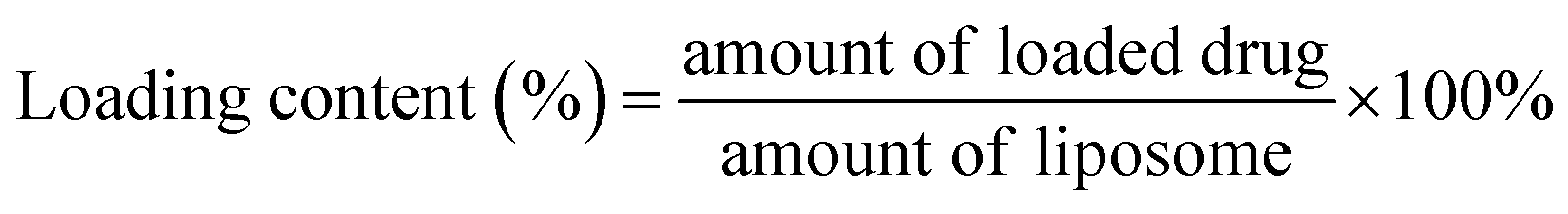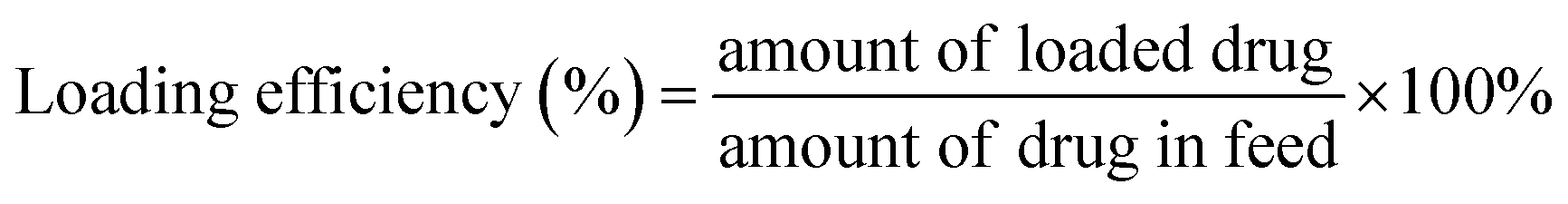DOI:
10.1039/C7RA02065A
(Paper)
RSC Adv., 2017,
7, 29618-29625
Non-thermal acoustic treatment as a safe alternative to thermosensitive liposome-involved hyperthermia for cancer therapy
Received
19th February 2017
, Accepted 28th May 2017
First published on 7th June 2017
Abstract
A heat-triggered drug release strategy based on ultrasound-assisted mild hyperthermia and thermosensitive liposomes has emerged as a promising option to enable spatiotemporally controlled, efficient drug delivery for cancer treatment. However, consequential thermal vascular damage may decrease drug extravasation into tumor tissue and affect the therapeutic efficacy of follow-up treatments. To overcome this limitation, we explored a non-thermal acoustic treatment. Doxorubicin (DOX), an anticancer drug, was encapsulated in fatty acid-conjugated, elastin-like peptide (FELP)-bearing thermosensitive liposomes (FTSLs) for comparison of two treatments and their therapeutic implications. DOX-FTSLs had an average hydrodynamic size of 134.9 nm with a unimodal distribution. Their thermosensitivity allowed the triggering of rapid DOX release at 42 °C, a mild-hyperthermia relevant temperature, with sustained DOX release at 37 °C. Interestingly, non-thermal acoustic treatment right after systemic administration of DOX-FTSLs into tumor-bearing mice led to higher tissue penetration without permanent vascular damage, greater intratumoral DOX accumulation, and similar therapeutic efficacy to thermal treatment. Overall, non-thermal acoustic treatment may be a safe alternative to thermosensitive liposome-involved hyperthermia for liposomal chemotherapy.
Introduction
Nanoparticles have been extensively studied as a delivery system for anticancer drugs to address pharmaceutical challenges1 such as dose-limiting systemic toxicity, rapid clearance2,3 and poor targeting of the tumoral region.4,5 That is primarily because nanoparticles selectively accumulate at tumor sites in a size-dependent manner through highly-permeable endothelial gaps existed over defective angiogenic tumor vasculature.6,7 A deficient tumor lymphatic drainage system allows the particles to be also retained more in tumors than in normal tissue. These features support the concept of passive cancer targeting through the enhanced permeability and retention effect.8,9
Of the nanoparticles, liposomes have attracted much attention as a versatile drug carrier because they are biocompatible, biodegradable, and amenable to incorporating biologically active drug molecules.10,11 However, the short blood circulation time and mechanism of sustained drug release through passive diffusion of liposomes may not bring cytotoxic local drug concentrations to tumors.11 Therefore, efforts have focused on developing stimuli-sensitive liposomes that can induce rapid, target-specific release of drugs in response to stimuli such as pH,12,13 temperature,14,15 ultrasound,16–18 and light.19–21 In particular, thermosensitive liposomes have been intensively investigated in the recent decades for heat-triggered drug release in conjunction with mild hyperthermia induced by ultrasound or radiofrequency.10,22,23 Mild hyperthermia causing local heating of tumor tissue to around 42 °C improves the efficiency of chemotherapy by temporarily increasing tumor blood flow and microconvection in the tumor interstitium.22,24 If thermosensitive liposomes are broken in mild hyperthermia conditions, a burst release of the drug can be induced selectively at the desired site.24,25 Recently, we demonstrated enhanced antitumor efficacy with a synergistic combination of mild hyperthermia and fatty acid-conjugated elastin-like polypeptide (FELP)-bearing thermosensitive liposomes (FTSLs) that have a long blood circulation time and selective collapse at or above 40 °C.4,26 However, after hyperthermia treatment by high intensity focused ultrasound (HIFU), thermal vascular damage has been observed as reported elsewhere.27 We hypothesized that although mild hyperthermia temporarily improves the efficacy of liposomal chemotherapy, the resulting irreversible thermal damage can inhibit extravasation of the drug to the tumor site. This consequence may decrease the overall therapeutic efficacy of follow-up treatments.
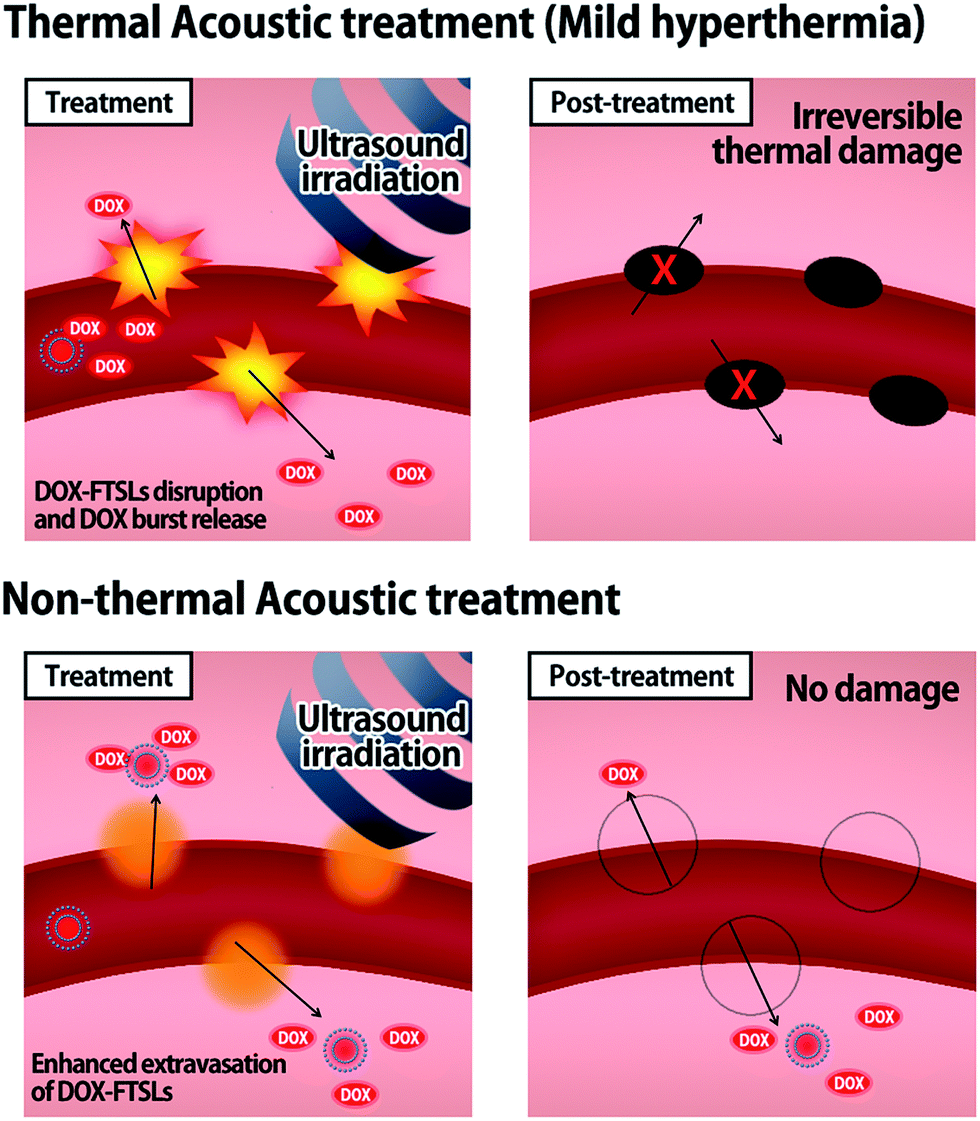
Herein, we devised non-thermal acoustic treatments by adjusting HIFU power, and compared its treatment effects on the in vivo behavior of doxorubicin (DOX)-loaded FTSLs (DOX-FTSLs) with thermosensitive liposome-involved hyperthermia. Thermal and non-thermal acoustic treatments were harnessed to improve the therapeutic efficacy of liposomal chemotherapy via different drug delivery mechanisms (Fig. 1). With ultrasound-induced hyperthermia, liposome collapse near tumor tissue triggers a burst release of drugs and irreversible damage occurs in the tumor tissue microvasculature.4,28 Unlike thermal ultrasound, non-thermal acoustic treatment temporarily increases the permeability of tumor vessels via an acoustic cavitation effect29 without a rise in temperature and liposome collapse so that undisrupted liposomes are expected to be efficiently extravasated into tumor tissue. To the best of our knowledge, this is the first report to demonstrate superior antitumor drug delivery potential and comparable therapeutic efficacy of the non-thermal acoustic treatment compared with the hyperthermia in vivo.
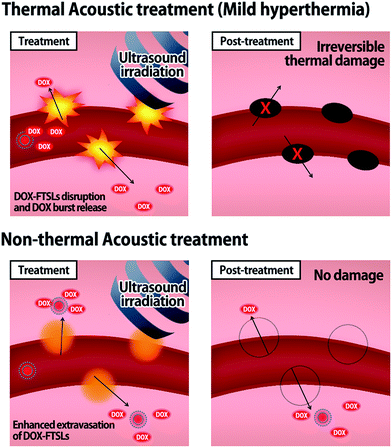 |
| | Fig. 1 Schematic illustration representing the drug delivery mechanisms of thermal and non-thermal acoustic treatment using DOX-FTSLs. | |
Experimental
Materials
1,2-Dipalmitoyl-sn-glycero-3-phosphocholine (DPPC), 1,2-distearoyl-sn-glycero-3-phospho-ethanolamine-N-[methoxy(polyethylene glycol)-2000] (DSPE-PEG), 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine (DPPE) and cholesterol were obtained from Avanti Polar Lipid, Inc. (Alabaster, USA). FELP (C18H37–[VPGVG]3–NH2) was purchased from American Peptide, Inc. (Sunnyvale, USA). The near-infrared fluorescent (NIRF) dye, Cy5.5 was obtained from Amersham Biosciences (Piscataway, USA). DOX was purchased from Sigma-Aldrich (St. Louis, USA). All other chemicals were analytical grade. The EMT6 cell line was purchased from the American Type Culture Collection (Rockville, USA). For cell culture, Dulbecco's modified Eagle medium (DMEM), Dulbecco's phosphate-buffered saline (PBS), trypsin–EDTA, and fetal bovine serum (FBS) were purchased from Invitrogen (Waltham, USA). All experiments involving live animals were performed in compliance with the relevant laws and institutional guidelines of Korea Institute of Sciences and Technology, and institutional committees approved all experimental protocols (KIST-2016-006).
Preparation of DOX-FTSLs
DOX-FTSLs were produced by a thin film hydration method as described previously.4,26 FTSLs were formulated with DPPC![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DSPE-PEG:cholesterol:FELP at a 55:2:15:0.4125 molar ratio. Lipids, cholesterol, and FELP were dissolved in chloroform, and solvent was evaporated to prepare a thin film, which was hydrated with 250 mM ammonium sulfate solution (pH 4.0) by rigorous vortexing and sonication. The resulting solution was extruded through a 100 nm pore polycarbonate membrane to facilitate formation of liposomes with homogeneous size distribution. The ammonium sulfate buffer was replaced with 25 mM Tris·HCl (pH 9.0) using a pre-packed Sephadex (G-25M) column. DOX was then loaded into the liposomes by adding it to liposome suspensions and incubating at 37 °C for 1 h. To remove free DOX and adjust pH to neutral, a pre-packed Sephadex (G-25M) column was used with PBS (pH 7.4) as eluent. Cy5.5-labeled DOX-FTSLs (Cy5.5-DOX-FTSLs) were also prepared by using Cy5.5-conjugated DPPE in the FTSLs formulation for in vivo NIRF imaging.
:DSPE-PEG:cholesterol:FELP at a 55:2:15:0.4125 molar ratio. Lipids, cholesterol, and FELP were dissolved in chloroform, and solvent was evaporated to prepare a thin film, which was hydrated with 250 mM ammonium sulfate solution (pH 4.0) by rigorous vortexing and sonication. The resulting solution was extruded through a 100 nm pore polycarbonate membrane to facilitate formation of liposomes with homogeneous size distribution. The ammonium sulfate buffer was replaced with 25 mM Tris·HCl (pH 9.0) using a pre-packed Sephadex (G-25M) column. DOX was then loaded into the liposomes by adding it to liposome suspensions and incubating at 37 °C for 1 h. To remove free DOX and adjust pH to neutral, a pre-packed Sephadex (G-25M) column was used with PBS (pH 7.4) as eluent. Cy5.5-labeled DOX-FTSLs (Cy5.5-DOX-FTSLs) were also prepared by using Cy5.5-conjugated DPPE in the FTSLs formulation for in vivo NIRF imaging.
Characterization of DOX-FTSLs
The hydrodynamic size and size distribution of DOX-FTSLs were measured by dynamic light scattering (Zetasizer Nano ZS90; Malvern Instruments, Worcestershire, UK) with a He–Ne laser operated at 633 nm and 25 °C in the 173° backscatter mode. Size measurements were triplicate. The concentration of DOX was determined by its absorbance at 480 nm using a UV-vis spectrometer (G1103A; Agilent, Worcestershire, USA) after dissolving in a water/tetrahydrofuran mixture (v/v). The DOX content and DOX loading efficiency of DOX-FTSLs were calculated by the following formulas:
In vitro DOX release test of DOX-FTSLs
The temperature-dependent drug release profiles of DOX-FTSLs were studied by measuring DOX absorbance with a UV-vis spectrometer (G1103A; Agilent, Worcestershire, USA). 0.5 mL of DOX-FTSLs solution (1 mg mL−1) was loaded into a dialysis membrane bag (MWCO = 3.5 kDa), immersed in 10 mL of 10% serum–PBS buffer (pH 7.4) and gently shaken in a water bath (37 °C and 42 °C, 100 rpm). At 5, 15, 30, 60, and 120 min, the amount of released DOX in medium was determined by measuring the absorbance at 480 nm. The separate sample groups were prepared for each pre-set time points.
Experimental settings for acoustic treatments
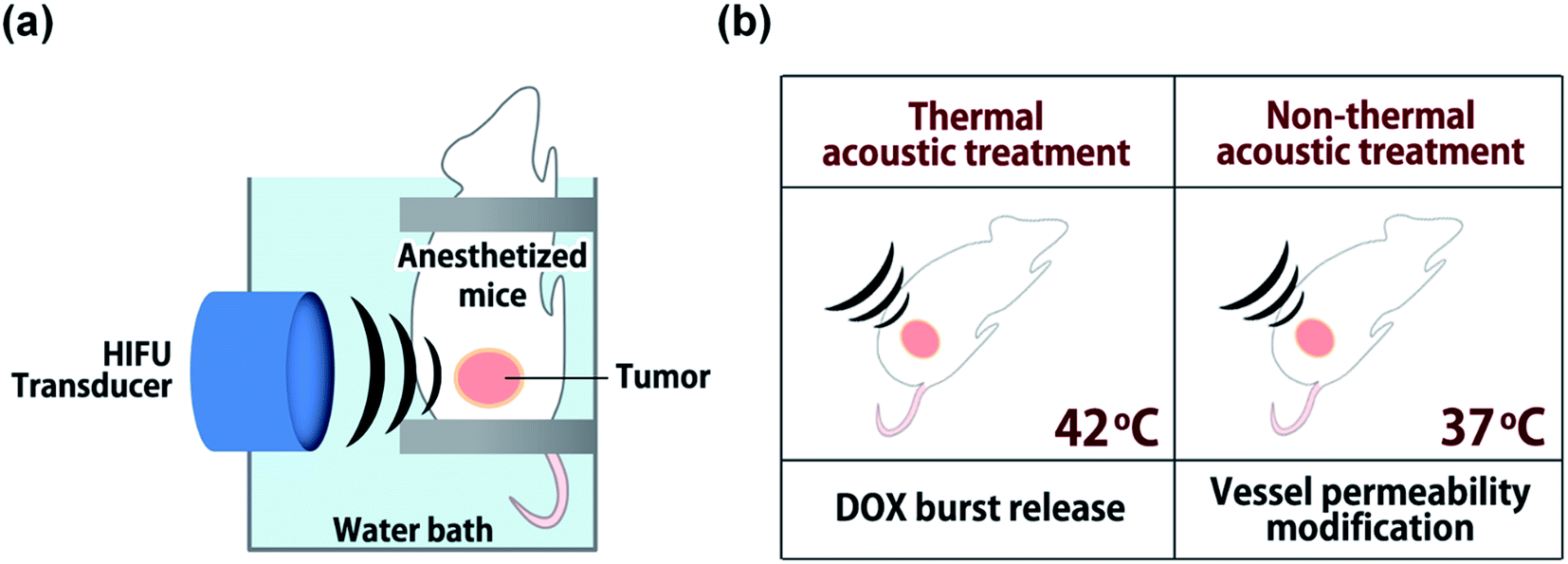
A small animal HIFU system (VIFU 2000, Alpinion, Seoul, Korea) was used for ultrasound treatments to tumor tissues (Fig. 2). Mice were anesthetized via intraperitoneal injection of avertin (2,2,2-tribromoethanol) and fixed on a custom-made holder for treatment. The transducer and target tumor were aligned in a water bath filled with degassed water and maintained at 37 °C. HIFU parameters were optimized by iterative adjustment based on direct in vivo temperature measurements using a thermocouple wire (50 μm in diameter, Physitemp Instrument Inc., Clifton, USA) inserted into tumor tissues. For thermal treatment, positions with seven or eight HIFU foci were preset on tumors at 2.5 mm intervals; each was sequentially exposed to HIFU for 300 s (power: 20 W, duty cycle: 15%, pulse repetition frequency: 15 Hz) to maintain tumor tissues at 42 °C. For non-thermal acoustic treatment, power was adjusted to 10 W to maintain tumor tissue at 37 °C while keeping other parameters the same as for thermal treatment. In an animal study (n = 3), DOX-FTSLs (5 mg DOX per kg) were systemically administered through the tail vein of mice, and HIFU (thermal or non-thermal acoustic treatment) was immediately used to irradiate tumors. To evaluate the therapeutic efficacy of DOX-FTSLs with thermal or non-thermal acoustic treatment, tumor areas were irradiated again under thermal acoustic treatment conditions at 12 hours post injection for burst release of DOX by disruption of FTSLs already deposited in tumor tissues.
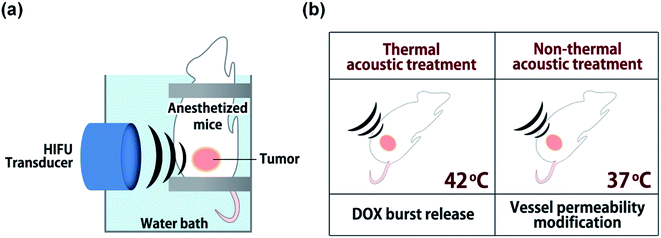 |
| | Fig. 2 Schematic representation of the experimental set up for acoustic treatments. (a) Alignment of HIFU transducer with murine tumor mass. (b) Experimental conditions for thermal or non-thermal acoustic treatment. | |
Tumor implantation
For animal studies, an allograft tumor model was established using EMT6 mouse mammary tumor cells with five-week-old male BALB/c nude mice. For cell cultivation, DMEM with 15% (v/v) FBS, 100 μg mL−1 penicillin, and 100 μg mL−1 streptomycin was used. Tumor models were prepared by subcutaneously injecting 200 μL of cell suspension (1 × 106 EMT6 cells for each mouse) into the left thigh. Tumors were grown for 10 days to 150–200 mm3.
Ex vivo histological analysis of tumor tissues
For histological staining, tumors were collected from mice groups 1 hour post acoustic treatment. Tissues were fixed with 4% paraformaldehyde and embedded into paraffin. Paraffin-embedded tumor tissues were sectioned in 4 μm slices, stained with hematoxylin and eosin (H&E), and observed by bright-field microscopy (BX 51; Olympus, Melville, USA).
In vivo biodistribution of DOX-FTSLs
In vivo biodistribution of DOX-FTSLs was evaluated for each treatment protocol using an eXplore Optix system (ART Advanced Research Technologies Inc., Montreal, Canada) with laser set to output power 10 μW for 0.3 s per point, after intravenous injection of Cy5.5-DOX-FTSLs (5 mg DOX per kg) into tumor-bearing mice. At predetermined time points, fluorescence images of mice were obtained with excitation at 635 nm and detection at 693 nm for Cy5.5. Quantification of NIRF images was done by OptiView software (ART Advanced Research Technologies Inc., Montreal, Canada).
In vivo fluorescence images of tumor vessels
To qualitatively and quantitatively assess extravasation of DOX and Cy5.5-DOX-FTSLs into tumors in mice models, NIRF images of tumor vessels were observed by an optical live imaging system (OV-100; Olympus Corp., Tokyo, Japan). Cy5.5-DOX-FTSLs (5 mg DOX per kg) were systemically injected into tail vein of tumor-bearing mice with immediate thermal or non-thermal acoustic treatment on tumor tissues. At 12 h post injection, tumoral skin of EMT-6-bearing mice was removed for fluorescent detection. Accumulation of Cy5.5-DOX-FTSLs and DOX around tumor vessels was visualized and quantified using an RFP filter (excitation = 535–555 nm; emission = 570–623 nm) and Cy5.5 filter (excitation = 620–650 nm and emission = 680–710 nm). NIRF intensities were quantitatively determined using Image-Pro Plus software 4.0 (Media Cybernetics, Rockville, USA).
Antitumor efficacy of DOX-FTSLs
Mice were divided into five groups: saline, free DOX, DOX-FTSLs, DOX-FTSLs (thermal), DOX-FTSLs (non-thermal). Tumor-bearing mice for control groups (saline, free DOX, and DOX-FTSLs) were not exposed to HIFU. DOX-FSTLs (thermal) group and DOX-FSTLs (non-thermal) group were irradiated by HIFU immediately after systemic injection of DOX-FSTLs (5 mg DOX per kg). After 12 hours, mice underwent thermal acoustic treatment to burst liposomes accumulated in tumors. Upon drug administration followed by HIFU treatment, tumor growth was recorded over 12 days. Tumor size was measured by Vernier calipers and calculated as: volume = (width of tumor)2 × (length of tumor) × π/6. Values were presented as mean with standard errors for groups of n = 3.
Statistical analysis
One way analysis of variance (ANOVA) was conducted to determine statistical significance of differences between groups, and marked with an asterisk (*) in figures if p value was less than 0.05.
Results and discussion
Preparation and characterization of DOX-FTSLs
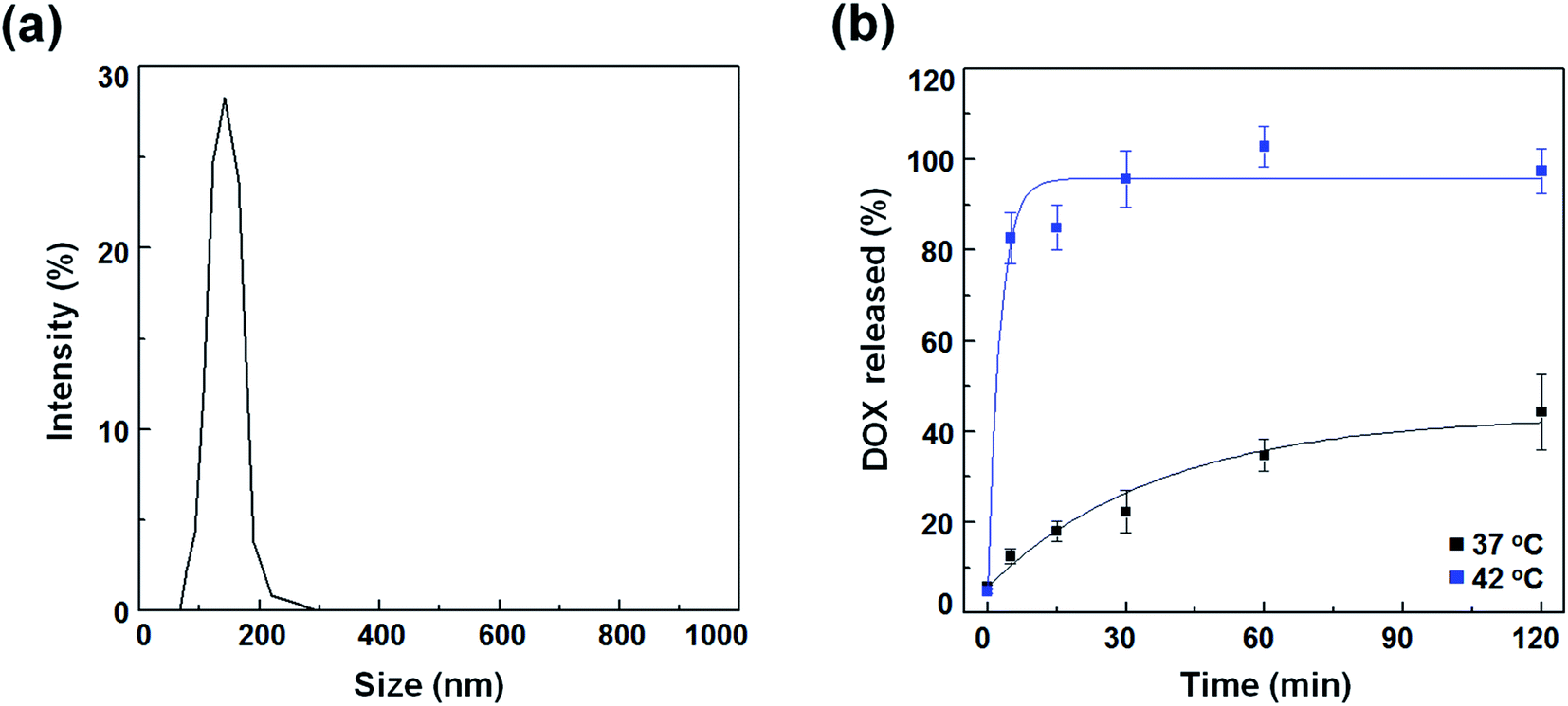
To compare the therapeutic potency of thermal acoustic treatment with a non-thermal alternative, we prepared DOX-FTSLs, which can be ruptured under thermal conditions. FTSLs were composed of two phospholipids (DPPC and DSPE-PEG)30 with cholesterol as the steric stabilizer of the bilayer31 and FELP as the transition temperature adjuster.32 DOX, a model chemotherapeutic drug, was encapsulated in FTSLs to produce DOX-FTSLs. After purification, DOX content of DOX-FTSLs was 9.3% and DOX loading efficiency was 93.2%. DOX-FTSLs had a mean diameter of 134.97 nm and polydispersity index value of 0.417 with a unimodal size distribution (Fig. 3a), consistent with our previous report.4
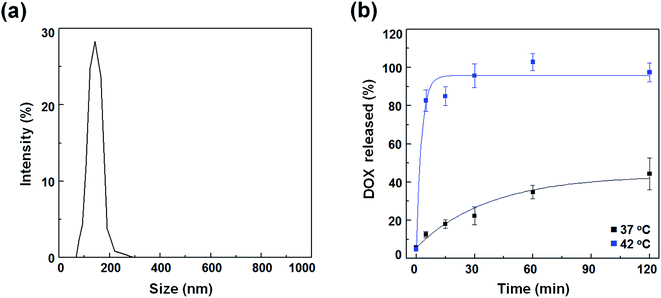 |
| | Fig. 3 Characterization of DOX-FTSLs. (a) Size distribution of DOX-FTSLs in PBS (pH 7.4). (b) In vitro DOX release profiles of DOX-FTSLs at 37 °C and at 42 °C. Error bars represent standard deviation (n = 3). | |
In vitro temperature-dependent release profile of DOX-FTSLs
DOX release profiles of DOX-FTSLs were obtained as a function of time at two temperatures mimicking body temperature (37 °C) and mild hyperthermia (42 °C) (Fig. 2b). At 37 °C, DOX-FTSLs released about 44% of encapsulated DOX within 2 h, exhibiting first-order release kinetics. The DOX release profiles of DOX-FTSLs were obtained as a function of time at two different temperatures mimicking body temperature (37 °C) and mild hyperthermia (42 °C) (Fig. 3b). At 37 °C, DOX-FTSLs released about 44% of the encapsulated DOX within 2 h, exhibiting first-order release kinetics. Conversely, at 42 °C, a substantial amount of DOX (82.75%) released from the DOX-FTSLs was observed within the initial 5 min, and the amount of DOX released was soon saturated to 96% of the total encapsulated DOX amount. The thermosensitive burst release under hyperthermia conditions might have been caused by the destabilization of the liposomal membrane with conformational changes of FELPs at 42 °C, which is higher than the transition temperature of the DOX-FTSLs (40 °C).13 The DOX-FTSLs maintained liposomal structure better at the non-thermal physiological temperature of 37 °C compared with the thermal temperature of 42 °C. Based on these results, in vivo ultrasound-assisted hyperthermia was also expected to trigger the exclusive release of DOX with collapse of DOX-FTSLs in the focal region, with non-thermal acoustic treatment reducing DOX leakage.
Evaluation of tumor tissue damage after acoustic treatments
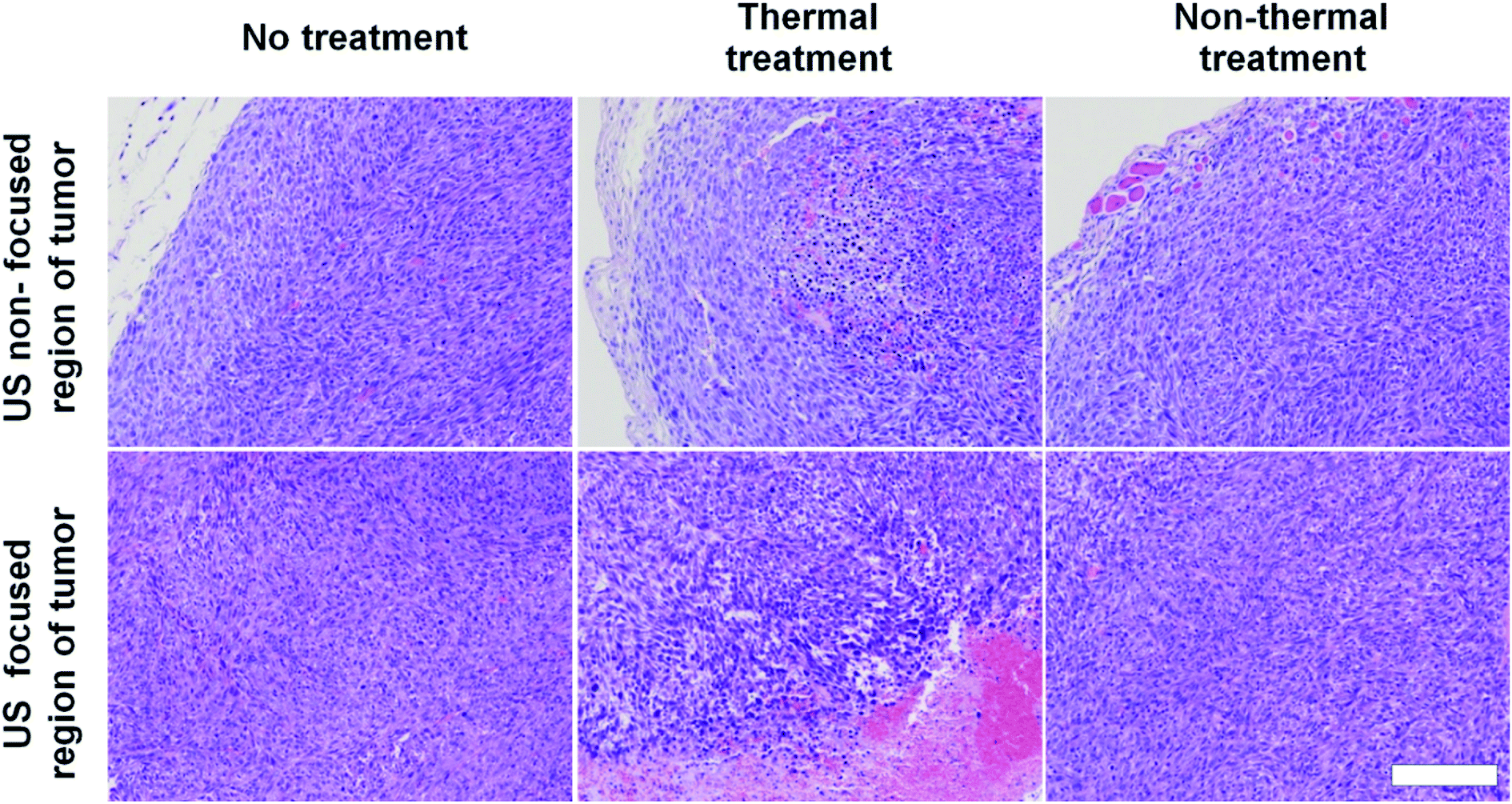
To assess damage to tumor tissues after thermal or non-thermal acoustic treatment, we tuned acoustic parameters to maintain tumor tissues around 42 °C for thermal and 37 °C for non-thermal. During ultrasound irradiation, we monitored tumor tissue temperatures to confirm proper control. Recorded average temperatures for thermal treatment were 42.5 ± 0.36 °C, sufficient to disrupt DOX-FTSLs. Average temperatures for non-thermal treatment were 37.7 ± 0.4 °C. We considered fluctuations in temperatures acceptable for experiments since differences from target temperatures were less than 1 °C, which still met the purpose of the specific temperatures. In histological analysis, only H&E-stained mouse tumor tissues from ultrasound focal regions treated under thermal conditions were swollen from edema, implying substantial damage (Fig. 4). No significant damage was observed in the control groups including the non-thermal treatment group.
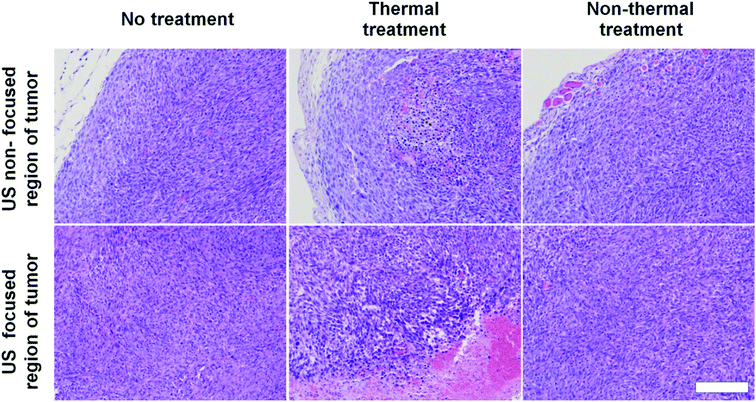 |
| | Fig. 4 H&E staining images of tumor tissues excised from treatment groups. Scale bar represents 200 μm. | |
In vivo biodistribution
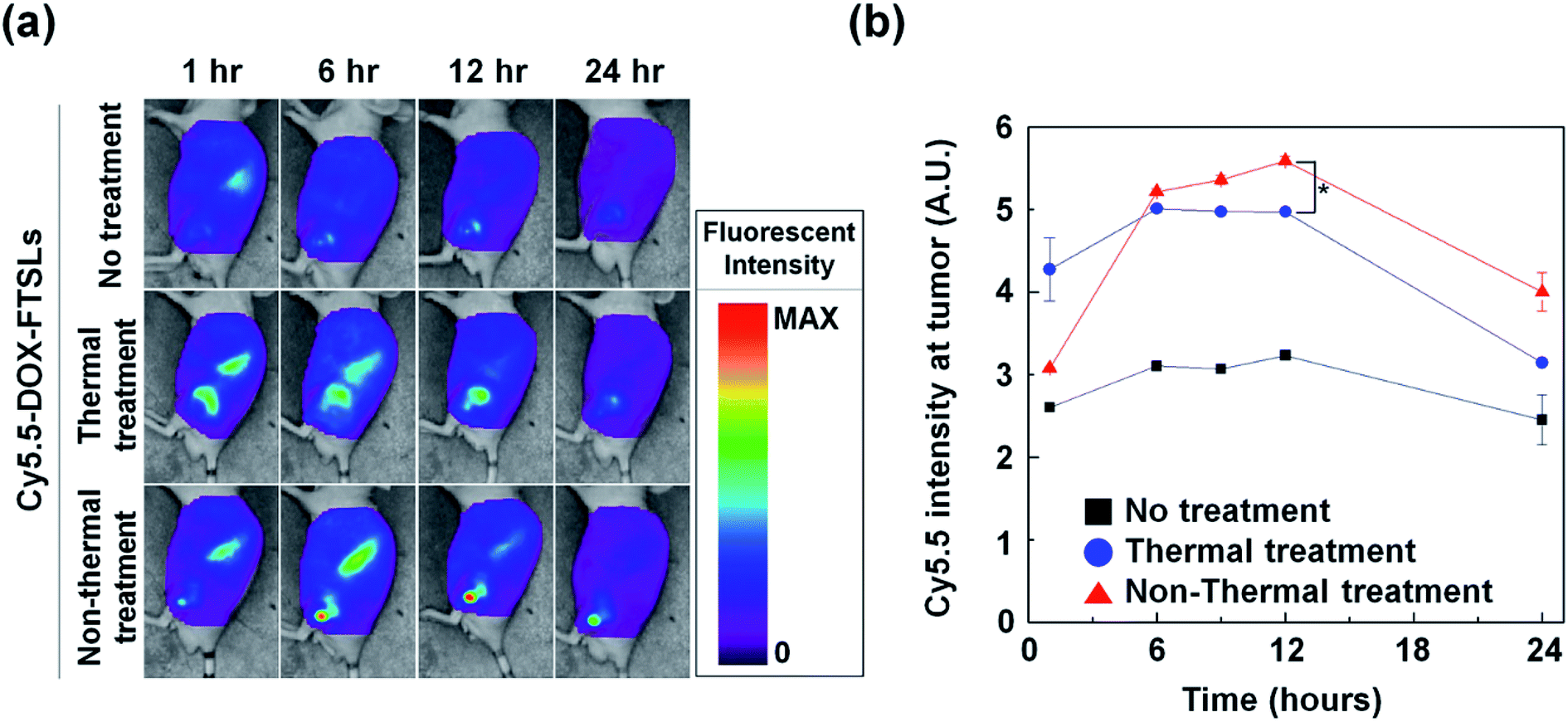
To compare the effects of acoustic treatment methods on tumor-targeting efficiency of DOX-FTSLs, in vivo biodistributions were investigated (Fig. 5). DOX-FTSLs were tracked using a NIRF imaging system via labeling with Cy5.5, systemic administration into EMT6 tumor-bearing mice, and monitoring for 24 h. Overall, thermal and non-thermal acoustic treatment improved intratumoral accumulation of Cy5.5-DOX-FTSLs. After the first 1 h, fluorescent signal intensity at tumor site of the mice received the thermal or non-thermal treatment was 1.64-fold or 1.18-fold higher, respectively, than that of the no treatment group. No groups showed drastic changes in signal intensity at tumor sites for the time period between 6 h and 12 h post injection. In the non-thermal group, tumor signal intensity increased to 1.75-fold higher than the no-treatment group, and was higher than the thermal group at 6 hours after Cy5.5-DOX-FTSL injection. The non-thermal group signal gradually increased until 12 hours post injection, showing significantly greater signal intensity than the thermal group. These results indicated that although thermal treatment resulted in higher intratumoral accumulation of Cy5.5-DOX-FTSLs than non-thermal treatment at 1 hour post injection, after five hours, non-thermal treatment had higher accumulation. Observed thermal damage on tumor tissue including vasculature shown in Fig. 4 might correlate with less improvement with thermal treatment on a tumor-specific biodistribution compared to non-thermal treatment.
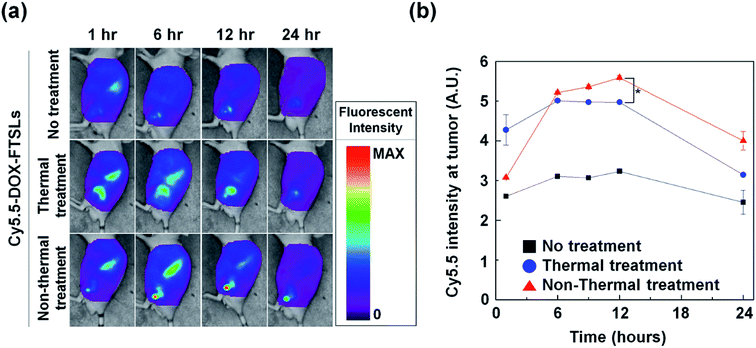 |
| | Fig. 5 (a) In vivo biodistribution of Cy5.5-labeled DOX-FTSLs by treatment group. (b) Normalized fluorescent intensities at tumor tissues as a function of time. Error bars represent standard error (n = 3; *p < 0.05 calculated by one-way ANOVA). | |
Evaluation of vessel permeability change after acoustic treatments
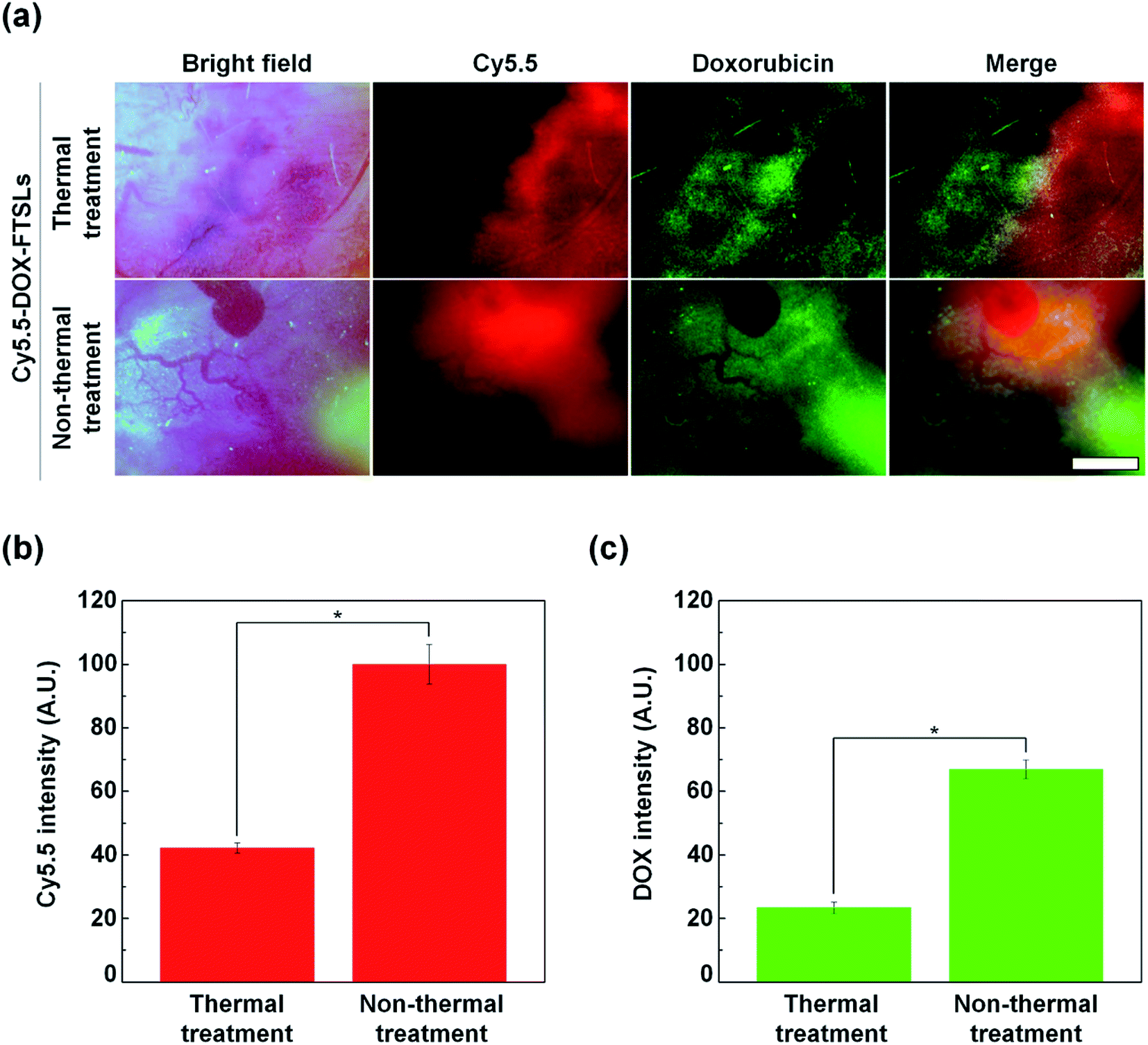
To compare the effect of thermal and non-thermal treatment on extravasation of the DOX-FTSLs and DOX, in vivo NIRF live imaging of tumor tissue was conducted at 12 h post injection of Cy5.5-DOX-FTSLs (Fig. 6a). We observed tumor blood vessels closely, and visualized DOX-FTSLs using Cy5.5 and DOX using its own fluorescence. By bright field image of the thermal treatment group, tumor vessels showed damage with risk of reducing or blocking tumor blood flow because of thrombosis. This effect would decrease the supply of drugs into tumors. A negligible amount of fluorescent signal corresponding to extravasated DOX-FTSLs was seen near damaged vessels while the fluorescent signal of released DOX was widespread. This is probably because the substantial amount of DOX is first released from the DOX-FTSLs upon liposome rupture near tumor tissue and then extravasation occurs. Conversely, no vessel damage was observed in tumor tissues receiving non-thermal acoustic treatment and the DOX and DOX-FTSL fluorescent signal was entirely dispersed and overlapping. These results suggested that tumor vessel damage from thermal treatment hindered accumulation of DOX-FTSLs while nonthermal treatment accommodated accumulation of both DOX and DOX-FTSLs without producing recognizable vessel damage. Quantitative analysis of intratumoral accumulation showed that, under nonthermal acoustic treatment conditions, 2.87-fold more DOX and 2.37-fold more DOX-FTSLs extravasated into tumoral regions compared with thermal conditions (Fig. 5b). These results verified our main hypothesis that thermal vascular damage under hyperthermia conditions inhibits extravasation of drug to tumor sites. Based on our experiments, non-thermal treatment was safer and a better tool for delivering drugs to tumoral regions.
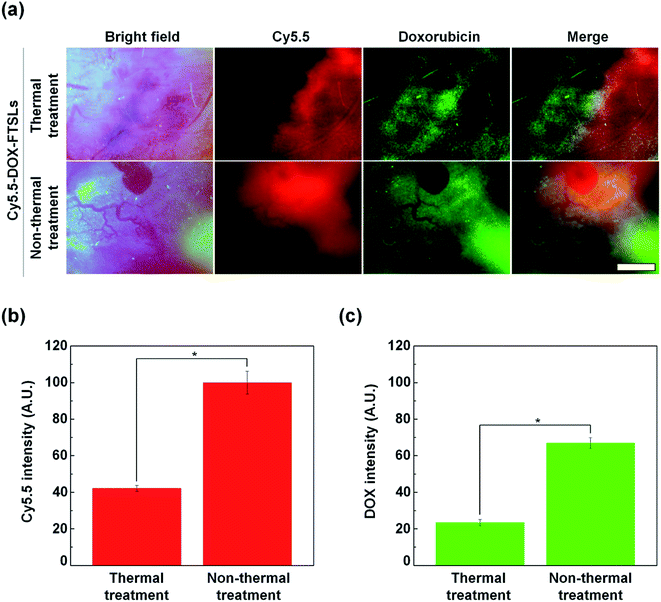 |
| | Fig. 6 (a) In vivo NIRF images of tumor vessels and tissues at 12 h after intravenous injection of Cy5.5-labeled DOX-FTSLs combined with thermal or non-thermal acoustic treatment (red: Cy5.5, green: DOX). Scale bar represents 150 μm. (b) Quantification of liposome accumulation at tumor tissues with Cy5.5 NIRF images. (c) Quantification of DOX accumulation at tumor tissues with DOX NIRF images. Error bars represent standard deviation (n = 3; *p < 0.05 calculated by one-way ANOVA). | |
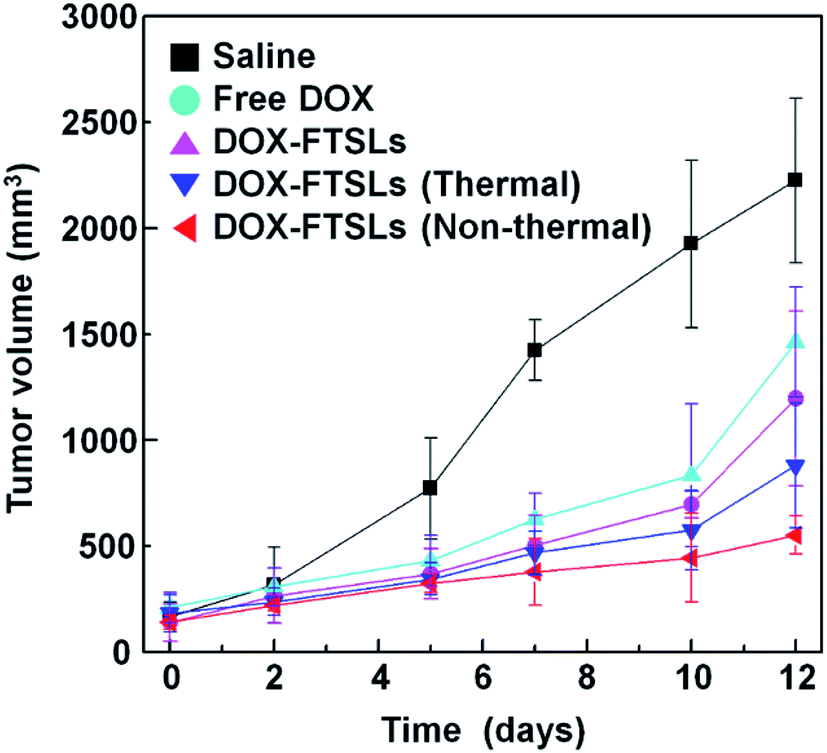
In vivo antitumor efficacy
Although non-thermal treatment has been favored for tumor-targeted drug delivery, whether it improves overall therapeutic efficacy compared with control groups still needed to be assessed. To evaluate the therapeutic efficacy of the treatment methods, in vivo tumor growth was examined by measuring tumor volumes over 12 days (Fig. 7). After administration of DOX-FTSLs, free DOX or saline at a dose of 5 mg DOX per kg, thermal or non-thermal acoustic treatment was immediately carried out as indicated. Quantified intratumoral DOX could be either encapsulated in the FTSLs or released from them. To reflect the drug action of total accumulated DOX, we needed to induce burst release of encapsulated DOX. Therefore, a second thermal acoustic treatment was applied to tumor tissues of acoustically-treated samples at 12 h post injection, when intratumoral DOX-FTSL accumulation was maximal and DOX-FTSLs were systemically circulating in the bloodstream (Fig. 5). Non-thermal acoustic treatment resulted in tumor growth inhibition that was comparable to thermal treatment with the lowest average final tumor volume; it was only 37.7% of the volume with DOX-FTSLs in the no acoustic treatment group and 62.7% of the volume with DOX-FTSLs in the thermal treatment group at the same dose. Thus, non-thermal acoustic treatment, which resulted in effective drug accumulation without irreversible vessel damage, may be a safe alternative to thermosensitive liposome-involved hyperthermia.
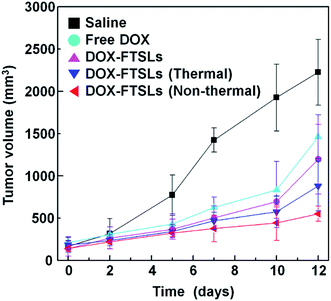 |
| | Fig. 7 Antitumor efficacy of DOX-FTSLs by treatment group. Error bars represent standard deviation (n = 3). | |
Conclusion
Hyperthermia-assisted liposomal chemotherapy is considered a synergistic combinational approach, since thermally triggered, tumor site-specific drug release is ideal for improving overall therapeutic efficacy while minimizing possible side effects of chemotherapeutic drugs. However, vascular damage often observed in HIFU focal regions of treated tumor tissues might be a limitation to necessary follow-up treatments that are common for conventional chemotherapy. We visualized tumor vessel damage caused by thermal acoustic treatment, and quantitatively assessed the resultant decrease in intratumoral accumulation of DOX-FTSLs and DOX compared with non-thermal treatment. Our results demonstrated the superior drug delivery potential and comparable therapeutic efficacy of non-thermal acoustic treatment compared with hyperthermia, suggesting the former as a safe alternative to partially replace hyperthermia in an overall treatment regimen. Though much work needs to be done to provide detailed guidelines for clinical applications, this simple strategy had the potential to improve clinical practice by offering more cancer treatment options.
Acknowledgements
This work was financially supported by the Basic Science Research Programs (NRF-2016R1A6A3A11932550 and NRF-2010-0027955), and the Global Research Laboratory Program (NRF-2016K1A1A2A02942563) through the National Research Foundation (NRF) of Korea. Additionally, it was supported by the Program for Returners into R&D (WISET-2016-253) through the NRF and the Center for Women in Science, Engineering and Technology.
References
- E. Blanco, H. Shen and M. Ferrari, Nat. Biotechnol., 2015, 33, 941–951 CrossRef CAS PubMed.
- J. Harris and B. Robert, Nat. Rev. Drug Discovery, 2003, 2, 214–221 CrossRef CAS PubMed.
- S. Jeon, H. Ko, R. N. Vijayakameswara, H. Y. Yoon, D. G. You, H. S. Han, W. Um, G. Saravanakumar and J. H. Park, RSC Adv., 2015, 5, 70352–70360 RSC.
- J. M. Cha, D. G. You, E. J. Choi, S. J. Park, W. Um, J. Jeon, K. Kim, I. C. Kwon, J. C. Park, H. R. Kim and J. H. Park, J. Biomed. Nanotechnol., 2016, 12, 1724–1733 CrossRef CAS.
- M. Swierczewska, H. S. Han, K. Kim, J. H. Park and S. Lee, Adv. Drug Delivery Rev., 2016, 99, 70–84 CrossRef CAS PubMed.
- Y. Matsumoto, J. W. Nichols, K. Toh, T. Nomoto, H. Cabral, Y. Miura, R. J. Christie, N. Yamada, T. Ogura, M. R. Kano, Y. Matsumura, N. Nishiyama, T. Yamasoba, Y. H. Bae and K. Kataoka, Nat. Nanotechnol., 2016, 11, 533–538 CrossRef CAS PubMed.
- H. Cabral, Y. Matsumoto, K. Mizuno, Q. Chen, M. Murakami, M. Kimura, Y. Terada, M. R. Kano, K. Miyazono, M. Uesaka, N. Nishiyama and K. Kataoka, Nat. Nanotechnol., 2011, 6, 815–823 CrossRef CAS PubMed.
- H. Maeda, J. Wu, T. Sawa, Y. Matsumura and K. Hori, J. Controlled Release, 2000, 65, 271–284 CrossRef CAS PubMed.
- H. Maeda, H. Nakamura and J. Fang, Adv. Drug Delivery Rev., 2013, 65, 71–79 CrossRef CAS PubMed.
- V. P. Torchilin, Nat. Rev. Drug Discovery, 2005, 4, 145–160 CrossRef CAS PubMed.
- A. Puri, K. Loomis, B. Smith, J.-H. Lee, A. Yavlovich, E. Heldman and R. Blumenthal, Crit. Rev. Ther. Drug Carrier Syst., 2009, 26, 523–580 CrossRef CAS PubMed.
- E. Yuba, Y. Kanda, Y. Yoshizaki, R. Teranishi, A. Harada, K. Sugimura, T. Izawa, J. Yamate, N. Sakaguchi, K. Koiwai and K. Kono, Biomaterials, 2015, 67, 214–224 CrossRef CAS PubMed.
- Y. Zhao, W. Ren, T. Zhong, S. Zhang, D. Huang, Y. Guo, X. Yao, C. Wang, W. Q. Zhang, X. Zhang and Q. Zhang, J. Controlled Release, 2016, 222, 56–66 CrossRef CAS PubMed.
- L. Li, T. Hagen, M. Hossann, R. Süss, G. Rhoon, A. Eggermont, D. Haemmerich and G. Koning, J. Controlled Release, 2013, 168, 142–150 CrossRef CAS PubMed.
- K. Kono, M. Takashima, E. Yuba, A. Harada, Y. Hiramatsu, H. Kitagawa, T. Otani, K. Maruyama and S. Aoshima, J. Controlled Release, 2015, 216, 69–77 CrossRef CAS PubMed.
- S. Dromi, V. Frenkel, A. Luk, B. Traughber, M. Angstadt, M. Bur, J. Poff, J. Xie, S. K. Libutti, K. C. P. Li and B. J. Wood, Clin. Cancer Res., 2007, 13, 2722–2727 CrossRef CAS PubMed.
- J. VanOsdol, K. Ektate, S. Ramasamy, D. Maples, W. Collins, J. Malayer and A. Ranjan, J. Controlled Release, 2017, 247, 55–63 CrossRef CAS PubMed.
- S. Rizzitelli, P. Giustetto, J. Cutrin, D. Castelli, C. Boffa, M. Ruzza, V. Menchise, F. Molinari, S. Aime and E. Terreno, J. Controlled Release, 2015, 202, 21–30 CrossRef CAS PubMed.
- T. Lajunen, L. Viitala, L. S. Kontturi, T. Laaksonen, H. Liang, E. Vuorimaa-Laukkanen, T. Viitala, X. Guével and M. Tliperttula, J. Controlled Release, 2015, 203, 85–98 CrossRef CAS PubMed.
- K. Carter, S. Shao, M. Hoopes, D. Luo, B. Ahsan, V. Grigoryants, W. Song, H. Huang, G. Zhang, R. Pandey, J. Geng, B. Pfeifer, C. Scholes, J. Ortega, M. Karttunen and J. Lovell, Nat. Commun., 2014, 3546 Search PubMed.
- G. Wu, A. Mikhailovsky, H. A. Khant, C. Fu, W. Chiu and J. A. Zasadzinski, J. Am. Chem. Soc., 2008, 130, 8175–8177 CrossRef CAS PubMed.
- G. Koning, A. Eggermont, L. Lindner and T. Hagen, Pharm. Res., 2010, 27, 1750–1754 CrossRef CAS PubMed.
- S. Mura, J. Nicolas and P. Couvreur, Nat. Mater., 2013, 12, 991–1003 CrossRef CAS PubMed.
- Z. S. Al-Ahmady, W. T. Al-Jamal, J. V. Bossche, T. T. Bui, A. F. Drake, A. J. Mason and K. Kostarelos, ACS Nano, 2012, 6, 9335–9346 CrossRef CAS PubMed.
- H. R. Kim, D. G. You, S.-J. Park, K.-S. Choi, W. Um, J.-H. Kim, J. H. Park and Y. S. Kim, Mol. Pharm., 2016, 13, 1528–1539 CrossRef CAS PubMed.
- S. M. Park, M. S. Kim, S.-J. Park, E. S. Park, K.-S. Choi, Y. S. Kim and H. R. Kim, J. Controlled Release, 2013, 170, 373–379 CrossRef CAS PubMed.
- F. Wua, W.-Z. Chena, J. Baia, J.-Z. Zoua, Z.-L. Wanga, H. Zhua and Z.-B. Wanga, Ultrasound Med. Biol., 2002, 28, 535–542 CrossRef.
- A. Schroeder, R. Honen, K. Turjeman, A. Gabizon, J. Kost and Y. Barenholz, J. Controlled Release, 2009, 137, 63–68 CrossRef CAS PubMed.
- V. Frenkel, Adv. Drug Delivery Rev., 2008, 60, 1193–1208 CrossRef CAS PubMed.
- H. Lee, H. R. Kim and J. C. Park, Phys. Chem. Chem. Phys., 2014, 16, 3763–3770 RSC.
- W. W. Sułkowski, D. Pentak, K. Nowak and A. Sułkowska, J. Mol. Struct., 2005, 744–747, 737–747 CrossRef.
- S. M. Park, J. M. Cha, J. Nam, M. S. Kim, S. J. Park, E. S. Park, H. Lee and H. R. Kim, PLoS One, 2014, 9, e103116 Search PubMed.
Footnote |
| † These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *ab
*ab