
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structure and dynamics processes in free-base chlorins controlled by chemical modifications of macroring and aryl groups in meso-positions†
J. Śniechowska,
P. Paluch and
M. J. Potrzebowski *
*
Centre of Molecular and Macromolecular Studies, Polish Academy of Sciences, Sienkiewicza 112, 90-363 Lodz, Poland. E-mail: marekpot@cbmm.lodz.pl
First published on 10th May 2017
Abstract
In this work we present the synthesis, detailed structural characterization and analysis of molecular motion for unsymmetrical pyrrolidine-fused chlorins employing NMR, UV spectroscopy and DFT theoretical calculations. In samples, the meso-rings were modified by substitution of hydrogen by fluorine in ortho 1 and meta positions 2. The sample with perfluorinated ring 3 and phenyl derivative 4 were used as reference species. The assignment of signals was performed employing 2D NMR techniques. The rotational dynamics was examined by means of 1H and 19F variable-temperature (VT) 1D NMR spectroscopy and 2D EXSY experiments. The synergism of steric effect between pyrrolidine ring and meso-rings is unambiguously proved. Models 1 and 3 behave very similar, aromatic rings are rigid in temperature range 233–373 K. For sample 2 and 4 the distinct molecular dynamics was revealed. The barrier of rotation depends on localization of ring in the chlorin structure. Those which are bonded in the neighborhood of pyrrolidine ring are more rigid compared to those localized on the opposite site. The temperature is a trigger which sequentially releases the rotation of aromatic group in the desired localization. Introduction of fluorine in labeled position has influence on static geometry defined by saddling angles.
Introduction
Chlorins (hydroporphyrins) belong to the big family of porphyrinoids, an intriguing class of macrocyclic heteroorganic compounds widely distributed in the nature. Due to unique physical and chemical properties porphyrinoids play a key role in important biological processes including cell respiratory, light harvesting, and electron transfer. They are also a meaningful class of compounds from the practical point of view, intensively studied for application in catalysis,1 electronics,2 supramolecular chemistry3 and materials science.4 In addition, they are used as photosensitizers5 and anticancer or antibacterial drugs6 in medicine. Excellent spectroscopic features of chlorins are reasons why these compounds found number of spectacular applications e.g. in photodynamic therapy, light-energy conversion systems or synthetic models of photosynthetic reaction centers.Chlorins strongly absorb light in the near infrared part of electromagnetic spectrum and are relatively photostable. Introduction of halogen atoms (bromine, chlorine, or fluorine) to the structure of tetrapyrrolic compounds enhances their photodynamic activity and controls chemical and spectroscopic properties.6–9 The most important chemical modification is replacement of hydrogen by fluorine atoms or fluorinated moieties in the aromatic residues for meso-position. Skillful introductions of fluorine substituents into chlorins may increase photostability, lipophilicity and high level of singlet oxygen generation.10 It is well established that photonic properties of porphyrinoids are modulated by first-order molecular structure and conformation of meso-aryl groups with respect to the macroring plane. When the aromatic groups of tetra-meso-aryl-substituted porphyrin are coplanar with the macrocycle then aryl ring would be a part of porphyrin chromophore. Great efforts are being concentrated on the improvement of their properties and what follows on their possible applications.
The porphyrinoid derivatives have recently attracted much attention as components of nanomolecular machines; rotors,11,12 wheels,13 and gyroscopes.14 Application of the porphyrinoids in the construction of different types of functional molecular systems was reviewed by Michl and coworkers,15 Kay, Leigh, Zerbetto16 and others.17 Both metal free, as well as metal complexes of porphyrins, are used as building units of advanced machines. For instance, molecular oscillator based on supramolecular complexes of metalloporphyrin and fullerenes was described by Tashiro et al.18 Molecular gear assembling metalloporphyrins and cavitand were reported by Kobayashi and coworkers.19 Systems based on tin(IV) porphyrins as stator bearing at the meso-position, a monodentate coordinating site and tridentate chelate on the different handles as rotors were shown.20,21 Dynamics processes of porphyrinoids are also key element in construction of molecular switches and sensors.22 Ishihara et al. have reviewed number of derivatives which can be used for this purpose.23 Unique complexation properties of cyclic zinc(II) bisporphyrin with flexible linker were employed for synthesis of molecular switches with controllable photo and electro properties.24 The photomolecular switches with strong absorption properties of porphyrinoids were also reported.25
The porphyrinoid derivatives can undergo complex motions including positional displacements of submolecular components and/or local molecular dynamics both in the liquid and solid states. Knowledge about the nature of these processes and tools for controlling the motion in molecular machines is a key information determining the synthetic strategy. The best recognized dynamics process in porphyrinoids is rotation of aromatic rings in meso-position. Gottwald and Ullman26 were the first to observe rotation of the phenyl rings in meso-tetraphenylporphyrin. This problem was discussed in detail for different porphyrin derivatives in review articles and textbooks.27–29
The local molecular motion of meso-aryl rings can be restricted or eliminated by hindrance effect. In the most common approach introducing the bulky group at β-position of pyrrolic ring and steric interactions with adjacent residue has influence on the rotation barrier. Such phenomenon was reported in detail by Noss et al.30 and by other authors for number of chemically modified porphyrins. To the best of our knowledge the literature about dynamics process in chlorins is rather limited. Stolzenberg et al.31 reported activation barriers for meso-aryl group rotation in metal-free and titanyl hydroporphyrins. It was found that activation barriers in these compounds ranged from 15.6 to 18.6 kcal mol−1.
In this paper we show how dynamic processes in the chlorins can be controlled and sequentially modulated by combination of steric effects in macrocyclic framework and aryl residues in meso-position. For this purpose we employ derivatives as shown in Fig. 1.
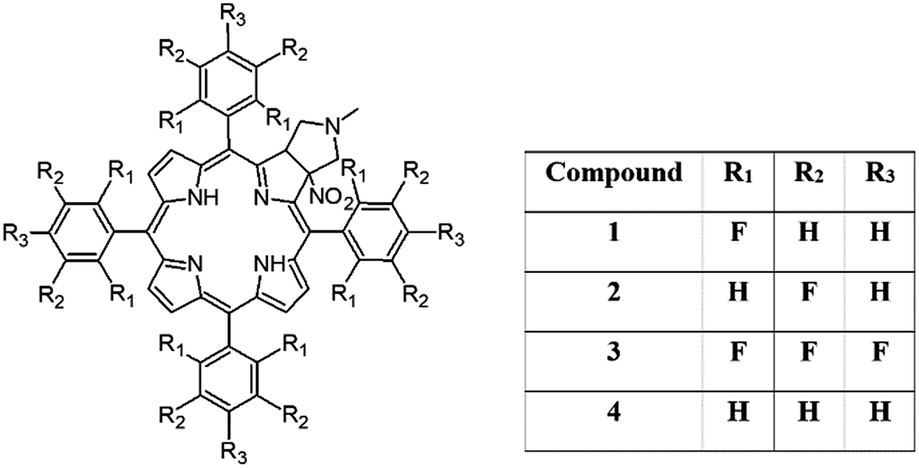 | ||
| Fig. 1 Structure of pyrrolidine-fused chlorins 1–4. | ||
Results and discussion
Synthesis of unsymmetrical chlorins
It is well-known that 1,3-dipolar cycloaddition32–36 is an useful method of transformation of porphyrins into chlorins. Based on Cavaleiro's37 discovery that porphyrins could act as dipolarophiles, we synthesized target compounds. The synthesis of compounds is a multi-step sequence and synthetic route is presented in Scheme 1. The starting porphyrins 5 and 6 were synthesized according to procedures described in the literature. Porphyrin 5 was prepared from aldehyde and pyrrole with a mixture of nitrobenzene and acetic acid38 as a solvent. For porphyrin 6 we adopted Adler and Longo procedure39 involving the heating of equimolar amounts of the fluorinated aldehyde and pyrrole in refluxing propionic acid. In both methods crude products were crystallized. Compounds 7 and 8 were purchased from PorphyChem. Treatment of free-base porphyrins 5–8 with cooper acetate in chloroform afforded the corresponding copper complex of porphyrins. Nitration at β-position was achieved by the reaction of copper(II) nitrate with complex 9–12 in mixture acetic acid/acetic anhydride in chloroform in reflux temperature. Subsequent demetallation under strong acid conditions afforded the expected β-nitro-tetra-aryloporphyrins 17–20. After that, the porphyrins were converted to pyrrolidine-fused chlorins by the reaction with azomethine ylide generated in situ from sarcosine and paraformaldehyde.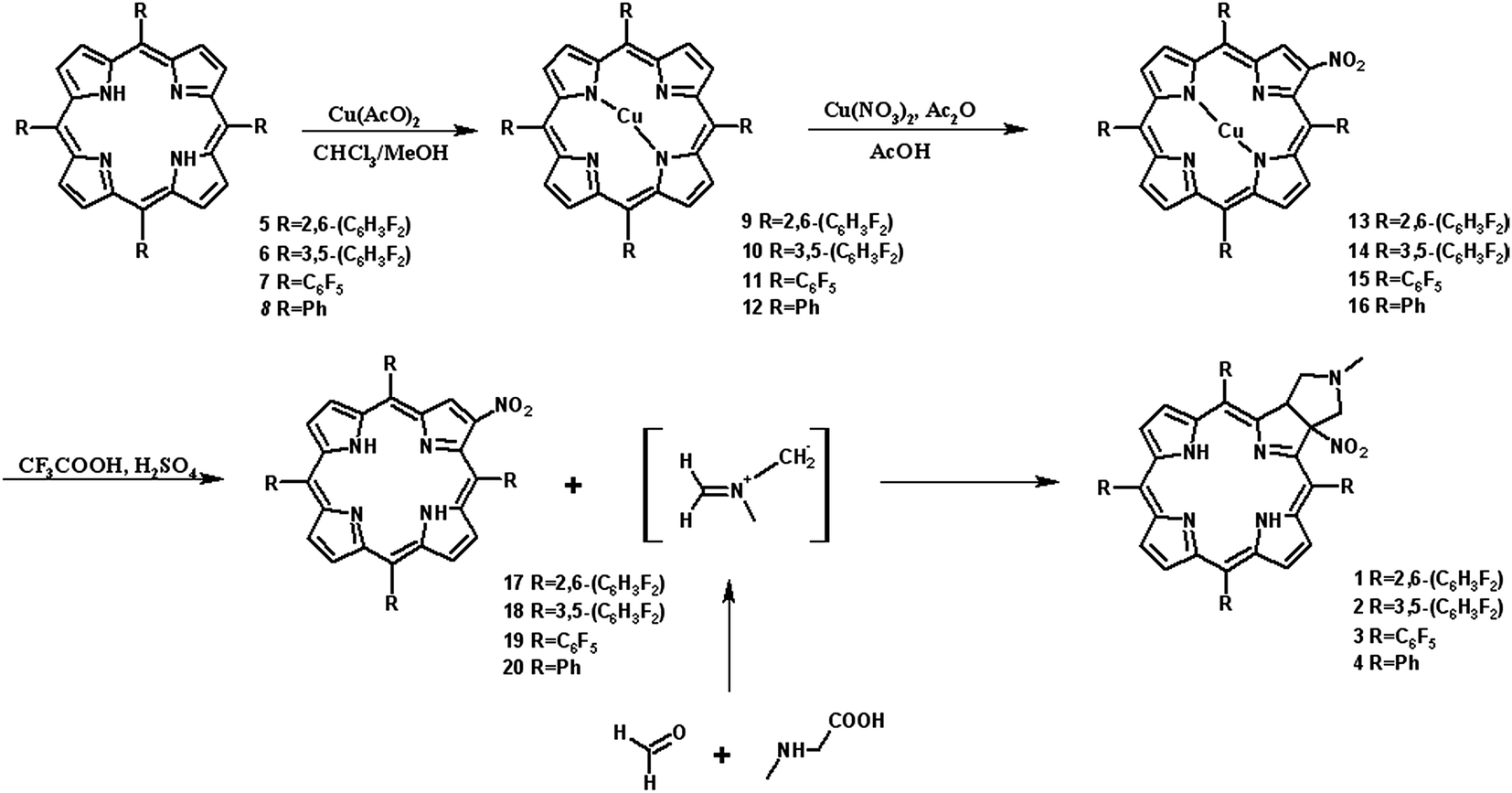 | ||
| Scheme 1 Synthetic route. | ||
Geometry of unsymmetrical chlorins – theoretical models
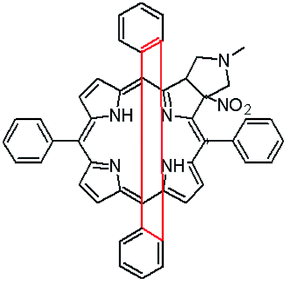
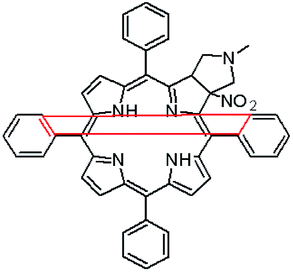
The starting point for discussion of structure and dynamic processes for derivatives under investigation is knowledge about preferable conformation. The static and dynamic aspects of the conformation of meso-tetra-arylporphyrinoids were discussed in number of papers where different methodologies were employed. In the case when the sample forms crystals with quality suitable for monocrystal studies the X-ray crystallography is mainly used for search of conformation. When the crystallization process fails, the alternative approaches are theoretical calculations. It is a case of samples researched in our project as we were not able to grow good quality crystals.Computer methods allow analyzing both the conformation of the meso-phenyl rings and the deformation of the porphyrinoid macrocycle, as well as the barriers of rotation of the meso-phenyl rings (atropisomerism). The measure of nonplanar distortions is shown by the saddling dihedral angles labelled as χ1, χ2, χ3, χ4 (for notation used see Table 1). It is interesting to note that values of these angles differ between derivatives. The largest values suggesting the macrocycle deformation are found for sample 2 and 4 while in contrast samples 1 and 3 are almost planar. Moreover, comparison of sample 1 and 2 reveals that the position of fluorine atoms in the aryl ring is a crucial factor influencing the nonplanarity/planarity of macroring. Ortho-fluorines strongly interact with macrocycle leading to the flattening of structure. This effect in pictorial form is shown in Fig. 2.
|
|
|
|
|
|
|
|---|---|---|---|---|---|---|
| Compound | χ1 | χ2 | χ3 | χ4 | χ5 | χ6 |
| 1 | 1.31 | 4.65 | 3.09 | 1.07 | 12.56 | 11.14 |
| 2 | 5.19 | 9.65 | 11.57 | 9.25 | 31.98 | 29.25 |
| 3 | 2.28 | 4.98 | 3.76 | 1.68 | 14.36 | 10.95 |
| 4 | 5.68 | 10.12 | 12.04 | 10.05 | 33.79 | 32.83 |
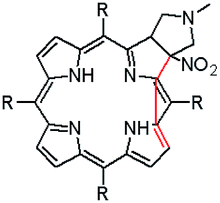
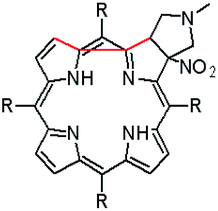
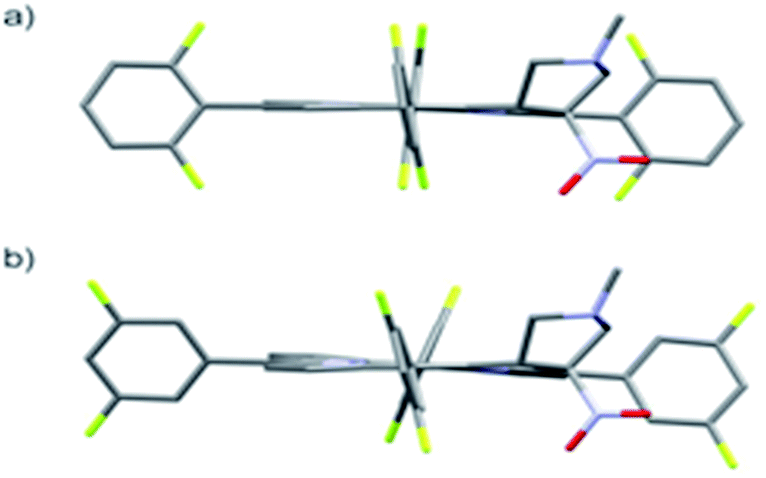 | ||
| Fig. 2 DFT (B3LYP/6-311++g(d,p)) optimized structures of chlorins 1 and 2. | ||
Another interesting structural feature is conformation of aryl rings with respect to the macroring plane. In each case aryl groups are in gauche conformation. However, more detailed analysis and measurement of the torsional angles between the planes of aryl groups shows intriguing features related to the position of fluorine in rings. The χ5, χ6 torsional angles are defined as shown in Table 1. As one can see the values for samples 2 and 4 again are very similar. It clearly proves that fluorine atoms which are not in direct contact with macroring weakly interact with it. During further comparison of the values of χ5 and χ6 we noted that the largest distinction is seen for compound 3. It suggest that bulky fluorine groups located in the neighbourhood of nitro residue “feel” the steric effect. Concluding, the theoretical calculations and computed models clearly show that position of substitution of fluorine in the meso-aromatic ring has strong influence on the preferable conformation. Thus we can anticipate that molecular dynamics of these groups will also be distinctive.
Correlation between molecular structure and optical properties
The absorption spectra of synthesized pyrrolidine-fused chlorins 1–4 were measured in dichloromethane and toluene. Fig. 3 shows the absorption spectra in toluene at room temperature and the band maxima of absorption and molar absorbance coefficients were summarized in Table 2.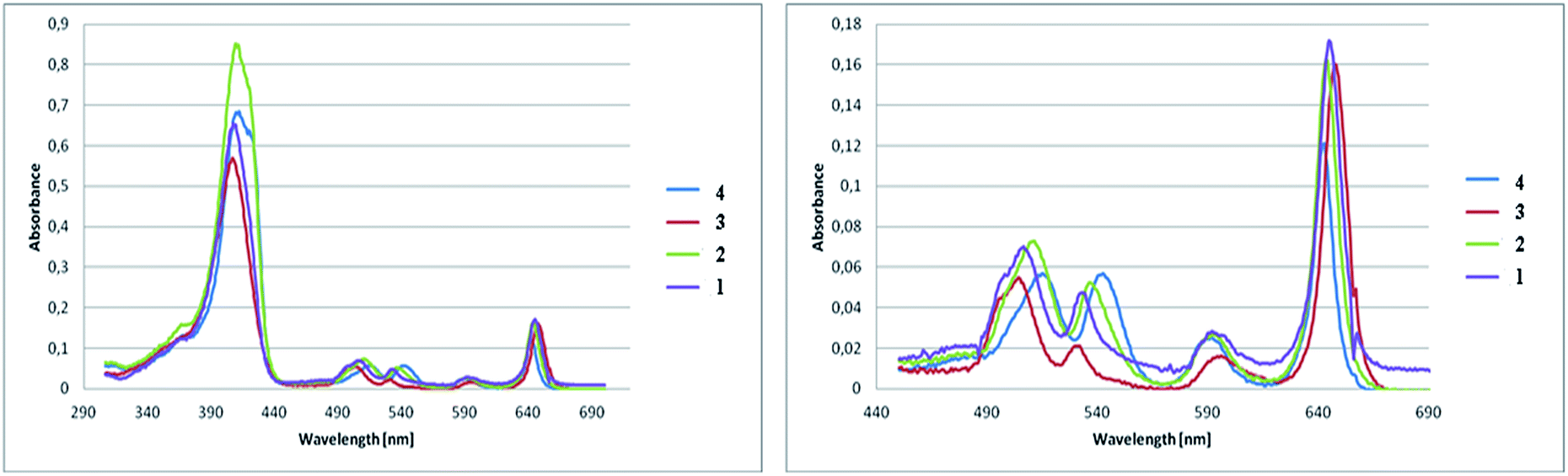 | ||
| Fig. 3 (Left) Absorption spectra at room temperature of pyrrolidine-fused chlorins 1–4 in toluene solution. (Right) Expansion of 440–690 nm region of spectra. Molar concentrations of the chlorins 1–4 were roughly the same. | ||
| Compound | Soret band [nm] | Molar absorbance coefficient [dm3 mol−1 cm−1] in toluene |
|---|---|---|
| 1 | 408 | 141![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 040 040 |
| 2 | 409 | 180726 |
| 3 | 408 | 120968 |
| 4 | 411 | 163822 |
The absorption spectra of 1–4 differ in respect of the positions and the intensity ratios of the bands as well as a molar absorbance coefficient. The positions of the Soret band are nearly identical with each other. Compared to the chlorin 4, the Soret bands for fluorinated analogues were moved, but the shifts are not significant. Major changes are noticeable for Q band. It is apparent from the absorption bands that small shifts in the absorption maxima occur as the results of substitution and amount of fluorine atoms in the chlorins. With the increase in the number of fluorine atoms in the molecule, the strongest absorption bands are shifted bathochromically compared to the non-fluorinated chlorin 4. The number and location of fluorine atoms affects also the intensity of the absorption. The highest value of molar absorbance coefficient at room temperature in toluene is related to chlorin 2, the next very close is for sample 4.
NMR studies of chlorins 1–4
The crucial information which is prerequisite for discussion of local molecular motion is the precise assignment of meso-aryl groups and their localization in the structure of chlorins. The notation which we used in this project for labeling of A, B, C, D aryls in meso-positions is shown in Scheme 2.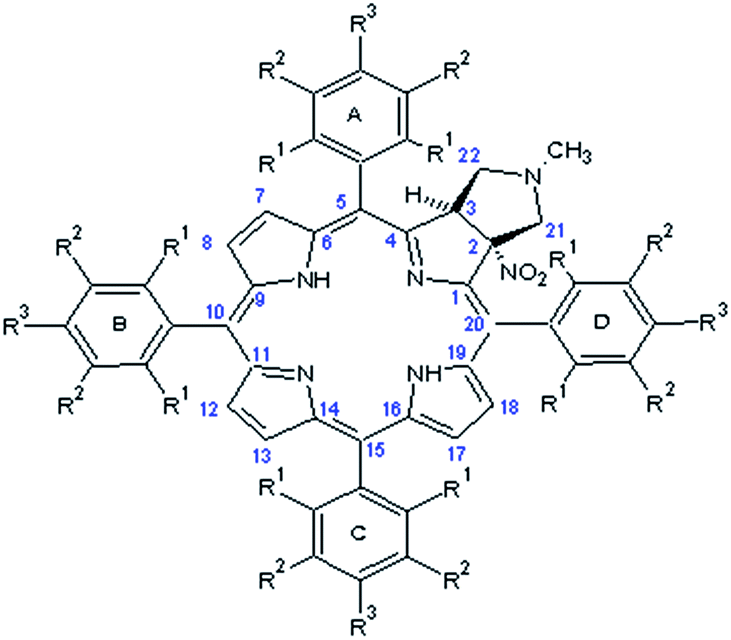 | ||
| Scheme 2 General labeling and numeration in chlorins. | ||
NMR spectroscopy is a technique which provides straightforward structural information about geometry of compounds. For chlorins 1–4 we have carried out NMR measurements at ambient temperature employing 1D and 2D homo- and heteronuclear correlations. In the case of samples 1–3 which are fluorine derivatives, 19F nucleus is an additional structural probe invaluable in further studies. Fig. 4 shows standard 1H and 19F NMR spectra for sample 1.
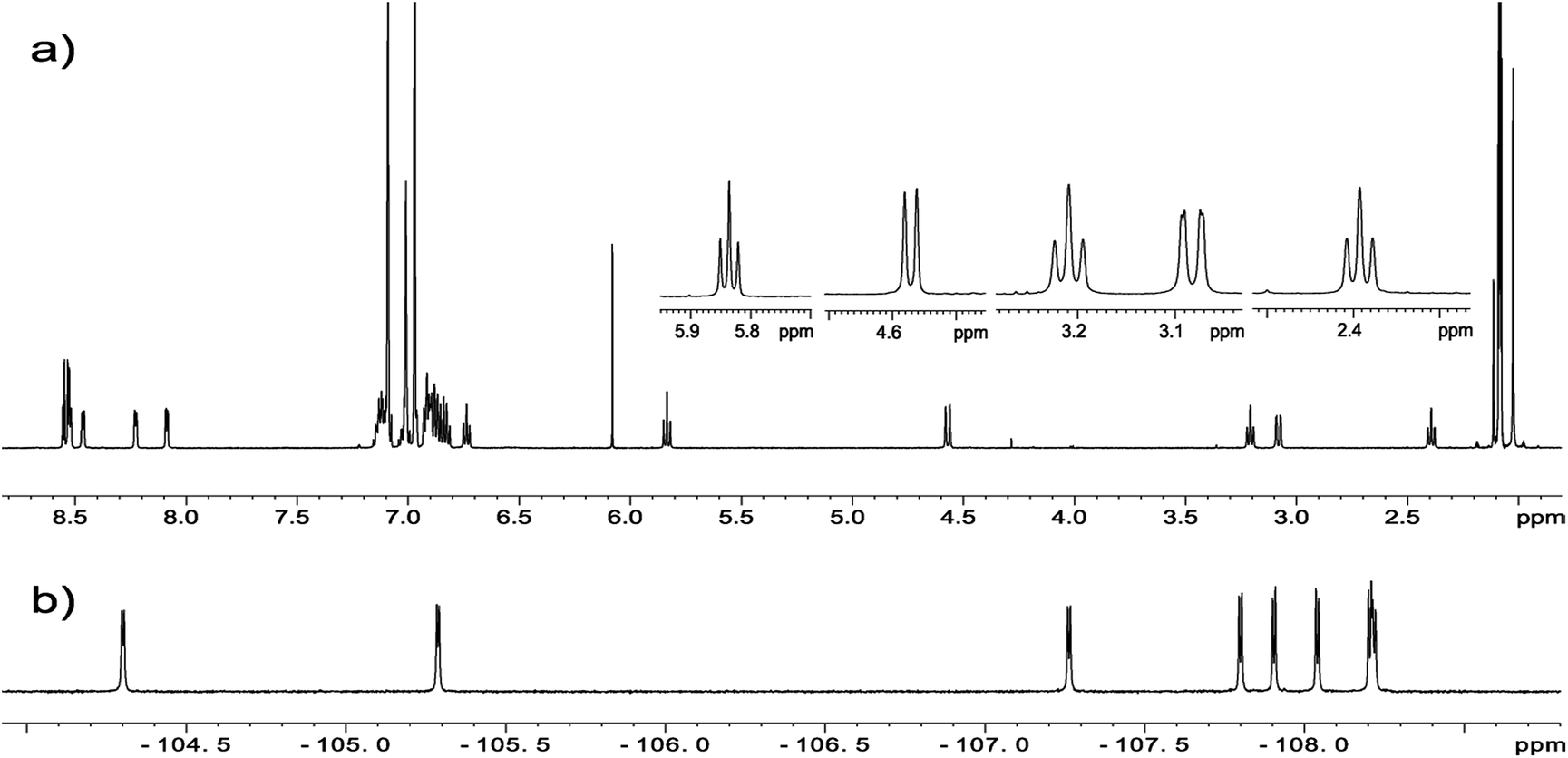 | ||
| Fig. 4 (a) 1H NMR spectrum of chlorin 1 and expansion of 2–6 ppm region showing H21/H21′, H22/H22′ and H23 protons. (b) 19F NMR spectrum of chlorin 1 recorded in toluene-d8 at 600 MHz. | ||
For each sample a broad singlet at −1.5 to −3.5 ppm representing inner pyrrolic NH groups was observed. In all compounds signals for H3, H21, H21′ and H22, H22′ found in the range 2–6 ppm are well resolved and can be unambiguously assigned (Fig. 4a). The spin system pattern clearly confirms that N-methylpyrrolidine ring is connected to C2 and C3 carbons. In other case, for substitution at C7/C8 or C12/C13 as well C17/C18 the six spin system should be observed.
The detailed description of strategy used for assignment of A, B, C, D aryl residues we show employing sample 1 and sample 4. In other cases the employed methodology is similar. To specify the describing of the spatial alignment for moieties under investigation we adopted the convention assuming that the pyrrolidine ring is above macroring plane while nitro group is below. Consequently, fluorine or hydrogen atoms in meso-aryl rings which are at the same side as the nitro substituent were denoted as down, and up when they are on the opposite side.
In analysis of fluorinated samples, homonuclear 1H–1H, 19F–19F 2D NMR correlations and experiments based on 1H–19F Heteronuclear Overhauser Effect (HOESY) were found to be the most informative. The 19F–19F COSY spectrum carried out with proton decoupling (Fig. 5) allowed us to assign pairs of fluorine atoms belonging to individual meso-aryl rings. As can be seen, the influence of pyrrolidine ring and nitro group on 19F chemical shifts of neighbouring aryls in meso-position is significant. The distinction of δ19F for up and down fluorine atoms is ca. 1 ppm.
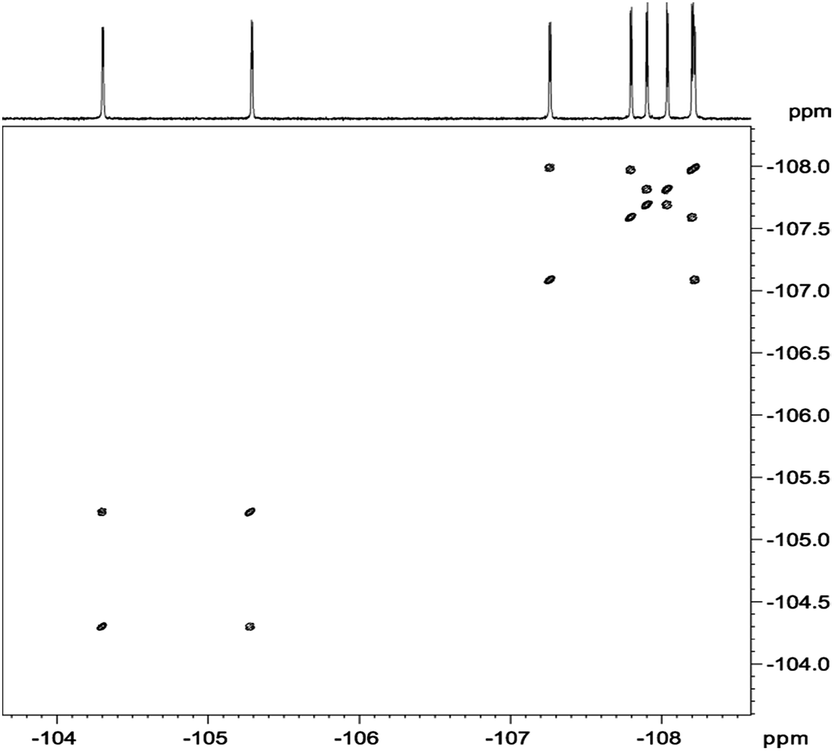 | ||
| Fig. 5 19F{1H}–19F{1H} DQF-COSY spectrum of chlorin 1 recorded in toluene-d8. The spectrum obtained at 600 MHz spectrometer. | ||
Next, we carried out 1H–19F HOESY experiment (Fig. 6) assuming that this correlation, showing dipolar interactions by space will enable us to distinguish spatial localization of fluorine atoms. The analysis began with H3, H21, H21′ and H22, H22′ signals for which the assignment is unquestionable. The position of FD-up is concluded on the base of correlation between H21 and H21′ (Fig. 6), while FA-up on correlation between H22 and H22′. FA-down shows the correlation peak with H3. FD-up and FD-down correlate with H18. In the same way H7 correlates with FA-up and FA-down. Correlation between H17 and two fluorine signals provides information about assignment of FC-up and FC-down. Position of FC-up and FC-down is assigned by correlation with H13. Assignment of FB-up and FB-down was not possible, but these two signals were assigned via correlation between H8 and H12.
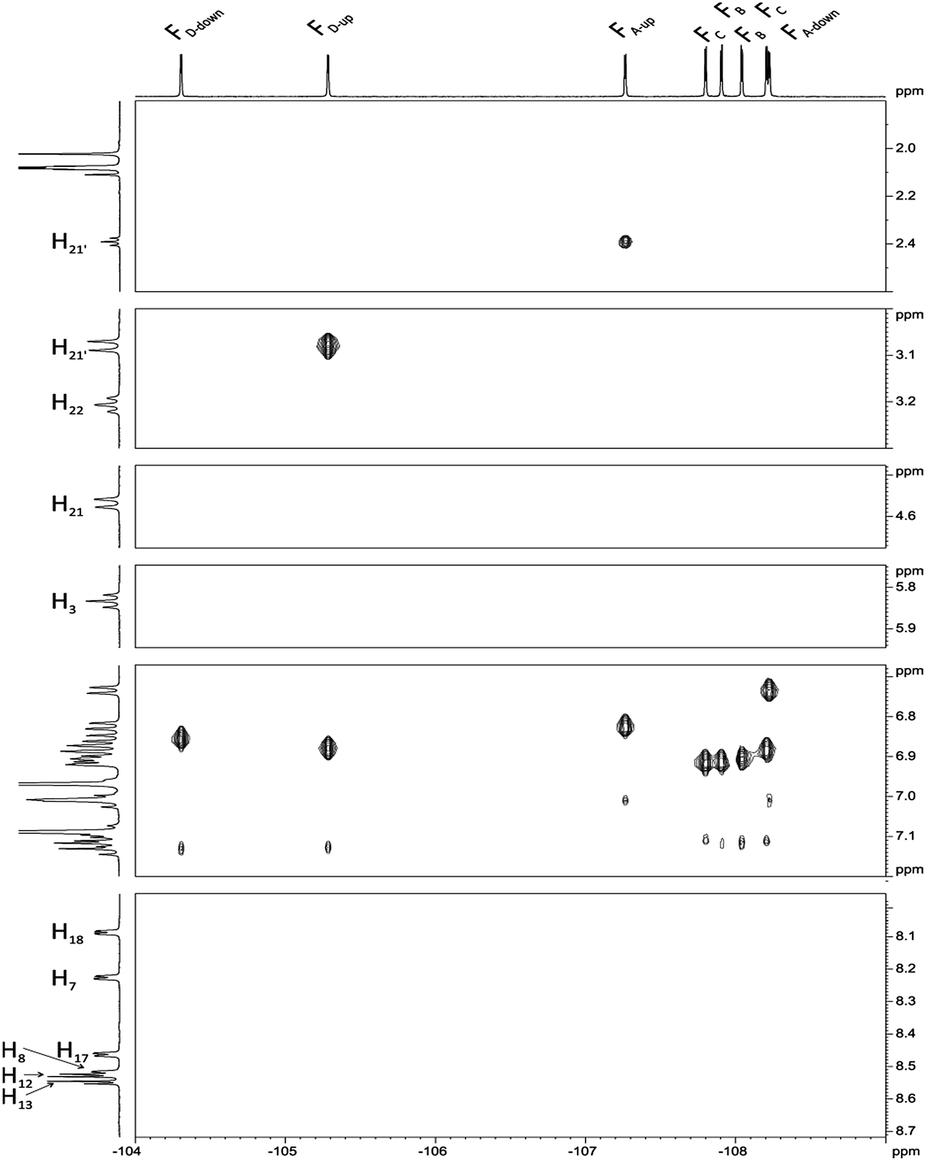 | ||
| Fig. 6 1H–19F HOESY spectrum of chlorin 1 recorded in toluene-d8. The spectrum obtained at 600 MHz spectrometer. | ||
Interesting effects were observed in 1H–19F HETCOR (Fig. 7) spectrum which shows a strong correlation between H21′–FD-up and weaker correlation between H22′–FA-up. At the first glance this correlation via 7 bonds is very surprising. However inspection of DFT optimized (at B3LYP/6-311++g(d,p) level) structure gives reasonable explanation of this effect. Distance between H21′–FD-up is 2.50 Å and H22′–FA-up is 3.22 Å. In this case the so called “J coupling through space” operates due to existence of an interaction between antibonding orbitals. Such coupling is well described in literature mostly in case of 19F–19F and 1H–19F coupling but also for 19F–15N, 19F–31P, 19F–77Se.40 H21′–FD-up interaction also causes additional (1.8 Hz) splitting of H21 signal in 19F non decoupled 1H spectrum. H22′–FA-up interaction is much weaker and causes only little line broadening in 19F non decoupled 1H spectrum (this coupling cannot be easily directly measured and we estimated it to be 0.5 Hz).
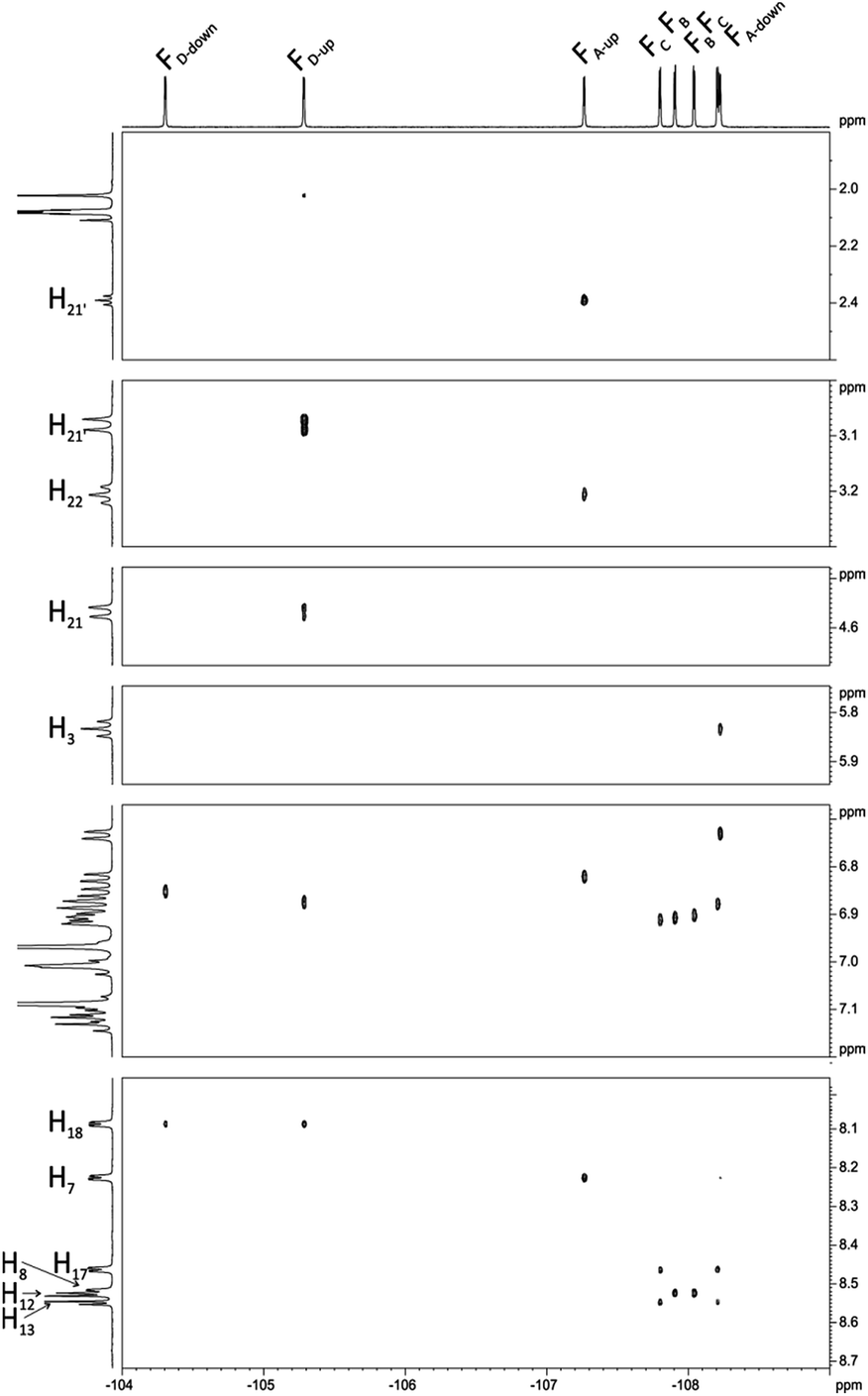 | ||
| Fig. 7 1H–19F HETCOR spectrum of chlorin 1 recorded in toluene-d8. The spectrum obtained at 600 MHz spectrometer. | ||
For structural assignment of sample 4 we employed classical NMR strategy based on the analysis of homonuclear 2D 1H–1H COSY (Fig. 8) and 1H–1H ROESY spectra (Fig. 9). As in the previous case we begun the structural study with diagnostic signals H3, H21, H21′ and H22, H22′. Knowing the chemical shifts of these signals it was possible to assign protons in β-pyrrolic position. Proton in o-HA-up position was assigned by correlation with H22 and H22′ protons in 1H–1H ROESY spectrum. Having in mind o-HA-up correlation it was further possible to assign H7 proton in β-pyrrolic positions. In the similar way we assigned H18 using correlation with o-HD-up. Correlation between H18 and o-HD-up as well H8 and H17 was done using 1H–1H COSY experiment and their correlation with previously assigned H7 and H18. It was impossible to assign positions of up and down o-protons in B and C rings. Both up/down protons were assigned by correlation with H8 or H17 for C and B rings respectively in 1H–1H ROESY spectrum. H12 and H13 signals were assigned by correlation with o-protons of C or B ring. Due to high overlapping even in 2D experiment, and additional complication caused by chemical exchange, it was impossible to assign meta and para protons of phenyl rings.
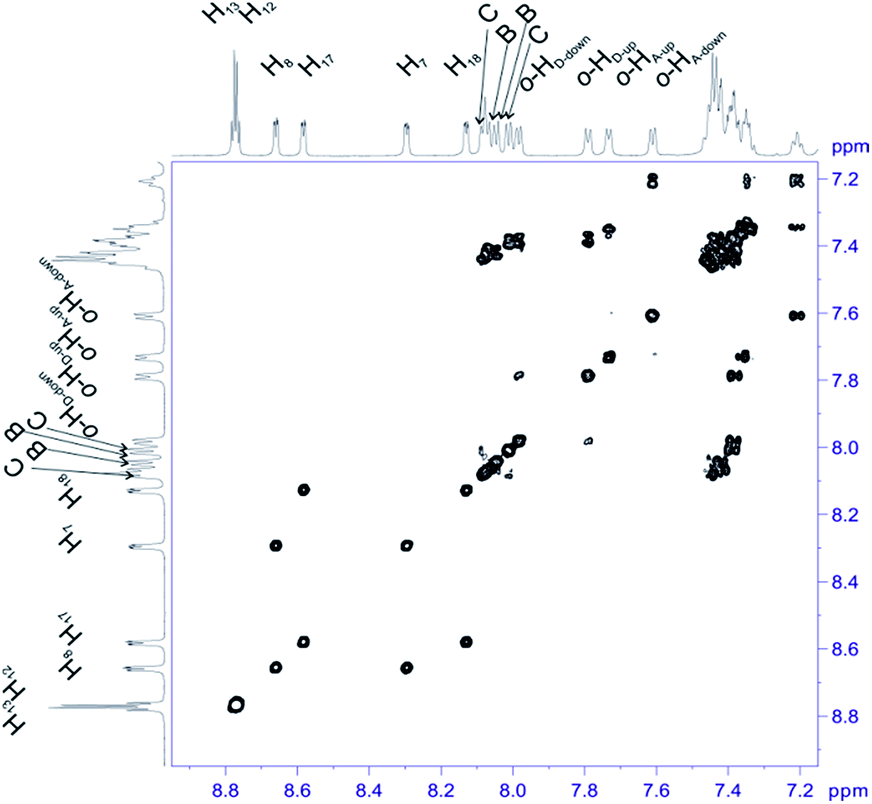 | ||
| Fig. 8 Most diagnostic part of 1H–1H COSY spectrum of compound 4 recorded at 243 K in toluene-d8. The spectrum obtained at 600 MHz spectrometer. | ||
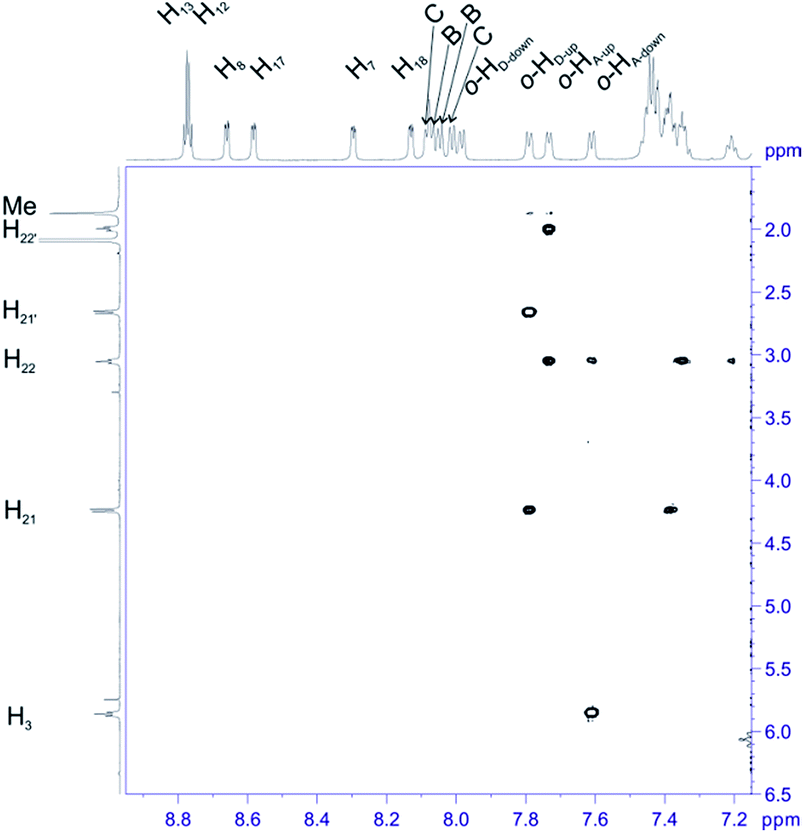 | ||
| Fig. 9 The most diagnostic part of 1H–1H ROESY spectrum of compound 4 recorded at 243 K in toluene-d8. The spectrum obtained at 600 MHz spectrometer. | ||
Dynamics of meso rings
Having the set of assigned 1H and 19F signals, we were able to investigate the molecular dynamics of meso-aryl rings in relation to their position in the structure and in function of temperature. In all cases we have carried out variable-temperature (VT) NMR measurements in the range of 233 to 353 K (sample 1 and 2) and in the range of 283–353 K (sample 3) employing toluene-d8 as solvent. Fig. 10 shows the 1D 19F VT NMR spectra for samples 1–3.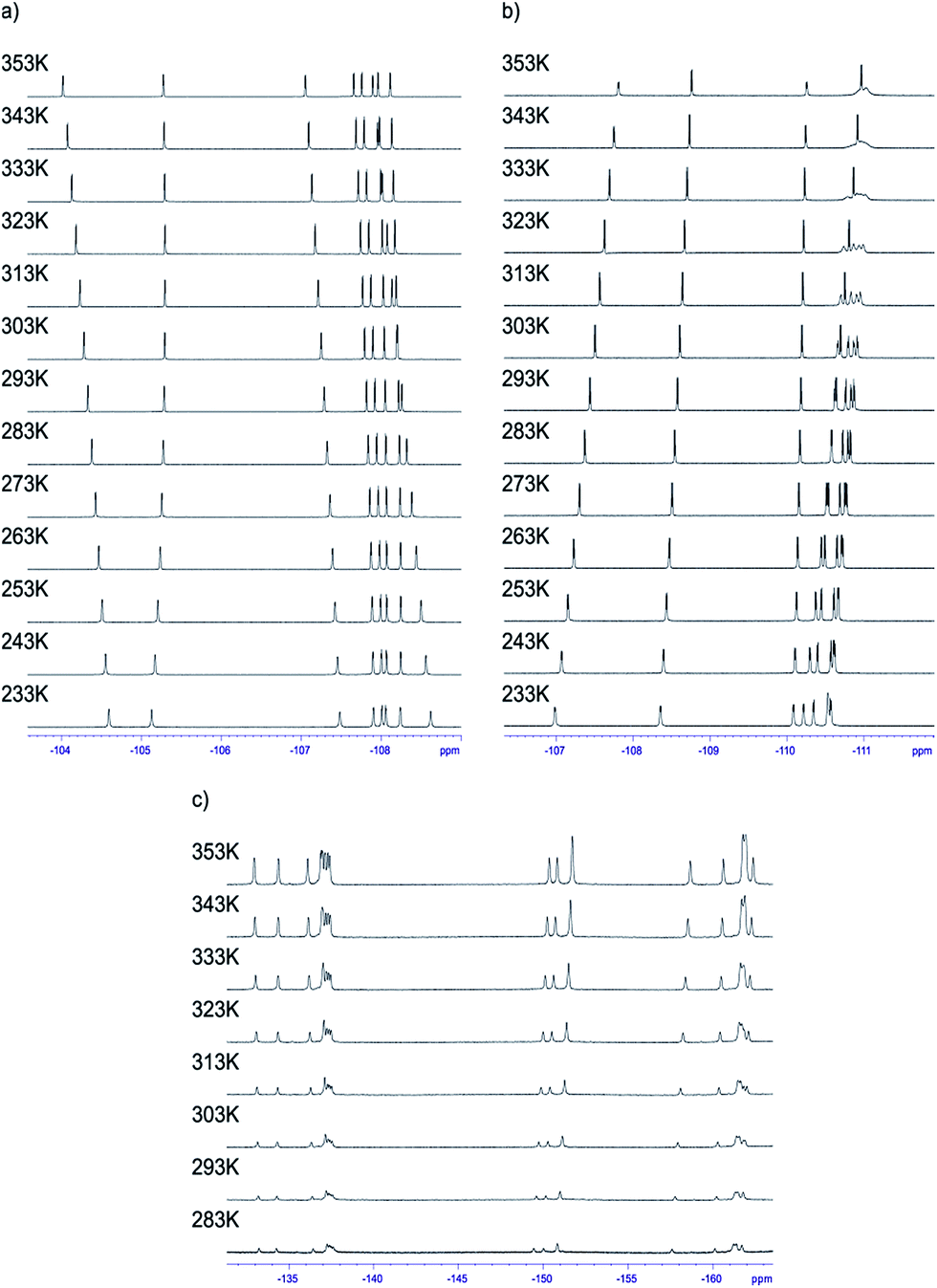 | ||
| Fig. 10 1D 19F VT NMR spectra in the range of 233–353 K of compound (a) 1 and (b) 2. In case compound 3 (c) due to low solubility of this compound spectra were measured starting from 283 K. The spectra were recorded in toluene-d8 at 600 MHz. | ||
In the case of sample 1 and sample 3 the line broadening of 19F signals caused by rotation of fluorophenyl rings in meso-positions is not observed. The only visible thermal effect is a small change of chemical shifts (temperature drift). This temperature shift effect may be caused by changing the solvation sphere and change of interactions between chlorin and toluene molecules. Sample 2 behaves differently when compared to 1 and 3. In this case we observed line shape effect for fluorine atoms representing B and C rings. This process begun at 303 K. Starting from 318 K it was not possible to observe multiplicity (doublet) of signals for B and C rings. Near temperature 338 K signals representing B and C rings are very broad. At 353 K we monitored two separated, quite broad signals for averaged B and C fluorine atoms. These signals sharpened when temperature was further increased. It is worth to express that line broadening was not observed for fluorine atoms representing A and D rings. It suggests that these residues are rigid in the broad range of temperature. Going further with analysis of molecular motion for B and C rings we calculated (from line shape fitting) exchange barriers in temperature range 308–373 K. Having these values it was possible to calculate the activation energy (Ea) of rotation from Arrhenius plot (Fig. 11). The Ea values were found to be 18.5 ± 0.5 and 17.7 ± 0.8 kcal mol−1 for B and C rings, respectively. These energies are very close to each other and similar to data available in literature for another porphyrinoids (subporphyrins,41 porphyrins, metalloporphyrins and metallo-chlorins15). Our studies unambiguously proved that the substitution in β-position have significant influence on molecular dynamics of meso-rings. This dynamic process can be controlled by synthesis of derivatives substituted by fluorine atom in assumed position of aromatic group.
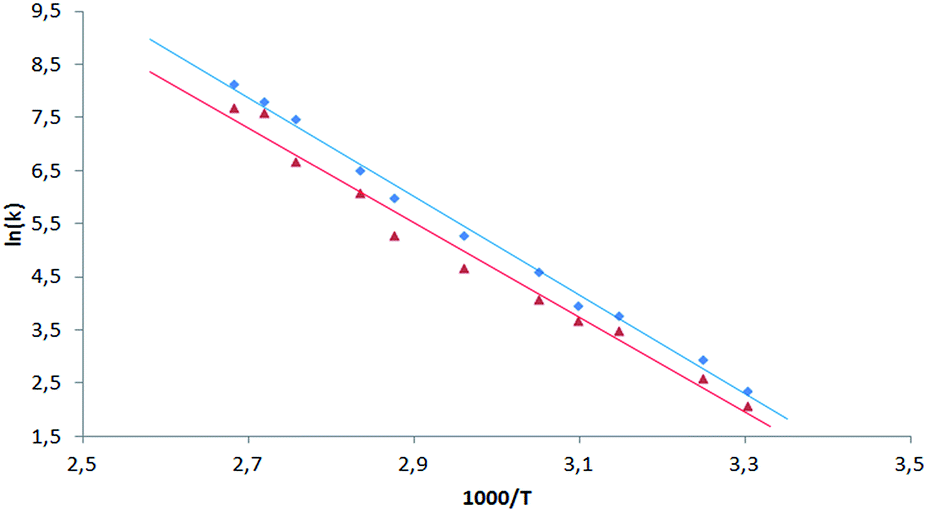 | ||
| Fig. 11 Arrhenius plot for exchange rate of 19F signals of B (blue square) and C (red triangle) rings for sample 2. | ||
Further evidence confirming the distinct molecular motion of meso aromatic groups for fluorine derivatives 1–3 was obtained by employing 19F{1H}–19F{1H} 2D EXSY experiment (Fig. 12). Using this technique it is possible to observe correlation between the exchanged signals even in slow chemical exchange regime.
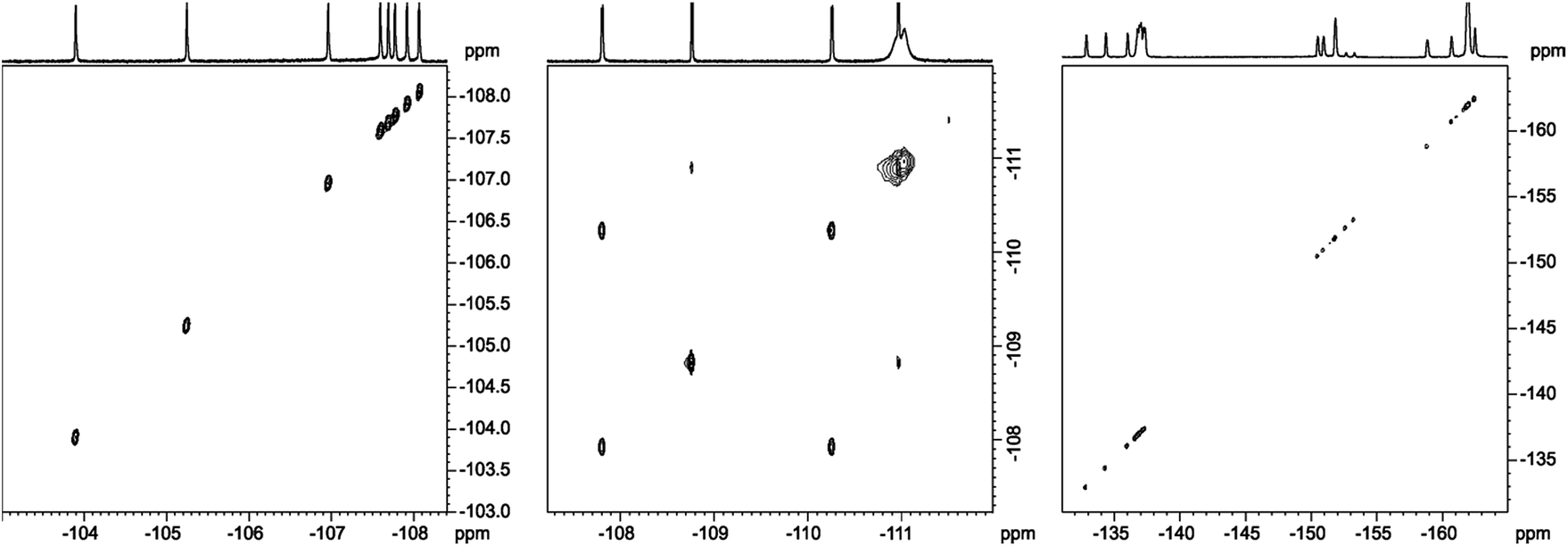 | ||
| Fig. 12 19F{1H}–19F{1H} 2D EXSY spectra recorded for compound 1 (left), 2 (middle), 3 (right) at temperature 373 K, 353 K, 373 K, respectively. The spectra obtained in toluene-d8 at 600 MHz spectrometer. | ||
For sample 1 and 3 even at 353 K it was not possible to observe correlation signals. It means that all rings are static in the 233–353 K temperature range. It is worth to express that in the case of 2,6-disubstituted phenyl rings, 1 steric hindrance caused by substitution of aromatic groups has great influence on the rotation barrier and excludes dynamic processes in each pyrrolidine-fused chlorin in meso-position.
The case of sample 2 is different. As we mentioned above, 1D NMR analysis proved the molecular motion of B and C meso-rings. To answer whether A and D rings are completely static in NMR time scale, or if they exhibited small mobility, we performed 19F{1H}–19F{1H} EXSY experiment at temperature 353 K. In this case it was easy to observe the exchange signals between FA-up–FA-down and FD-up–FD-down, so these signals were in slow exchange limit. It means that B and C rings are much more mobile than A and D rings, but all of the rings exhibit mobility at 353 K in NMR time scale.
In the case of sample 4 we do not have such an excellent NMR probe for analysis of molecular motion as fluorine nucleus. Thus the basic source of information is 1H NMR spectroscopy. Fig. 13 shows the aromatic region of meso-residues in the 233–353 K temperature range.
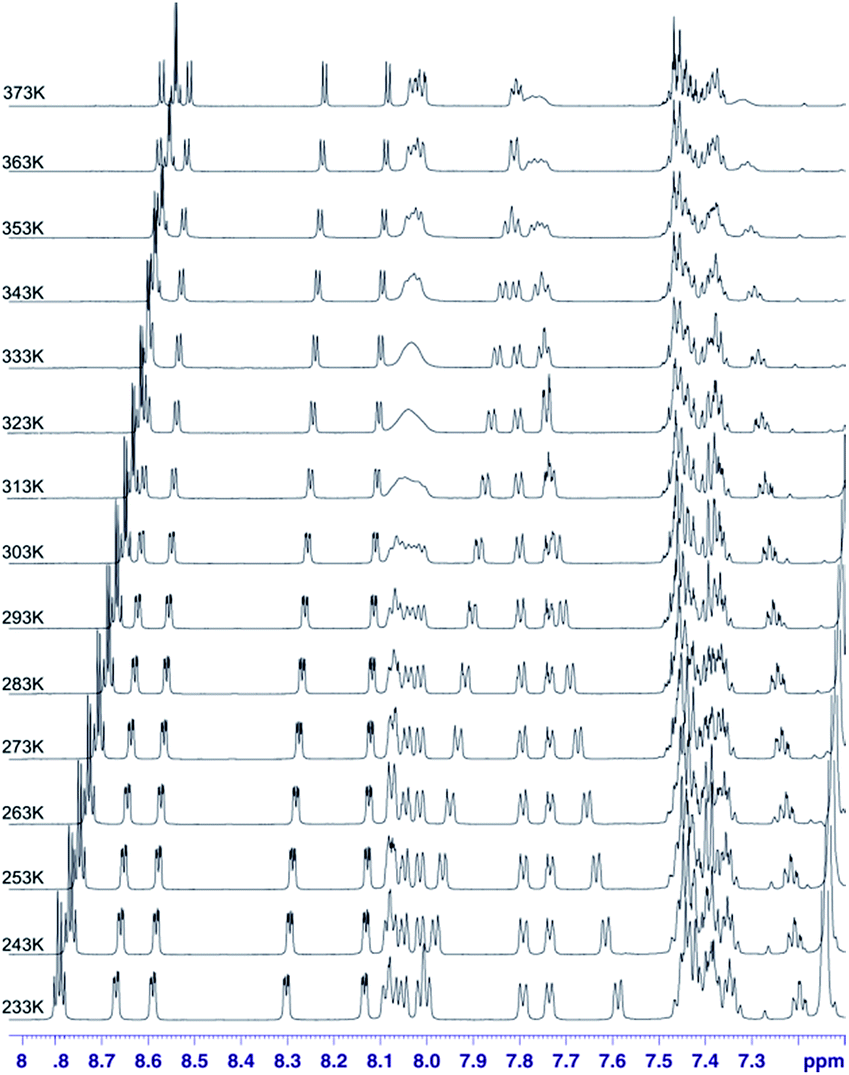 | ||
| Fig. 13 1H VT spectra in the 233–373 K temperature range recorded in toluene-d8 for chlorin 4. The spectra obtained at 600 MHz spectrometer. | ||
In this case it was possible to observe lineshape effect for ortho signals of B and C ring starting from temperature 293 K. Coalescence of HB-up–HB-down and HC-up–HC-down pairs of signals were observed at temperature 333 K. Further increasing of temperature caused reducing of line widths. For A ring it was possible to observe lineshape effect for protons in ortho position in A ring starting from 333 K, but it was not possible to observe coalescence. It was not possible to observe lineshape effect for D ring signals. Unfortunately, it was not possible to determine the precise value of rotation barrier due to complex 5 spin system for each ring, additionally complicated by strong overlapping of meta and para signals. In this case we estimated only Gibbs free energy from coalescence temperature. It was ca. −16 kcal mol−1 for B and C rings. In this case we observed similar behaviour of meso-rings like in case 3,5-difluorophenyl analogue – B and C rings have the smallest rotational barrier, A ring has higher rotational barrier and D ring has the highest rotational barrier. This observation is easy to understand – D ring has higher activation energy than A ring due to additional hindrance caused by nitro group. Of course both of these rings have larger rotation activation energy then B and C ring, caused by strong influence of additional N-methylpyrrolidine ring near to A and D rings. It means that substitution at meta position has no significant steric or electronic effect for rotation barrier.
Conclusions
The major aim of our investigation is to understand and explain the influence of modifications of the structures of fluorinated pyrrolidine-fused chlorins on the dynamic processes. These compounds were synthesized and characterized in detail. The rotational dynamic of aryl rings in meso-position of chlorins was examined by VT 1H and 19F 1D NMR spectroscopy and 19F{1H}–19F{1H} 2D EXSY experiments. The calculated activation energies for aryl substituents in our compounds were very similar to other porphyrinoids. We proved that even small modification in aryl rings may have an effect on rotation of these rings. Our results show that introduction of atoms even as small as fluorine in ortho position in aryl rings causes an inhibitation of rotation. This is due to the increased steric bulk of the hydrogens in β-pyrrolic positions and fluorine atoms. Natural Bond Orbital (NBO) analysis did not reveal specific interactions between the electron-lone-pairs of fluorine atoms and the π-cloud of the macroring (see ESI†).46 In nonfluorinated chlorin a rotational dynamic of aryl ring can be controlled by temperature.Experimental details
General information
The chlorins 1–4 were prepared using chemicals which were purchased from Sigma Aldrich, TCI and used without further purification. Reagent grade solvents were purchased from Polish Chemicals Reagents and were distilled prior to use. Silica gel columns for chromatography were prepared with silica (Kieselgel 60, 200–400 mesh). All NMR experiments were run on a 600 MHz Bruker Avance III spectrometer equipped with BBOF probhead operating at 600.13, 564.69, 150.92 MHz for 1H, 19F and 13C nuclei, respectively. 2D 1H–19F HOESY, 19F–19F EXSY and 2D 1H–1H ROESY experiments were measured with 200 ms mixing time. 1H–19F 2D HETCOR correlation was optimized for 10 Hz 1H–19F coupling constants. VT experiment was carried automatic with automatic mode (using modified au zgvt script) with at least 20 min stabilization of temperature at target temperature with automatic shimming and probhead tuning after temperature equilibration. The following abbreviations were used to describe peak splitting patterns when appropriate: br = broad singled, s = singlet, d = doublet, t = triplet, q = quartet, dd = doublet of doublets, bm = broad multiplet and m = multiplet. Coupling constants J are reported in Hz. UV-VIS spectra were obtained on Specord S600 “Analytik jena AG”. Mass spectra of obtained compounds were registered by MALDI-TOF Mass Spectrometer – PerSeptive. All calculations were carried out using the Gaussian09 program package.42 For geometry optimization of all structures B3LYP and basis set 6-311++ g(d,p) have been used.General procedures for synthesis of copper complexes of porphyrins43
A mixture of porphyrin (0.45 mmol) in chloroform (34 mL) and Cu(OAc)2·H2O (2.25 mmol) in methanol (17 mL) was stirred for 2 h at room temperature. Reaction was monitored by TLC (hexane:chloroform 4:1) after which the reaction mixture was poured onto water and extracted with chloroform. The organic layer was dried over Na2SO4. After evaporation of solvent to dryness afforded dark pink solid of: compound 9 (0.34 g, yield: 93%), MS (MALDI TOF) m/z = 821.5 [M + H]+. Compound 10 (0.33 g, yield: 90%), MS (MALDI TOF) m/z = 821.5 [M + H]+. Compound 11 (0.41 g, yield: 89%), MS (MALDI TOF) m/z = 1037.1 [M + H]+. Compound 12 (0.28 g, yield: 92%), MS (MALDI TOF) m/z = 677.3 [M + H]+.
General procedures for the nitration complexes of porphyrins44
In round bottom flask cooper complex of porphyrin (0.4 mmol) was dissolved in chloroform (275 mL) and the solution is heated to reflux. Afterwards, Cu(NO3)2·2.5H2O (2 mmol) in mixture of acetic acid (5.5 mL) and acetic anhydrate (27.5 mL) was added. The reaction was stirred for 2 h in reflux with TLC monitoring after which it was washed with water, saturated solution of sodium biscarbonate and the organic layer was dried with MgSO4/Na2CO3. After evaporating the solvent, the products were isolated by column chromatography (using a chloroform/n-hexane as the eluent) to give: compound 13: 0.28 g, yield 80%, MS (MALDI TOF) m/z = 866.2 [M + H]+. Compound 14 0.26 g, yields 78%, HRMS (ES+/TOF) m/z: [M + H]+ calcd for C44H22F8N5O2CuNa 887.0605; found 887.0582, MS (MALDI TOF) m/z = 866.2 [M + H]+. Compound 15 0.32 g, yield 75%, MS (MALDI TOF) m/z = 1082.1 [M + H]+. Compound 16 0.23, yield 80%, MS (MALDI) m/z = 722.3 [M + H]+.General procedures of demetallation of copper complex45
:chloroform/n-hexane 1:3). Compound 17: 0.23 g, yield: 95%, 1H NMR (600 MHz, DMF-d7) δ −2.61 (bs, 2H), 7.73–7.76 (m, 2H), 7.86–7.90 (m, 6H), 8.21–8.30 (m, 8H), 9.15 (q, J = 4.8 Hz, 2H), 9.42 (d, J = 4.8 Hz, 1H) 9.47–9.51 (m, 3H), 9.79 (s, 1H), 13C NMR (151 MHz, DMF-d7): δ 111.73–112.26, 115.52, 117.03–117.65, 128.40, 129.14, 129.40, 130.56, 130.66, 131.19, 133.00–133.31, 133.87, 135.76, 156.15, 161.53, 163.16, 163.55. HRMS (ES+/TOF) m/z: [M + H]+ calcd for C44H22F8N5O2 804.1646; found 804.1639, MS (MALDI TOF) m/z = 804.7 [M + H]+. Compound 18: 0.22 g. Yield: 93%, 1H NMR (600 MHz, CDCl3) δ −2.76 (bs, 2H), 7.27–7.29 (m, 2H), 7.32–7.38 (bm, 3H), 7.77–7.82 (m, 8H), 8.78 (q, J = 4.7 Hz, 2H), 9.00 (t, J = 5.1 Hz, 2H), 9.04 (d, J = 4.9 Hz, 1H), 9.12 (d, J = 5.0 Hz, 1H), 9.13 (s, 1H); 13C NMR (151 MHz, CDCl3): δ 104.06, 104.23, 104.37, 104.39, 104.473, 104.64, 104.70, 104.80, 117.85, 117.97, 118.10, 118.39–118.41, 118.61, 118.65–118.69, 118.74, 118.79, 120.69, 128.39, 129.05, 129.65, 130.09, 131.78, 135.24, 142.22, 142.46, 143.64, 143.86, 144.12, 145.62, 153.20, 160.53–160.76, 162.20–162.42. HRMS (ES+/TOF) m/z: [M + H]+ calcd for C44H22F8N5O2 804.1646; found 804.1632, MS (MALDI TOF) m/z = 804.7 [M + H]+.:chloroform/n-hexane 1:3). Compound 19: yield: 0.28 g, 90%, 1H NMR (600 MHz, CDCl3) δ −2.78 (s, 2H), 8.82 (q, J = 4.7 Hz, 2H), 9.03–9.08 (bm, 4H), 9.18 (s, 1H), 13C NMR (151 MHz, CDCl3): δ 103.27, 104.17, 104.45, 106.36, 113.31, 114.40–115.09, 128.42, 128.62, 129.68, 131.20, 135.36, 136.83, 138.52, 140.51, 140.79, 141.69, 141.88, 142.63, 143.38, 143.61, 143.82, 145.67, 145.97, 147.29, 147.64, 153.67, 156.86, 162.57, 169.56, MS (MALDI TOF) m/z = 1020.5 [M + H]+. Compound 20: 0.19 g, yield: 95%, 1H NMR (600 MHz, CDCl3) δ −2.58 (bs, 2H), 7.73–7.83 (bm, 12H), 8.21–8.27 (m, 6H), 8.28 (d, J = 7.2 Hz, 2H), 8.75 (q, J = 4.6 Hz, 2H), 8.92 (t, J = 4.6 Hz, 2H), 8.97 (d, J = 4.9 Hz, 1H), 9.04 (d, J = 4.9 Hz, 1H), 9.08 (s, 1H), 13C NMR (151 MHz, CDCl3): δ 120.10, 120.58, 120.85, 123.01, 126.88, 126.91, 126.96, 127.07, 128.01, 128.23, 128.40, 128.54, 128.92, 129.47, 129.91, 131.86, 134.56, 134.70, 134.71, 135.03, 135.05, 135.42, 137.97, 139.33, 140.20, 140.38, 141.11, 141.33, 141.57, 146.04, 153.09, 156.37, 156.57, MS (MALDI TOF) m/z = 661.7 [M + H]+.General procedure for the 1,3-dipolar cycloadditions
The synthetic procedure has been reported by Cavaleiro et al.32 with slight modification. In round bottom flask a toluene (20 mL) solution of the β-nitro-5,10,15,20-tetraarylporphyrin (0.01 mmol), sarcosine (2 equiv), and paraformaldehyde (5 equiv) was heated at reflux for 5 h under a nitrogen atmosphere. After 2.5 h the additional portions of sarcosine (2 equiv), and paraformaldehyde (5 equiv) were added and the reaction mixture was refluxed for another 2.5 h period. After being cooled to room temperature and evaporated, the reaction mixture was applied on the top of a silica gel column. Compound 1: eluent:chloroform, 0.047 g yield 43%, 1H NMR (600 MHz, CDCl3) δ −1.97 (bs, 2H), 2.32 (s, 3H), 2.41 (t, J = 9.2 Hz, 1H), 3.06 (d, J = 11.4 Hz, 1H), 3.33 (t, J = 8.4 Hz, 1H), 4.37 (d, J = 11.7 Hz, 1H), 5.70 (t, J = 8.9 Hz, 1H), 7.26–7.36 (bm, 9H), 7.71–7.78 (bm, 4H), 8.32 (d, J = 3.0 Hz, 4H), 8.51 (s, 1H), 8.59 (d, J = 7.3 Hz, 2H), 8.74 (d, J = 3.2 Hz, 1H), 8.80 (d, J = 3.5 Hz, 1H). 13C NMR (151 MHz, CDCl3): δ 41.06, 62.30, 64.06, 64.63, 98.95, 105.75, 108.93, 110.18, 110.97, 111.11–111.41, 111.57, 111.59, 111.72, 111.87, 112.31, 112.45, 116,12, 116.26, 116.40, 118.03, 118.17, 118.22, 118.33, 118.50, 118.58, 124.16, 124.96, 127.64, 128.19, 128.32, 129.00, 131.03, 131.44, 132.31, 132.61, 133.14, 135.44, 136.29, 140.01, 153.15, 154.11, 156.96, 160.85, 161.23, 161.32, 161.7, 161.82, 162.23, 162.51, 163.00, 163.35, 163.45, 163.90. HRMS (ES+/TOF) m/z: [M + H]+ calcd for C47H29F8N6O2 861.2224; found 861.2207, MS (MALDI TOF) m/z = 861.8 [M + H]+. Compound 2: eluent: CHCl3, 0.059 g yield 55%, 1H NMR (600 MHz, CDCl3) δ −2.23 (s, 2H), 2.02 (t, J = 9.0 Hz, 1H), 2.31 (s, 3H), 2.85 (d, J = 11.4 Hz, 1H), 3.29 (t, J = 9 Hz, 1H), 4.17 (d, J = 11.4 Hz, 1H), 5.67 (t, J = 8.8 Hz, 1H), 7.21–7.29 (m, 7H), 7.44 (d, J = 8.4 Hz, 1H), 7.51 (d, J = 8.4 Hz, 1H), 7.57 (d, J = 8.4 Hz, 1H), 7.64 (d, J = 8.4 Hz, 2H), 7.68 (dd, J = 7.8 Hz, 17.4 Hz, 2H), 8.26 (dd, J = 1.6 Hz, 4.8 Hz, 1H), 8.45 (dd, J = 1.6 Hz, 4.8 Hz, 1H), 8.55 (q, J = 4.5 Hz, 2H), 8.72 (dd, J = 1.3 Hz, 4.7 Hz, 1H), 8.77 (dd, J = 1.1 Hz, 4.8 Hz, 1H). 13C NMR (151 MHz, CDCl3): δ 41.08, 63.52, 64.29, 66.58, 103.82, 104.2, 104.90, 105.28, 110.27, 110.70, 115.80, 117.02–117.442, 120.76, 121.67, 125.11, 125.36, 128.09, 128.58, 132.98, 133.41, 135.13, 135.74, 140.14, 140.35, 141.25, 144.15–144.34, 152.67, 153.42, 156.24, 159.49, 160.56–160.65, 161.30, 161.80, 162.22–162.31, 162.80, 163.00, 163.46. HRMS (ES+/TOF) m/z: [M + H]+ calcd for C47H29F8N6O2 861.2224; found 861.2216, MS (MALDI TOF) m/z = 861.8 [M + H]+. Compound 3: eluent: diethyl ether:chloroform 1:5, 0.058 g yield 55%, 1H NMR (600 MHz, CDCl3) δ −2.14 (d, J = 16.5, 2H), 2.38–2.41 (m, 4H), 3.00 (d, J = 11.4 Hz, 1H), 3.37–3.40 (t, J = 8.4 Hz, 1H), 4.45 (d, J = 11.5 Hz, 1H), 5.69 (t, J = 9.0 Hz, 1H), 8.37 (dd, J = 4.8 Hz, 1.5 Hz, 1H), 8.55 (d, J = 4.8 Hz, 1H), 8.58 (q, J = 4.6 Hz, 1H), 8.78 (d, J = 4.2 Hz, 1H), 8.84 (d, J = 4.7 Hz, 1H). 13C NMR (151 MHz, CDCl3): δ 40.90, 61.94, 63.69, 64.09, 96.50, 96.73, 69.50, 69.73, 105.47, 106.50, 107.63, 112.90, 114.85–115.37, 124.59, 125.30, 128.03, 128.67, 133.03, 133.523, 135.51, 136.37, 136.71, 137.40, 138.42, 139.11, 140.09, 140.28, 141.44, 142.18, 143.14, 143.96, 145.05, 145.50, 145.67, 146.70, 147.11, 147.34, 148.34, 153.25, 154.13, 157.51, 161.77. HRMS (ES+/TOF) m/z: [M + H]+ calcd for C47H17F20N6O2 1077.1094; found 1077.1082. MS (MALDI TOF) m/z = 1077.7 [M + H]+. Compound 4: eluent: diethyl ether:chloroform 1:5, 0.062 g yield 57%, 1H NMR (600 MHz, CDCl3) δ −1.95 (bs, 2H), 2.21 (t, J = 6 Hz, 1H), 2.28 (s, 3H), 2.90 (d, J = 12 Hz, 1H), 3.24 (t, J = 9.0 Hz, 1H), 4.05 (d, J = 11.4 Hz, 1H), 5.71 (t, J = 9.0 Hz), 7.18–7.30 (m, 3H), 7.64–7.78 (m, 13H), 7.93–7.940 (m, 1H), 8.00 (d, J = 7.2 Hz, 1H), 8.08–8.25 (m, 6H), 8.45 (d, J = 4.2 Hz, 1H), 8.59 (q, J = 4.2 Hz, 2H), 8.71 (d, J = 4.2 Hz, 1H), 8.77 (d, J = 4.8 Hz, 1H). 13C NMR (151 MHz, CDCl3): δ 41.14, 63.79, 64.67, 66.99, 105.53, 112.16, 112.54, 122.98, 123.98, 124.85, 125.15, 125.26, 126.64, 126.70, 126.79, 127.43, 127.79, 127.80, 127.92, 127.93, 128.10, 128.18, 128.44, 128.46, 128.67, 128.99, 132.27, 132.77, 133.15, 133.23, 133.84, 133.91, 133.98, 134.09, 135.60, 136.19, 137.81, 138.72, 140.60, 140.86, 141.59, 141.62, 141.75, 153.07, 153.84, 156.37, 159.57, 717.8, MS (MALDI TOF) m/z = [M + H]+.
Acknowledgements
This research has been financially supported by the Polish National Center of Sciences (Grant No. 2013/11/N/ST5/02040). We gratefully thank the Tomasz Pawlak for help with NBO analysis.References
- C. M. Che, V. K. Y. Lo, C. Y. Zhou and J. S. Huang, Chem. Soc. Rev., 2011, 40, 1950 RSC.
- M. I. J. Stich, L. H. Fischer and O. S. Wolfbeis, Chem. Soc. Rev., 2010, 39, 3102 RSC.
- C. M. Drain, K. C. Russell and J. M. Lehn, Chem. Commun., 1996, 337 RSC.
- L. R. Milgrom, The Colours of life, Oxford University, Oxford, U. K., 1997, ch. 7 Search PubMed.
- K. Licha, Top. Curr. Chem., 2002, 222, 1 CrossRef CAS.
- T. Gośliński and J. Piskorz, J. Photochem. Photobiol., C, 2011, 12, 304 CrossRef.
- E. G. Azenha, A. C. Serra, M. Pineiro, M. M. Pereira, J. S. d. Melo, L. G. Arnaut, S. J. Formosinho and A. M. de’A. Rocha Gonsalves, Chem. Phys., 2002, 280, 177 CrossRef CAS.
- A. Serra, M. Pineiro, C. I. Santos, A. M. d’A. Rocha Gonsalves, M. Abrantes, M. Laranjo and M. F. Botelho, Photochem. Photobiol., 2010, 86, 206 CrossRef CAS PubMed.
- C. J. P. Monteiro, J. Pina, M. M. Pereira and L. G. Arnaut, Photochem. Photobiol. Sci., 2012, 11, 1233 CAS.
- M. M. Pereira, C. J. P. Monteiro, A. V. C. Simões, S. M. A. Pinto, A. R. Abreu, G. F. F. Sá, E. F. F. Silva, L. B. Rocha, J. M. Dąbrowski, S. J. Formosinho, S. Simões and L. G Arnaut, Tetrahedron, 2010, 66, 9545 CrossRef CAS.
- A. Kimura, K. Funatsu, T. Imamura, H. Kido and Y. Sasaki, Chem. Lett., 1995, 207 CrossRef CAS.
- N. Kariya, T. Imamura and Y. Sasaki, Inorg. Chem., 1998, 37, 1658 CrossRef CAS.
- J. Li, A. Ambroise, S. I. Yang, J. R. Diers, J. Seth, C. R. Wack, D. F. Bocian, D. Holten and J. S. Lindsey, J. Am. Chem. Soc., 1999, 121, 8927 CrossRef CAS.
- B. Boitrel, A. Lecas, Z. Renko and E. Rose, Chem. Commun., 1985, 1820 RSC.
- G. S. Kottas, L. I. Clarke, D. Horinek and J. Michl, Chem. Rev., 2005, 105, 1281 CrossRef CAS PubMed.
- E. R. Kay, D. A. Leigh and F. Zerbetto, Angew. Chem., Int. Ed., 2007, 46, 72 CrossRef CAS PubMed.
- S. Erbas-Cakmak, D. A. Leigh, C. T. McTernan and A. L. Nussbaumer, Chem. Rev., 2015, 115(18), 10081 CrossRef CAS PubMed.
- K. Tashiro, Y. Hirabayashi, T. Aida, K. Saigo, K. Fujiwara, K. Komatsu, S. Sakamoto and K. Yamaguchi, J. Am. Chem. Soc., 2002, 124, 12086 CrossRef CAS PubMed.
- M. Nakamura, K. Kishimoto, Y. Kobori, T. Abe, K. Yoza and K. Kobayashi, J. Am. Chem. Soc., 2016, 138, 12564 CrossRef CAS PubMed.
- T. Lang, A. Guenet, E. Graf, N. Kyritsakas and M. W. Hosseini, Chem. Commun., 2010, 46, 3508 RSC.
- T. Lang, E. Graf, N. Kyritsakas and M. W. Hosseini, New J. Chem., 2013, 37, 112 RSC.
- A. B. C. Deutman, T. Woltinge, J. M. M. Smits, R. de Gelder, J. A. A. W. Elemans, R. J. M. Nolte and A. E. Rowan, Molecules, 2014, 19, 5278 CrossRef PubMed.
- S. Ishihara, J. Labuta, W. van Rossom, D. Ishikawa, K. Minami, J. P. Hill and K. J. Ariga, Phys. Chem. Chem. Phys., 2014, 16, 9713 RSC.
- P. Mondal and S. P. Rath, Chem.–Eur. J., 2016, 22, 5607 CrossRef CAS PubMed.
- P. A. Liddell, G. Kodis, A. L. Moore, T. A. Moore and D. Gust, J. Am. Chem. Soc., 2002, 124, 7668 CrossRef CAS PubMed.
- L. K. Gottwald and E. F. Ullman, Tetrahedron Lett., 1969, 36, 3071 CrossRef.
- C. J. Medforth, in The Porphyrin Handbook, ed. K. M. Kadish, K. M. Smith and E. Guilard, Academic Press, New York, 2000, vol. 5, pp. 3–74 Search PubMed.
- H. Scheer and J. J. Katz, in Porphyrins and Metalloporphyrins, ed. K. M. Smith, Elsevier, Amsterdam, 1975, p. 399 Search PubMed.
- T. Janson and J. J. Katz, in The Porphyrins, ed. D. Dolphin, Academic Press, New York, 1979, IV, ch. 1 Search PubMed.
- L. Noss, P. A. Liddell, A. L. Moore, T. A. Moore and D. Gust, J. Phys. Chem. B, 1997, 101, 458 CrossRef CAS.
- A. M. Stolzenberg and G. S. Haymond, Inorg. Chem., 2002, 41, 300 CrossRef CAS PubMed.
- A. M. G. Silva, A. C. Tome, M. G. P. M. S. Neves, A. M. S. Silva and J. A. S. Cavaleiro, J. Org. Chem., 2005, 70, 2306 CrossRef CAS PubMed.
- J. Flemming and D. Dolphin, Tetrahedron Lett., 2002, 43, 7281 CrossRef CAS.
- M. Gałęzowski and D. T. Gryko, J. Org. Chem., 2006, 71, 5942 CrossRef PubMed.
- X. F. Li, J. P. Zhuang, Y. L. Li, H. B. Liu, S. Wang and D. B. Zhu, Tetrahedron Lett., 2005, 46, 1555 CrossRef CAS.
- X. G. Liu, Y. Q. Feng, X. F. Hu and X. G. Li, Synthesis, 2005, 20, 3632 Search PubMed.
- A. M. G. Silva, A. C. Tome, M. G. P. M. S. Neves, A. M. S. Silva and J. A. S. Cavaleiro, Chem. Commun., 1999, 1767 RSC.
- A. M. d’A. Rocha Gonçalves, J. M. T. B. Varejão and M. M. Pereira, J. Heterocycl. Chem., 1991, 28, 635 CrossRef.
- A. D. Adler, F. R. Longo, J. D. Finarelli, J. Goldmacher, J. Assour and L. Korsakoff, J. Org. Chem., 1967, 32, 476 CrossRef CAS.
- J. C. Hierso, Chem. Rev., 2014, 114, 4838 CrossRef CAS PubMed.
- K. Yoshida, G. Copley, M. Mori and A. Osuka, Chem.–Eur. J., 2014, 20, 10065 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision B. 01, Gaussian, Inc., Wallingford, CT, 2010 Search PubMed.
- B. P. Bandgar and P. Gujarathi, J. Chem. Sci., 2008, 120, 259 CrossRef CAS.
- R. Prasath, R. Butcher and P. Bhavana, Spectrochim. Acta, Part A, 2012, 87, 258 CrossRef CAS PubMed.
- P. Wyrębek and S. Ostrowski, J. Porphyrins Phthalocyanines, 2007, 11, 822 CrossRef.
- E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales, and F. Weinhold, NBO 50. Theoretical Chemistry Institute, University of Wisconsin, Madison, WI, 2001 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra, assignment of pyrrolidine-fused chlorine 2 and 3, absorption spectroscopy, HRMS analysis of compounds, Cartesian coordinates of computed structures of pyrrolidine-fused chlorines, NBO analysis. See DOI: 10.1039/c7ra02217d |
| This journal is © The Royal Society of Chemistry 2017 |