
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceComputational insights into substrate binding and catalytic mechanism of the glutaminase domain of glucosamine-6-phosphate synthase (GlmS)†
Wanlei Weia,
Gerald Monardb and
James W. Gauld *a
*a
aDepartment of Chemistry and Biochemistry, University of Windsor, Windsor, Ontario N9B 3P4, Canada. E-mail: gauld@uwindsor.ca; Tel: +1 253 3000 ext. 3992
bUniversité de Lorraine, UMR 7565 SRSMC, Boulevard des Aiguillettes B.P. 70239, F-54506 Vandoeuvre-les-Nancy, France
First published on 7th June 2017
Abstract
Glucosamine-6-phosphate synthase (GlmS) is a key enzyme in the biosynthesis of hexosamine across a variety of species including Escherichia coli, fungi, and humans. In particular, its glutaminase domain catalyzes the conversion of glutamine to glutamic acid with the release of ammonia. A catalytically important cysteinyl (Cys1) has been suggested to act as the mechanistic nucleophile after being activated by the N-terminal amine of the glutaminase domain (i.e., its own α-amine). Using molecular dynamics (MD) and quantum mechanics/molecular mechanics (QM/MM) computational methods, we have investigated the active site of the glutaminase domain, the protonation state of its N-terminal amine, substrate binding, and catalytic mechanism. In addition, the potential for an active site histidyl (His71) to alternatively act as the required base was examined. The N-terminal amine is concluded to have a reduced pKa due to being buried within the enzyme and the nearby presence of a protonated arginyl residue. Previous suggestions that this was due in part to hydrogen bonding with the hydroxyl of Thr606 is not supported; such an interaction is not consistent, and accounts for only 4% of the total duration of the MD simulation. The most feasible enzymatic pathway is found to involve a neutral N-terminal Cys1 α-amine acting as a base and directly deprotonating (i.e., without the involvement of a water, the Cys1SH thiol). The tetrahedral oxyanion intermediate formed during the mechanism is stabilized by a water and two enzyme residues: Asn98 and Gly99. Furthermore, the overall rate-limiting step of the mechanism is the nucleophilic attack of a water on the thioester cross-linked intermediate with a barrier of 74.4 kJ mol−1. An alternate mechanism in which His71 acts as the nucleophile-activating base, and which requires the Cys1 α-amine to be protonated, is calculated to be enzymatically feasible but to have a much higher overall rate-limiting barrier of 93.7 kJ mol−1.
Introduction
The class II glutamine-dependent amidotransferase (Gn-AT) family of enzymes is central to a variety of important physiological processes in a range of organisms from bacteria to mammals. They are multi-active site enzymes; each contains a functionally conserved glutaminase domain but possess a unique synthase domain. At least four known homologous proteins are included in this family of enzymes,1 which are essential for the biosynthesis of purines,2 asparagine,3 glutamate,4,5 and hexosamine.6–8 A crucial Gn-AT enzyme involved in the latter pathway is glucosamine-6-phosphate synthase (GlmS). More specifically, it is responsible for synthesizing D-glucosamine-6-phosphate (GlcN6P) from glutamine and D-fructose-6-phosphate.6GlcN6P is a key metabolic precursor for a plethora of important macromolecules in different organisms. For example, in humans and other mammals it is involved in the synthesis of a variety of glycoproteins, and as a sensory molecule of glucose uptake.8 Indeed, when adipocytes and fibroblasts were exposed to GlcN6P they developed insulin resistance9 and expressed phenotypes similar to type II diabetes in transgenic mice.7 Subsequent work on the inhibition of GlmS showed that insulin resistance could be prevented,10 leading to a number of proposed enzyme inhibitors.11 In bacteria, GlcN6P is the precursor of peptidoglycan and lipopolysaccharides that are the building blocks of their cell walls. Thus, there is an interest in the development of different inhibitors against GlmS as potential antibiotics.12–15 Similarly, in fungi, inhibitors of GlmS have gained interest because GlcN6P is the required substrate for chitin formation.14 Experimentally, it has been shown that inhibition of this enzyme in fungi for even a short period of time is lethal. In contrast, short-term inhibition of human GlmS is not lethal because it is quickly re-expressed and also its hexosamine products have reasonably long lifespans.15
Unlike other Gn-ATs, GlmS cannot use exogenous ammonia as a source of nitrogen.9 That is its overall synthetic role is critically dependent on the hydrolytic deamination of glutamine in its glutaminase domain to produce free ammonia.8 The latter is then shuttled to the synthase domain, over 18 Å away, via a hydrophobic channel that helps prevent its loss to the solvent.16,17 In the synthase domain the NH3 is reacted with D-fructose-6-phosphate (F6P) to produce GlcN6P. Protein crystallization with intermediate analogues have shown that GlmS catalysis is modular and occurs in a specific order.16 F6P binding in the synthase domain triggers the glutaminase domain to bind L-glutamine and the formation of the ammonia channel, though it is blocked by the Q-loop secondary structure. However, the sealing of the glutaminase domain after L-glutamine binding allows the tunnel to connect. After the products are formed in both domains, the L-glutamic acid leaves first, followed by the glucose-6-phosphate (G6P). This tight regulation also helps ensure that the ammonia from the glutaminase domain is not lost to the medium.16
Site-directed mutagenesis and kinetics experiments have suggested that for the catalytic mechanism of the glutaminase domain the thiolate derivative of a conserved N-terminal cysteinyl (Cys1) acts as a nucleophile and attacks the amide carbon centre of the substrate as shown in Scheme 1.16 Indeed, inhibition and mutational studies have shown that GlmS can be inactivated by covalent modification of the thiol of Cys1 by the glutamine analogue N3-(4-methoxyfumaroyl)-l-2,3-diaminopropanoic acid (FMDP).14,16 A previous study using in part the program PROPKA18 suggested that the buried N-terminal amine of Cys1 (Cys1NH2) has a markedly reduced pKa and as a result is likely neutral at physiological pH.16 Based on X-ray crystallographic structures it has been suggested that Cys1NH2 may be hydrogen bonded to the side-chain hydroxyl of Thr606, thus stabilizing the neutral form.6 In addition, experimental mutational and kinetic studies observed that glycylation of the Cys1NH2 moiety of the glutaminase domain drastically reduced the catalytic activity of GlmS.16 Consequently, the glutaminase domain has been proposed16 to use a mechanism analogous to that of other N-terminal nucleophile hydrolases;19 the α-amine of Cys1 acts as a base to activate (deprotonate) the thiol of Cys1 as shown in Scheme 1.
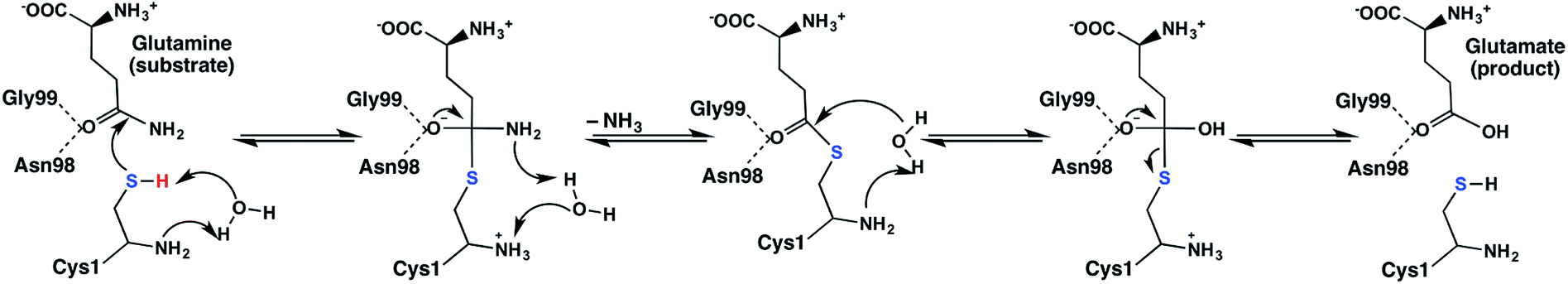 | ||
| Scheme 1 Proposed catalytic mechanism of the glutaminase domain of GlmS.16 | ||
More specifically, it has been proposed that the neutral α-amine of Cys1 helps deprotonate the thiol of Cys1 via a water molecule that bridges the two groups. The resulting thiolate is then able to nucleophilically attack the glutamine substrate to form a covalently cross-linked tetrahedral oxyanion intermediate. It should be noted that Asn98 and Gly99 may form an oxyanion hole and thus help stabilize the negative charge buildup on the substrate's oxygen.20 Subsequent collapse of the tetrahedral intermediate occurs with proton transfer, via the active site water, from the Cys1NH3+ group onto the leaving ammonia (NH3) derived from the glutamine substrate. The second half of the mechanism is hydrolysis of the thioester bond and in many aspects is the reverse of the first-half. Indeed, in the next step a water nucleophilically attacks the thioester intermediate at its carbonyl carbon (Ccarb) to form a second covalently cross-linked tetrahedral oxyanion intermediate. This step is facilitated by the once again neutral Cys1NH2 group which also accepts a proton from the reacting water. The oxyanion's Ccarb–SCys1 bond then cleaves resulting in formation of glutamic acid and regeneration of the active site Cys1 thiol (Scheme 1). The exact details by which the Cys1 thiolate is ultimately neutralized without the involvement of the water consumed during the mechanism is unclear.
Unfortunately, however, many of the exact details of the mechanism remain unclear. For instance, covalent modifications could impact catalytic efficiency via disruption of the active site structure due to possible steric clashes with neighboring residues.21 In addition, while its glutaminase activity was observed to be markedly reduced by covalent modifications, it was not eliminated. This may indicate that another active site residue could act as the base that deprotonates the Cys1 thiol. In contrast, Teplyakov et al.9 have suggested that an ammonium–thiolate ion pair may in fact be the actual resting state of the enzyme.
In this present study we have used molecular dynamics (MD) simulations and quantum mechanical (QM)-chemical cluster computational approaches to investigate the protonation state of key active site functional groups, e.g., the α-amine of Cys1, and subsequent substrate (glutamine) binding. An ONIOM(QM/MM) approach was then used to elucidate possible catalytic mechanisms of the glutaminase domain of glucosamine-6-phosphate synthase (GlmS) in which either a neutral N-terminus Cys1NH2 group or active site histidyl (His71) acts as the base that deprotonates the thiol of Cys1.
Computational methods
Preparation of the sample
The X-ray crystal structure of a homodimer of glucosamine-6-phosphate synthase (GlmS) from Escherichia coli (PDB ID: 2J6H), in complex with glucose-6-phosphate (Glc6P) and 5-oxo-L-norleucine (DON), was used as the initial template structure for the computational studies.6 This structure was selected because it was previously concluded to represent an active conformation of GlmS as both of its active sites contain bound ligands that are either product analogs or ultimately a mimic of a putative mechanistic intermediate, and a rotation has occurred of a tryptophan residue (Trp74) that opens the ammonia channel.6,16 Modification of DON to glutamine and Glc6P to fructose-6-phosphate (F6P) in the glutaminase and synthase domains respectively, was manually performed. More specifically, the covalent link between Cys1 and the delta carbon of DON was broken. In addition, the delta carbon was mutated to a nitrogen and protons were added according to the hybridization states of atoms.The enzyme was hydrogenated in accordance with PROPKA,18,22–24 with histidyls protonated according to their polar environment. The tleap module of AMBER14 (ref. 25) was used to build the topology and coordinate files. The enzyme was solvated with 87921 TIP3P26 water molecules, which resulted in cubic boxes with an edge length of ∼141.1 Å. In total, the system modeled consisted of 282![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 727 atoms.
727 atoms.
Molecular dynamics (MD) simulations
The AMBER14 program25 with the CUDA-enabled graphics processing units (GPUs) version of pmemd27,28 was used for all MD simulations. Proteins and waters were described using the ff14SB29 and TIP3P26 force fields, respectively. The neutral N-terminal cysteinyl (Cys1), glutamine, and F6P ligands were built with the antechamber30,31 program using the ff14SB force field and RESP atomic charges. It should be noted that the charges of Cys1 and F6P fragments, capped by methyl groups, were derived from gas phase optimizations at the HF/6-31G(d) level of theory using the Gaussian09 program.32 Periodic boundary conditions and an NVT ensemble were applied,33 while a cut-off of 8 Å was applied in real space for long-range electrostatic interactions using the Particle-Mesh Ewald (PME) procedure. A timestep of 2 fs was used for both equilibration and production MD's by restricting bond stretches associated with hydrogen atoms through the SHAKE algorithm. The equilibrations were conducted in five stages after energy minimization: (1) proper geometry of the hydrogen atoms, all heavy atoms including water oxygens, were restrained with a harmonic potential of 50 kcal mol−1 Å−2 for 100 ps, at 10 K; (2) an identical potential and conditions were applied for an additional 100 ps without restraining the water oxygens so as to ensure optimized positions of the waters with respect to the protein environment; (3) the harmonic potential restraint on the protein heavy atoms was decreased to 5 kcal mol−1 Å−2 for 100 ps; (4) the harmonic potential restraint was removed for 100 ps; and finally (5) the system was gradually heated to 300 K over a time period of 2000 ps. The velocities were randomly updated every 10 steps for equilibration stages 1–4 and every 100 steps for stage 5. The production run was conducted for 150 ns following equilibration.QM/MM calculations
All QM/MM calculations were performed within the ONIOM formalism as implemented in the Gaussian09 program.32 A cluster analysis of the MD simulation was done using the RMSD's of all residue Cα atoms that lie within 20 Å of the glutamine substrate in the crystal structure. A suitable, representative structure was chosen from the most frequent RMSD population. Due to our interest in studying the deamination reaction of GlmS, our model was extended by two layers of surrounding residues from the deamination active site, and included 1512 atoms. For the mechanism involving N-terminal Cys1NH2 group acting as the initial base, the QM region included Cys1, the –CαH2NH– groups of Gly2 and Gly99, Asn98 (except for its α-amino), and the glutamine substrate (except its α-amino and carboxylate). All other atoms were placed in the low (MM) layer, with regions on the exterior being restrained at their Cα atoms to keep the native conformation of the protein. It should be noted that the Cα atoms were held fixed at their position in the above selected representative structures. Unless otherwise noted, changes observed in the MM region upon going from the MD to QM/MM levels of theory were small to negligible. Optimized geometries of all energy minima (i.e., reactant, intermediate, and product complexes) and transition structures were obtained at the ONIOM(M062X/6-31G(d,p):AMBER96) level of theory within the mechanical embedding (ME) formalism, as were harmonic vibrational frequencies and thus Gibbs free energy corrections.34–36 Relative free energies were obtained by performing single point calculations on the above optimized structures at the ONIOM(M062X/6-311++G(2d,p):AMBER96)-ME level of theory, with inclusion of the appropriate Gibbs free energy correction. The M062X functional was chosen due to its ability to reliably reproduce experimental kinetic values,37–40 its improved descriptions of non-covalent interactions,41 and successful application to other biocatalysts.42 In addition, we note that similar ONIOM(QM/MM) approaches have been successfully applied to the study of mechanistically-related enzymes.43Results and discussion
Fully-bound GlmS–ligand complexes with a protonated or neutral Cys1 α-amine
The root-mean-square deviation (RMSD) of all residue Cα atoms within 20 Å of the glutamine substrate bound within the glutaminase domain's active site, relative to their initial crystal structure positions, was monitored over the course of 150 ns MD simulations on fully-bound GlmS with either a protonated or neutral Cys1 amino group. The resulting plots are shown in Fig. 1.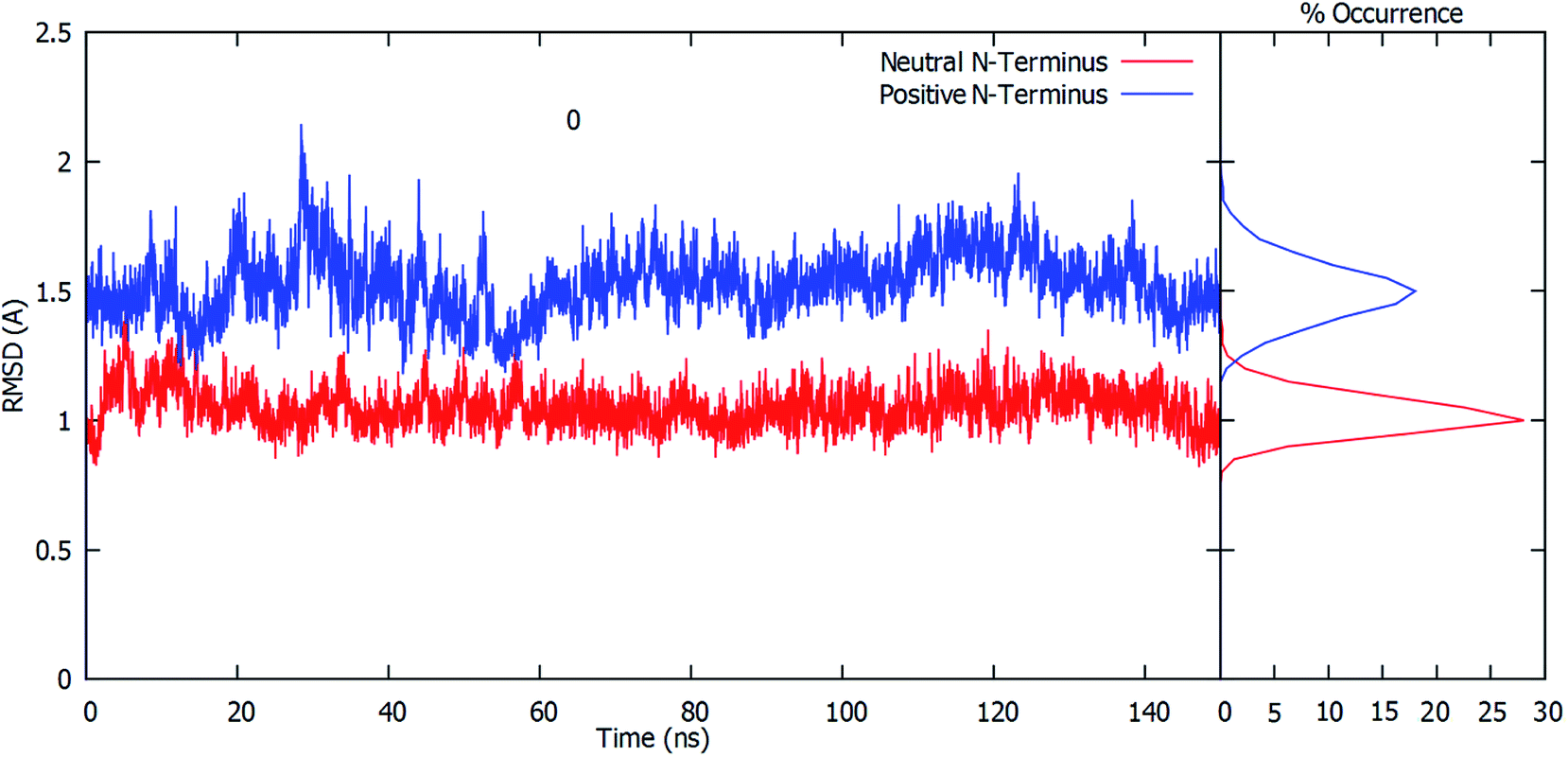 | ||
| Fig. 1 RMSDs (Å) plots of all residue Cα atoms within 20 Å of the active site-bound glutamine substrate, relative to their initial crystal structure positions, versus time (ns) when the Cys1 amino group is protonated (blue) or neutral (red), while the frequency of occurrence is shown on the right side of the graph. Note: the Cα atoms used are those that in the crystal structure were within 20 Å of the bound substrate (see Computational Methods). | ||
As can be seen, when the N-terminus cysteinyl α-amine is neutral (i.e., Cys1NH2) the observed RMSDs all lie reasonably consistently within a low, narrow range of approximately 0.8–1.2 Å. Indeed, further analysis of the calculated RMSD values indicates that the highest percent occurrence occurs for ∼1.0 Å. Hence, there appears to be close agreement with the initial starting structure derived from the reference crystal structure (see Computational Methods). In contrast, when the Cys1 α-amine is protonated (i.e., Cys1NH3+) the calculated RMSD values over the course of the simulation are decidedly higher and more broadly spread between approximately 1.2–1.9 Å, with some peaks outside this range. This is also clearly seen by the decidedly broader spread and lower percent occurrence for the corresponding calculated RMSD values, with the maximum occurrence now at ∼1.5 Å. This observed disparity in the RMSD's is due in part to small differences in the positioning of the active site residues (see below).
For both enzyme–substrate complexes (i.e., when the Cys1 α-amine is neutral or protonated, a representative structure corresponding to the most populated RMSD value (i.e., 1.0 and 1.5 Å, respectively) was obtained from the MD simulations. For each of these complexes the bound position of the glutamine substrate and key active site residues, as well as selected distances in Angstroms, are shown in Fig. 2. As can be seen, there appear to be distinct differences in the preferred structure of the active site and positioning of the substrate glutamine between when the Cys1 α-amine is neutral and protonated.
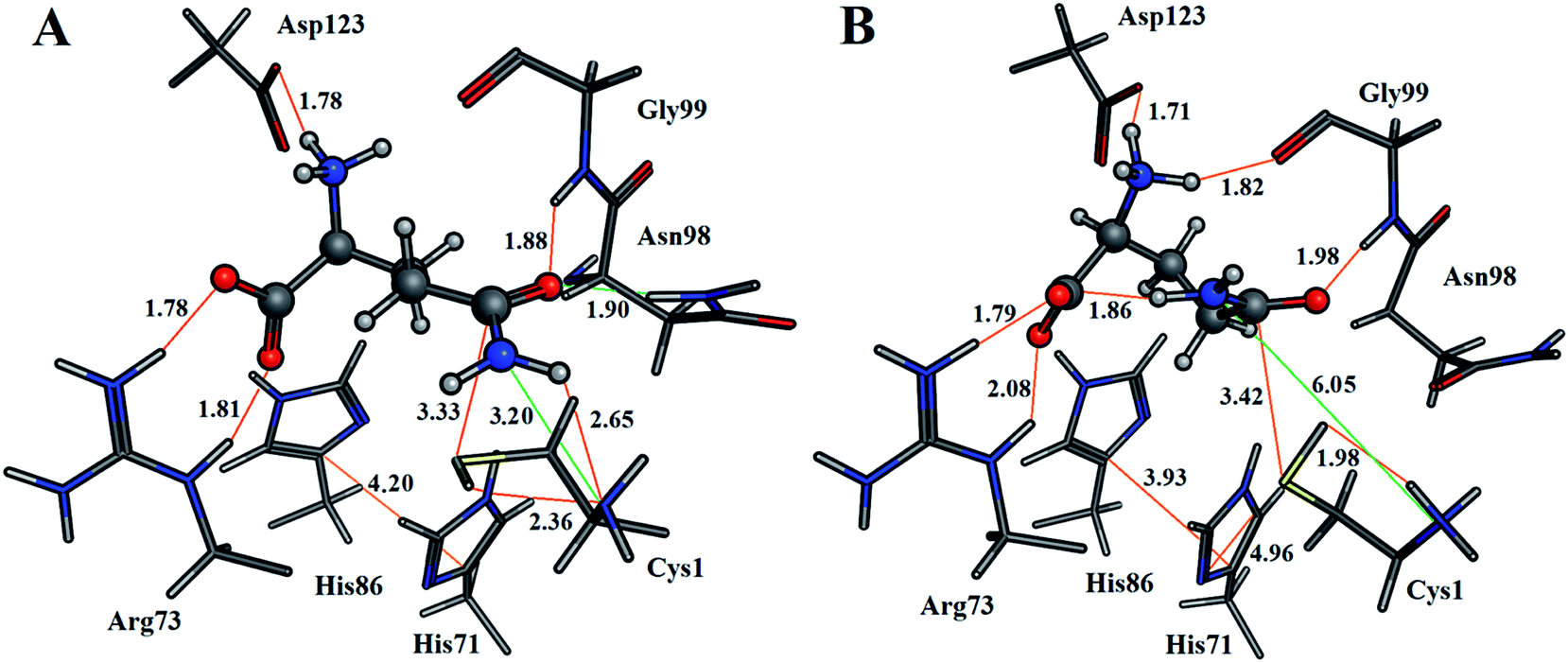 | ||
| Fig. 2 Representative structures showing the bound glutamine substrate and key active site moieties of the most populated RMSD values, with selected distances (Angstroms), obtained from the MD simulations for when the Cys1 α-amine is (A) neutral, or (B) protonated. | ||
To gain further insight into the bound-substrates conformational range in each complex, the RMSD's of the glutamine's heavy atoms over the course of the simulation were determined and shown in Fig. 3. When the Cys1 α-amine is neutral (Cys1NH2), only two distinct populations for the bound glutamine are observed. The most prevalent population has a maximum occurrence of slightly greater than 30% with an RMSD of ∼0.6 Å, while the second population has a maximum occurrence of ∼13% with an RMSD of 0.1 Å. These populations correspond to the glutamine conformation shown in Fig. 2A (conf. A), with φ(Cα–Cβ–Cγ–Cδ) and ψ(Cβ–Cγ–Cδ–Nε) dihedral angles of approximately 187° ± 23 and 193° ± 63, respectively. However, when the Cys1 α-amine is protonated (Cys1NH3+) three distinct populations are observed (see Fig. 3). Two of these appear to correspond to those previously observed when the Cys1 α-amine is neutral but now with a much lower occurrence of 3% or less. Instead, the most populated conformer with a 20% occurrence has a markedly higher RMSD of 1.7 Å with φ and ψ dihedral angles of approximately 342° ± 22 and 77° ± 22 and corresponds to that shown in Fig. 2B (conf. B). This suggests that if the Cys1 α-amine in fully-bound GlmS is protonated, the distinctly preferred conformation of the substrate is conf. B, although conf. A is possible.
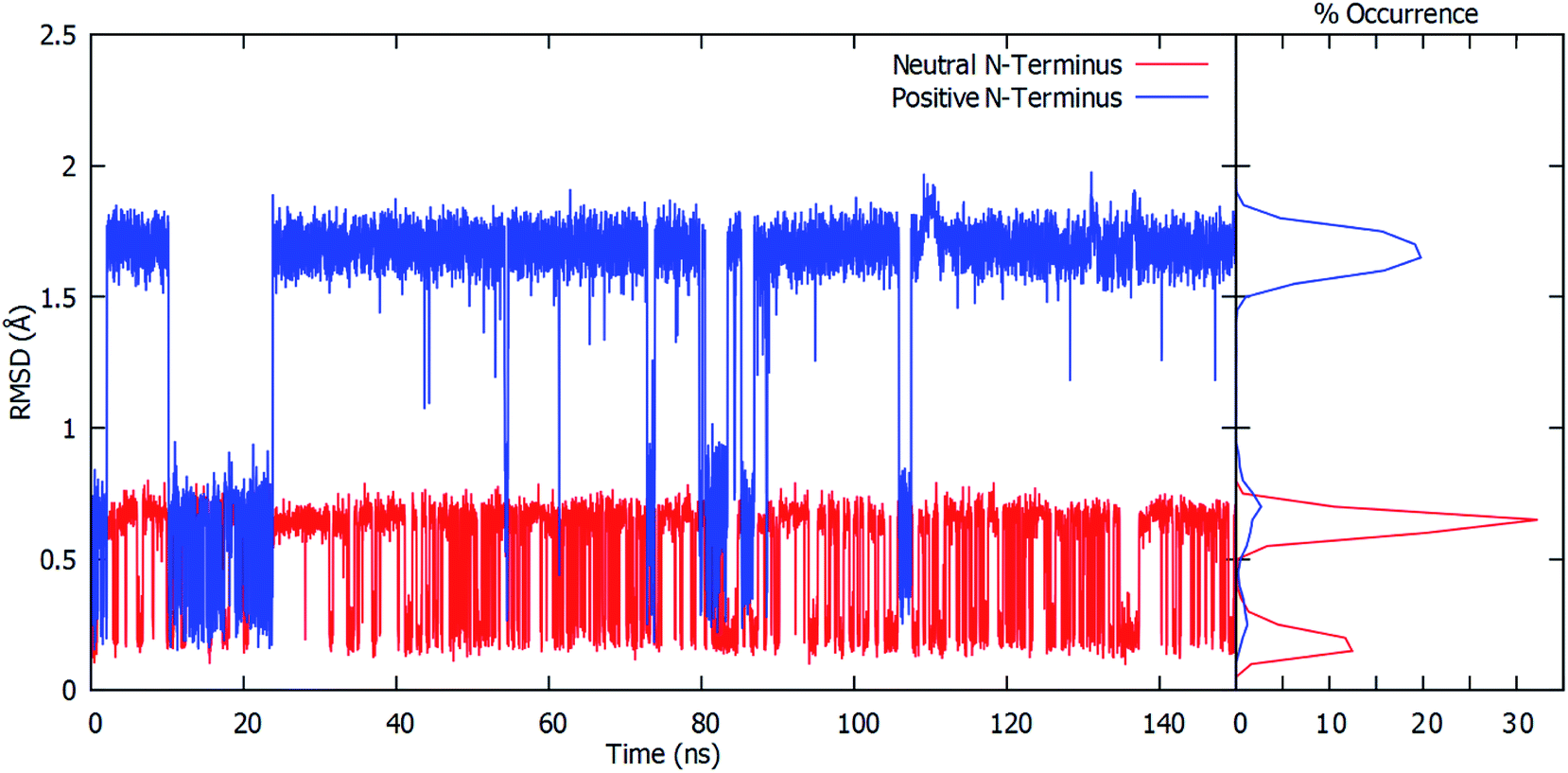 | ||
| Fig. 3 Plots of calculated RMSDs (Å) over the course of the MD simulations for all glutamine ligand heavy atoms relative to the initial conformation (see Computational Methods) versus time (ns) when the Cys1 α-amine is protonated (blue) or neutral (red). | ||
The enzyme–substrate hydrogen bond interactions were monitored throughout the MD simulations, with their individual % occurrence determined, and are shown in Fig. 4. One noted difference between the bound active sites for when Cys1 α-amine is neutral or protonated is the consistent formation (76% occurrence) in the latter of an intramolecular hydrogen bond between the substrates amide –NsubH2 group and one of its carboxylate oxygens, which is not observed when the Cys1 α-amine is neutral. This could be in part due to electrostatic interactions between the enzyme's Cys1NH3+ moiety and the substrates –NsubH2 group, causing the latter to shift. Indeed, it is noted that the distance between Cys1 α-amine nitrogen and the substrate's amide side chain nitrogen (Nsub) is significantly smaller in conf. A (3.20 Å) than in conf. B (6.05 Å), Fig. 2.
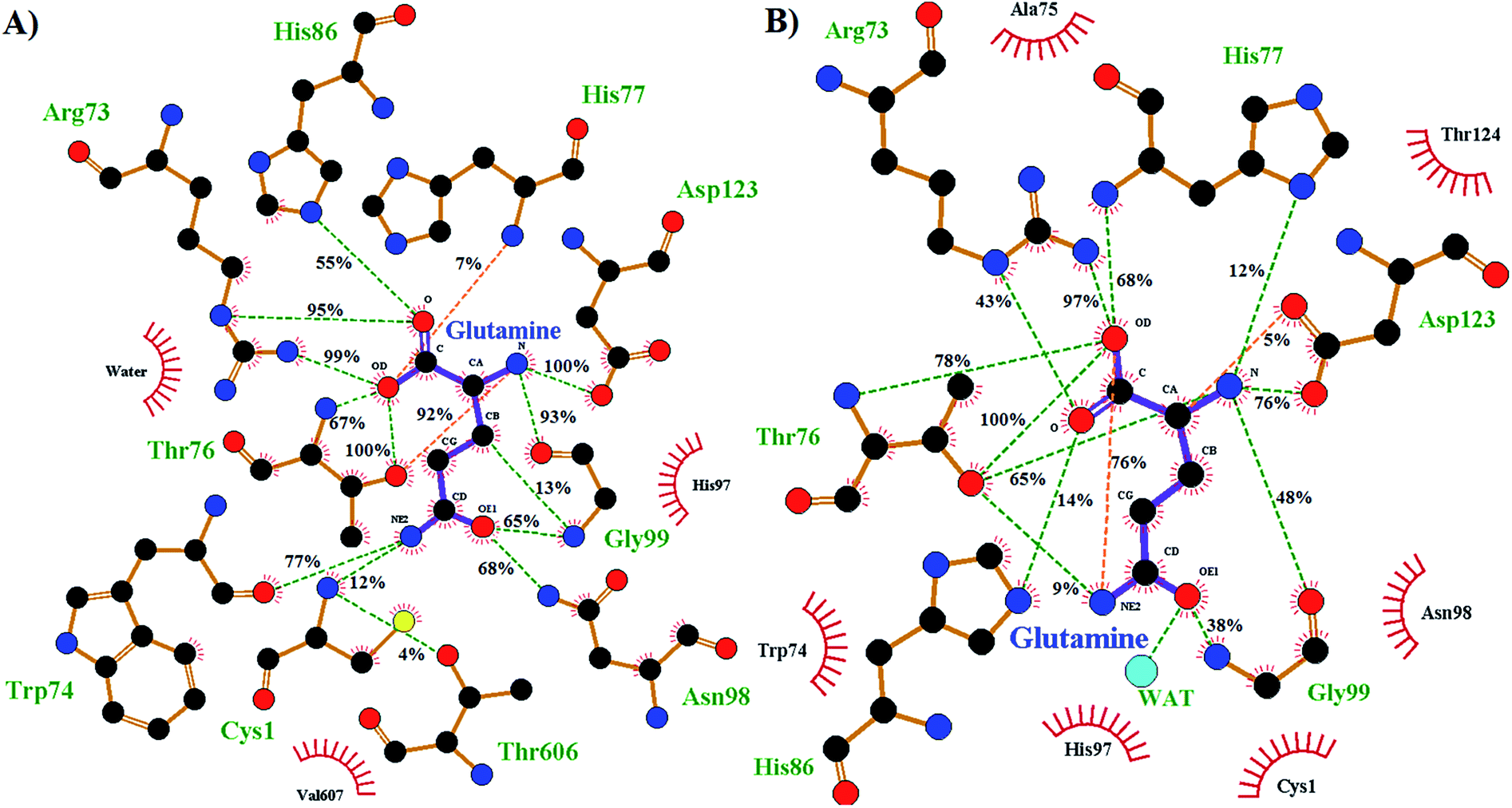 | ||
| Fig. 4 Ligand–enzyme hydrogen bond interaction maps of active site-bound glutamine, with percentage occurrence shown, for GlmS with a (A) neutral or (B) protonated Cys1 α-amine. | ||
During the MD simulation of GlmS with a neutral Cys1 α-amine (Cys1NH2), strong consistent hydrogen bonds between the substrate's side chain amide carbonyl oxygen (Osub) and –NH– and –NH2– groups of Gly99 (65%) and Asn98 (68%) respectively are observed (Fig. 4). Notably, these two residues have been previously suggested to form an oxyanion hole to stabilize the negative charge build up on Osub during the reaction.20 In contrast, when the Cys1 α-amine is protonated (Cys1NH3+) the Osub⋯HNGly99 hydrogen bond is considerably less consistent with only a 38% occurrence. Meanwhile, no Osub⋯H2NAsn98 hydrogen bond is observed as the Asn98 side-chain has rotated away from the substrate. Indeed, a 180° rotation of Asn98 along the dihedral angle of Cα–Cβ–Cγ–Nδ would be required in order for it to hydrogen bond with Osub. In place of Asn98, Osub instead forms a hydrogen bond with an active site water (see Fig. 4).
Arg26 has been suggested to be functionally important, helping to position the cys1NH2 group for catalysis.16 In the present MD simulations the Cys1N⋯Arg26Cζ distance for the neutral and protonated Cys1 α-amine complexes varied between 4–5 Å and 5–8 Å, respectively. No hydrogen bond between Arg26 and the Cys1 α-amine was observed in either case. Instead, the Arg26 side-chain interacts with the side-chain carboxylate of Asp192. Thus, it is possible that the role of the Arg26 is to provide an environment that favors a neutral Cys1NH2 group. Indeed, our present PROPKA analysis on the average GlmS structure with a neutral Cys1NH2 group, as well as a previous18 analysis, predicts the Cys1 α-amine to have a markedly reduced pKa due to both being buried and the nearby presence of Arg26. This is in contrast to previous suggestions that the reduced pKa of Cys1NH2 may be due in part to it hydrogen bonding with Thr606.16 Indeed, as seen in Fig. 4 such a hydrogen bonding interaction only occurred 4% of the time in the MD simulation on the fully-bound enzyme–ligand complex containing a neutral Cys1NH2. Regardless, the present results suggest that the Cys1 α-amine is predominantly neutral in the fully-bound complex, and that in such a complex the active site residues and bound glutamine substrate are well positioned for the subsequent reaction steps.
As detailed above, it is known that during the mechanism the sulfur of Cys1 nucleophilically attacks the glutamine substrates side chain amide carbon centre (Csub). However, for this to occur the Cys1 thiol needs to be activated, deprotonated, by a base. In Fig. 2A we can see that when Cys1NH2 is neutral the distance between Cys1's thiol hydrogen and Cys1NH2 nitrogen is only 2.36 Å, with the thiol and amine forming a direct weak Cys1N⋯HSCys1 hydrogen bond. While proton transfer between these groups has been suggested to possibly occur via a water bridge, based in part on a water occupying such a position in a ligand-free crystal structure of GlmS,16 no suitably positioned water(s) were observed during the simulation. In addition, it is noted that the mechanistically relevant Cys1S⋯(O)Csubstrate distance is 3.33 Å (Fig. 2A). In the alternate scenario where the Cys1 α-amine is protonated the Cys1S⋯(O)Csubstrate distance is only slightly longer at 3.42 Å (Fig. 2B). However, the Cys1NH3+ group is no longer able to directly activate the Cys1 thiol. The His71 imidazole (His71Im) is the nearest alternative functional group and residue that may be able to deprotonate the Cys1 thiol but from the MD simulations is on average ∼4.7 Å from the Cys1SH proton (Fig. S1†).
Hence, to gain further insights we examined both possible catalytic mechanisms in which either a neutral Cys1NH2 or, if the Cys1 α-amine is protonated, the His71Im activates the Cys1 thiol.
Catalytic mechanism with neutral Cys1NH2 as the activating base
The experimentally proposed mechanism20 suggested that the neutral N-terminal amino group of Cys1 (Cys1NH2) is able to act as the base that activates (deprotonates) the Cys1 thiol (Cys1SH), albeit via a water molecule bridge between Cys1SH and Cys1NH2 (see Scheme 1). Furthermore, this occurs concomitantly with nucleophilic attack of the resulting Cys1 thiolate sulfur on the substrates side chain carbonyl carbon (Csub). However, the present MD results found that the thiol and neutral α-amine of Cys1 are able to directly hydrogen bond with each other (Fig. 2A). While this does not absolutely preclude the possibility that a water could facilitate proton transfer between them, it does suggest that it could be possible without water. Hence, we examined possible catalytic mechanisms in which Cys1NH2 directly acts as the initial base that activates the Cys1 thiol. The free energy surface obtained is shown in Fig. 5. The corresponding optimized structures (i.e., minima and transition structures) with selected distances are shown in Fig. 6. It is noted that His71Im was kept neutral throughout this mechanism.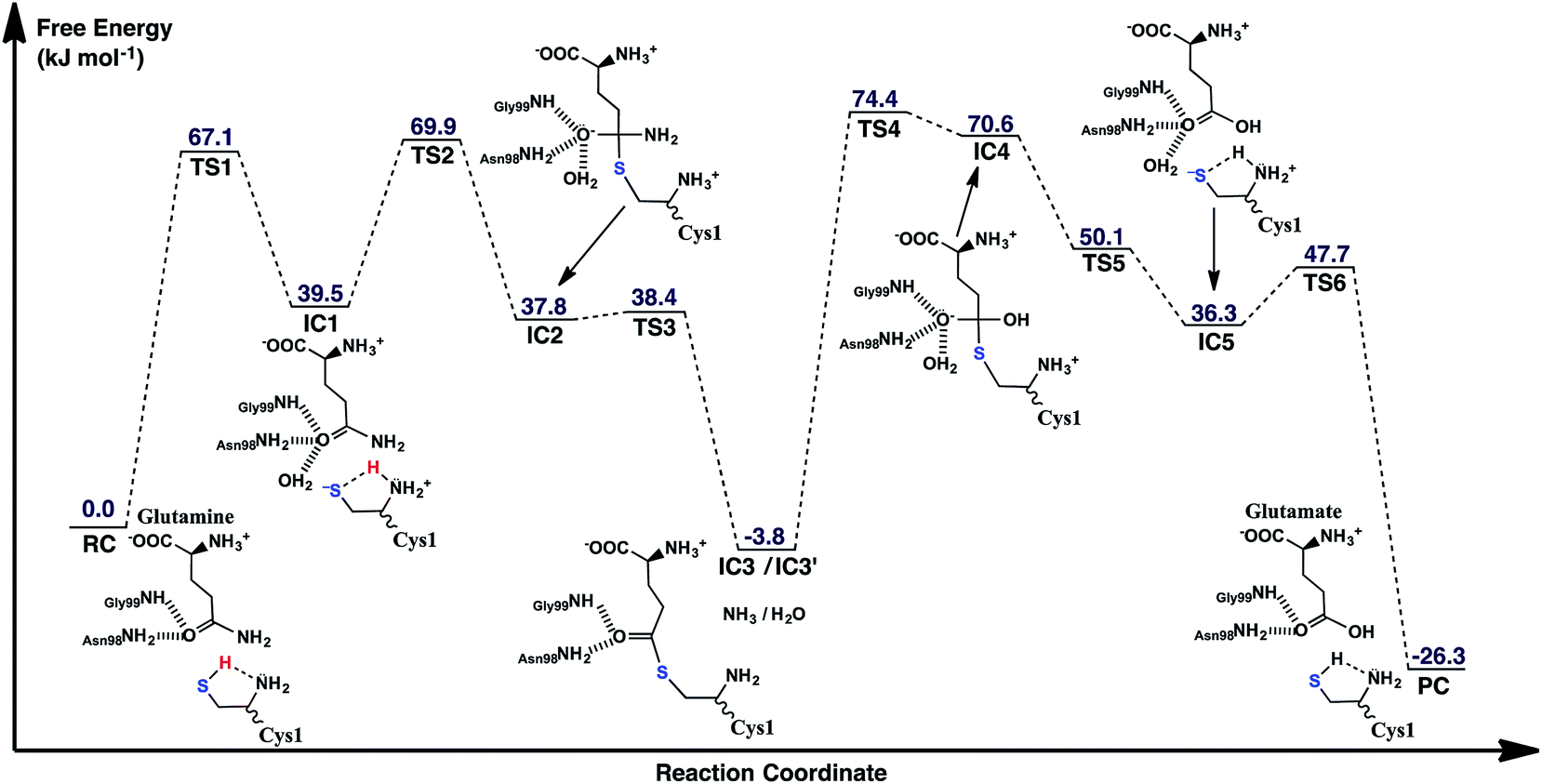 | ||
| Fig. 5 The free energy surface (in kJ mol−1) obtained for the conversion of glutamine to glutamic acid with release of ammonia in which the N-terminal Cys1 amine of GlmS acts as the mechanistic base. | ||
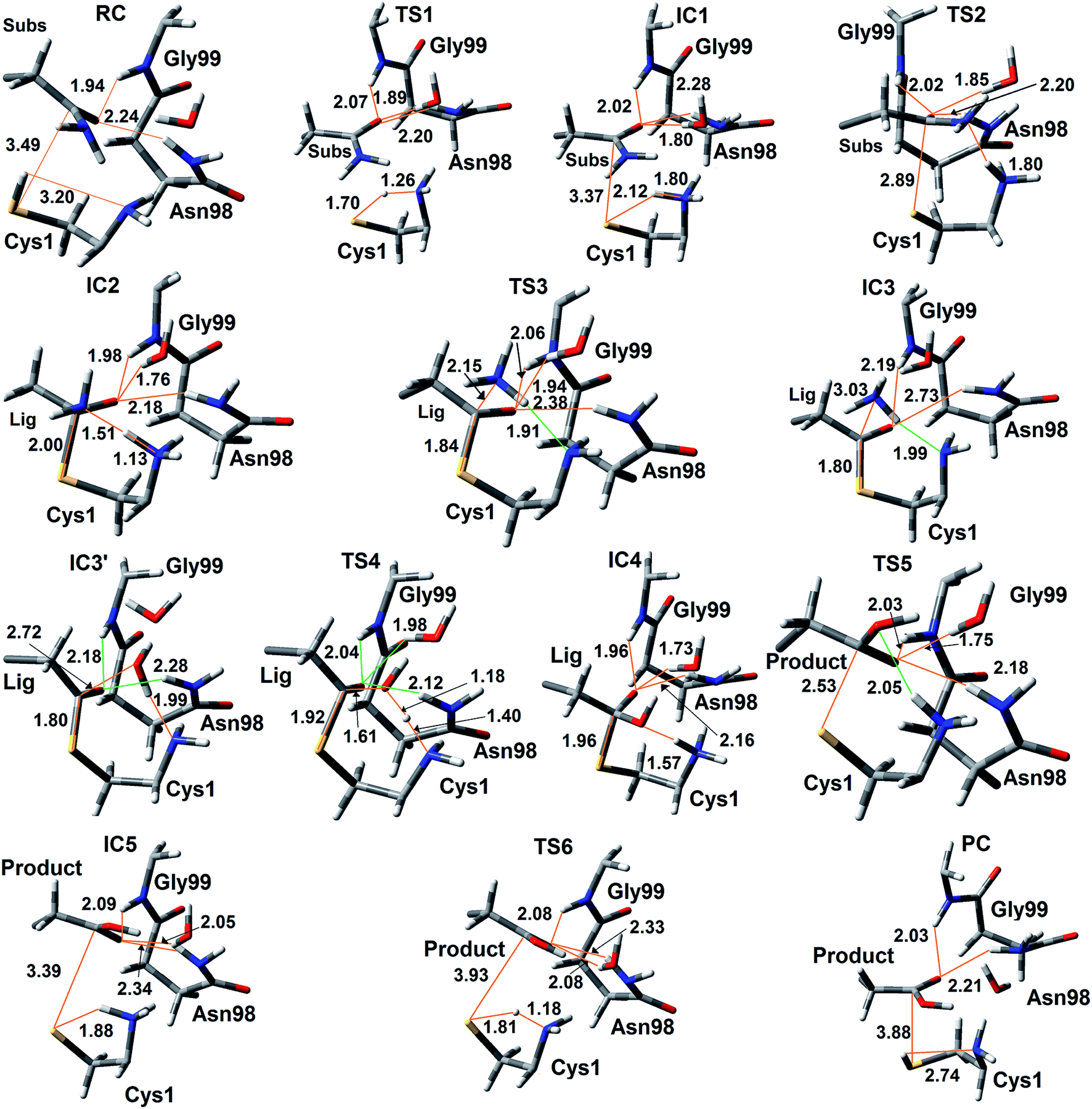 | ||
| Fig. 6 Optimized structures (see Computational Methods) of the reactant (RC), intermediate (IC), and product (PC) complexes, as well as transition structures (TS) obtained for the catalytic mechanism of the GlmS glutaminase domain in which the N-terminal Cys1NH2 group acts as the base. For clarity only key active site components and ligand, with selected bond lengths (Angstroms), are shown. | ||
The present results suggest that activation of the Cys1 thiol and nucleophilic attack of Cys1S− on the substrates side chain carbonyl carbon (Csub) occurs via a stepwise process. More specifically, as seen in Fig. 5, in the first step Cys1SH directly transfers a proton to Cys1NH2 via TS1 with a free energy barrier of only 67.1 kJ mol−1 relative to the initial reactant complex (RC). In the resulting activated intermediate, IC1, lying 39.5 kJ mol−1 higher in free energy than RC, the Cys1S− thiolate is stabilized by hydrogen bonds with both the now formally protonated Cys1NH3+ moiety, r(Cys1S−⋯HNCys1) = 2.12 Å and His71Im (Fig. 6). It is noted that in the MD simulations the latter group, His71Im, was observed to form a weak hydrogen bond with the Cys1 sulfur (i.e., Cys1S⋯HNεHis71) approximately 4.4% of the time; suggesting that it may also have a small role in orienting the Cys1 thiol for proton abstraction. This stabilization also likely helps facilitate the proton transfer as the same interactions are observed in TS1. The higher relative free energy of IC1 with respect to RC likely in part reflects the formation of the energetically less favoured thiolate anion Cys1S−. In IC1 the key Cys1S⋯Csub distance has shortened to 3.37 Å, a decrease of 0.12 Å from that observed in RC (Fig. 6). Furthermore, in addition to the backbone –NH– of Gly99 and side chain amide of Asn98, an active site water has also moved into hydrogen bonding distance of the glutamine substrates side chain carbonyl oxygen.
The next step is nucleophilic attack of the Cys1S− moiety on the substrates side chain carbonyl carbon (Csub). This occurs via TS2 at a cost of just 30.4 kJ mol−1 relative to IC1, or 69.9 kJ mol−1 with respect to RC. In fact this is the rate-limiting step for the first half of the overall mechanism; the deamination of glutamine. In the resulting tetrahedral oxyanion intermediate formed (IC2), the Cys1S–Csub bond has essentially been formed with a length of 2.0 Å. This is slightly longer than might be expected for a typical of S–C bond (∼1.8 Å). This is due in part to the fact that the Csub–O bond has elongated from 1.2 to 1.3 Å, indicating decreased double-bond character as a result of increased negative charge on the oxygen. However, this is still shorter than that of a typical C–O single bond indicating that the oxyanion has only been partially stabilized by the hydrogen bonds it forms with the side chain –NH2 of Asn98, the –NH– backbone of Gly99, and an active site water.
It is important to note that the substrate's side chain amide subNH2 group is now pyramidal, indicating loss of the double-bond character of the substrate's former side chain amide group upon going from IC1 to IC2 (Fig. 6). Significantly, it now forms strong hydrogen bonds with the protonated α-amine of Cys1 with a subN(H2)⋯+H3NCys1 distance of 1.51 Å. That is, IC2 is now appears nicely arranged for the subsequent required intramolecular proton transfer from Cys1NH3+ to the subN(H2) moiety, and that this may occur directly.
Indeed, the next step is collapse of the tetrahedral oxyanion intermediate IC2 with cleavage of the Csub–NH2 bond and concomitant transfer of a proton from Cys1NH3+ onto the leaving subNH2 group (Fig. 5). This occurs via the seven-membered ring transition structure TS3 with a free energy barrier of just 0.6 kJ mol−1 with respect to IC2, or 38.4 kJ mol−1 relative to RC. The resulting thioester intermediate IC3 formed with loss of NH3, lies slightly lower in energy than RC by 3.8 kJ mol−1. That is, formation of IC3 is exergonic with respect to RC. It is noted that with loss of the NH3 the Csub–SCys1 bond has shortened to 1.80 Å while the Csub centre has regained sp2 character with reformation of the Csub![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O carbonyl.
O carbonyl.
The structures RC to IC3 represent the first half of the deamination reaction. It is known from previous studies of GlmS and other amidotransferases that the free NH3 exits the glutaminase domain through an ammonia channel to the synthase site.9 Consequently, in order to complete the overall reaction, the free NH3 in IC3 was replaced by a water to give IC3′. The second-half of the mechanism is thus hydrolysis of the Csub–SCys1 bond, which is in some aspects the reverse of the first-half mechanism but with several key differences.
First, the active site water nucleophilically attacks the Csub centre of the covalently cross-linked enzyme-intermediate complex. This is facilitated by the N-terminus Cys1NH2 group which helps to activate the water by concomitantly accepting one of its protons. This step proceeds via the seven-membered ring transition structure TS4 with a free energy barrier of 78.2 kJ mol−1 with respect to IC3′. Furthermore, it is in fact the overall rate-limiting step of the mechanism. The resulting tetrahedral oxyanion intermediate formed, IC4, lies 70.6 kJ mol−1 higher in energy than RC. It is noted that this is decidedly higher in energy than the corresponding amine analogue IC2 (37.8 kJ mol−1; Fig. 5). Structurally, similar bond lengths and interactions are observed as in IC2. For example, in IC4, the Csub centre has changed from sp2 to sp3 hybridization, while the oxyanion centre is again stabilized by hydrogen bonds with the side chain amide of Asn98, the backbone –NH– of Gly99, and an active site water. In addition, the Csub–SCys1 bond has elongated by 0.16 Å to 1.96 Å and the Cys1 α-amine group has now become protonated (Fig. 6).
The apparent instability of the oxyanion intermediate is also suggested by that fact that it collapses without a barrier (TS5) to give the enzyme⋯glutamic acid product complex IC5 lying 40.1 kJ mol−1 higher in energy than IC3′, or 36.3 kJ mol−1 with respect to RC (Fig. 5). In IC5 the Cys1S–Csub bond has been cleaved as indicated by its significantly increased distance of 3.39 Å (Fig. 6). While the glutamic acid product now formed is bound within the active site. The Cys1S− thiolate formed is stabilized by a hydrogen bond with His71Im and, of particular note, a strong hydrogen bond with the now protonated Cys1NH3+ group with a length of 1.88 Å. Indeed, the final step, regeneration of a neutral Cys1SH thiol, can occur quite readily by direct transfer of a proton from Cys1NH3+ onto the Cys1S− thiolate. This step proceeds via TS6 with a free energy barrier of just 11.4 kJ mol−1 relative to IC5. The final enzyme⋯product complex (PC), with the product glutamic acid bound within the regenerated active site, lies 22.5 and 26.3 kJ mol−1 lower in free energy than IC3′ and RC respectively, indicating that hydrolysis of the thioester intermediate as well as the overall mechanism are exergonic.
Catalytic mechanism with His71 as the activating base
As noted above, in the substrate bound active site of the reactant complex RC, the His71Im group is positioned near the thiol of Cys1. Thus, given its possible potential to be the activating base, we also examined alternate mechanisms in which it plays this role instead of Cys1NH2. It should be noted, however, that to do so the N-terminus amine of Cys1 was protonated as no other acidic functional group appeared to be suitably positioned to protonate the leaving amine of the glutamine substrate. The free energy surface obtained is shown in Fig. 7 while the optimized structures of the corresponding reactant, intermediate, and product complexes, as well as transition structures are provided in the ESI, Fig. S2.†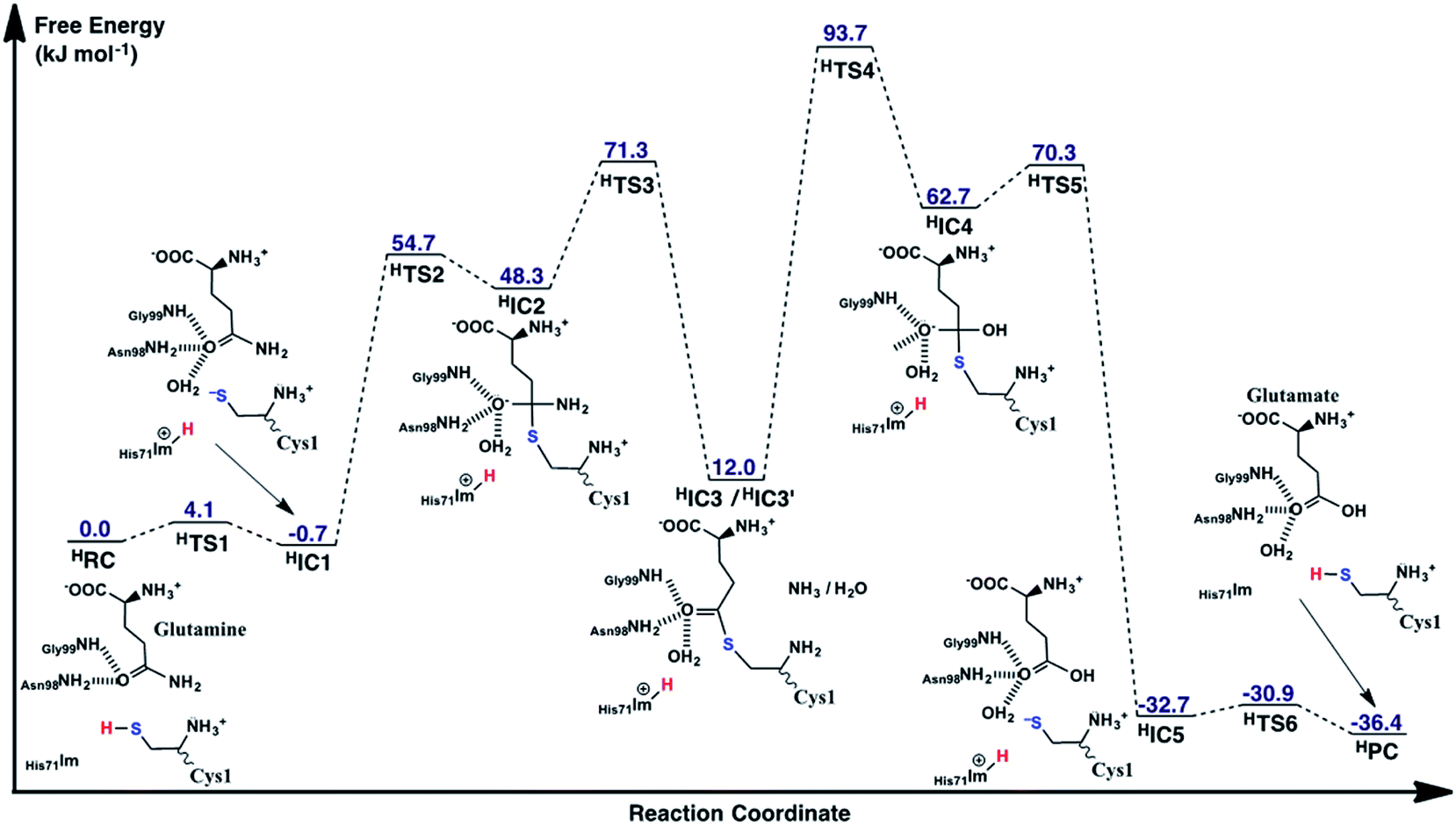 | ||
| Fig. 7 The free energy surface (in kJ mol−1) obtained for conversion of glutamine to glutamic acid with release of ammonia, when His71 acts as the base that activates the Cys1 thiol. | ||
While the overall mechanism is similar to that detailed above for the case of Cys1NH2 acting as the initial mechanistic base, there are a number of important differences. For instance, as seen in Fig. 7, the first-half of the reaction follows the same general sequence: (i) Cys1SH thiol activation, (ii) nucleophilic attack of the resulting Cys1S− thiolate on Csub to form a tetrahedral cross-linked oxyanion intermediate, and (iii) its subsequent collapse with loss of the side chain amine of the substrate glutamine.
Now, however, the initial activation of the Cys1 thiol occurs via proton transfer (HTS1) to His71Im with a very low free energy barrier of just 4.1 kJ mol−1 relative to HRC. The resulting intermediate HIC1, contains a protonated His71 (His71Im-H+) and Cys1S− thiolate, and lies marginally lower in energy than the initial reactant complex by 0.7 kJ mol−1. Subsequent nucleophilic attack of the Cys1S− sulfur at the substrate glutamine's carbonyl centre (Csub) proceeds via HTS2 at a cost of 54.7 kJ mol−1 relative to HRC. This barrier, relative to the initial reactant complex, is lower than that observed for the analogous step in the case of Cys1NH2 acting as the base (69.9 kJ mol−1; Fig. 5). This reflects in part the lower relative energy of HIC1 with respect to the initial reactant complex, i.e., greater stabilization of the Cys1S− thiolate by the His71Im-H+ and Cys1NH3+ groups with which it is hydrogen bonded. Indeed, considering just the individual step itself (i.e., HIC1 to HIC3 via HTS2) the barrier is 25.0 kJ mol−1 higher than for the analogous step where Cys1NH2 was the base (i.e., IC1 to IC3 via TS2), again emphasizing the stabilization of the thiolate ion in HIC1.
The resulting tetrahedral cross-linked oxyanion intermediate HIC2 lies 48.3 kJ mol−1 higher in energy than the initial reactant complex HRC. This is slightly higher than calculated for the stability of IC2 relative to its corresponding reactant complex RC (37.8 kJ mol−1). It is noted that the oxyanion centre of HIC2 is only stabilized by two hydrogen bonds; one with the side chain amide of Asn98 and the other with the backbone –NH– of Gly99. As a result, the Csub–O bond has less single-bond character which in turn causes the length of the newly formed Csy1S–Csub bond in HIC2 (2.19 Å; Fig. S1†) to be notably longer than observed in IC2 (2.00 Å; Fig. 6).
As for the case of Cys1NH2 acting as the initial base (see Fig. 5), collapse of the oxyanion intermediate HIC2 occurs via loss of the glutamine substrate's amide –NH2, with a concomitant proton transfer from the Cys1NH3+ group (Fig. 7). However, this now proceeds via HTS3 with a much larger barrier of 23.0 kJ mol−1 relative to HIC2, or 71.3 kJ mol−1 relative to the initial reactant complex HRC. In contrast, for the analogous step via TS3 the barrier was just 0.6 kJ mol−1 (see Fig. 5). Importantly, when His71 acts as the mechanistic base, collapse of the oxyanion intermediate is now the rate-limiting step in the first-half reaction of the mechanism, i.e., for just deamination of the glutamine substrate. The barrier for this rate-limiting step is also slightly higher in energy by 1.4 kJ mol−1 than the rate-limiting step of the first half reaction when Cys1NH2 acted as the initial base (i.e., formation of the tetrahedral oxyanion intermediate IC2; Fig. 5).
Unlike that observed when Cys1NH2 acted as the initial base, the resulting deaminated cross-linked intermediate (HIC3) is destabilized relative to initial reactant complex (HRC) by 12.0 kJ mol−1. These higher relative energies are at least partly due to a shift in the position of the enzyme–ligand covalent adduct as indicated by the considerably longer distances between the CsubO oxygen and both the Asn98 side chain amide, and backbone –NH– of Gly99. Specifically, in HIC3 these distances are 3.88 and 3.96 Å respectively (Fig. S1†), while in IC3 the corresponding distances are 2.73 and 2.19 Å (Fig. 6).
The second half of the mechanism, hydrolysis and breaking of the Csy1S–Csub bond, follows the same general process describe for Cys1NH2 as the initial base. More specifically, an active site water nucleophilically attacks the Csub centre of HIC3′ with concomitant transfer of one of its protons onto Cys1NH2. Again, this step is the rate-limiting step of the overall mechanism of the glutaminase domain of GlmS and proceeds via HTS4 with a barrier of 81.7 kJ mol−1 relative to HIC3′. The barrier for this individual step is only 3.5 kJ mol−1 higher than that for the corresponding step for Cys1NH2 as the base (i.e., IC3′ via TS4 to IC4). Importantly, however, HIC3′ lies 15.8 kJ mol−1 higher in energy compared to IC3′ (see Fig. 5). Thus, HTS4 is in fact 93.7 kJ mol−1 higher in energy than the initial reactant complex HRC. In contrast, when Cys2NH2 acted as the initial base, the analogous step (i.e., conversion of IC3′ to IC4 via TS4), which was also the overall rate limiting step of the mechanism, had a barrier of 74.4 kJ mol−1 relative to its corresponding reactant complex RC. Thus, while the overall mechanism in which His71 acts as the activating base may be enzymatically feasible, it will likely occur much more slowly than when Cys1NH2 plays the same role.
The resulting tetrahedral oxyanion intermediate HIC4 lies 62.7 kJ mol−1 higher in energy than HRC. The oxyanion centre itself is stabilized by hydrogen bonds with the amide side chain of Asn98, backbone –NH– of Gly99 and an active site water. The Cys1S–Csub bond has also lengthened to 2.02 Å with the Cys1S centre now being 2.40 Å from the His71Im-H+ proton. Subsequent cleavage of the Cys1S–Csub bond occurs via HTS5 with a barrier of just 7.6 kJ mol−1 to give the thiolate containing enzyme⋯product complex HIC5. In the latter complex, lying 32.7 kJ mol−1 lower in energy the HRC, the glutamic acid product has been formed while the Cys1S− thiolate is stabilized by moderately strong hydrogen bonds with the protonated imidazole of His71 and N-terminus amine of Cys1 with distances of 2.21 and 2.30 Å, respectively (Fig. S1†). Neutralization of Cys1S− can then occur quite readily through HTS6 at a cost of just 1.8 kJ mol−1, to give the final enzyme⋯product complex HPC lying slightly lower in energy than HIC5 by −3.7 kJ mol−1 (i.e., −36.4 kJ mol−1 relative to HRC). It is noted that it is also 10.1 kJ mol−1 lower in relative energy than the enzyme⋯product complex PC formed when Cys1NH2 was the activating base.
Due to the conserved nature of the glutaminase domain in class II amidotransferases,1 the present results obtained may be applicable to other related family members. Class I amidotransferases, on the other hand, differ structurally and in the activation of the nucleophilic cysteinyl thiol group.1 In the present study on Cys1NH2 acting as the thiol-activating base, the three highest barrier reaction steps lie within just 7.5 kJ mol−1 of each other with respect to RC (Fig. 5). Thus, potentially, more extensive computational models, or slight differences amongst active sites of different but related enzymes, may impact specific details but the general overall mechanism is likely the same. For example, we recently42 computationally examined Streptococcus pneumoniae nicotinamidase (SpNic) which hydrolyzes the amide bond in nicotinamide. It also uses an active site cysteinyl as its nucleophile that must be activated by a nearby base, but the base is now an aspartyl; Asp9. Importantly, SpNic uses an overall mechanism analogous to that of the glutaminase domain of GlmS. In particular, the first half reaction is also deprotonation of the cysteinyl thiol and formation of a covalently cross-linked enzymeCys–ligand intermediate. This was calculated, using a methodology similar to that employed herein, to occur in two steps with a rate limiting step of 65–69 kJ mol−1. The present study predicts a similar mechanism and barrier in GlmS; the barrier for the third step is negligible at 0.6 kJ mol−1 (see Fig. 5). In addition, the subsequent hydrolysis of the cross-linked thioester intermediate in SpNic was predicted to occur in one step, again similar to that found in the present study, but with a barrier of 57–62 kJ mol−1. Thus, the overall mechanism suggested herein for the glutaminase domain of GlmS does appear to share general features with those of some other relevant enzymes.
Conclusion
In this present study we have computationally investigated the structure and catalytic mechanism of the glutaminase domain, conserved amongst class II amidotransferases, of the physiologically important enzyme glucosamine-6-phosphate synthase (GlmS). More specifically, we have complementarily applied molecular dynamics (MD) simulations and quantum mechanics/molecular mechanics (QM/MM) methods to investigate the protonation state of the crucial N-terminus amino group of the Cys1 residue (Cys1NH2), and its influence on the active site and substrate (glutamine) binding. An ONIOM(QM/MM) approach was used to elucidate catalytic mechanisms in which either a neutral N-terminus Cys1NH2 group or nearby histidyl (His71) acts as the required initial base that activates (deprotonates) the thiol of Cys1.MD simulations were performed on fully-bound enzyme⋯substrate reactant complexes in which the N-terminus amino Cys1NH2 was neutral or protonated. Closest agreement with the experimental X-ray crystallographic structure, with RMSDs of just 0.8–1.2 Å, were obtained when the N-terminus amine was neutral (i.e., Cys1NH2). The bound glutamine substrate was also observed to be better-positioned within the active site for the subsequent catalytic deamination. Its side chain carbonyl oxygen hydrogen bonds with both the side chain amide of Asn98 and backbone –NH– of Gly99; the putative functional groups of the oxyanion hole. Meanwhile, the distance between the substrate's side chain carbonyl carbon (Csub), the site of nucleophilic attack, and the Cys1S nucleophile is 3.3 Å. In contrast, when the Cys1 α-amine is protonated (i.e., Cys1NH3+) the RMSDs are larger at 1.2–1.8 Å. Furthermore, the interactions between the substrates carbonyl oxygen and oxyanion hole functional groups are disrupted, while the Csub⋯SCys1 distance increased slightly to 3.4 Å.
The present results suggest that the Cys1 α-amine has a significantly reduced pKa, and is thus likely to be neutral at physiological pH, due to both being buried within the enzyme and the nearby protonated guanidinium of the active site aringyl, Arg26. In contrast, the previous suggestion that neutralization of the Cys1NH2 group may be due at least in part to hydrogen bonding with the Thr606 hydroxyl is concluded to be unlikely. Such an interaction is only observed to occur 4% of the time during the MD simulations.
In addition, at no time during the MD simulations is a water observed to form a previously suggested hydrogen bond bridge between the Cys1NH2 and Cys1SH groups.
Hence, the most likely catalytic mechanism is determined to involve a neutral Cys1NH2 group acting as the base that activates the Cys1 thiol. In particular it is observed that: (i) transfer of the proton from Cys1SH to Cys1NH2 can occur directly (i.e., without an intermediary H2O) with a free energy barrier of 67.1 kJ mol−1 relative to the initial reactant complex (RC); (ii) the rate-limiting step of the mechanisms first stage (deamination of the glutamine substrate with formation of the cross-linked enzyme–ligand intermediate (IC3/IC3′)) is nucleophilic attack of the Cys1S− thiolate on the substrate's Csub center with a barrier of 69.9 kJ mol−1 relative to RC; (iii) the overall rate-limiting step of the mechanism occurs in stage 2 (hydrolysis of the Cys1S–Csub bond and product formation) and is nucleophilic attack of a H2O at the Csub center of IC3′ with a barrier of 74.4 kJ mol−1 with respect to RC; (iv) the oxyanion centers of the tetrahedral intermediates formed in both stage 1 and stage 2 of the mechanism are stabilized by hydrogen bonds with the side chain amide of Asn98, backbone –NH– of Gly99, and an active site water; (v) the overall mechanism is exergonic with the product complex (PC) lying 26.3 kJ mol−1 lower in energy than RC. Although an alternative reaction mechanism whereby a nearby His71 extracts the proton of the Cys1 thiol group is possibly enzymatically feasible, it seems unlikely based on the protonation states of GlmS. Furthermore, the barrier for its rate limiting step, nucleophilic attack of a water on the Csub centre of the Csub–SCys1 covalently cross-linked intermediate, is significantly higher at 93.7 kJ mol−1.
Funding sources
The authors thank the Natural Sciences and Engineering Research Council (NSERC) for funding, MITACS Globalink Research Award-Campus France (WW) for an award, and Compute Canada, Sharcnet, and Université de Lorraine for high-performance computational resources.Conflict of interest
The authors declare no competing financial interest.Acknowledgements
We acknowledge helpful discussions with past and present members of the Gauld group and computational groups at Université de Lorraine.References
- F. Massiere and M. A. Badet-Denisot, The mechanism of glutamine-dependent amidotransferases, Cell. Mol. Life Sci., 1998, 54, 205–222 CrossRef CAS PubMed.
- P. Boer and O. Sperling, Role of Cellular Ribose-5-Phosphate Content in the Regulation of 5-Phosphoribosyl-1-Pyrophosphate and De-Novo Purine Synthesis in a Human Hepatoma-Cell Line, Metab., Clin. Exp., 1995, 44, 1469–1474 CrossRef CAS PubMed.
- E. E. Palmer, J. Hayner, R. Sachdev, M. Cardamone, T. Kandula, P. Morris, K. R. Dias, J. Tao, D. Miller, Y. Zhu, R. Macintosh, M. E. Dinger, M. J. Cowley, M. F. Buckley, T. Roscioli, A. Bye, M. S. Kilberg and E. P. Kirk, Asparagine Synthetase Deficiency causes reduced proliferation of cells under conditions of limited asparagine, Mol. Genet. Metab., 2015, 116, 178–186 CrossRef CAS PubMed.
- M. A. Vanoni and B. Curti, Structure-function studies of glutamate synthases: A class of self-regulated iron-sulfur flavoenzymes essential for nitrogen assimilation, IUBMB Life, 2008, 60, 287–300 CrossRef CAS PubMed.
- R. H. H. van den Heuvel, D. I. Svergun, M. V. Petoukhov, A. Coda, B. Curti, S. Ravasio, M. A. Vanoni and A. Mattevi, The active conformation of glutamate synthase and its binding to ferredoxin, J. Mol. Biol., 2003, 330, 113–128 CrossRef CAS PubMed.
- S. Mouilleron, M. A. Badet-Denisot and B. Golinelli-Pimpaneau, Glutamine binding opens the ammonia channel and activates glucosamine-6P synthase, J. Biol. Chem., 2006, 281, 4404–4412 CrossRef CAS PubMed.
- L. F. Hebert Jr, M. C. Daniels, J. Zhou, E. D. Crook, R. L. Turner, S. T. Simmons, J. L. Neidigh, J. S. Zhu, A. D. Baron and D. A. McClain, Overexpression of glutamine: fructose-6-phosphate amidotransferase in transgenic mice leads to insulin resistance, J. Clin. Invest., 1996, 98, 930–936 CrossRef CAS PubMed.
- A. Teplyakov, G. Obmolova, M. A. Badet-Denisot and B. Badet, The mechanism of sugar phosphate isomerization by glucosamine 6-phosphate synthase, Protein Sci., 1999, 8, 596–602 CrossRef CAS PubMed.
- A. Teplyakov, C. Leriche, G. Obmolova, B. Badet and M. A. Badet-Denisot, From Lobry de Bruyn to enzyme-catalyzed ammonia channelling: molecular studies of D-glucosamine-6P synthase, Nat. Prod. Rep., 2002, 19, 60–69 RSC.
- S. Marshall, V. Bacote and R. R. Traxinger, Discovery of a metabolic pathway mediating glucose-induced desensitization of the glucose transport system. Role of hexosamine biosynthesis in the induction of insulin resistance, J. Biol. Chem., 1991, 266, 4706–4712 CAS.
- S. Milewski, A. Janiak and M. Wojciechowski, Structural analogues of reactive intermediates as inhibitors of glucosamine-6-phosphate synthase and phosphoglucose isomerase, Arch. Biochem. Biophys., 2006, 450, 39–49 CrossRef CAS PubMed.
- M. A. Khan, Y. Gopel, S. Milewski and B. Gorke, Two Small RNAs Conserved in Enterobacteriaceae Provide Intrinsic Resistance to Antibiotics Targeting the Cell Wall Biosynthesis Enzyme Glucosamine-6-Phosphate Synthase, Front. Microbiol., 2016, 7, 908 Search PubMed.
- S. L. Bearne and C. Blouin, Inhibition of Escherichia coli glucosamine-6-phosphate synthase by reactive intermediate analogues. The role of the 2-amino function in catalysis, J. Biol. Chem., 2000, 275, 135–140 CrossRef CAS PubMed.
- M. Wojciechowski, S. Milewski, J. Mazerski and E. Borowski, Glucosamine-6-phosphate synthase, a novel target for antifungal agents. Molecular modelling studies in drug design, Acta Biochim. Pol., 2005, 52, 647–653 CAS.
- S. L. Bearne, Active site-directed inactivation of Escherichia coli glucosamine-6-phosphate synthase. Determination of the fructose 6-phosphate binding constant using a carbohydrate-based inactivator, J. Biol. Chem., 1996, 271, 3052–3057 CAS.
- S. Mouilleron, M. A. Badet-Denisot, B. Badet and B. Golinelli-Pimpaneau, Dynamics of glucosamine-6-phosphate synthase catalysis, Arch. Biochem. Biophys., 2011, 505, 1–12 CrossRef CAS PubMed.
- N. Floquet, S. Mouilleron, R. Daher, B. Maigret, B. Badet and M. A. Badet-Denisot, Ammonia channeling in bacterial glucosamine-6-phosphate synthase (Glms): molecular dynamics simulations and kinetic studies of protein mutants, FEBS Lett., 2007, 581, 2981–2987 CrossRef CAS PubMed.
- M. H. M. Olsson, C. R. Sondergaard, M. Rostkowski and J. H. Jensen, PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical pK(a) Predictions, J. Chem. Theory Comput., 2011, 7, 525–537 CrossRef CAS PubMed.
- (a) H. J. Duggleby, S. P. Tolley, C. P. Hill, E. J. Dodson, G. Dodson and P. C. E. Moody, Penicillin acylase has a single-amino-acid catalytic centre, Nature, 1995, 373, 264–268 CrossRef CAS PubMed; (b) C. E. McVey, M. A. Walsh, G. G. Dodson, K. S. Wilson and J. A. Brannigan, Crystal structures of penicillin acylase enzyme-substrate complexes: structural insights into the catalytic mechanism, J. Mol. Biol., 2001, 313, 139–150 CrossRef CAS PubMed; (c) C. Oinonen and J. Rouvinen, Structural comparison of Ntn-hydrolases, Protein Sci., 2000, 9, 2329–2337 CrossRef CAS PubMed.
- S. Mouilleron and B. Golinelli-Pimpaneau, Domain motions of glucosamine-6P synthase: comparison of the anisotropic displacements in the crystals and the catalytic hinge-bending rotation, Protein Sci., 2007, 16, 485–493 CrossRef CAS PubMed.
- J. M. Word, R. C. Bateman, B. K. Presley, S. C. Lovell and D. C. Richardson, Exploring steric constraints on protein mutations using MAGE/PROBE, Protein Sci., 2000, 9, 2251–2259 CrossRef CAS PubMed.
- D. C. Bas, D. M. Rogers and J. H. Jensen, Very fast prediction and rationalization of pK(a) values for protein-ligand complexes, Proteins: Struct., Funct., Bioinf., 2008, 73, 765–783 CrossRef CAS PubMed.
- H. Li, A. D. Robertson and J. H. Jensen, Very fast empirical prediction and rationalization of protein pK(a) values, Proteins: Struct., Funct., Bioinf., 2005, 61, 704–721 CrossRef CAS PubMed.
- C. R. Sondergaard, M. H. M. Olsson, M. Rostkowski and J. H. Jensen, Improved Treatment of Ligands and Coupling Effects in Empirical Calculation and Rationalization of pK(a) Values, J. Chem. Theory Comput., 2011, 7, 2284–2295 CrossRef CAS PubMed.
- D. A. Case, J. T. Berryman, R. M. Betz, D. S. Cerutti, T. E. Cheatham III, T. A. Darden, R. E. Duke, T. J. Giese, H. Gohlke, A. W. Goetz, N. Homeyer, S. Izadi, P. Janowski, J. Kaus, A. Kovalenko, T. S. Lee, S. LeGrand, P. Li, T. Luchko, R. Luo, B. Madej, K. M. Merz, G. Monard, P. Needham, H. Nguyen, H. T. Nguyen, I. Omelyan, A. Onufriev, D. R. Roe, A. Roitberg, R. Salomon-Ferrer, C. L. Simmerling, W. Smith, J. Swails, R. C. Walker, J. Wang, R. M. Wolf, X. Wu, D. M. York and P. A. Kollman, AMBER 2015, University of California, San Francisco, 2015 Search PubMed.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, Comparison of Simple Potential Functions for Simulating Liquid Water, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- A. W. Gotz, M. J. Williamson, D. Xu, D. Poole, S. Le Grand and R. C. Walker, Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 1. Generalized Born, J. Chem. Theory Comput., 2012, 8, 1542–1555 CrossRef CAS PubMed.
- R. Salomon-Ferrer, A. W. Gotz, D. Poole, S. Le Grand and R. C. Walker, Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 2. Explicit Solvent Particle Mesh Ewald, J. Chem. Theory Comput., 2013, 9, 3878–3888 CrossRef CAS PubMed.
- J. A. Maier, C. Martinez, K. Kasavajhala, L. Wickstrom, K. E. Hauser and C. Simmerling, ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB, J. Chem. Theory Comput., 2015, 11, 3696–3713 CrossRef CAS PubMed.
- J. M. Wang, W. Wang, P. A. Kollman and D. A. Case, Automatic atom type and bond type perception in molecular mechanical calculations, J. Mol. Graphics Modell., 2006, 25, 247–260 CrossRef CAS PubMed.
- J. M. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, Development and testing of a general amber force field, J. Comput. Chem., 2004, 25, 1157–1174 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- H. C. Andersen, Molecular-Dynamics Simulations at Constant Pressure and-or Temperature, J. Chem. Phys., 1980, 72, 2384–2393 CrossRef CAS.
- A. D. A. Becke, New Mixing of Hartree-Fock and Local Density-Functional Theories, J. Chem. Phys., 1993, 98, 1372–1377 CrossRef CAS.
- D. A. Case, T. E. Cheatham, T. Darden, H. Gohlke, R. Luo, K. M. Merz, A. Onufriev, C. Simmerling, B. Wang and R. J. Woods, The Amber biomolecular simulation programs, J. Comput. Chem., 2005, 26, 1668–1688 CrossRef CAS PubMed.
- Y. Zhao and D. G. Truhlar, The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals, Theor. Chem. Acc., 2008, 120, 215–241 CrossRef CAS.
- D. A. Parkhomenko, M. V. Edeleva, V. G. Kiselev and E. G. Bagryanskaya, pH-sensitive C-ON bond homolysis of alkoxyamines of imidazoline series: a theoretical study, J. Phys. Chem. B, 2014, 118, 5542–5550 CrossRef CAS PubMed.
- M. Kumar, D. H. Busch, B. Subramaniam and W. H. Thompson, Role of tunable acid catalysis in decomposition of alpha-hydroxyalkyl hydroperoxides and mechanistic implications for tropospheric chemistry, J. Phys. Chem. A, 2014, 118, 9701–9711 CrossRef CAS PubMed.
- P. Alarcon, B. Bohn, C. Zetzsch, M. T. Rayez and J. C. Rayez, Reversible addition of the OH radical to p-cymene in the gas phase: multiple adduct formation. Part 2, Phys. Chem. Chem. Phys., 2014, 16, 17315–17326 RSC.
- B. Denegri, M. Matic and O. Kronja, A DFT-based model for calculating solvolytic reactivity. The nucleofugality of aliphatic carboxylates in terms of Nf parameters, Org. Biomol. Chem., 2014, 12, 5698–5709 CAS.
- Y. Zhao and D. G. Truhlar, The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06 functionals and twelve other functionals, Theor. Chem. Acc., 2008, 120, 215–241 CrossRef CAS.
- (a) H. M. Dokainish and J. W. Gauld, Formation of a stable iminol intermediate in the redox regulation mechanism of protein tyrosine phosphatase 1B (PTP1B), ACS Catal., 2015, 5, 2195–2202 CrossRef CAS; (b) K. Swiderek, S. Marti, I. Tuñón, V. Moliner and J. Bertran, Peptide bond formation mechanism catalysed by ribosome, J. Am. Chem. Soc., 2015, 137, 12024–12034 CrossRef CAS PubMed.
- B. F. Ion, E. Kazim and J. W. Gauld, A multi-scale computational study on the mechanism of Streptococcus pneumoniae Nicotinamidase (SpNic), Molecules, 2014, 19, 15735–15753 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Images of optimized structures for the second potential energy surface. See DOI: 10.1039/c7ra04906d |
| This journal is © The Royal Society of Chemistry 2017 |