
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Scrutiny of electrostatic-driven conformational ordering of polypeptide chains in DMSO: a study with a model oligopeptide†
Kinshuk Raj Srivastava‡
 *,
Bhupesh Goyal§
,
Anil Kumar¶
and
Susheel Durani
*,
Bhupesh Goyal§
,
Anil Kumar¶
and
Susheel Durani
Department of Chemistry, Indian Institute of Technology Bombay, Mumbai-400076, India. E-mail: kinshuks@umich.edu; kinshukraj1@gmail.com; Tel: +91-734-834-8017
First published on 26th May 2017
Abstract
The physicochemical effects of the solvent DMSO on protein confirmation remain enigmatic despite its diverse applications in the proteomics field. Many attempts to understand the effects of DMSO have focused on the unfolding of α-helical-rich proteins; however, the cause of the profound stability of β-sheets in DMSO remains to be elucidated. Therefore, we designed an octapeptide as a β-hairpin fold to serve as a model β-sheet; we then performed combined experimental and simulation studies to investigate the effects of DMSO on the structure and stability of the β-hairpin. We compared the results of the designed octapeptide with its cognate polyalanine model to directly analyze the side chain interactions responsible for ordering the octapeptide in a specific conformation. NMR and simulation results established the ordering of the octapeptide as a β-hairpin fold, while simulations manifested the unfolded conformation of the cognate polyalanine in DMSO. It appears that owing to their weaker dielectric and strong dipolar strengths, DMSO abolishes the α-conformation as well as the solvated backbone amidic NH groups through hydrogen bonds; therefore, it destabilizes the intramolecular backbone hydrogen bonds, which leads to the unfolding of polyalanine peptides. Furthermore, our results conform to the possibility that DMSO stabilizes electrostatic and quadrupolar interactions among polar side chain atoms due to its low dielectric strength. Accordingly, we propose that the molecular mechanism of DMSO-induced stabilization of β-sheets is a combination of polar electrostatic interactions among the side chains and backbone desolvation through bulky side chains, which promotes backbone hydrogen bonding.
Introduction
Protein folding is a very complex physiochemical process whose underlying mechanisms in different environments remain obscure.1–17 The native structure of a protein is the result of a critical interplay of intramolecular interactions, i.e. hydrophobic effects, hydrogen bonding, salt bridges, and disulfide bonds, as well as interactions of the protein with its solvent environment.18–25 Water is a canonical solvent for biological systems, and its role has been widely explored in protein folding.26–31 While hydrophobic effects are the major driving force behind protein folding in aqueous solvent, electrostatic interactions are important in folding, stability, and molecular recognition. The strength of electrostatic interactions is inversely proportional to the dielectric constant of the environment. Therefore, electrostatic interactions are stronger in the buried solvent-excluded core of a protein where the dielectric constant is low (ε = 4 to 10) compared to the solvent-exposed surface, which experiences dielectric constants of ε ∼ 78.5 (that of water).32 Furthermore, the dielectric constants of non-aqueous solvents are lower than that of water, such as ε ∼ 33 for methanol and ε ∼ 46 for DMSO; therefore, it is believed that electrostatic interactions drive the folding and stability of proteins in non-aqueous environments.33 The role that non-aqueous solvents play in the folding and stability of proteins is not fully understood; therefore, further investigations along this line will facilitate the use of non-aqueous solvents in diverse applications in the biotech and pharma industries.Non-aqueous solvents have been extensively used in proteomic research; among these, dimethyl sulfoxide (DMSO) is unique due to its capacity to solubilize peptides and act as a denaturant, activator, cryoprotectant, inhibitor, and molecular chaperone.34–41 Furthermore, DMSO has drawn major attention in the study of transmembrane peptides/proteins due to its amphipathic nature and good membrane-mimicking properties, which are attributed to its lower dielectric constant (ε = 46) and high dipolar strength (dipole moment = 4.0 D).25,33,42–57 Although DMSO is the most commonly used co-solvent, the understanding of its physicochemical effects on protein folding and structure remains elusive. A high concentration of DMSO in aqueous solvent has been reported to unfold peptides or proteins by disrupting intra-peptide hydrogen bonds. With a dipole strength of 3.96 D, compared to 1.85 D for water and 1.70 D for methanol, against 3.50 D of the peptide, DMSO has a stronger dipole than the peptide, while water has a much weaker dipole.33 DMSO is a much stronger acceptor of hydrogen bonds from peptide-NH groups than water, according to quantum mechanical calculations;58 therefore, proteins unfold by establishing hydrogen bonds with the oxygen atoms of DMSO and their backbone amides.25 Recently, DMSO-induced unfolding of α-helical rich proteins has been extensively studied; this is primarily governed by preferential solvation of exposed hydrophobics through the methyl groups of DMSO, followed by solvation of exposed backbone NHs via the oxygen atoms of DMSO.44,47–50 Contrastingly, short β-hairpin peptides and β-sheet rich proteins were found to be stable in DMSO.56,59 Furthermore, β-barrel motifs, e.g. ion channels, have been found to be very stable in membranes.60–71 The exceptional stability of β-sheets in the low dielectric environment of DMSO and in membranes motivated us to design a β-hairpin peptide as a model of β-sheets and to investigate its conformational stability in DMSO. We believe that the results will provide a better molecular-level understanding of the interactions responsible for ordering octapeptides as β-hairpins in DMSO. We designed the octapeptide by grafting hydrophobic and polar residues onto two different faces of a β-hairpin to achieve amphipathicity. The octapeptide was subjected to combined experimental and simulation studies in DMSO. Additionally, a simulation study of a cognate polyalanine peptide was performed, and the results were compared with that of the octapeptide to address the role of side chain interactions in conformational ordering. NMR and simulation results have established that the octapeptide folds as a well-ordered β-hairpin in DMSO, whereas the simulation results affirm the unfolded conformation of the polyalanine model. Further analysis of the simulation results was conducted to elucidate the physicochemical factors responsible for the ordering of the octapeptide in DMSO.
Results
We designed an octapeptide with a full complement of side chains as a β-hairpin and performed combined experimental and simulation studies in DMSO to investigate the physiochemical factors responsible for the folding of the polypeptide chain in the low dielectric environment of DMSO. The peptide design was accomplished by harnessing noncovalent interactions between strands, such as salt bridges and hydrogen bonding, as well as weakly polar interactions, i.e. aromatic–aromatic, cation–π, and CH–π interactions. The octapeptide (Ac–Glu1–Arg2–Tyr3–DAsn4–DPhe5–Gln6–Trp7–Lys8–NH2) was designed as a type I′ β-hairpin, as presented in Fig. 1. There are several conformational promoting effects; for example, Trp7, Phe5, and Arg2 were grafted on one face of the β-hairpin plane to maximize the possible combination of π–π and cation–π interactions required for stabilizing the β-hairpin conformation. Moreover, on the other face of the β-hairpin, Lys8 and Glu1 were planned to stabilize the hairpin through salt bridge interactions. Additionally, Tyr3 and Gln6 were grafted to further stabilize the β-hairpin through a network of hydrogen bonding interactions involving Glu1. D-Amino acid residues (DAsn4–DPhe5) were used in the middle to nucleate the type I′ turn in the targeted β-hairpin design. The peptide was synthesized by solid phase Fmoc chemistry and was found to be pure by analytical HPLC, eluting as a sharp peak. In the MALDI-MS peak shown in Fig. S1 (ESI†), the peptide displays a molecular ion peak at 1212, corresponding to MH+ against Mcalc 1211.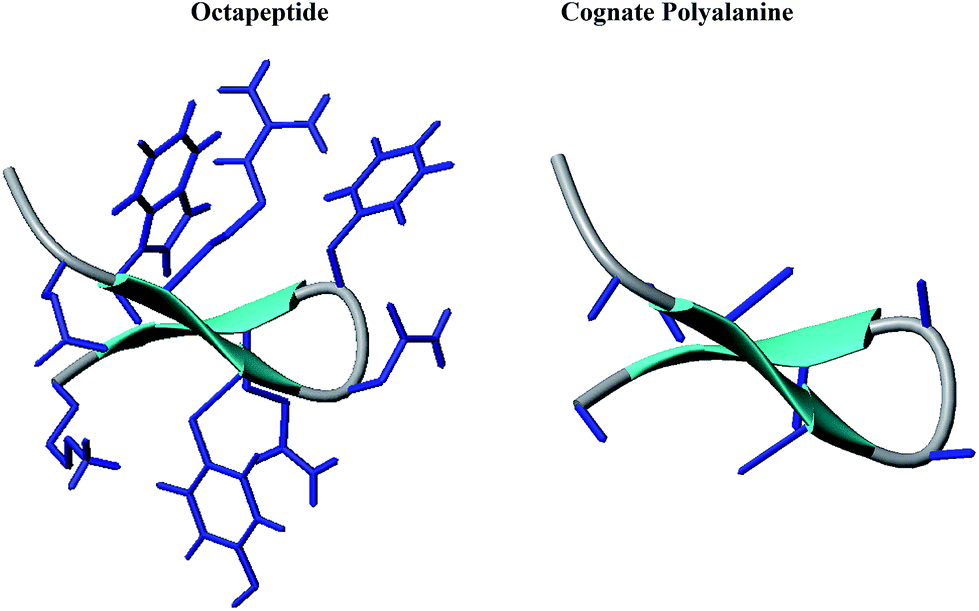 | ||
| Fig. 1 Cartoon representations of the designed octapeptide (Ac–Glu1–Arg2–Tyr3–DAsn4–DPhe5–Gln6–Trp7–Lys8–NH2) and cognate polyalanine. | ||
Experimental NMR characterization
The 1H NMR spectrum of the octapeptide was recorded at 2.5 mM; the trace is shown in Fig. S2 (ESI†). We observed no noticeable concentration-dependent variations in the chemical shifts or line widths in spectra recorded at 2.5 mM and 0.25 mM concentrations in DMSO (results not shown). Presumably, the peptides are free of aggregation under the NMR concentration regime. Assignment of chemical shifts, as presented in Table 1, for the β-hairpin octapeptide was achieved by the combined use of TOCSY and NOESY.72 Some illustrative results as portions of the 2D spectra are given in Fig. S3 (see ESI†) for the β-hairpin octapeptide with labeled cross peaks. The 3JNH–CαH observed coupling constraints in the 1D spectra are listed in Table 1. The J values are >7 Hz, suggesting a general bias towards β-sheet type ϕ-torsional angles in DMSO.| Residue | 3JNH–CαH | NH | CαH | CβH | CγH/CδH | Others |
|---|---|---|---|---|---|---|
| Glu-1 | 7.2 | 8.097 | 4.218 | 1.725, 1.872 | 2.232 | |
| Arg-2 | 7.2 | 8.052 | 4.258 | 1.654 | 1.441, 2.996 | 7.456 |
| Tyr-3 | 8.8 | 7.871 | 4.409 | 2.849, 2.689 | 6.620, 6.967 | |
| DAsn-4 | 5.6 | 8.261 | 4.569 | 2.512, 2.312 | 7.386, 6.963 | |
| DPhe-5 | 7.2 | 8.070 | 4.405 | 2.814, 2.996 | 7.206, 7.149, 7.073 | |
| Gln-6 | — | 8.167 | 4.182 | 1.650, 1.814 | 1.952 | 6.812, 7.232 |
| Trp-7 | 7.2 | 8.088 | 4.498 | 2.996, 3.156 | 7.586, 7.320, 7.056, 6.974, 7.180, 10.789 | |
| Lys-8 | 8.0 | 7.912 | 4.165 | 1.663 | 1.258, 1.503 | 2.750, 7.656 |
The peptide was examined for NOEs; the observed NOEs between the side chains are presented in Fig. 2, while the NOEs between main chain atoms are shown in Fig. S4 (ESI†). A full listing of the observed NOEs and relative distances between interacting protons derived from duly calibrated NOE volumes are listed in Table ST1 (see the ESI†). The octapeptide features NOEs that support its folded conformation. In addition to sequential backbone NH–NH and NH–CαH connectivity, the peptide also features short (i → i ± 2) and medium (i → i ± 3) range interactions, e.g. NH(Tyr3)–Hα(Gln6), SC(Tyr3)–SC(Gln6), NH(Tyr3)–NH(Phe5), and Hα(Tyr3)–SC(Gln6), indicating the existence of a turn conformation between Tyr3–DAsn4–DPhe5–Gln6. The NOEs that appear between the Arg2–Trp7, Arg2–Phe5, Phe5–Trp7 and Arg2–Asn4 side chain atoms suggest their spatial proximity, especially the NOEs observed between Arg2(Hε)–Trp7(Hη/Hζ), Arg2(Hε)–Phe5(Hδ), Arg2(Hβ)–Phe5(Hδ/ζ), Phe5(Hε)–Trp7(Hε), and Asn4(Hδ)–Phe5(Hδ/ζ), which suggest the likelihood of π–π and CH–π, NH–π type interactions. The observed NOEs between Arg2(Hβ)–Trp7(Hδ) and Arg2(Hε)–Phe5(Hδ) suggest their possible interaction through cation–π interactions. The observed SC–SC interactions between the residue pairs Glu1–Lys8, Arg2–Lys8, Tyr3–Lys8, and Tyr3–Gln6 suggest that the octapeptide adopts a β-hairpin conformation in DMSO.
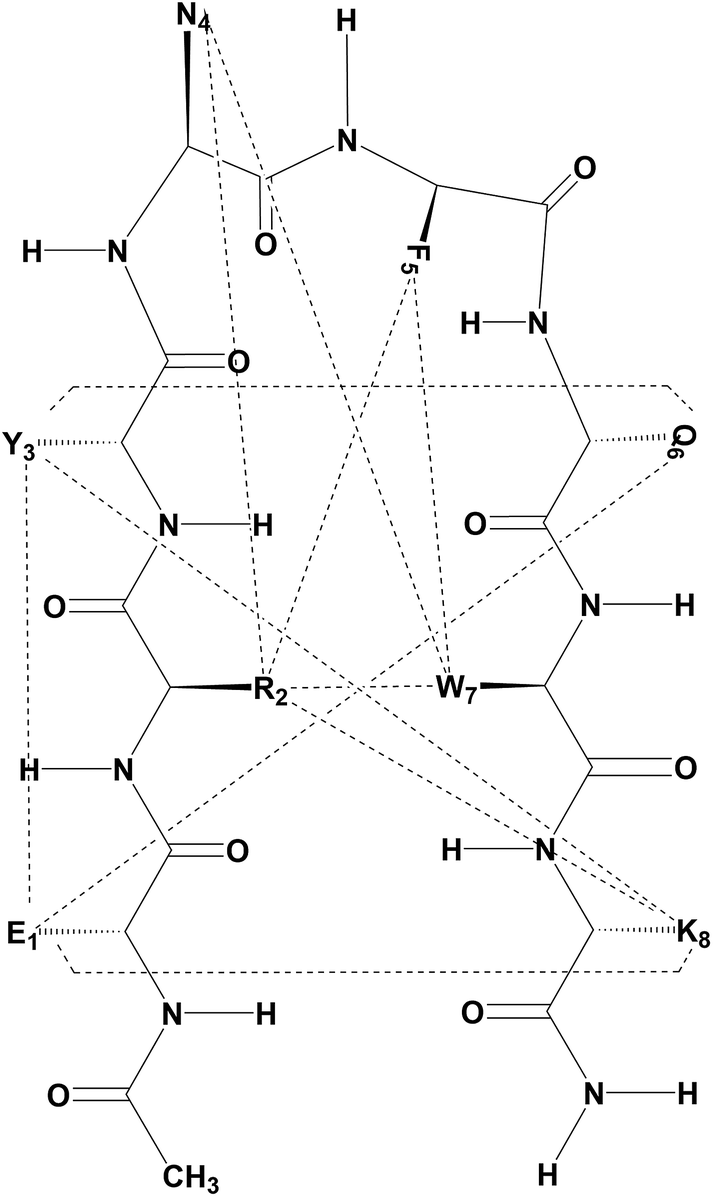 | ||
| Fig. 2 Schematic of selected long range NOEs observed for the octapeptide in DMSO-d6. | ||
The conformation modeling was approached with CYANA 2.1 (ref. 73 and 74) using NMR-derived distance restraints calibrated to NOE volumes and ϕ constraints derived from 3JNH–CαH values, depending upon the residue stereochemistry. The modeling with CYANA 2.1 was implemented with total 101 distance restraints (40 intra, 31 sequential, 5 short, 11 medium, and 14 long-range) which resulted in a convergence of conformations as a β-hairpin fold. The ten lowest energy CYANA structures, shown in Fig. 3, displayed pairwise RMSD values for backbone atoms of 0.73 ± 0.28 Å for the top ten structures and 0.68 ± 0.27 Å for the mean structure. The mean structure was further energy minimized with GROMACS75,76 using the GROMOS force field and is presented in Fig. S5 (see the ESI†). The close proximity of the residues Arg2, Asn4, Phe5, and Trp7 on one face combined with the proximity of Glu1 and Lys8 on another face of this energy-minimized structure further supports the hypothesis that salt bridges, hydrogen bonding, and polar interactions (π–π and cation–π) drive the conformational ordering of the octapeptide into a β-hairpin fold.
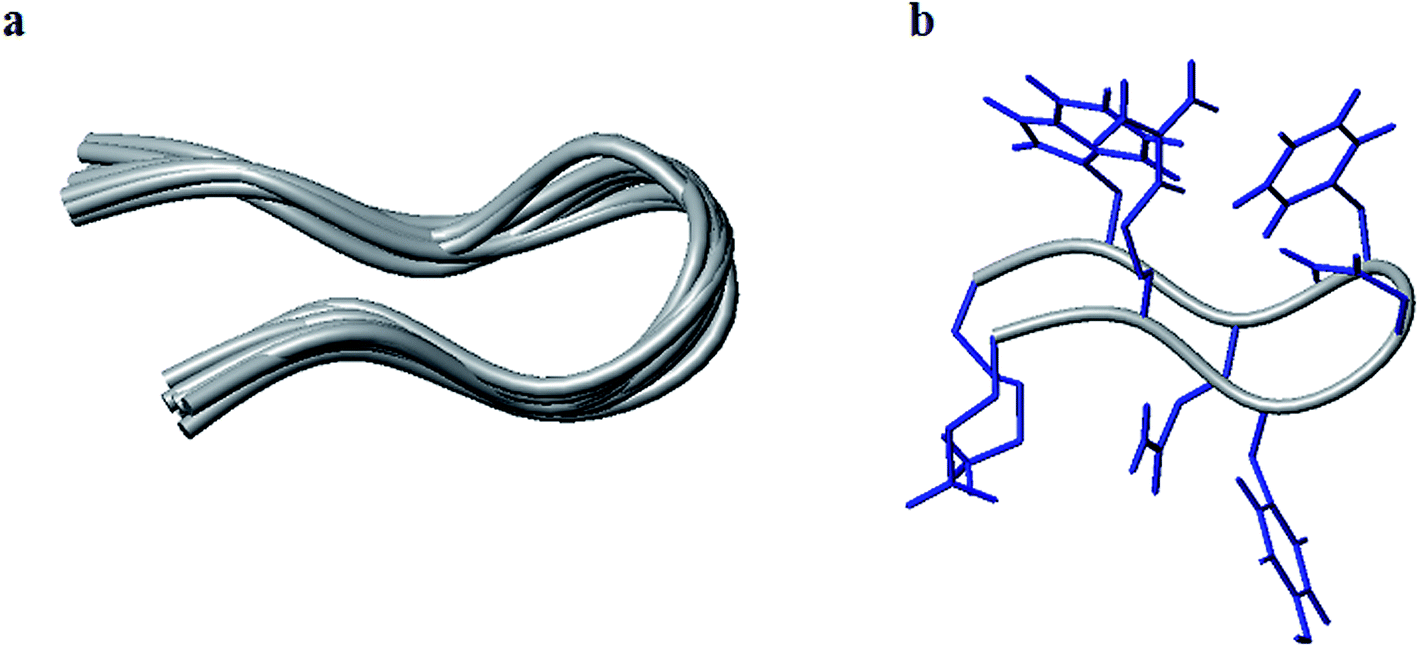 | ||
| Fig. 3 Tube representation of the ten lowest energy structures modeled with CYANA (a) and the lowest energy models with the full complement of side chains (b). | ||
Computational characterization: simulation of sequence effects in folding
To investigate the sequence-dependent ordering of the octapeptide, we performed simulations of the octapeptide and its cognate polyalanine in the explicit solvent DMSO using the GROMOS 43A1 force field of GROMACS75,76 at 298 K under NVT conditions. The ensembles were prepared with Molecular Dynamics (MD) simulations over 250 ns. The peptide structures of entire ensembles were clustered at 0.15 nm RMSD over backbone atoms which results in different microstates. The time-dependent asymptotic growth of microstates suggests an approximation of equilibrium for peptides conformations. The macrostates were evaluated in the time course of microstate evolution during MD; the results, given in Fig. 4, imply that the simulations attained an asymptote in their microstate evolution. Therefore, we assume each ensemble is a reasonable model for the equilibrium of interest. According to the results given in Table 2, the octapeptide achieves equilibrium earlier and populates 8 times fewer microstates than the cognate polyalanine. Therefore, the presence of side chains appears to restrict conformational freedom, which results in shrinkage of the octapeptide conformational landscape. Conformational equilibria were analyzed in the radius-of-gyration (Rg) over the peptide main chain. The results, summarized in Table 2 and Fig. 5, established that the octapeptide order has a more compact conformational fold than the cognate polyalanine. Bimodal Rg distribution of the octapeptide over the main chain atoms indicates the existence of two major conformations in the entire equilibria. The first maxima correspond to the first microstate (M1), comprising ∼51% of the entire ensemble population, which is ordered as a β-hairpin fold as shown in Fig. 6. Furthermore, we calculated the Rg values of the side chain atoms, presented in Fig. 8, to investigate their close proximity and their possible mutual interactions leading to the conformational ordering of the octapeptide. The calculated Rg distributions of Arg2–Phe5 and Arg2–Trp7 pair display first maxima at 0.36 nm and 0.31, respectively, implying a similar affinity of Arg2 with both Phe5 and Trp7. The central member of M1, Fig. 6, suggests that the Arg side chain is sandwiched between the aromatic rings of the Trp and Phe side chains, in which its guanidinium group is oriented laterally with regard to Trp and diagonally with regard to the Phe aromatic rings. The Rg values of Arg2 with both Phe5 and Trp7 feature bimodal distribution, which is indicative of two different types of conformations that the octapeptide can adopt in the entire canonical ensemble. The Rg values of the side chain pairs towards another face of the designed β-hairpin, such as Tyr3–Lys8, Glu1–Tyr3, and Glu1–Lys8, were found to be 0.33 nm, 0.31 nm, and 0.25 nm, respectively, suggesting their clustering and possible mutual interactions in the conformational ordering of the octapeptide. The broader Rg distribution of the Asn4–Phe5 and Phe5–Trp7 pair suggests the non-specific nature of their side chain interactions. The Rg and NOE results suggest the role of polar electrostatic interactions (salt bridges, hydrogen bonding, dispersions such as cation–π and CH–π) among the side chains drives the conformational ordering of the octapeptide as a β-hairpin fold in DMSO.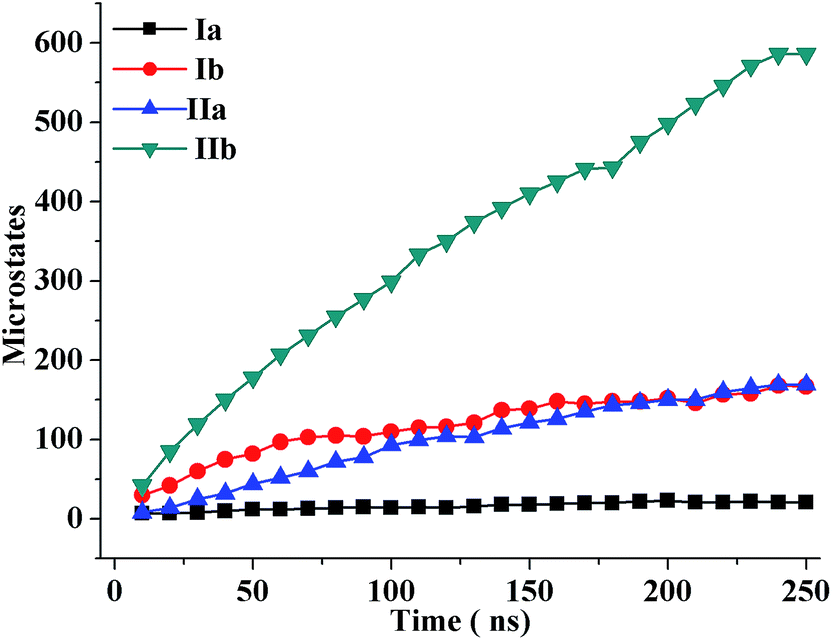 | ||
| Fig. 4 Time evolution of the microstates of the oligopeptide structures during molecular dynamics simulations. | ||
| Model system | No. of conformers | No. of microstates | % population in microstate 1 | Radius of gyration (nm) | |
|---|---|---|---|---|---|
| Macrostate | Microstate 1 | ||||
| Octapeptide | 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
21 | 52 | 0.51 ± 0.05 | 0.47 ± 0.02 |
| Polyalanine | 25000 |
167 | 19 | 0.70 ± 0.07 | 0.75 ± 0.03 |
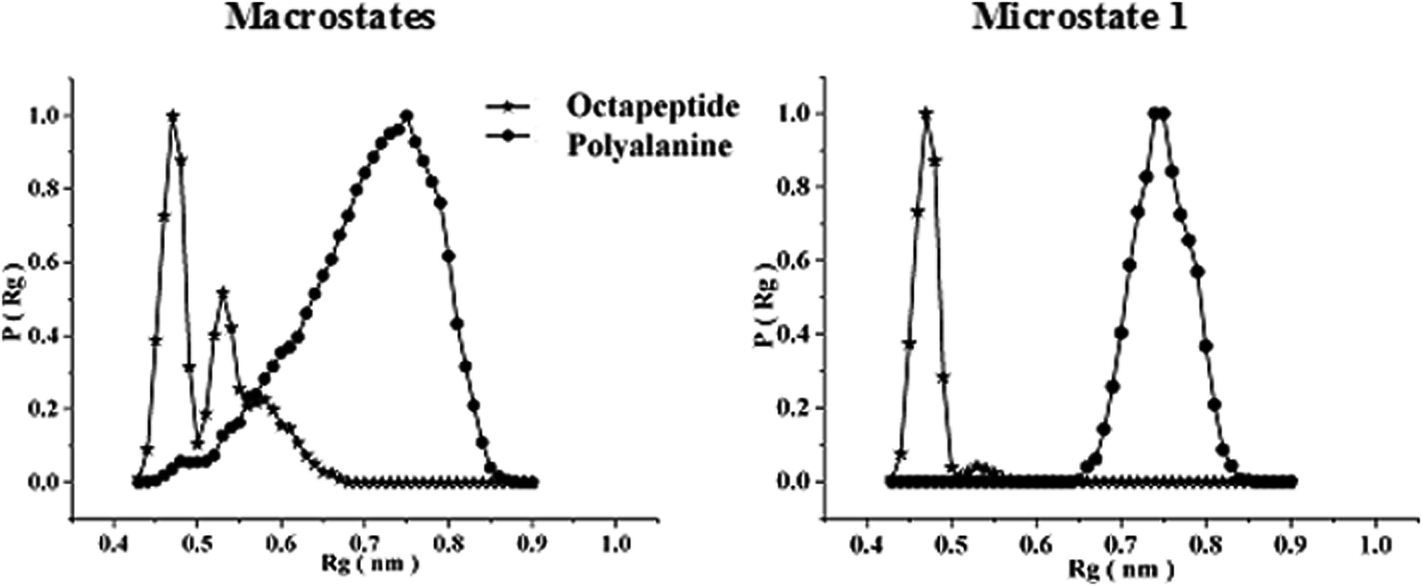 | ||
| Fig. 5 Radius of gyration (Rg) distributions over the main chain atoms of the octapeptide and the cognate polyalanine over the entire macrostate as well as the first microstate only. | ||
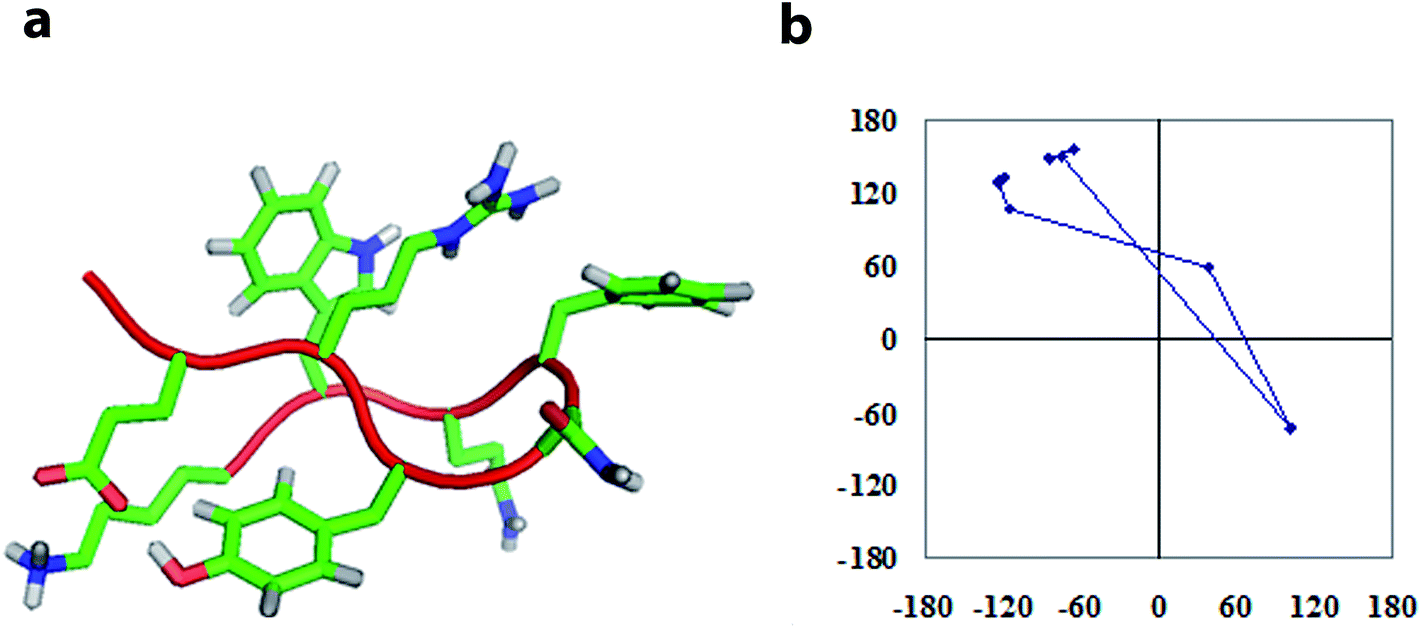 | ||
| Fig. 6 Tube and stick representations of the central members of the top microstate of the octapeptide with the percentage population in parentheses (a) and the residue-by-residue meander in ϕ, ψ space (b). | ||
A half-ensemble lower in free energy was accessed to obtain statistics for the intra-peptide hydrogen bonds involving the main chain (MC) and side chain (SC) atoms; these are reported in Table ST2 (see the ESI†). For the octapeptide, we observed 2.55 MC–MC hydrogen bonds, 5.44 per conformer, which includes MC–MC, SC–SC, and MC–SC hydrogen bonds, whereas the cognate polyalanine is devoid of intramolecular hydrogen bonds. The total of 5.44 intramolecular hydrogen bonds per conformation provides further evidence for a folded conformation of the octapeptide. We further analyzed the patterns of the hydrogen bonds to anticipate the conformation characteristics of the octapeptide. The presence of both medium (i → i ± 3) and long-range (i ≥ i ± 6) MC–MC as well as both short-range (i → i ± 2) and long-range (i ≥ i ± 6) SC–SC hydrogen bonds strongly suggests that the octapeptide is a β-hairpin fold.
We sought to analyze the ϕ, ψs distributions of the equilibrium ensemble in the α-, β-, and PPII-basins to further understand the conformational characteristics of peptides in DMSO. We calculated the percentage population in each basin for all residues along with the residue-wise distributions; the results are given in Table ST2, Fig. S6 (see ESI†). The observed occupancy of D-amino acids in the α-basin and L-amino acids in the β-sub-basin (β + PPII) suggests ordering of the octapeptide in a β-hairpin conformation and provides support for our design. Contrastingly, the cognate polyalanine is present in only the β-sub-basin; this is indicative of the unfolded nature of the conformational ensemble. The residue-wise distribution of DAsn4–DPhe5 in D-specific α-basins implies their ordering in a turn conformation and further advocates the role of stereochemistry in the nucleation of the hairpin.
The central members of microstates which populate half of the entire equilibrium ensemble that is lower in free energy are presented in Fig. 6 for the octapeptide and Fig. 7 for the cognate polyalanine. The first microstate of the octapeptide, comprised of a ∼51% population of ensembles, is folded in a type I′ β-hairpin conformation as per the sequential ϕ, ψs distribution and hydrogen bonding pattern. However, the cognate polyalanine top 5 microstates populate the half ensemble, and all are unfolded conformations in which the first microstate populates only 18% of the total ensemble. The polyalanine peptide features kinks or curvature in its backbone due to the presence of D-amino acid residues in the middle of the peptide sequence, which suggests that stereochemical constraints are aiding the nucleation of a type I′ turn even in the absence of side chains. Without side chains, the microstates are essentially unfolded; meanwhile, with a complement of side chains, the octapeptide is well ordered due to the apparent interactions between the specific side chains.
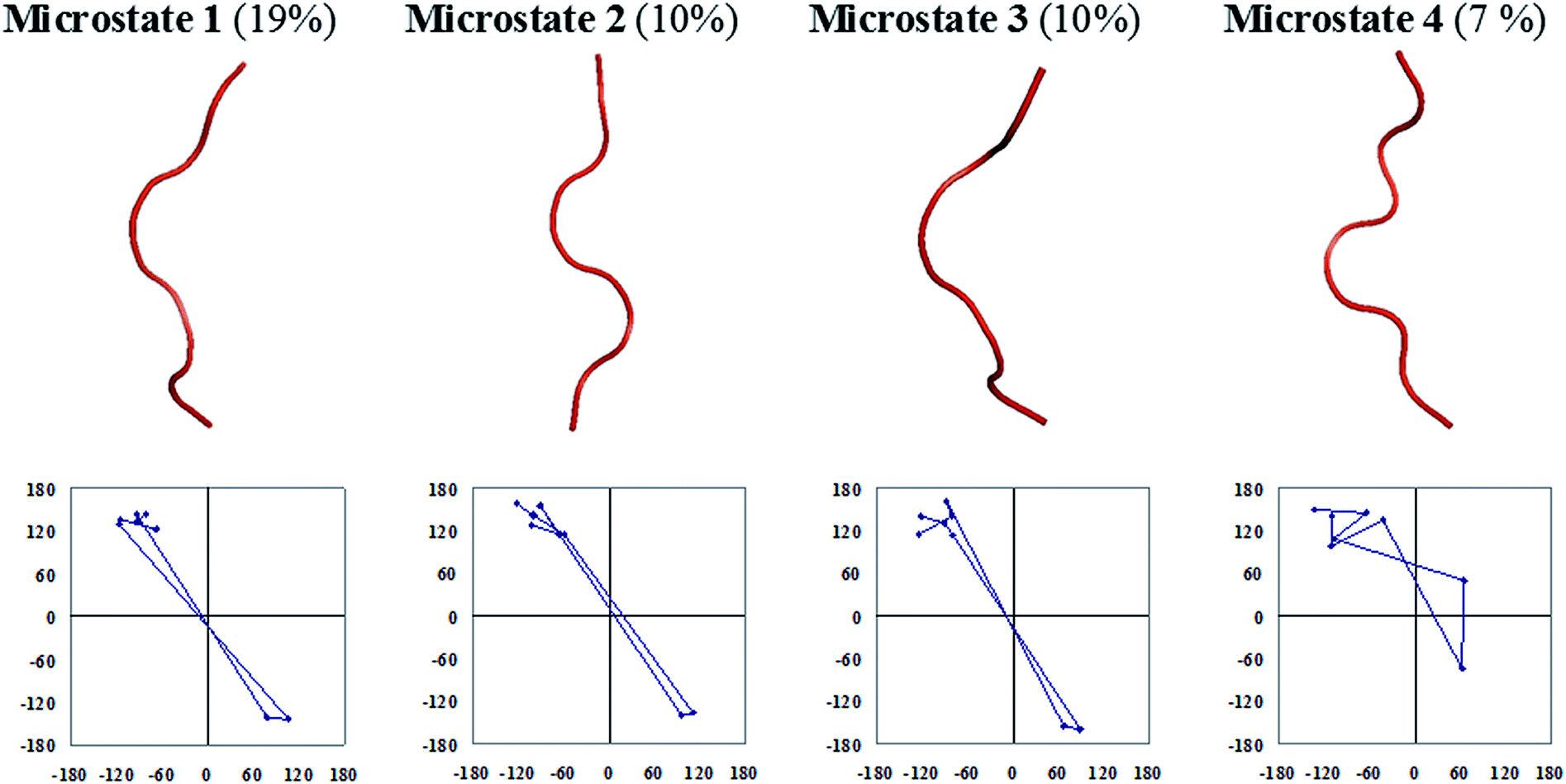 | ||
| Fig. 7 Backbone trace representations and residue-by-residue meanders in ϕ, ψ space of a few top microstates of the cognate polyalanine with their percentage populations given in parentheses. | ||
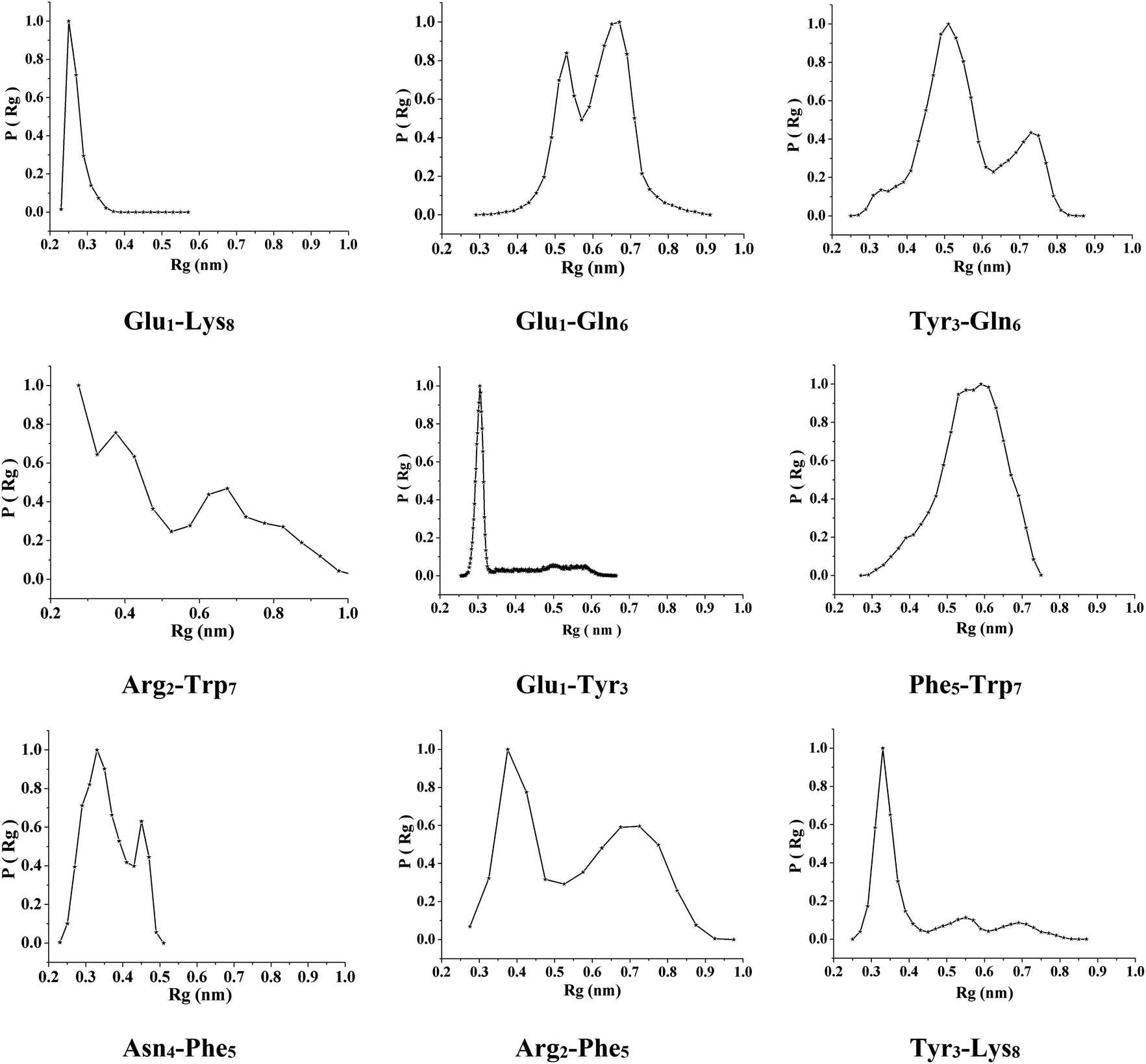 | ||
| Fig. 8 Distributions of the radius of gyration of the octapeptide side chain atoms over the entire macrostate. | ||
Solvation shell analysis
We calculated the pairwise radial distribution function (RDF) of solvent atoms around peptide atoms to gain insight about the local concentration and orientation of DMSO molecules around the peptide; this will enable us to interpret the role of DMSO as a solvent in the conformational folding–unfolding of the polypeptide chain. The RDF data, presented in Fig. S7 and S8 for the octapeptide and in Fig. S9 for the polyalanine peptide (see ESI†), were calculated for specific atoms of the peptide over the entire conformational ensemble populated in the simulated trajectories. The results show that the density of solvent DMSO atoms varies significantly from that of bulk solvent and also between different atoms of the peptides, which is indicative of the specific solvent structure around the peptide atoms of interest. For the octapeptide, the first RDF maximum of oxygen against the NH groups of the backbone is at 0.29 nm, which corresponds to hydrogen bonding [NH⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S(CH3)2] distances. The peptidic carbonyl and carboxylate oxygen feature RDF maxima at 0.32 nm against the DMSO methyls, which suggests solvation of the peptidic oxygens by the DMSO methyls. However, the intensity of the first RDF maxima of the solvent DMSO atoms against the backbone amides and carbonyls are significantly smaller compared to the side chain atoms, which suggests shielding of the octapeptide backbone from DMSO. Contrastingly, the RDF maxima of the DMSO atoms for the cognate polyalanine atoms are higher than for the octapeptide, which suggests that the polyalanine backbone is more solvent-exposed and the octapeptide backbone atoms are preferably solvent sequestered. These data clearly indicate that the unfolding of polyalanine peptides is due to preferential solvation of their backbones, while sequestration of the octapeptide backbone from the solvent could promote intramolecular hydrogen bonding and therefore contribute to the stabilization of the folded state of the octapeptide. Furthermore, we propose that side chain-mediated shielding of the octapeptide can promote intramolecular backbone hydrogen bonding and hence its ordering in a compact conformation.
S(CH3)2] distances. The peptidic carbonyl and carboxylate oxygen feature RDF maxima at 0.32 nm against the DMSO methyls, which suggests solvation of the peptidic oxygens by the DMSO methyls. However, the intensity of the first RDF maxima of the solvent DMSO atoms against the backbone amides and carbonyls are significantly smaller compared to the side chain atoms, which suggests shielding of the octapeptide backbone from DMSO. Contrastingly, the RDF maxima of the DMSO atoms for the cognate polyalanine atoms are higher than for the octapeptide, which suggests that the polyalanine backbone is more solvent-exposed and the octapeptide backbone atoms are preferably solvent sequestered. These data clearly indicate that the unfolding of polyalanine peptides is due to preferential solvation of their backbones, while sequestration of the octapeptide backbone from the solvent could promote intramolecular hydrogen bonding and therefore contribute to the stabilization of the folded state of the octapeptide. Furthermore, we propose that side chain-mediated shielding of the octapeptide can promote intramolecular backbone hydrogen bonding and hence its ordering in a compact conformation.
The intensities of the RDF maxima of DMSO atoms against the side chain terminal atoms are higher; this is indicative of their preferential solvation, which aids in solubilizing the peptide in DMSO. Arg Nη,ε and Trp Nε display first RDF maxima against the oxygen atoms of DMSO at 0.29 nm, which highlight their solvation through hydrogen bonding. Similarly, the Asn and Gln side chain amides display peaks at 0.29 nm, which is indicative of their solvation by the oxygen of DMSO. The RDF maxima of the side chain carbonyl oxygens of Asn and Gln against the DMSO methyls features first maxima at 0.31 nm; this suggests solvation by the methyls of DMSO, presumably through CH⋯O interactions. Furthermore, the side chain carboxylate oxygen of Glu displays a similar trend of RDF maxima at 0.30 nm, and the results suggest its solvation by the methyls of DMSO via CH⋯O interactions. The Tyr phenolic oxygen features first RDF maxima against the DMSO oxygens at 0.29 nm, suggesting their solvation through hydrogen bonding [OH⋯OS(CH3)2]. Interestingly, we observed a higher intensity of the first RDF maximum of the DMSO methyls against the carbonyl oxygen atoms of the ASN and Gln side chains compared to the carboxylate oxygen of Glu and the phenolic oxygen of the Tyr side chain. These data further support the observed computed Rg values and suggest that the Glu and Tyr side chains are involved in side chain interactions, whereas the Asn and Gln side chains are preferably more solvated. Furthermore, we observed that the intensities of the RDF maxima for the aromatic carbons of the Trp, Tyr, and Phe residues, as well as the methylenes of the Arg, Lys, Glu, and Gln side chains, display very low magnitudes, close to the value of the bulk solvent. The lower magnitude of the RDF maxima of the DMSO atoms against the aromatic and methylene groups of the octapeptide suggests their possible involvement in mutual side chain interactions which can potentially contribute to the stabilization of the octapeptide as a β-hairpin fold. Our solvation shell analysis further supports the amphipathic nature of the DMSO solvation of peptides and proteins.
Discussion
In the present study, we investigated the effects of DMSO, a non-aqueous solvent, on the conformational ordering of an octapeptide as a β-hairpin conformation. To directly study the role of the side chains, we compared the equilibrium ensemble properties of the octapeptide with its cognate polyalanine peptide, studied herein. The exceptional stability of β-sheets in the low dielectric environment of DMSO motivated us to de novo design an octapeptide as a β-hairpin and investigate the physicochemical factors responsible for stabilization of the β-hairpin fold in DMSO. The destabilizing effects of DMSO on α-helical structures have been explored thoroughly; however, the exceptional stability of β-sheets in DMSO remains enigmatic. DMSO is reported to denature α-helical rich proteins through preferential solvation of exposed hydrophobic surfaces.42–45,47,48,50 It is important to note that compared to β-sheets, the backbones of α-helices are more exposed; additionally, its hydrophobic surface can therefore easily be attacked by the solvent DMSO.77 To develop a molecular-level understanding of the physicochemical factors, we performed combined experimental and simulation studies for the octapeptide, while the cognate polyalanine was investigated by simulations only. We matched the experimentally observed and calculated (simulation) equilibrium ensemble properties to verify the simulation results, followed by analysis of the conformational ensembles obtained from the simulation, to elucidate the interactions responsible for the conformational ordering of peptides in DMSO.The higher dipole strength of DMSO compared to the peptide groups makes it a better acceptor of backbone NH groups (μDMSO = 3.96; μwater = 1.85; peptide group μpeptide = 3.50).25,33 Additionally, DMSO is a weak dielectric solvent compared to water (εDMSO = 46; εwater = 80) and cannot screen the local repulsive interactions between peptide dipoles; therefore, it indirectly promotes unfolding of the polypeptide chain.25,78 Thus, unfolding of the protein main chain appears to be achieved through a complex interplay of both the dipolar and dielectric effects of DMSO.25 Consistent with our previous studies of octaalanine and lysine-solubilized polyalanine in DMSO, the cognate polyalanine studied herein was found to be unfolded. However, the lower magnitudes of the RDF maxima of the DMSO oxygen around the octapeptide backbone amides compared to those of the polyalanine indicates steric blocking of DMSO access to the octapeptide backbone through the side chains. Protrusion of the side chains from both the faces away from the β-hairpin plane appears to sterically block the access of DMSO to the backbone NH groups. Furthermore, the octapeptide was found to populate 2.5 hydrogen bonds per conformer between the backbone atoms, whereas no hydrogen bond was detected for the polyalanine peptide in the entire simulated ensemble; this suggests that shielding of the octapeptide backbone promotes intramolecular hydrogen bonding. The low density of solvent atoms around the aromatic and methylene atoms of the side chains suggests their desolvation and possible involvement in mutual interactions to stabilize the folded conformation of the octapeptide. The ϕ, ψ distributions and number and pattern of backbone hydrogen bonds, along with the observed NOEs between the backbone atoms and the 3JNH–CαH values, provide evidence of ordering of the octapeptide as a β-hairpin conformation. Moreover, the observed NOEs and calculated Rg distributions between the side chain atoms demonstrate stable interactions, such as salt bridges (Glu1–Lys8), hydrogen bonding (Tyr3 with both Glu1 and Lys8) and cation–π (Arg2 with Trp7 and Phe5), among the side chains of the octapeptide; this appears to provide further stabilization to the β-hairpin conformation. The observation that electrostatic interactions are major contributing factors in the folding of the octapeptide in the low dielectric environment of DMSO further advocates the importance of electrostatic effects on the folding and stability of proteins.25,78–86
The present study reveals many important physicochemical effects of DMSO as a solvent. The results of the present study will not only aid understanding of DMSO effects in the ordering of an octapeptide as a β-hairpin, but will also allow us to infer information about the stability of β-sheets of larger proteins in DMSO. The stability of polar electrostatic interactions in DMSO, observed in the present study, further explains why DMSO is reported to enhance the self-association of tubulin with the microtubules.87,88 The tubulin self-assembly process, which is primarily driven by electrostatic interactions between the charged tubulin monomers, may be further promoted by the presence of DMSO. Interestingly, the profound stability and amphipathic nature of the octapeptide studied herein can offer advantages for its use in several other biotechnological applications, such as antimicrobial activity, as an inhibitor against transmembrane protein diseases, and as an amphiphilic reagent for protecting membrane proteins, to facilitate their structural and functional studies.89 Furthermore, similar to other reported studies,71 we believe that our designed octapeptide can serve as a good model to investigate the folding mechanism of β-sheets in the form of β-barrel motifs inside membranes.
Conclusion
We performed a comparative study of an octapeptide and its cognate polyalanine to investigate the physicochemical factors responsible for conformational ordering of the octapeptide as a β-hairpin fold in the low dielectric environment of DMSO. To achieve this objective fold, we de novo designed the octapeptide with a full complement of side chains by maximizing interactions along the strand to stabilize the octapeptide in a β-hairpin conformation. The designed octapeptide was then subjected to combined experimental and simulation studies, whereas only simulation studies were performed with the cognate polyalanine in DMSO. Data from solvation shell analysis, hydrogen bond statistics, and ϕ, ψ distributions demonstrate unfolding of the polyalanine peptide, which could be due to preferential solvation of the backbone atoms by DMSO. Contrastingly, NOEs, Rg values, backbone dihedral angle distributions, hydrogen bond statistics, and solvation shell analysis demonstrate ordering of the octapeptide in a β-hairpin conformation. These results affirm that the combination of intramolecular hydrogen bonding, promoted by side chain-mediated steric blocking of DMSO access to the octapeptide backbone, along with electrostatic interactions between the side chain atoms, is responsible for the stability of the β-hairpin conformation of the octapeptide. Based on the understanding gained from the present study, we can draw inferences about the possible mechanism of DMSO-induced stabilization of the β-sheets; this can be mainly attributed to backbone desolvation, which promotes backbone hydrogen bonding. The insights gained in the present study further advance our current understanding of the physicochemical effects of DMSO and therefore will facilitate the use of DMSO in diverse pharmaceutical and biotechnological applications.Materials and methods
Peptide synthesis
The synthesis was performed manually on Rink Amide AM resin using standard Fmoc chemistry and HOBt/DIC as coupling reagents. Each coupling, monitored with Kaiser and chloranil tests, typically required about 6 hours. Deprotections were carried out with 30% (v/v) piperidine–DMF. The N-terminus was acetylated (–NHCOCH3) with Ac2O:DIPEA:DMF in a 1:2:20 ratio. The cleavage of the final polypeptide and deprotection of the side chains were achieved together with reagent K (82.5% TFA/5% dry-phenol/5% thioanisole/2.5% ethanedithiol/5% water). The product was precipitated with anhydrous diethyl ether and was then lyophilized from a 1:4 ratio of H2O:tBuOH solution to obtain a white powder. Peptide purity was assessed with HPLC over RP-C18 (10 μM, 10 mm × 250 mm; Merck) eluting with CH3CN\H2O (0.1% TFA) 0% to 100% gradients.
MALDI-MS measurements
Mass spectra were recorded in MALDI-TOF mode on a duly calibrated AXIMA-CFR Kratos instrument. Positive ions were detected in linear/reflectron mode.Nuclear magnetic resonance
1H NMR spectra were recorded in 100% DMSO-d6 at 2.5 mM and 0.25 mM concentrations at 298 K on a 800 MHz Bruker instrument; VT spectra were recorded on a 400 MHz Varian NMR instrument using a cryoprobe. Phase-sensitive TOCSY90 and NOESY72 experiments were performed for conformational characterization of the peptides. The solvent was suppressed with a pre-saturation or WATERGATE sequence, as provided by Bruker or VARIAN. The 800 MHz NMR data were processed with TOPSPIN, provided by Bruker, or CARA (Computer Aided Resonance Assignment). Typically, sine-squared window function phases shifted by 70° were applied in both dimensions with the data zero filled to 2 K × 1 K or 4 K × 4 K before the Fourier transformations. J values were extracted directly from the 1D 1H NMR traces. Structure calculation was performed with CYANA 2.1.73 Dihedral angle restraints and lower limit distance restraints for hydrogen bonding were used for structure refinement. D-Amino acid residues were introduced in the CYANA library under the guidance of the developer. Representative structures were energy minimized using the GROMACS75,76 software package. All structural figures presented in this report were made using either Pymol or MolMol.Preparation of equilibrium ensembles
Molecular dynamics simulations were performed with the GROMOS-96 43A1 force field in the GROMACS 3.3.3 package75,76 in a periodic box of explicit solvent, using the DMSO model of Geerke et al.91 The simulation was performed under NVT conditions,92 viz., a fixed number of particles and constant volume and temperature. The non-bonded list cutoff was 1.4 nm with a shift at 0.8 nm. The integration step was 2 fs. The initial velocities were drawn from the Maxwellian distribution. The temperature was coupled to an external bath with a relaxation time constant of 0.1 ps. Bond lengths were constrained with SHAKE93 to a geometric accuracy of 10−4. The electrostatics were treated in this case with Particle Mesh Ewald (PME)94,95 methods implementing a Coulomb cutoff of 1.4 nm, a Fourier spacing of 0.12 nm, and an interpolation order of 4. The oligopeptides were modeled with our in-house software, CAPM (Computer Aided Peptide Modeling), and PDBmake and cartoon representations were created with either MOLMOL or Pymol. Oligopeptides constrained to the center of a periodic cubic box with an edge length of 5 nm were soaked in DMSO, which was added to 1 atm density at 298 K. The box was rescaled to maintain a density of 1095 kg m−3, corresponding to the experimental density of DMSO.91 First, the solute was energy minimized; then, the solvent was energy minimized while restraining the solute, and finally both were energy minimized after removing restraints. MD was initialized, and an 3 ns trajectory was exempted from the analysis as a pre-equilibration period. The trajectory was sampled thereafter at 10 ps intervals.Characterization of polypeptide macrostates and microstates
The peptide conformers were clustered in Cartesian space to ≤0.15 nm RMSD cutoff over backbone atoms using an algorithm by Daura et al.96 This gives microstates in a diminishing population, viz., diminishing thermodynamic stability. The Helmholtz free energy was calculated from the relative probabilities pA and pB of finding the system in microstates A and B as ΔFA–B = −RTlnpB/pA, where R is the gas constant, T is the temperature, and pA and pB are the number of members in microstates A and B.
Solvation shell analysis
Radial and spatial distribution functions of specific solvent atoms were calculated over the most populous polypeptide microstate in each ensemble using the g_rdf and g_spatial utilities in GROMACS.Acknowledgements
We acknowledge DST (09DST028), Government of India, for financial support and IIT Bombay for the computing facility “Corona”. KRS, BG, and AK were recipients of fellowships from the Council of Scientific and Industrial Research (CSIR). We acknowledge TIFR, Mumbai for help with the NMR experiments.References
- C. B. Anfinsen, Science, 1973, 181, 223–230 CAS.
- R. L. Baldwin, Adv. Protein Chem., 2002, 62, 361–367 CrossRef CAS PubMed.
- R. L. Baldwin, J. Biol. Chem., 2003, 278, 17581–17588 CrossRef CAS PubMed.
- R. L. Baldwin, J. Mol. Biol., 2007, 371, 283–301 CrossRef CAS PubMed.
- R. P. Bywater, J. Biomol. Struct. Dyn., 2013, 31, 351–362 CAS.
- Y. Cote, G. G. Maisuradze, P. Delarue, H. A. Scheraga and P. Senet, J. Phys. Chem. Lett., 2015, 6, 1082–1086 CrossRef CAS PubMed.
- V. Daggett and A. Fersht, Nat. Rev. Mol. Cell Biol., 2003, 4, 497–502 CrossRef CAS PubMed.
- K. A. Dill, Biochemistry, 1990, 29, 7133–7155 CrossRef CAS PubMed.
- K. A. Dill and J. L. MacCallum, Science, 2012, 338, 1042–1046 CrossRef CAS PubMed.
- K. A. Dill, S. B. Ozkan, M. S. Shell and T. R. Weikl, Annu. Rev. Biophys., 2008, 37, 289–316 CrossRef CAS PubMed.
- S. W. Englander and L. Mayne, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 15873–15880 CrossRef CAS PubMed.
- A. Onofrio, G. Parisi, G. Punzi, S. Todisco, M. A. Di Noia, F. Bossis, A. Turi, A. De Grassi and C. L. Pierri, Phys. Chem. Chem. Phys., 2014, 16, 18907–18917 RSC.
- J. N. Onuchic and P. G. Wolynes, Curr. Opin. Struct. Biol., 2004, 14, 70–75 CrossRef CAS PubMed.
- M. B. Prigozhin and M. Gruebele, Phys. Chem. Chem. Phys., 2013, 15, 3372–3388 RSC.
- G. D. Rose, P. J. Fleming, J. R. Banavar and A. Maritan, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 16623–16633 CrossRef CAS PubMed.
- S. Sacquin-Mora, J. R. Soc., Interface, 2015, 12, 20150876 CrossRef PubMed.
- P. G. Wolynes, Biochimie, 2015, 119, 218–230 CrossRef CAS PubMed.
- D. R. Canchi and A. E. Garcia, Annu. Rev. Phys. Chem., 2013, 64, 273–293 CrossRef CAS PubMed.
- J. L. England and G. Haran, Annu. Rev. Phys. Chem., 2011, 62, 257–277 CrossRef CAS PubMed.
- Q. R. Johnson, R. B. Nellas and T. Shen, Biochemistry, 2012, 51, 6238–6245 CrossRef CAS PubMed.
- A. Merlino, N. Pontillo and G. Graziano, Phys. Chem. Chem. Phys., 2016, 19, 751–756 RSC.
- S. Murakami and M. Kinoshita, J. Chem. Phys., 2016, 144, 125105 CrossRef PubMed.
- H. Oshima and M. Kinoshita, J. Chem. Phys., 2015, 142, 145103 CrossRef PubMed.
- P. Ren, J. Chun, D. G. Thomas, M. J. Schnieders, M. Marucho, J. Zhang and N. A. Baker, Q. Rev. Biophys., 2012, 45, 427–491 CrossRef PubMed.
- K. R. Srivastava, A. Kumar, B. Goyal and S. Durani, J. Phys. Chem. B, 2011, 115, 6700–6708 CrossRef CAS PubMed.
- R. L. Baldwin and G. D. Rose, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 12462–12466 CrossRef CAS PubMed.
- M. C. Bellissent-Funel, A. Hassanali, M. Havenith, R. Henchman, P. Pohl, F. Sterpone, D. van der Spoel, Y. Xu and A. E. Garcia, Chem. Rev., 2016, 116, 7673–7697 CrossRef CAS PubMed.
- J. Grdadolnik, F. Merzel and F. Avbelj, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 322–327 CrossRef CAS PubMed.
- D. J. Huggins, J. Struct. Biol., 2016, 196, 394–406 CrossRef CAS PubMed.
- F. Palazzesi, M. Salvalaglio, A. Barducci and M. Parrinello, J. Chem. Phys., 2016, 145, 121101 CrossRef PubMed.
- J. B. Udgaonkar, Arch. Biochem. Biophys., 2013, 531, 24–33 CrossRef CAS PubMed.
- L. Li, C. Li, Z. Zhang and E. Alexov, J. Chem. Theory Comput., 2013, 9, 2126–2136 CrossRef CAS PubMed.
- C. Reichardt, Solvents and solvent effects in organic chemistry, WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim, 2003 Search PubMed.
- V. I. Gordeliy, M. A. Kiselev, P. Lesieur, A. V. Pole and J. Teixeira, Biophys. J., 1998, 75, 2343–2351 CrossRef CAS PubMed.
- Q. Guo, Q. Wu, D. Bai, Y. Liu, L. Chen, S. Jin, Y. Wu and K. Duan, Antimicrob. Agents Chemother., 2016, 60, 7159–7169 CAS.
- S. W. Jacob, M. Bischel and R. J. Herschler, Curr. Ther. Res., 1964, 6, 193–198 CAS.
- J. E. Lovelock and M. W. Bishop, Nature, 1959, 183, 1394–1395 CrossRef CAS PubMed.
- H. Mi, D. Wang, Y. Xue, Z. Zhang, J. Niu, Y. Hong, K. Drlica and X. Zhao, Antimicrob. Agents Chemother., 2016, 60, 5054–5058 CrossRef CAS PubMed.
- N. C. Santos, J. Figueira-Coelho, J. Martins-Silva and C. Saldanha, Biochem. Pharmacol., 2003, 65, 1035–1041 CrossRef CAS PubMed.
- A. C. Williams and B. W. Barry, Adv. Drug Delivery Rev., 2004, 56, 603–618 CrossRef CAS PubMed.
- Z. W. Yu and P. J. Quinn, Biosci. Rep., 1994, 14, 259–281 CrossRef CAS PubMed.
- A. N. Batista, J. M. Batista Jr, L. Ashton, V. S. Bolzani, M. Furlan and E. W. Blanch, Chirality, 2014, 26, 497–501 CrossRef CAS PubMed.
- A. N. Batista, J. M. Batista Jr, V. S. Bolzani, M. Furlan and E. W. Blanch, Phys. Chem. Chem. Phys., 2013, 15, 20147–20152 RSC.
- S. Bhattacharjya and P. Balaram, Proteins: Struct., Funct., Genet., 1997, 29, 492–507 CrossRef CAS.
- W. Dzwolak, J. Kalinowski, C. Johannessen, V. Babenko, G. Zhang and T. A. Keiderling, J. Phys. Chem. B, 2012, 116, 11863–11871 CrossRef CAS PubMed.
- M. Jackson and H. H. Mantsch, Biochim. Biophys. Acta, 1991, 1078, 231–235 CrossRef CAS.
- S. Roy and B. Bagchi, J. Phys. Chem. B, 2014, 118, 5691–5697 CrossRef CAS PubMed.
- S. Roy, B. Jana and B. Bagchi, J. Chem. Phys., 2012, 136, 115103 CrossRef PubMed.
- T. Tretyakova, M. Shushanyan, T. Partskhaladze, M. Makharadze, R. van Eldik and D. E. Khoshtariya, Biophys. Chem., 2013, 175–176, 17–27 CrossRef CAS PubMed.
- I. K. Voets, W. A. Cruz, C. Moitzi, P. Lindner, E. P. Areas and P. Schurtenberger, J. Phys. Chem. B, 2010, 114, 11875–11883 CrossRef CAS PubMed.
- T. Arakawa, Y. Kita and S. N. Timasheff, Biophys. Chem., 2007, 131, 62–70 CrossRef CAS PubMed.
- M. Bellanda, E. Peggion, R. Burgi, W. van Gunsteren and S. Mammi, J. Pept. Res., 2001, 57, 97–106 CrossRef CAS PubMed.
- A. M. Duarte, C. P. van Mierlo and M. A. Hemminga, J. Phys. Chem. B, 2008, 112, 8664–8671 CrossRef CAS PubMed.
- A. M. Duarte, C. J. Wolfs, N. A. van Nuland, M. A. Harrison, J. B. Findlay, C. P. van Mierlo and M. A. Hemminga, Biochim. Biophys. Acta, 2007, 1768, 218–227 CrossRef CAS PubMed.
- I. L. Karle, J. L. Flippen-Anderson, K. Uma and P. Balaram, Biopolymers, 1993, 33, 827–837 CrossRef CAS PubMed.
- R. Mahalakshmi, S. Raghothama and P. Balaram, J. Am. Chem. Soc., 2006, 128, 1125–1138 CrossRef CAS PubMed.
- A. Giugliarelli, M. Paolantoni, A. Morresi and P. Sassi, J. Phys. Chem. B, 2012, 116, 13361–13367 CrossRef CAS PubMed.
- Y. J. Zheng and R. L. Ornstein, J. Am. Chem. Soc., 1996, 118, 4175–4180 CrossRef CAS.
- S. Rana, B. Kundu and S. Durani, Chem. Commun., 2004, 2462–2463 RSC.
- J. U. Bowie, Nature, 2005, 438, 581–589 CrossRef CAS PubMed.
- H. Hong, Arch. Biochem. Biophys., 2014, 564, 297–313 CrossRef CAS PubMed.
- S. H. White and W. C. Wimley, Annu. Rev. Biophys. Biomol. Struct., 1999, 28, 319–365 CrossRef CAS PubMed.
- T. Doherty, A. J. Waring and M. Hong, Biochim. Biophys. Acta, 2006, 1758, 1285–1291 CrossRef CAS PubMed.
- S. J. Ganesan, H. Xu and S. Matysiak, Phys. Chem. Chem. Phys., 2016, 18, 17836–17850 RSC.
- G. Manzo, M. A. Scorciapino, P. Wadhwani, J. Burck, N. P. Montaldo, M. Pintus, R. Sanna, M. Casu, A. Giuliani, G. Pirri, V. Luca, A. S. Ulrich and A. C. Rinaldi, PLoS One, 2015, 10, e0116379 Search PubMed.
- M. Meier and J. Seelig, J. Mol. Biol., 2007, 369, 277–289 CrossRef CAS PubMed.
- M. Meier and J. Seelig, J. Am. Chem. Soc., 2008, 130, 1017–1024 CrossRef CAS PubMed.
- M. A. Scorciapino, G. Pirri, A. V. Vargiu, P. Ruggerone, A. Giuliani, M. Casu, J. Buerck, P. Wadhwani, A. S. Ulrich and A. C. Rinaldi, Biophys. J., 2012, 102, 1039–1048 CrossRef CAS PubMed.
- M. Tang and M. Hong, Mol. BioSyst., 2009, 5, 317–322 RSC.
- J. Thundimadathil, R. W. Roeske and L. Guo, Biochem. Biophys. Res. Commun., 2005, 330, 585–590 CrossRef CAS PubMed.
- W. C. Wimley, K. Hristova, A. S. Ladokhin, L. Silvestro, P. H. Axelsen and S. H. White, J. Mol. Biol., 1998, 277, 1091–1110 CrossRef CAS PubMed.
- A. Kumar, R. R. Ernst and K. Wuthrich, Biochem. Biophys. Res. Commun., 1980, 95, 1–6 CrossRef CAS PubMed.
- P. Guntert, C. Mumenthaler and K. Wuthrich, J. Mol. Biol., 1997, 273, 283–298 CrossRef CAS PubMed.
- K. Wüthrich, NMR of Proteins and Nucleic Acids, 1986 Search PubMed.
- E. Lindahl, B. Hess and D. van der Spoel, J. Mol. Model., 2001, 7, 306–317 CrossRef CAS.
- W. F. Van Gunsteren, S. R. Billeter, A. A. Eising, P. H. Hünenberger, P. Krüger, A. E. Mark, W. R. P. Scott and I. G. Tironi, Biomolecular Simulation: The GROMOS96 Manual and User Guide, 1996 Search PubMed.
- N. Juranic, S. Macura and F. G. Prendergast, Protein Sci., 2003, 12, 2633–2636 CrossRef CAS PubMed.
- A. Kumar, V. Ramakrishnan, R. Ranbhor, K. Patel and S. Durani, J. Phys. Chem. B, 2009, 113, 16435–16442 CrossRef CAS PubMed.
- F. Avbelj, Biochemistry, 1995, 34, 755–764 CrossRef CAS PubMed.
- F. Avbelj and R. L. Baldwin, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 1309–1313 CrossRef CAS PubMed.
- E. G. Baker, G. J. Bartlett, M. P. Crump, R. B. Sessions, N. Linden, C. F. Faul and D. N. Woolfson, Nat. Chem. Biol., 2015, 11, 221–228 CrossRef CAS PubMed.
- J. Batra, H. Tjong and H. X. Zhou, Protein Eng., Des. Sel., 2016, 29, 301–308 CrossRef CAS PubMed.
- S. Kumar and R. Nussinov, ChemBioChem, 2002, 3, 604–617 CrossRef CAS PubMed.
- S. Kumar and R. Nussinov, ChemBioChem, 2004, 5, 280–290 CrossRef CAS PubMed.
- H. Nakamura, Q. Rev. Biophys., 1996, 29, 1–90 CrossRef CAS PubMed.
- M. Y. Tsai, W. Zheng, D. Balamurugan, N. P. Schafer, B. L. Kim, M. S. Cheung and P. G. Wolynes, Protein Sci., 2016, 25, 255–269 CrossRef CAS PubMed.
- E. Hamel, C. M. Lin, S. Kenney and P. Skehan, Arch. Biochem. Biophys., 1991, 286, 57–69 CrossRef CAS PubMed.
- J. Robinson and Y. Engelborghs, J. Biol. Chem., 1982, 257, 5367–5371 CAS.
- H. Tao, S. C. Lee, A. Moeller, R. S. Roy, F. Y. Siu, J. Zimmermann, R. C. Stevens, C. S. Potter, B. Carragher and Q. Zhang, Nat. Methods, 2013, 10, 759–761 CrossRef CAS PubMed.
- B. Borah, J. S. Cohen and A. Bax, Biopolymers, 1985, 24, 747–765 CrossRef CAS PubMed.
- D. P. Geerke, C. Oostenbrink, N. F. A. Van Der Vegt and W. F. Van Gunsteren, J. Phys. Chem. B, 2004, 108, 1436–1445 CrossRef CAS.
- V. Ramakrishnan, R. Ranbhor, A. Kumar and S. Durani, J. Phys. Chem. B, 2006, 110, 9314–9323 CrossRef CAS PubMed.
- J. Ryckaert, G. Ciccotti and H. Berendsen, J. Comput. Phys., 1977, 23, 327–341 CrossRef CAS.
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS.
- U. Essmann, L. Perera, M. L. Berkowitz, T. Darden, H. Lee and L. G. Pedersen, J. Chem. Phys., 1995, 103, 8577–8593 CrossRef CAS.
- X. Daura, W. F. Van Gunsteren and A. E. Mark, Proteins: Struct., Funct., Genet., 1999, 34, 269–280 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Fig. S1–S8 and Tables ST1 and ST2. See DOI: 10.1039/c7ra02137b |
| ‡ Life Science Institute, University of Michigan, Ann Arbor, MI, USA 48105. |
| § Department of Chemistry, School of Basic and Applied Sciences, Sri Guru Granth Sahib World University, Fatehgarh Sahib-140406, Punjab, India. |
| ¶ Department of Chemistry, University of Toronto, Toronto, ON, M5S 3H6. |
| This journal is © The Royal Society of Chemistry 2017 |