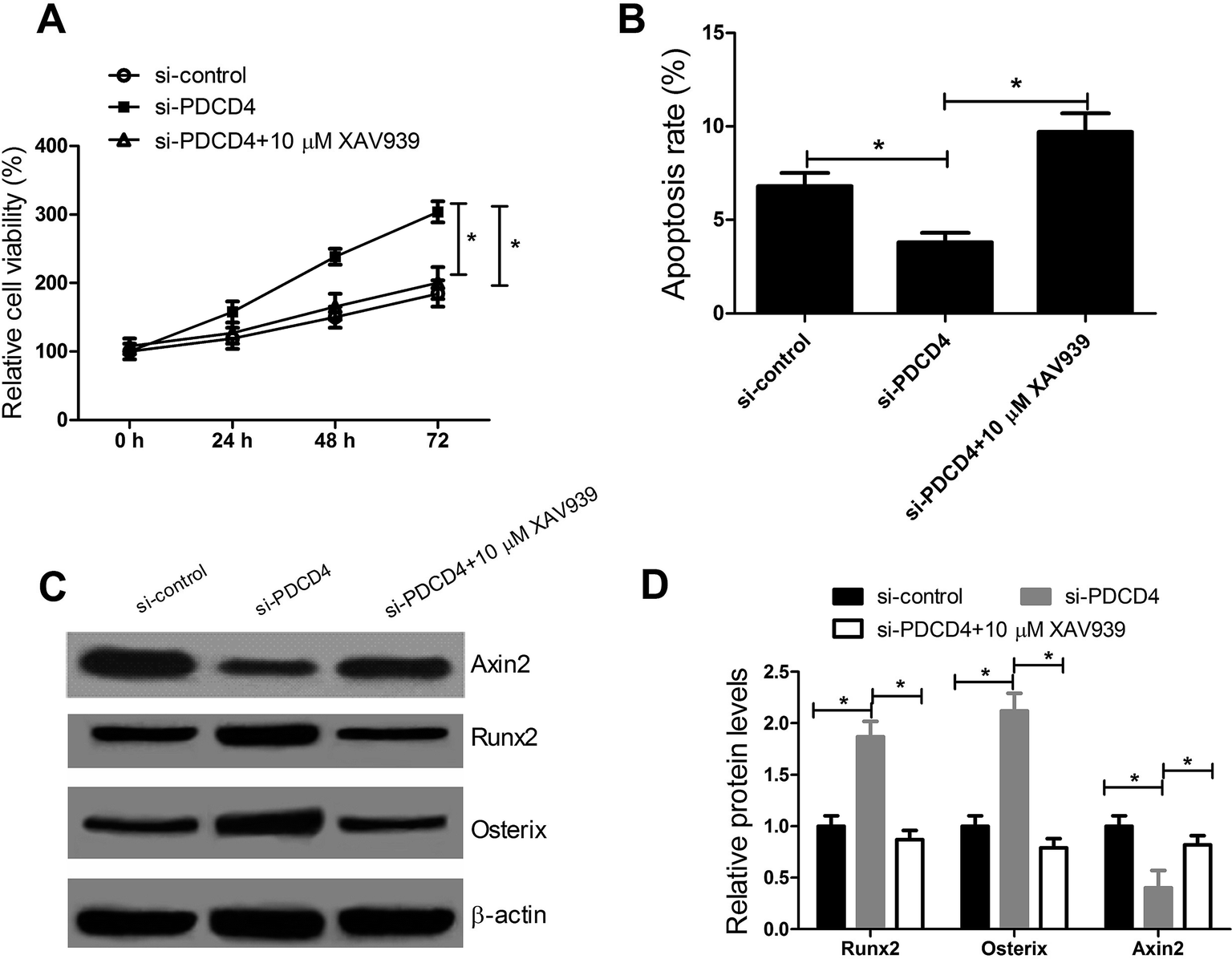 | ||
| Fig. 5 PDCD4 inhibits proliferation and differentiation and induces apoptosis of OP-hMSCs via inactivation of Wnt/β-catenin pathway. (A) CCK-8 assay was applied to test cell viability after OP-hMSCs were treated with si-PDCD4 or si-PDCD4 + XAV939 (10 μM). (B) Cell apoptosis was detected and calculated by flow cytometry analysis after OP-hMSCs were treated with si-PDCD4 or si-PDCD4 + XAV939 (10 μM) for 48 h. (C and D) Western blot was performed to measure the Runx2, Osterix and Axin2 expressions after OP-hMSCs were treated with si-P DCD4 or si-PDCD4 + XAV939 (10 μM) for 48 h. *P < 0.05 vs. controls. | ||
4. Discussion
Based on their multi-potent and undifferentiated character, MSCs may be a powerful cell-based therapeutic option for tissue regeneration in some pathological conditions. In the present study, we found that PDCD4 inhibits proliferation and differentiation and induces apoptosis of MSCs through blocking Wnt/β-catenin pathway, which may provide a promising therapeutic target for osteoporosis.PDCD4, as a tumor suppressor, frequently exhibits downregulated expression and exerts different functions in many types of cancers. For example, PDCD4 inhibited transcriptional activity of NF-κB and then suppressed the expression of invasion related NF-κB target genes by direct interaction with p65 in glioblastoma cells.33 PDCD4 directly interacted with the DNA binding domain of transcription factor Twist1 to consequently down-regulate proliferation related Twist1 target gene Y-box binding protein-1.34 Moreover, PDCD4 suppressed colon carcinoma cell invasion by down-regulating the transcription of mitogen-activated protein kinase 1 (MAP4K1).35 A previous study suggested that PDCD4 overexpression induced apoptosis of breast cancer cells via a caspase-dependent mechanism.36 Ectopic expression of PDCD4 significantly inhibited ovarian cancer cell proliferation by up-regulating the cell cycle regulators p27 and p21.37 Consistent with these findings, our study revealed that PDCD4 was significantly upregulated in OP-hMSCs compared with N-hMSCs. Moreover, PDCD4 knockdown improved the proliferation and differentiation and suppressed apoptosis of OP-hMSCs, while PDCD4 overexpression repressed the proliferation and differentiation and induced apoptosis of N-hMSCs. These results clarified that PDCD4 inhibited proliferation and differentiation and promoted apoptosis of hMSCs, providing a potential therapy target for patients with osteoporosis. All these studies together with our findings support that PDCD4 play important role in the proliferation and apoptosis of cells. Besides, the apoptosis rate in OP-hMSCs is higher than N-hMSCs, which was mainly duo to the osteoporosis-derived in OP-hMSCs.38
Wnt/β-catenin signaling is a well-known signaling pathway involved in osteogenesis.38,39 It has been reported that Wnt signaling could increase osteogenesis by regulating RUNX2 expression.18 Noncanonical Wnt pathway is also involved in the osteogenic differentiation of MSCs through the Wnt/calcium.19 A previous study demonstrated that Wnt antagonists, such as Wnt inhibitory factor 1 (WIF-1) and Sclerostin, could interact with the Wnt proteins or Wnt receptors, inhibiting Wnt activity and differentiation and maturation of osteoblasts.40,41 Moreover, Berberine was demonstrated to promote osteogenic differentiation of BM-MSCs via canonical Wnt/β-catenin signaling pathway.42 Furthermore, a previous study confirmed that Osterix-expressing cells produced their own Wnts, which in turn activated Wnt signaling response, thereby regulating their proliferation and differentiation.43 However, how Wnt/β-catenin pathway was regulated in MSCs have not been well elucidated. In the present study, enforced expression of PDCD4 blocked the expressions of Wnt/β-catenin pathway related proteins β-catenin, C-myc, CyclinD1, Wnt1 and Lgr5 in N-hMSCs, while si-PDCD4-mediated silencing showed a contrary effect in OP-hMSCs. However, the Axin2 level was elevated in N-hMSCs with PDCD4 overexpression and lowered in si-PDCD4 transfecting OP-hMSCs.
According to the previous studies44,45, XAV939 inhibits the growth of the cells was not due to the cytotoxicity of the XAV939, because the LDH (lactate dehydrogenase) activity levels were reported to be the same across the treatment groups. In this study, XAV939 suppressed cell viability, reduced expression of osteogenic markers Runx2 and Osterix, and induced apoptosis of hMSCs. Furthermore, XAV939 reversed the increased cell viability, reduced apoptosis rate and upregulated expression of Runx2 and Osterix in OP-hMSCs caused by PDCD4 knockdown. Taken together, PDCD4 inhibits proliferation and differentiation and induces apoptosis of hMSCs via suppressing Wnt/β-catenin pathway.
5. Conclusions
In summary, the present study clearly reveals that PDCD4 suppresses proliferation and differentiation and induces apoptosis of hMSCs by inactivation of Wnt/β-catenin pathway. Our findings indicated that PDCD4 may serve as a promising therapeutic target for bone disease such as osteoporosis.Conflicts of interest
There is no conflict of interest regarding the publication of this paper.Authors' contribution
Yang Liu designed and performed the experiment. Dongyue Su analyzed the data and wrote the manuscript. Tao Song supervised the study and reviewed the manuscript.Acknowledgements
This work was supported by Science and Technology Research Project of Henan Province (Grant No. 162102310076).References
- R. Vijayakumar and D. Büsselberg, Journal of Local and Global Health Science, 2016, 2 CrossRef.
- M. Prideaux, D. M. Findlay and G. J. Atkins, Curr. Opin. Pharmacol., 2016, 28, 24–30 CrossRef CAS PubMed.
- K. H. Nguyen, F. Xu, S. Flowers, E. A. Williams, J. C. Fritton and E. Moran, Stem Cells, 2015, 33, 3028–3038 CrossRef CAS PubMed.
- Y. Jiang, B. N. Jahagirdar, R. L. Reinhardt, R. E. Schwartz, C. D. Keene, X. R. Ortiz-Gonzalez, M. Reyes, T. Lenvik, T. Lund and M. Blackstad, Nature, 2002, 418, 41–49 CrossRef CAS PubMed.
- V. Egea, L. Von Baumgarten, C. Schichor, B. Berninger, T. Popp, P. Neth, R. Goldbrunner, Y. Kienast, F. Winkler and M. Jochum, Cell Death Differ., 2011, 18, 853–863 CrossRef CAS PubMed.
- B. Mahon, Clin. Exp. Immunol., 2007, 149, 353 CrossRef PubMed.
- G. D. Wu, J. A. Nolta, Y. S. Jin, M. L. Barr, H. Yu, V. A. Starnes and D. V. Cramer, Transplantation, 2003, 75, 679–685 CrossRef PubMed.
- T. C. Mackenzie and A. W. Flake, Blood Cells, Mol., Dis., 2001, 27, 601–604 CrossRef CAS PubMed.
- R. Manuguerra-GagnÉ, P. R. Boulos, A. Ammar, F. A. Leblond, G. Krosl, V. Pichette, M. R. Lesk and D. C. Roy, Stem Cells, 2013, 31, 1136–1148 CrossRef PubMed.
- F. Yang, V. Y. Leung, K. D. Luk, D. Chan and K. M. Cheung, Mol. Ther., 2009, 17, 1959–1966 CrossRef CAS PubMed.
- C. Zhao, C. Zhang, S. Zhou, Y. Xie, Y. Wang, H. Huang, Y. Shang, W. Li, C. Zhou and M. Yu, Cytotherapy, 2007, 9, 414–426 CrossRef CAS PubMed.
- T. P. Martens, A. F. Godier, J. J. Parks, L. Q. Wan, M. S. Koeckert, G. M. Eng, B. I. Hudson, W. Sherman and G. Vunjak-Novakovic, Cell Transplant., 2009, 18, 297–304 Search PubMed.
- M. H. Mankani, S. A. Kuznetsov, R. M. Wolfe, G. W. Marshall and P. G. Robey, Stem Cells, 2006, 24, 2140–2149 CrossRef PubMed.
- A. Caplan, J. Pathol., 2009, 217, 318–324 CrossRef CAS PubMed.
- H. Clevers, Cell, 2006, 127, 469–480 CrossRef CAS PubMed.
- J. J. Westendorf, R. A. Kahler and T. M. Schroeder, Gene, 2004, 341, 19–39 CrossRef CAS PubMed.
- C. Y. Logan and R. Nusse, Annu. Rev. Cell Dev. Biol., 2004, 20, 781–810 CrossRef CAS PubMed.
- T. Gaur, C. J. Lengner, H. Hovhannisyan, R. A. Bhat, P. V. Bodine, B. S. Komm, A. Javed, A. J. Van Wijnen, J. L. Stein and G. S. Stein, J. Biol. Chem., 2005, 280, 33132–33140 CrossRef CAS PubMed.
- S. S. Lin, S. W. Ueng, C. C. Niu, L. J. Yuan, C. Y. Yang, W. J. Chen, M. S. Lee and J. K. Chen, Stem Cell Res. Ther., 2014, 12, 260–274 CAS.
- H. Yun, K. Park, T. Quang, H. Oh, J. Hong, Y. Kim and E. Kim, Cell Death Dis., 2015, 6, e1819 CrossRef CAS PubMed.
- B. Osta, G. Benedetti and P. Miossec, Front. Immunol., 2014, 5, 48 Search PubMed.
- J. H. Chang, Y. H. Cho, S. Y. Sohn, J. M. Choi, A. Kim, Y. C. Kim, S. K. Jang and Y. Cho, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 3148–3153 CrossRef CAS PubMed.
- C. Suzuki, R. G. Garces, K. A. Edmonds, S. Hiller, S. G. Hyberts, A. Marintchev and G. Wagner, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 3274–3279 CrossRef CAS PubMed.
- S. Zhang, J. Li, Y. Jiang, Y. Xu and C. Qin, J. Exp. Clin. Cancer Res., 2009, 28, 71 CrossRef PubMed.
- R. Nieves-Alicea, N. H. Colburn, A. M. Simeone and A. M. Tari, Breast Cancer Res. Treat., 2009, 114, 203–209 CrossRef CAS PubMed.
- B. A. Walter, G. Gómez-Macias, V. A. Valera, M. Sobel and M. J. Merino, J. Cancer, 2011, 2, 67–75 CrossRef PubMed.
- X. Wei, W. Wang, L. Wang, Y. Zhang, X. Zhang, M. Chen, F. Wang, J. Yu, Y. Ma and G. Sun, Cancer Med., 2016, 693–702 CrossRef PubMed.
- Y. Zhen, D. Li, W. Li, W. Yao, A. Wu, J. Huang, H. Gu, Y. Huang, Y. Wang and J. Wu, Oncol. Res., 2016, 23, 61–68 CrossRef PubMed.
- T. Schmid, A. P. Jansen, A. R. Baker, G. Hegamyer, J. P. Hagan and N. H. Colburn, Cancer Res., 2008, 68, 1254–1260 CrossRef CAS PubMed.
- A. Hayashi, S. Aishima, Y. Miyasaka, K. Nakata, K. Morimatsu, Y. Oda, E. Nagai, Y. Oda, M. Tanaka and M. Tsuneyoshi, Hum. Pathol., 2010, 41, 1507–1515 CrossRef CAS PubMed.
- K. Yuan, X. Yu, Y. Zhang, Z. Sun, K. Wu, L. Wang, S. Huang, Y. Qiu and T. Zeng, Int. J. Clin. Exp. Pathol., 2016, 9, 8617–8621 Search PubMed.
- G. Lluri, G. D. Langlois, P. D. Soloway and D. M. Jaworski, Exp. Cell Res., 2008, 314, 11–24 CrossRef CAS PubMed.
- S. K. Hwang, A. R. Baker, M. R. Young and N. H. Colburn, Carcinogenesis, 2014, 35, 1469–1480 CrossRef CAS PubMed.
- M. Shiota, H. Izumi, A. Tanimoto, M. Takahashi, N. Miyamoto, E. Kashiwagi, A. Kidani, G. Hirano, D. Masubuchi and Y. Fukunaka, Cancer Res., 2009, 69, 3148–3156 CrossRef CAS PubMed.
- H. S. Yang, C. P. Matthews, T. Clair, Q. Wang, A. R. Baker, C. C. H. Li, T. H. Tan and N. H. Colburn, Mol. Cell. Biol., 2006, 26, 1297–1306 CrossRef CAS PubMed.
- O. Afonja, D. Juste, S. Das, S. Matsuhashi and H. H. Samuels, Oncogene, 2004, 23, 8135–8145 CrossRef CAS PubMed.
- N. Wei, S. S. Liu, T. H. Leung, K. F. Tam, X. Y. Liao, A. N. Cheung, K. K. Chan and H. Y. Ngan, Cancer Res., 2010, 70, 5006 CrossRef PubMed.
- P. Andreopoulou and R. S. Bockman, Annu. Rev. Med., 2015, 66, 329–342 CrossRef CAS PubMed.
- A. Ahmadzadeh, F. Norozi, S. Shahrabi, M. Shahjahani and N. Saki, Cell Tissue Res., 2016, 363, 321–335 CrossRef CAS PubMed.
- B. L. Vaes, K. J. Dechering, E. P. van Someren, J. M. Hendriks, C. J. van de Ven, A. Feijen, C. L. Mummery, M. J. Reinders, W. Olijve and E. J. van Zoelen, Bone, 2005, 36, 803–811 CrossRef CAS PubMed.
- X. Li, Y. Zhang, H. Kang, W. Liu, P. Liu, J. Zhang, S. E. Harris and D. Wu, J. Biol. Chem., 2005, 280, 19883–19887 CrossRef CAS PubMed.
- K. Tao, D. Xiao, J. Weng, A. Xiong, B. Kang and H. Zeng, Toxicol. Lett., 2016, 240, 68–80 CrossRef CAS PubMed.
- S. H. Tan, K. Senarath-Yapa, M. T. Chung, M. T. Longaker, J. Y. Wu and R. Nusse, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, E5262–E5271 CrossRef CAS PubMed.
- M. Ono, P. Yin, A. Navarro, M. B. Moravek, S. A. Druschitz, C. J. Gottardi and S. E. Bulun, Fertil. Steril., 2014, 101, 1441–1449 CrossRef CAS PubMed.
- E. Esen, J. Chen, C. M. Karner, A. L. Okunade, B. W. Patterson and F. Long, Cell Metab., 2013, 17, 745–755 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |