
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Urea-assisted synthesis of hydroxyapatite nanorods from naturally occurring impure apatite rocks for biomedical applications†
W. P. S. L. Wijesinghe *abc,
M. M. M. G. P. G. Mantilakaad,
R. M. G. Rajapakse*ab,
H. M. T. G. A. Pitawalaae,
T. N. Premachandraf,
H. M. T. U. Herathg,
R. P. V. J. Rajapaksef and
K. G. Upul Wijayanthah
*abc,
M. M. M. G. P. G. Mantilakaad,
R. M. G. Rajapakse*ab,
H. M. T. G. A. Pitawalaae,
T. N. Premachandraf,
H. M. T. U. Herathg,
R. P. V. J. Rajapaksef and
K. G. Upul Wijayanthah
aPostgraduate Institute of Science, University of Peradeniya, Peradeniya, 20400, Sri Lanka
bDepartment of Chemistry, Faculty of Science, University of Peradeniya, Peradeniya, 20400 Sri Lanka. E-mail: slwijesinghe@gmail.com; rmgr@pdn.ac.lk; Tel: +94 81 2394435
cDepartment of Physical Sciences, Faculty of Applied Sciences, Rajarata University of Sri Lanka, Sri Lanka
dSri Lanka Institute of Nanotechnology, Nanotechnology and Science Park, Mahenwatte, Pitipana, Homagama, Sri Lanka
eDepartment of Geology, Faculty of Science, University of Peradeniya, Peradeniya, 20400, Sri Lanka
fDepartment of Veterinary Pathobiology, Faculty of Veterinary Medicine, University of Peradeniya, Peradeniya, 20400, Sri Lanka
gDepartment of Medical Laboratory Science, Faculty of Allied Health Sciences, University of Peradeniya, Peradeniya, 20400, Sri Lanka
hDepartment of Chemistry, Loughborough University, Loughborough, LE11 3TU, UK
First published on 9th May 2017
Abstract
Hydroxyapatite (HA) nanoparticles are heavily used materials in biomedical applications. Therefore, identification of cheap and readily available raw-materials for the synthesis of HA nanoparticles is very important to fulfill the current demand. Herein, for the first time, we have developed a novel method to convert readily available, extensively distributed, naturally occurring apatites into nontoxic hydroxyapatite nanoparticles for biomedical applications. In this method, powdered apatite is digested and combusted to produce calcium phosphate nanoparticles and hydrothermally treated to convert them into high purity HA. HA nanoparticles are characterized using X-ray diffraction (XRD), Fourier Transform Infrared (FT-IR) spectroscopy, Scanning Electron Microscopy (SEM) and Transmission Electron Microscopy (TEM). Synthesized HA nanoparticles are nontoxic according to the cytotoxicity results which confirm their potential usage in biomedical applications. Therefore, this method is very important to fulfill the current demand of HA nanoparticles and for value-addition to natural apatite.
Introduction
Hydroxyapatite (HA) nanoparticles are a very attractive and heavily used material in many fields due to their broad range of applications including as a coating material in bone implants, in photo-catalysis, drug delivery systems, bone cement, bone fillers, ceramic implants, in protein chromatography, water purification and catalysis.1–6 Among these applications, HA nanoparticles are especially synthesized to recover damaged human bones and teeth as they are mainly composed of natural hydroxyapatite.7–9 Therefore, nontoxic HA nanoparticles are excessively synthesized for the fulfillment of current demand mainly in biomedical fields. Furthermore, HA nanoparticles and their nanocomposites are marketed with high-costs.HA nanoparticles are mainly synthesized through chemical synthetic routes starting from chemicals including calcium nitrate, calcium oxide and diammoniumhydrogenorthophosphate.10–14 However, in the preparation of HA nanoparticles via chemical synthetic routes, reaction conditions and amounts of reactants have to be very carefully maintained. Otherwise, the final product would contain impurities such as calcium phosphate and un-reacted reactants which will undoubtedly reduce the quality of the final HA product.15 Hence, the product will not be suitable for its end-use. Furthermore, the crystalline structure of final HA product can be affected significantly with the variation of conditions and it is yet another crucial factor that diminishes the value and applications of HA. Therefore, alternative methods and simple techniques are to be developed to synthesize HA nanoparticles in industrial-scale with good quality, uniformity and perfect crystal structure.
Naturally occurring apatite has widely been distributed with large minable quantities throughout the globe.16 However, applications of most of these deposits are limited to production of fertilizers and more value-added products should be introduced for the value-addition to these natural apatite deposits. There are three main forms of apatite; fluoroapatite, chloroapatite and hydroxyapatite; based on their chemical composition and also their mixed forms readily exist naturally.16–18 Furthermore, naturally occurring apatite could be an economical source of HA when novel cost-effective methods are developed to produce high purity HA. Therefore, conversion of naturally occurring apatite into high purity HA will be a great effort and a timely needed requirement in order to fulfill the current demand of HA as well as for the value-addition to such natural apatite deposits. Synthesis of HA nanomaterials will further increase the value addition of natural apatite deposits since HA nanoparticles are much more expensive than conventional HA micro- and millimeter-sized particles. Furthermore, non-cytotoxic, biologically safe HA nanoparticles are very valuable and are marketed at high price. However, most of apatite deposits contain significant amount of impurities which have limited their use for the synthesis of high-purity HA and their nanoparticles.17,18 Therefore, novel techniques are required to synthesize high-purity HA from natural apatite. There are a few methods have been introduced to convert naturally occurring apatite into HA nanoparticles.19,20 However, to the best of our knowledge, cytotoxicity of converted HA nanoparticles from naturally occurring apatite has not been studied.
In this paper, we describe a novel method to convert naturally occurring apatite into high-purity, nontoxic HA nanoparticles. In this method, first, powdered apatite is digested in concentrated nitric acid, and then, the digested product is combusted using urea as a fuel in order to produce a mixture of different types of calcium phosphate nanoparticles. Finally, these precursor calcium phosphate nanoparticles are hydrothermally treated in order to convert them into high-purity HA nanoparticles. The cytotoxicity experiments of synthesized HA nanoparticles show that the HA nanoparticles are indeed nontoxic. This confirms the potential use of synthesized HA nanoparticles in biomedical applications. This technique is very important on value-addition to natural apatite and to fulfill the current demand of HA nanoparticles.
Experimental
Materials
Natural apatite (NAP) crystals were collected from apatite deposit located at the village of Eppawala in north-central part of Sri Lanka. The deposit covers approximately 6 km2 land area with six hillocks. Apatite crystals were collected from various locations of the deposit to represent whole deposit. All samples were crushed, ground, ball-milled and mixed together. Finally, the mixed powder was sieved to separate different size fractions. The sizes of less than 100 μm sieved fraction was used for the synthesis of HA nanoparticles and larger sizes were further ground and milled to obtain the right sizes. Chemicals used for the synthesis were urea (99% purity), calcium oxide (99% purity), and nitric acid (70% purity). These chemicals were purchased from Sigma-Aldrich.Synthesis of HA nanoparticles from apatite
The powdered apatite sample was heated, at 1100 °C, for 3 h, in a furnace to produce calcined apatite. The calcined apatite sample (2.0 g) was dissolved in 15 M HNO3 (10.0 mL) acid. The mixture was placed in a ceramic crucible and required amount of urea fuel was added to the mixture until the mole ratio of urea to NO3− is 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. The whole mixture was heated at 300 °C to start recrystallization process via combustion. 1.0 g of sample prepared from combustion method was placed in Teflon-lined stainless steel autoclave with 30 mL of distilled water for hydrothermal treatment at pH 10, at 150 °C, for 3 h. The precipitate was washed with distilled water for 3 times and was allowed to dry under ambient laboratory conditions. Another 1 g of sample prepared from combustion method was hydrothermally treated by adding 1 g of CaO into the reaction medium at pH 10 at 150 °C for 3 h. This method was triplicated to check reproducibility.
:1. The whole mixture was heated at 300 °C to start recrystallization process via combustion. 1.0 g of sample prepared from combustion method was placed in Teflon-lined stainless steel autoclave with 30 mL of distilled water for hydrothermal treatment at pH 10, at 150 °C, for 3 h. The precipitate was washed with distilled water for 3 times and was allowed to dry under ambient laboratory conditions. Another 1 g of sample prepared from combustion method was hydrothermally treated by adding 1 g of CaO into the reaction medium at pH 10 at 150 °C for 3 h. This method was triplicated to check reproducibility.
Characterization of materials
Crystalline phases of natural apatite sample, precursor phases and the final product were mainly characterized using X-ray diffraction (XRD) technique with the aid of Siemens D5000 X-ray powder diffractometer (Cu Kα radiation of wavelength 0.154 nm and scanning rate of 1 min−1). The obtained XRD patterns were analyzed using ICDD PDF 2 database with X-powder 12 software. Fourier Transform Infrared (FT-IR) spectra of materials were recorded on a Shimadzu IR-Prestige 21 instrument with the KBr pellet method. Herein, samples and fused KBr were thoroughly dried, in a vacuum oven, at 60 °C, for 12 h. Each well dried sample and fused KBr were mixed in 1:40 mass ratio and pressed at 5 tons using a standard pellet press machine to obtain required pellets for the FT-IR analysis. The high resolution transmission electron microscopy (HRTEM) was conducted using a JEOL 2100 transmission electron microscope (TEM) for final HA product to further confirm crystalline phases of the product based on selective area electron diffraction (SAED) pattern. The morphologies of samples were analyzed using the scanning electron microscopy (SEM). The elemental composition of prepared HA samples were analyzed using energy-dispersive X-ray (EDX) spectroscopic technique and Electron Energy Loss Spectroscopy (EELS) of TEM. The TEM analyses were conducted using a JEOL, JEM-2000FX Electron Microscope. SEM and EDX analyses were conducted using Zeiss Sigma VP field emission scanning electron microscope coupled with an EDX detector. Amounts of fluoride and chloride of the final product were measured by means of Hach DR/2400 spectrophotometer.
In vitro cytotoxicity evaluation of HA nanoparticles
HOS cells were cultured and the preparation was carried out under sterile conditions using a class 2 safety cabinet (Herasafe, Kendro Laboratory products, Germany).21,22 The cell viability and cytotoxic effects of the material was determined by performing MTT assay. The Dulbecco's Modified Eagle's Medium (DMEM) and 70% ethanol were used as negative and positive controls, respectively. Extracts were prepared by eluting test HA nanoparticles in complete medium at 37 °C for 1, 5, 7, 14, 21, 28 and 35 days. Then HOS cells were cultured for 24 h and further 4 h incubation in the presence of 10% MTT [(3-(4,5)-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] (Sigma Aldrich Ltd, UK).21 Finally, Absorbance was measured on Dynatech MR 700 micro plate reader, at a test wavelength of 570 nm and a reference wavelength of 630 nm.21,23Results and discussion
Characterization of synthesized HA products
According to geochemical studies of selected NAP deposit located at Eppawala, Sri Lanka, it contains high degree of fluoride and chloride ions together with hydroxyl groups.17,18 Therefore, fluoride and chloride ions should be removed to synthesize required hydroxyapatite as a biomaterial. The XRD peaks (Fig. 1) of NAP raw-sample found at 2θ values from 10° to 60° are compatible with standard XRD patterns of hydroxyapatite (JCPDS card no. 72-1243), fluoroapatite (FA) (JCPDS card no. 73-1727) and chloroapatite (CA) (JCPDS card no. 73-1728). There is no significant change of heat-treated NAP sample at 1100 °C as found out from XRD pattern (ESI Fig. S1a†). However, increase in peak intensities of XRD pattern of heat treated NAP excluding the reduction of intensity at (300) plain may be due to the removal of chloride ions from the crystal lattice during the heat treatment. Fig. 1b shows the XRD pattern of the synthesized products prepared via combustion method which contains diffraction peaks of apatites, tricalcium phosphate (TCP) and calcium pyrophosphate (Ca2P2O7) (CPP). Herein, heat-treated NAP reacts with the HNO3 acid in order to produce Ca(NO3)2. During the combustion, Ca(NO3)2 acts as an oxidant and urea acts as a fuel. Self-propagating redox reaction is identified along with the decomposition of nitrate and oxidation of urea, which releases a large amount of energy as heat. The combustion reaction has been initiated at low temperature and after the initiation, the reaction supplies required amount of heat to the reaction medium in order to continue the combustion. Hence, no external heat source is needed and this leads the formation of HA (Reaction S1 in the ESI†).24 Also, in mild acidic media (about pH = 5), there is a possibility to precipitate monetite (CaHPO4·2H2O) (Reaction S2 in the ESI†).25 During the combustion process, monetite can be converted into the CPP (Reaction S3 in the ESI†) due to the excessive amount of heat.25,26 XRD peaks of CPP are found at 2θ values of 18.0°, 20.0°, 23.5°, 25.2°, 26.9°, 27.7°, 33.7°, 34.6°, 36.5°, 37.0°, 39.9°, and 45.5° (JCPDS card no. 72-1651) (Fig. 2c). In addition, formation of TCP can be identified during the combustion of heat treated NAP with the aid of XRD pattern (Fig. 1b) with peaks at 2θ values of 31.0°, 34.3°, 37.8° and 51.4°. HA then reacts with CPP in order to form TCP with the generated heat from the combustion (Reaction S4 in the ESI†).25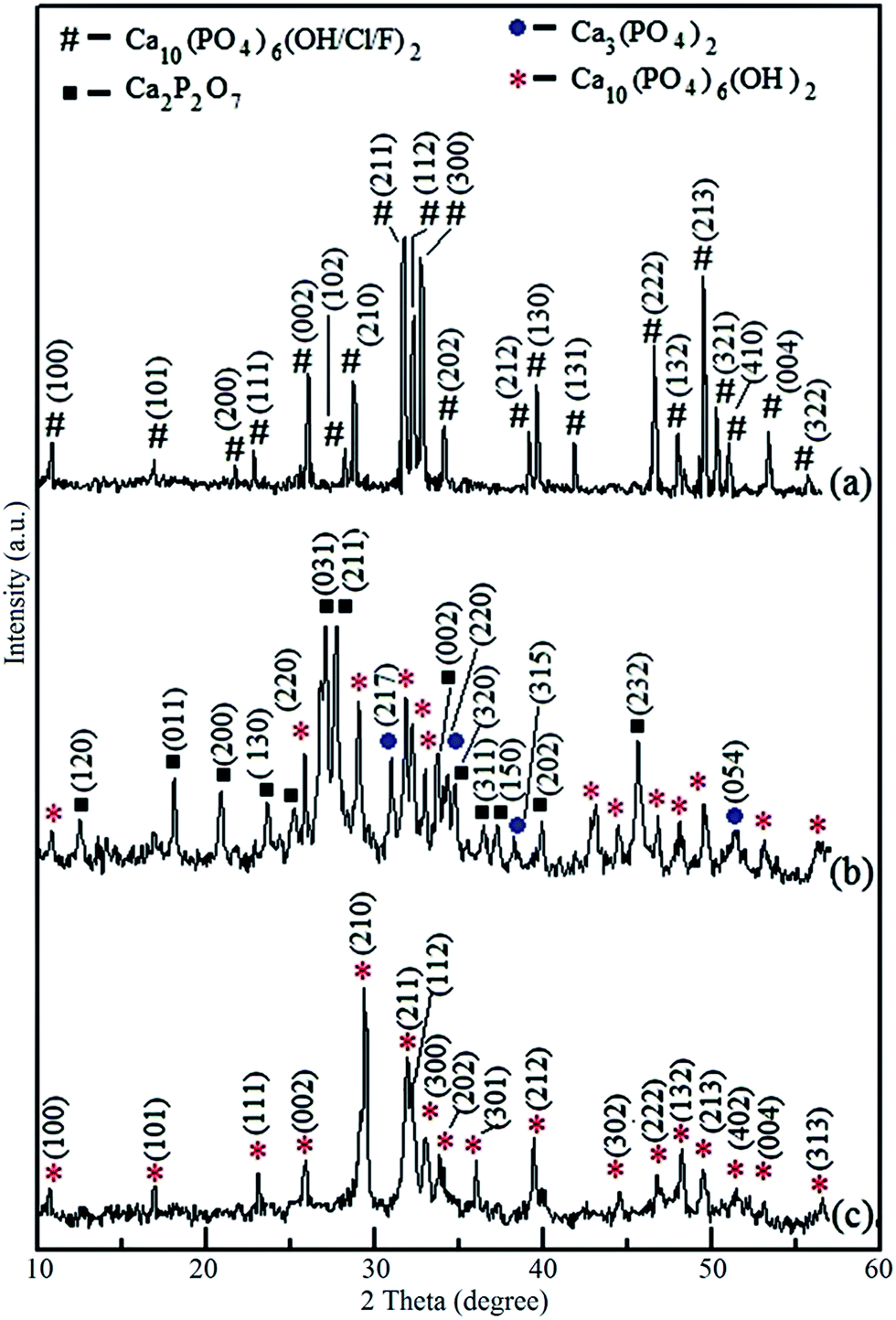 | ||
| Fig. 1 XRD patterns of (a) NAP sample, (b) synthesized product via combustion method, (c) hydrothermally treated final HA nanoparticles at pH 10 with CaO. | ||
 | ||
| Fig. 2 FTIR spectra of (a) Eppawala NAP (b) synthesized product via combustion method and (c) hydrothermally treated final HA nanoparticles at pH 10 with CaO. | ||
During the process of combustion, a mixture of calcium phosphates has been obtained from NAP as a result of conversion of TCP and CPP into the HA. (Reactions S5 and S6 in the ESI†). Therefore, the final product obtained from the urea-assisted combustion of NAP is hydrothermally treated in order to convert the mixture of calcium phosphates into HA nanoparticles. XRD peaks of crystal planes of TCP have completely been removed (ESI Fig. S1b†) during the hydrothermal treatment at pH 10. Crystal plane of HA at (102) has become dominant compared to the other planes. Therefore, conversion of TCP into HA has been occurred at pH 10. However, there is no phase transformation of CPP at different pH conditions through the hydrothermal process. However, transformation of CPP into HA has been occurred with CaO (Reaction S6 in the ESI†). The characteristic XRD peaks of HA are found at correct positions of final HA product prepared through hydrothermal synthesis with CaO at pH 10 (Fig. 1c) (JCPDS card no. 72-1243). However, there is a small difference in (210) basal plane of the synthesized product. Intensity of that peak has become higher than the other peaks as well as different from the standard data. This is due to the, over growth of HA crystals along the (210) plane during hydrothermal treatment. According to the Debye–Scherrer formula, which is applied to (210) and (211) planes, the responsible crystallite sizes are 43 nm and 12 nm, respectively. Hence, formation of needle-like HA crystals can be identified in SEM and TEM images (vide infra).
XRD results are further confirmed by FTIR studies (Fig. 2). The sharp band centered at 1644 cm−1 and the broad band around 3400 cm−1 represent the stretching vibration of combined water in the HA lattice. However, NAP sample contains less amount of combined water in the HA lattice because there is no more intense bands at the required positions in FTIR spectrum as shown in Fig. 2a. The bands centered at 950 cm−1 and 1046 cm−1 are obtained due to the symmetric (ν1) and anti-symmetric (ν3) stretching vibrations of the P–O bonds which are presented in PO43− groups of NAP. Similarl y, 606 cm−1, 567 cm−1 bands reflect the (ν4) anti-symmetric bending of the phosphate group of NAP as well as the bands at 431 cm−1 and 492 cm−1 are also related to the (ν2) symmetric bending of the phosphate group present in NAP.27,29 Characteristic bands for the carbonated apatite are found at 1380 cm−1 and 1447 cm−1 in these samples which can be attributed to the carbonate (–CO3) group.27–30 This suggests that some of the hydroxyl groups in NAP lattice have been replaced by the carbonate ions. However, the characteristic FTIR band of carbonate groups at 860 cm−1 has been masked due to high absorbance of bands of phosphate groups. The FTIR spectrum of synthesized product via combustion method is shown in Fig. 2b. The presence of pyrophosphate groups in the synthesized samples can be identified with the broad IR absorbance at around 1200 cm−1 position.31,32 Narrow IR absorption band at 750 cm−1 is also attributed to the pyrophosphate of synthesized samples.31,32 Incorporation of H2O into the synthesized crystals from combustion method can be identified with the highest appearance of bands at 1644 cm−1 and the broad band around 3400 cm−1.27 The band at 3549 cm−1 is related to the bending vibration of the hydroxyl group (–OH) of HA.27,28 Similarly, during the combustion process, incorporation of carbonate groups in the synthesized products can be identified with the increase in intensities of the bands at 1380 cm−1 and 1447 cm−1.30 According to the FTIR spectra, the presences of CPP and calcium phosphates have been found in the synthesized products via combustion method. XRD result of final HA product is also confirmed by FTIR spectrum of final HA product (Fig. 2c). As previously described, all the characteristic IR absorption bands of HA have been appeared in the IR spectrum of final HA product.
The crystalline phase and morphology of final HA product is characterized using TEM, HRTEM and SAED studies as shown in Fig. 3. The final HA product is in needle-like morphology. SAED result is in good agreement with XRD and FT-IR results. SAED shows that the final product only contains crystalline hydroxyapatite at the electron diffraction planes at (111), (102), (202), (302), (132), (331), (215) and (352). The d-spacing of the lattice fringe of final HA product according to the HRTEM images is found to be 2.6 Å (ESI Fig. S2†).
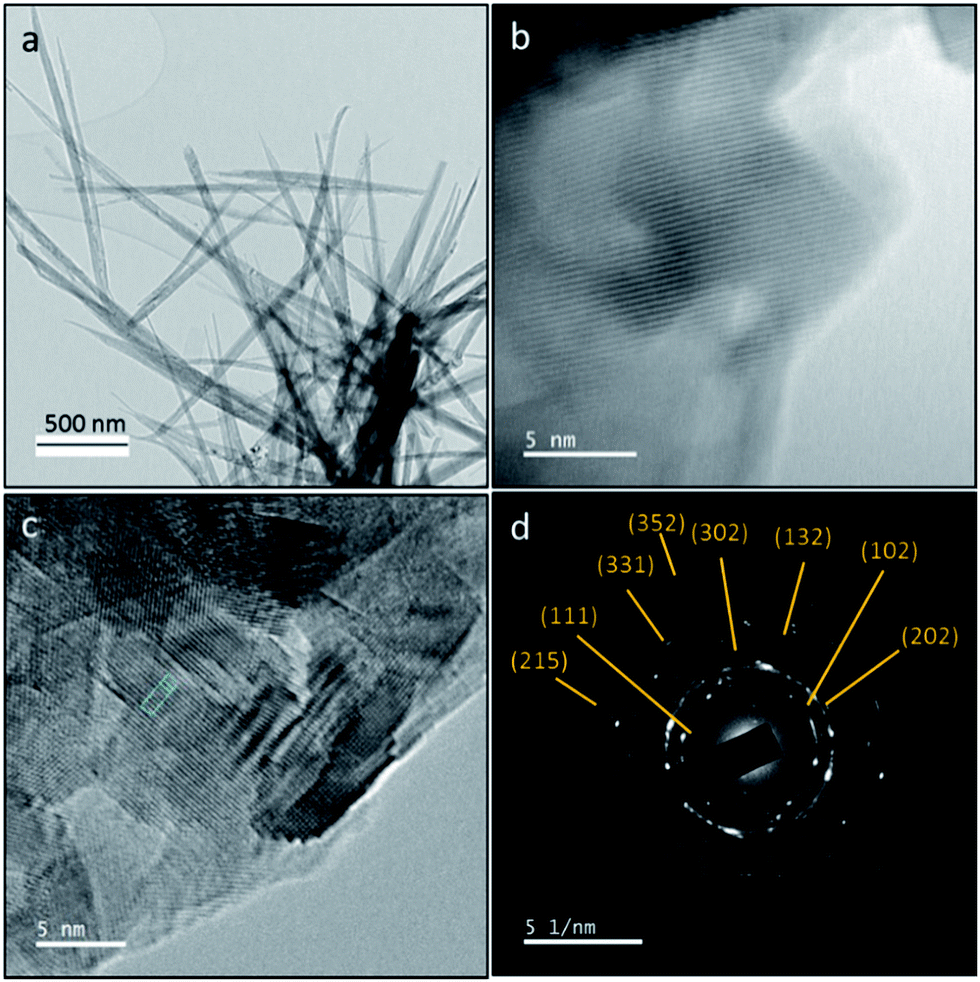 | ||
| Fig. 3 (a) TEM image (b) and (c) HRTEM images and (d) SAED pattern of synthesized HA nanorods. | ||
SEM images of formed products from NAP at the end of combustion are shown in Fig. 4. Rod-like particles are found in these SEM images. The average length and width of rod-like particles are about 100 nm and 50 nm, respectively. Herein, synthesized calcium phosphate and pyrophosphate particles have been aggregated to form irregular shapes and dumbbell-like morphology in micrometer range. SEM image of synthesized final HA product from NAP is shown in Fig. 4b. Needle-like particles have been found in SEM images. The average width and length of needle like particles is about 80 and 750 nm in size, respectively. As described earlier, morphology of such synthesized HA nanoparticles is well-matched with the XRD data and calculated crystallite sizes because those data also describes the formation of needle-like HA crystals.
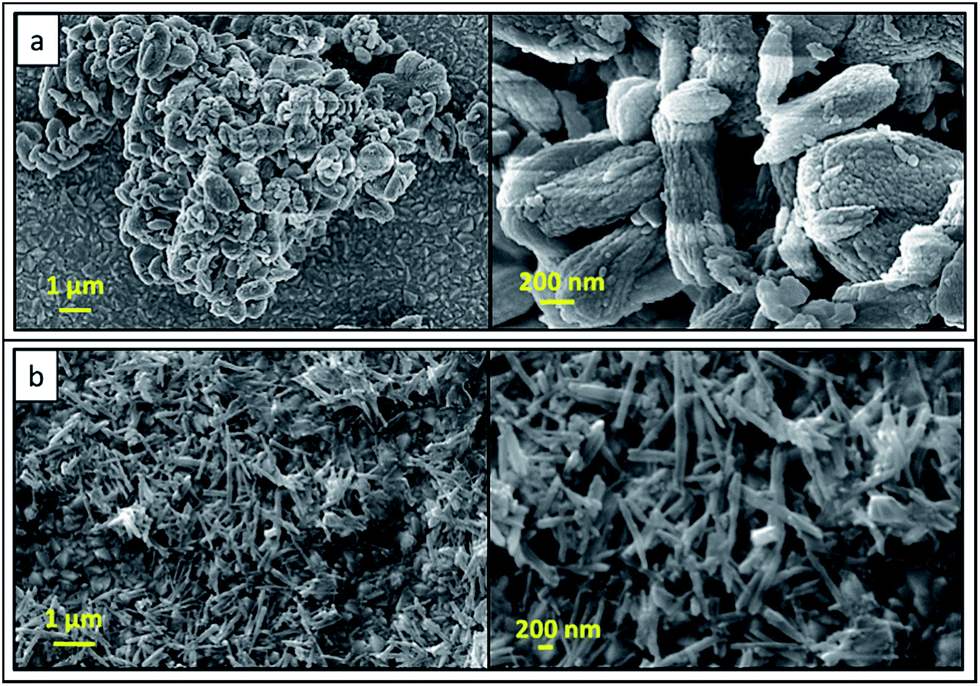 | ||
| Fig. 4 SEM images of (a) precursor product after combustion with urea-combustion (b) final HA nanorods. | ||
Morphologies and sizes of the HA nanoparticles synthesized by combustion method depend on the various parameters. The flame temperature (highest temperature generated in the combustion) directly affects on the characteristics of synthesized powder. In addition, other reaction parameters such as nature of fuel, fuel-to-oxidizer ratio and the initial temperature of the furnace are factors affecting on properties of the resulting powder. Another important feature in this process is the prevention of over growth of particles by cooling the product effectively through evolution of gases during the combustion process. Aghayan et al. (2012), have studied the morphological changes of HA nanoparticles with the ratio of urea to HNO3. In their study, rod shaped nanoparticles were formed when the ratio between urea to HNO3 is around one.24 This is due to the complete oxidation of urea by releasing more energy during the process. Moreover, the acidity of the solution is increased strongly with increasing amount of HNO3 and the reactivity of phosphate ions is decreased. Therefore, the amount of HNO3 leads to diminution of TCP while increasing the amount of HA.
In this study, the ratio between urea to HNO3 is 1:1. Therefore, during the combustion, large amount of energy is released while forming rod shaped HA, CPP and TCP. Hydrothermal method has been exploited to prepare 1D nanosized HA, owing to its capability to induce 1D growth.33–36 Therefore in the present study, we have used hydrothermal method to convert both CPP and CP into HA nanoparticles. Rod-shaped particles are elongated through [210] direction while forming needle-like HA crystals due to the high pressure and temperature during the hydrothermal treatment.
Fig. 5 shows the EDX and EELS spectra of pure NAP and final HA nanoparticles. The EDX spectrum in Fig. 5a clearly indicates the presence of F− and Cl− in pure NAP samples. However, there are no peaks in EDX spectrum (Fig. 5b) for F and Cl due to the removal of F− and Cl− during the synthesis process of HA nanoparticles. This is the important point of this conversion method of NAP to the HA nanoparticles because there is no published report in literature on removal of incorporated F− and Cl− from NAP. This removal process occurs during the process of combustion. This novel process contains interesting findings in order to synthesize high purity HA nanoparticles by removing harmful impurities from HA nanoparticles. According to the quantitative analysis of spectrophotometry, the final HA product does not contain F and Cl species. Furthermore, weight percentages of Si and Sr impurities of final HA nanoparticles based on EDX and EELS results are 0.29% and 0.29%, respectively. No other impurities are found in HA nanoparticles. Therefore HA nanoparticles are of more than 99% purity. This means that the devised method is very successful in the synthesizing high purity HA from impure apatite rocks. Furthermore, such small amount of Sr would not contribute to any toxicity to HA nanoparticles.
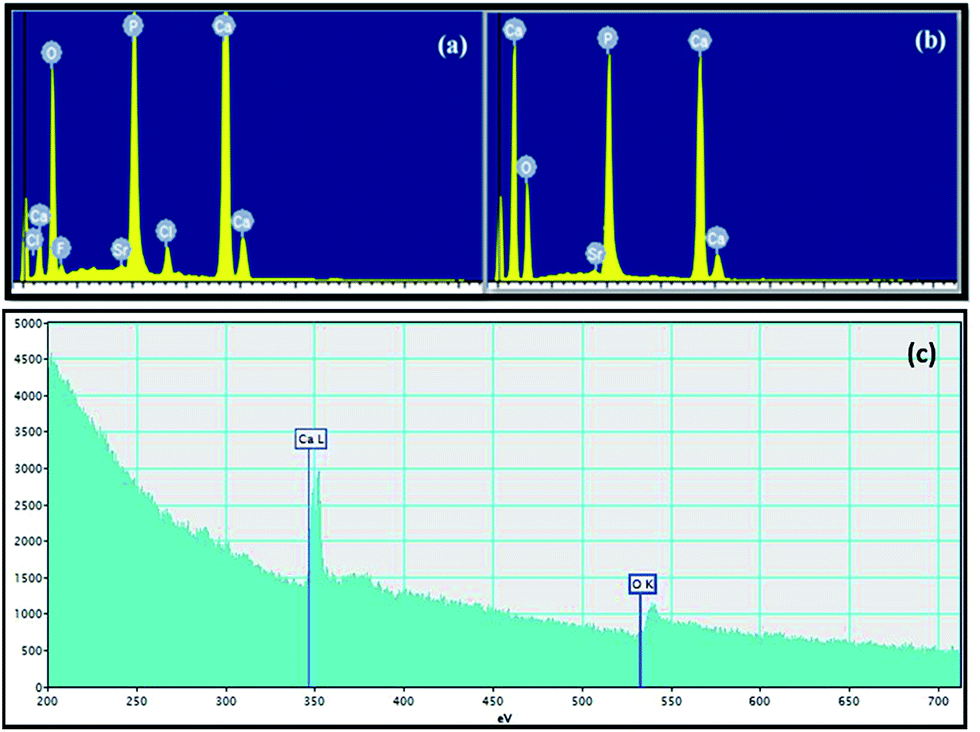 | ||
| Fig. 5 EDX spectra of (a) pure NAP and (b) finally synthesized HA nanoparticles and (c) EELS spectrum of HA nanorods. | ||
Evaluation of cytotoxicity of HA nanorods for biomedical applications
Absorption values of MTT test elution and controls are shown in Fig. 6. The results of the MTT assay, which measures the test materials, do not obtain any toxic leachable substance which would cause a deleterious effect to the cells and hereby do not affect cellular metabolism. Results of the test materials are slightly lower than those of the negative control that should be mentioned, the fact that the synthesized HA nanoparticles are less performing than the negative control. However, these results do not show any significant difference in the response (within the range of experimental error), at days 1, 7, 14, 28, and 35 of incubation and they are comparable to the negative control (p < 0.05). Therefore, the long term evolution of the response of osteoblast-like cells to the synthesized HA nanoparticles, confirm the non-cytotoxicity of the material. Also, reduction of the metabolic activity with the positive toxic control can be observed. In addition, the percentage viability of cells can be calculated using equation which is given in our previous publication.21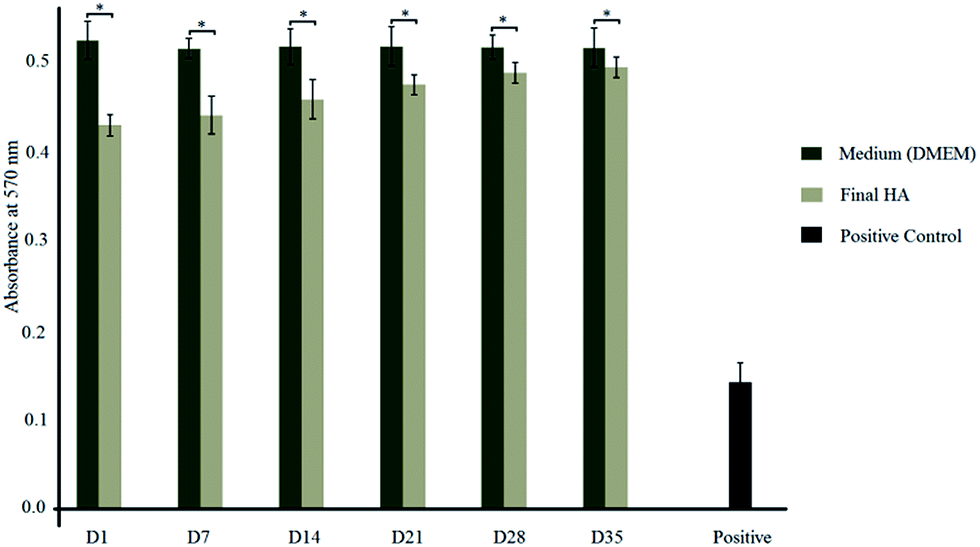 | ||
| Fig. 6 Absorption values of synthesized HA nanoparticles and DMEM resulted from MTT assay (*p < 0.05). | ||
The toxicity level of synthesized HA nanoparticles can be identified by considering standard percentage viability range.21,37–40 Those standard toxicity levels and calculated experimental data are tabulated in Table 1. Because of these tested samples having toxicity level 1 for all 6 elution (day 1, 7, 14, 21, 28 and 35), we can say that those can be considered as nontoxic to the human body.
| Standard viability data33,34 | Experimental data | |||
|---|---|---|---|---|
| Cell viability (%) | Toxicity level | Sample | Cell viability (%) | Toxicity level |
| ≥100 | 0 | Day 1 | 81.9 | 1 |
| 75–99 | 1 | Day 7 | 85.5 | 1 |
| 50–74 | 2 | Day 14 | 88.6 | 1 |
| 25–49 | 3 | Day 21 | 91.8 | 1 |
| 1–24 | 4 | Day 28 | 94.5 | 1 |
| 0 | 5 | Day 35 | 95.8 | 1 |
Conclusions
Naturally occurring apatite has successfully been converted into needle-like hydroxyapatite nanoparticles in nanometer range through combustion method followed by hydrothermal treatment. Mixture of rod-like and dumbbell-like phosphates including calcium pyrophosphate and tricalcium phosphate have been obtained after combustion of digested apatite with urea. The mixture of apatite have directly been converted into needle-like HA nanoparticles after the hydrothermal treatment at pH 10. Both fluoro- and chloro-apatites in the natural apatite sample have also been converted into HA nanoparticles. The final HA nanoparticles are of more than 99% purity and quality. The synthesized HA nanoparticles are suitable for biomedical applications as they are nontoxic. The reported novel method is up-scalable to industrialize the process. This effort is very useful for the value-addition to the readily available extensive deposits of apatite throughout the globe and to fulfill the current high demand of HA nanoparticles.Acknowledgements
The authors are grateful to the financial support by the National Research Council (Grant no. 11-46), Sri Lanka. The authors also thank Dr T. B. N. S. Madugalla for providing NAP samples.Notes and references
- Z. Ahmad, E. S. Thian, J. Huang, M. J. Edirisinghe, S. M. Best, S. N. Jayasinghe, W. Bonfield, R. A. Brooks and N. J. Rushton, J. Mater. Sci., 2008, 19, 3093 CAS.
- G. Munir, J. Huang, M. J. Edirisinghe, G. Koller, L. Di Silvio and W. J. Bonfield, J. R. Soc., Interface, 2011, 8, 678 CrossRef CAS PubMed.
- D. Gopi, E. Shinyjoy, M. Sekar, M. Surendiran, L. Kavitha and T. S. S. Kumar, Corros. Sci., 2013, 73, 321 CrossRef CAS.
- R. A. Surmenev, Surf. Coat. Technol., 2012, 206, 2035 CrossRef CAS.
- S. Onder, F. N. Kok, K. Kazmanli and M. Urgen, Mater. Sci. Eng., C, 2013, 33, 4337 CrossRef CAS PubMed.
- J. Chakraborty, N. Daneu, A. Recnik, M. Chakraborty, S. Dasgupta, J. Ghosh, S. Sengupta, S. Mazumdar, M. K. Sinha and D. J. Basu, J. Taiwan Inst. Chem. Eng., 2011, 42, 682 CrossRef CAS.
- Y. Wu and S. Bose, Langmuir, 2005, 21, 3232 CrossRef CAS PubMed.
- Y. Han, S. Li, X. Wang, I. Bauer and M. Yin, Ultrason. Sonochem., 2007, 14, 286 CrossRef CAS PubMed.
- B. Cengiz, Y. Gokce, N. Yildiz, Z. Aktas and A. Calimli, Colloids Surf., A, 2008, 322, 29 CrossRef CAS.
- S. Pramanik, A. K. Agarwal, K. N. Rai and A. Garg, Ceram. Interfaces, 2007, 33, 419 CrossRef CAS.
- R. A. Ayers, D. E. Burkes, G. Gottoli, H. C. Yi, F. Zhim, L. H. Yahia and J. J. Moore, J. Biomed. Mater. Res., Part A, 2007, 81, 634 CrossRef PubMed.
- A. Fahami, R. Ebrahimi-Kahrizsangi and B. Nasiri-Tabrizi, Solid State Sci., 2011, 13, 135 CrossRef CAS.
- A. Afshar, M. Ghorbani, N. Ehsani, M. R. Saeri and C. C. Sorrell, Mater. Des., 2003, 24, 197 CrossRef CAS.
- L. B. Kong, J. Ma and F. Boey, J. Mater. Sci., 2002, 37, 1131 CrossRef CAS.
- M. S. Shojai, M. T. Khorasani, E. D. Khoshdargi and A. Jamshidi, Acta Biomater., 2013, 9, 7591 CrossRef PubMed.
- J. M. Hughes and J. Rakovan, Rev. Mineral. Geochem., 2002, 48, 1 CrossRef CAS.
- A. Pitawala, M. Schidlowski, K. Dahanayake and W. Hofmeister, Miner. Deposita, 2003, 38, 505 CrossRef CAS.
- A. Pitawala and B. G. Lottermoser, Mineral. Petrol., 2012, 105, 57 CrossRef CAS.
- S. Saoiabi, S. EL Asri, A. Laghzizil, T. Coradin and K. Lahlil, Mater. Lett., 2010, 64, 2679 CrossRef CAS.
- S. El Asri, A. Laghzizil, A. Saoiabi, A. Alaoui, K. El Abassi, R. M’hamdi and T. Coradin, Colloids Surf., A, 2009, 350, 73 CrossRef CAS.
- W. P. S. L. Wijesinghe, M. M. M. G. P. G. Mantilaka, K. G. Chathuranga Senarathna, H. M. T. U. Herath, T. N. Premachandra, C. S. K. Ranasinghe, R. P. V. J. Rajapakse, R. M. G. Rajapakse, M. Edirisinghe, S. Mahalingam, I. M. C. C. D. Bandara and S. Singh, Mater. Sci. Eng., C, 2016, 63, 172 CrossRef CAS PubMed.
- H. M. T. U. Herath, Novel Ceramics and Ceramic Structures: An In-vitro Study of Osteoblast Response, Ph.D thesis, University of London, 2006.
- H. M. T. U. Herath, L. Disilvio and J. R. G. Evans, J. Biomed. Mater. Res., Part A, 2010, 94, 130 CrossRef CAS PubMed.
- M. A. Aghayan and M. A. Rodríguez, Mater. Sci. Eng., C, 2012, 32, 2464 CrossRef CAS.
- A. M. E. Kady, K. R. Mohamed and G. T. E. Bassyouni, Ceram. Int., 2009, 35, 2933 CrossRef.
- T. Hattori and Y. Lwadate, J. Am. Ceram. Soc., 1990, 73, 1803 CrossRef CAS.
- W. P. S. L. Wijesinghe, M. M. M. G. P. G. Mantilaka, E. V. A. Premalal, H. M. T. U. Herath, S. Mahalingam, M. Edirisinghe, R. P. V. J. Rajapakse and R. M. G. Rajapakse, Mater. Sci. Eng., C, 2014, 42, 83 CrossRef CAS PubMed.
- P. Wang, C. Li, H. Gong, X. Jiang, H. Wang and K. Li, Powder Technol., 2010, 203, 315 CrossRef CAS.
- C. Gu, D. R. Katti and K. S. Katti, Spectrochim. Acta, Part A, 2013, 103, 25 CrossRef CAS PubMed.
- P. Moghimian, A. Najafi, S. Afshar and J. Javadpour, Adv. Powder Technol., 2012, 23, 744 CrossRef CAS.
- T. V. Safronova, V. I. Putlyaev, M. A. Shekhirev and A. V. Kuznetsov, Glass Ceram., 2007, 64, 102 CrossRef CAS.
- H. Zhao, W. He, Y. Wang, Y. Yue, X. Gao, Z. Li and X. Zhang, Mater. Chem. Phys., 2008, 111, 265 CrossRef CAS.
- S. Murakami, K. Kato, Y. Enari, M. Kamitakahara, N. Watanabe and K. Ioku, Ceram. Int., 2012, 38, 1649 CrossRef CAS.
- J. S. Earl, D. J. Wood and S. J. Milne, J. Phys.: Conf. Ser., 2006, 26, 268 CrossRef.
- M. Kamitakahara, C. Ohtsuki and G. Kawachi, J. Ceram. Soc. Jpn., 2008, 116, 6 CrossRef CAS.
- S. Manafi and M. R. Rahimipour, Chem. Eng. Technol., 2011, 34, 972 CrossRef CAS.
- A. Jankovic, S. Erakovic, M. V. Sekulic, V. M. Stankovic, S. J. Park and K. Y. Rhee, Prog. Org. Coat., 2015, 83, 1 CrossRef CAS.
- M. Mumjitha and V. Raj, J. Mech. Behav. Biomed. Mater., 2015, 46, 205 CrossRef CAS PubMed.
- S. Fujibayashi, M. Neo, H. Kim, T. Kokubo and T. Nakamura, Biomaterials, 2003, 24, 1349 CrossRef CAS PubMed.
- L. Zhang, S. Zhu, Y. Han, C. Xiao and W. Tang, Mater. Sci. Eng., C, 2014, 43, 86 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Two XRD patterns and chemicals reactions those are related to this synthesis process are included in supplementary document. See DOI: 10.1039/c7ra02166f |
| This journal is © The Royal Society of Chemistry 2017 |