
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePressure-induced phase transition in N–H⋯O hydrogen-bonded crystalline malonamide†
Tingting Yan *a,
Dongyang Xib,
Zhenning Maa,
Xun Wanga,
Qingjie Wanga and
Qiang Lia
*a,
Dongyang Xib,
Zhenning Maa,
Xun Wanga,
Qingjie Wanga and
Qiang Lia
aSchool of Science, Shenyang Jianzhu University, Shenyang 110168, China
bSchool of Material Science and Engineering, Shenyang Jianzhu University, Shenyang 110168, China
First published on 20th April 2017
Abstract
In this study, malonamide (C3H6N2O2) was compressed under up to 10.4 GPa of pressure in a diamond anvil cell at room temperature. Pressure-induced structural evolutions were monitored with in situ Raman spectroscopy. The significant changes in Raman spectra at 2.1 GPa provided evidence for a phase transition. The variations in the Raman spectra were discussed, including the disappearance of original modes, appearance of new modes, and abrupt changes in the pressure dependence of Raman modes under different pressures. Ab initio calculations were employed to account for the changes in molecular arrangements and hydrogen-bonded networks. Hirshfeld surfaces and fingerprint plots were employed for the direct comparison of the variations in packing patterns and intermolecular interactions. Based on the calculated results, variations in the NH2 stretching Raman vibrations, and degree of freedom of the molecules, the rearrangements of the hydrogen-bonded networks likely caused the phase transition of crystalline malonamide.
Introduction
The hydrogen bond is a crucial and intensively studied intermolecular interaction because of its specificity, directionality, and reversibility.1–5 High-pressure techniques can be used to explore the behavior of hydrogen-bonded molecular crystals.6,7 Pressure can efficiently shorten the intermolecular distance of materials and tune the strength of hydrogen bonds.8–11 Pressure-induced changes in hydrogen bonds, combined with the effect of electrostatic interactions, π–π stacking, and van der Waals interactions, can cause the transition of crystal symmetry.12–15 Variations in hydrogen bonds and the cooperativity of various intermolecular interactions under pressure play a significant role in the structural stability of materials.Hydrogen-bonded molecular crystals under high pressure are the subject of extensive research and are ubiquitous phenomena.16–23 Amides are of particular research interest because they are widespread in nature and have many technological applications.24,25 Amides have the smallest peptide bonds and are model compounds for the investigation of hydrogen bonds in biologically active complex peptides. The phase transition of formamide, the simplest amide, under 5 GPa was first reported in 1988.26 Subsequently, Katrusiak observed that the N–H⋯O bonded sheets in compressed formamide are reconstructed due to the formation of pressure-promoted C–H⋯O hydrogen bonds. Furthermore, the isochoric freezing of formamide under high pressure induces a new phase.27 Similarly, oxamide transitions into a high-pressure phase at pressures above 9.6 GPa.28 Moreover, acrylamide polymerizes under pressures that exceed 4 GPa; the strong N–H⋯O bond of the initial phase breaks and the relatively weak N–H⋯O bond in the new phase forms under high pressure.29 Therefore, studies on hydrogen bonds in amides under high pressure are fundamental to pave a new way for molecular construction in crystal engineering.
Malonamide can be used as a model system to study hydrogen bonding interactions in amides under pressure. Malonamide, which is used to synthesize pharmaceuticals and pesticides, is equivalent to a glycine residue with an inverted peptide group.30,31 Malonamide derivatives are of considerable interest given their use in peptidomimetic compound synthesis.32 The crystal and molecular structures of malonamide at ambient conditions were determined by Chieh, Subramanian, and Trotter in 1970.33 They reported that malonamide crystallizes into a monoclinic structure with a P21/c space group. The unit cell parameters of malonamide are a = 13.07 Å, b = 9.45 Å, c = 8.04 Å, β = 73.0(0)°, V = 949.642 Å3, and Z = 8. As shown in Fig. 1, malonamide has a nonplanar crystal molecule because it contains two symmetry-unrelated molecules with different orientations. The two molecules in the asymmetric unit have similar conformations and dimensions. The amide groups are rotated out of the central CCC plane and the amide groups are almost perpendicular to each other in each molecule. The central CCC plane of the molecule is parallel to (010) and the C–H bond of the radical is parallel to the c-axis. Moreover, malonamide has ordinary three-dimensional hydrogen-bonded networks. The molecules are held together by eight hydrogen bonds that involve all the amino-hydrogen atoms. Each oxygen atom acts as an acceptor in two hydrogen bonds. In addition, one hydrogen bond from each molecule together with a pair of symmetry-related bonds joins the molecules into dimers. All the other intermolecular distances correspond to van der Waals interactions.
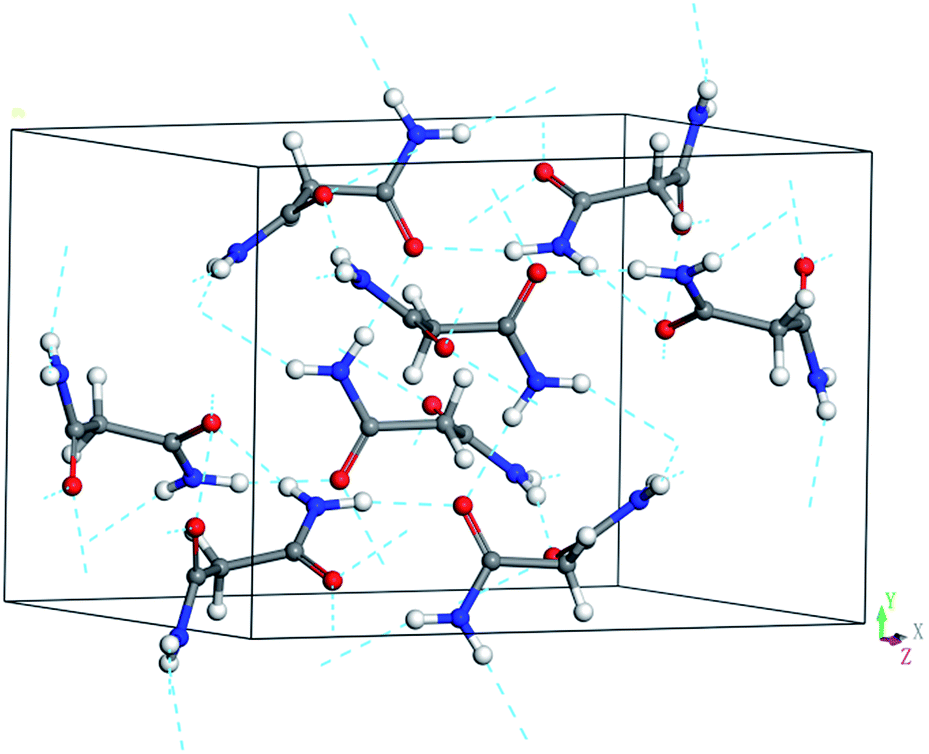 | ||
| Fig. 1 Ambient unit cell and hydrogen-bonded networks of malonamide. The dashed lines stand for N–H⋯O hydrogen bonds. | ||
In this work, we report in situ Raman-scattering measurements on malonamide in a diamond anvil cell (DAC) under pressures of up to 10.4 GPa at room temperature. The reversible phase transition at 2.1 GPa has been evidenced. Both external (intermolecular) and internal (intramolecular) modes are detected and analyzed in Raman spectra to provide detailed descriptions of the molecular fragment and hydrogen bond motions. Ab initio calculations provide valuable information on hydrogen-bonded networks and phase transition mechanisms. Hirshfeld surfaces and the fingerprint plots allow for the direct comparison of the variations in packing patterns. Structural changes and cooperative effects between hydrogen bonding and van deer Waals interactions are analyzed. The present study improves the current understanding of hydrogen bond behavior and explores the effects of intermolecular interactions on the stability of hydrogen-bonded molecular crystals.
Experimental section
Malonamide was purchased from Aldrich Co. (purity 99%) and used without further purification. High-pressure experiments were conducted using a symmetric diamond anvil cell (DAC) with a pair of type Ia diamonds with a 400 μm culet. A T301 steel gasket was preindented to a thickness of 40 μm. A hole with a diameter of 130 μm was drilled in the gasket as the sample chamber. Subsequently, the powder sample was placed in the hole together with one or two small ruby chips as the pressure calibrant via the R1 ruby fluorescent technique at an uncertainty of ±0.05 GPa.34 Liquid nitrogen was used as the pressure-transmitting medium (PTM). The ruby lines were sharp and well separated under the highest pressure in this study. All experiments were performed at room temperature.In situ high-pressure Raman measurements were conducted with the LabRAM HR Evolution Raman system. The sample was excited with the 532 nm line of a diode laser at a laser output power of 10 mW. The Raman system adopted a backscattering configuration. Notch filters were utilized to remove Rayleigh scattering. Raman signals were recorded using a liquid-nitrogen-cooled CCD detector at a spectral resolution of 1 cm−1. The standard silicon line was used for calibration prior to each experiment. When necessary, the Raman profiles were fit using a combination of Gaussian and Lorentzian functions.
Ab initio pseudopotential plane-wave density functional method combined in the program Materials Studio 7.0 was used. Our simulations were conducted at 0 K and equivalent hydrostatic pressure conditions. The high-pressure structural optimizations in this study, including the optimization of atomic positions and lattice constants, were performed by the BFGS algorithm. This algorithm provides a fast way of finding the lowest energy structure and the optimized cell. Exchange–correlation effects were treated in the scheme of Perdew–Burke–Ernzerhof GGA. The optimization was not completed until the forces in the atoms were less than 0.01 eV Å−1. Convergence tests provided a grid spacing of 0.03 Å−1 spacing and energy cutoff of 450 eV. The k-point was of fine quality at 0.05 Å−1 separation.
Results and discussion
The point group symmetry of the malonamide crystal (Z = 8) is C2h (2/m). The mechanical representation of this symmetry is| M = 78Ag + 78Au + 78Bg + 78Bu |
| Γacoustic = Au + 2Bu |
| Γoptic = 78Ag + 77Au + 78Bg + 76Bu |
The group theoretical classification of the 309 optical modes shows that the Raman-active modes belong to 78Ag + 78Bg symmetry. The remaining infrared-active modes belong to 77Au + 76Bu symmetry. Some of the Raman modes cannot be observed in our experiments because of their very weak intensities and limited splitting between correlation components.
Assignments for the Raman modes of malonamide are based on reported literature.32 In this work, the high-pressure Raman spectra of malonamide are measured under pressures of up to 10.4 GPa. Fig. 2 illustrates the Raman patterns in the frequency range of 30–305 cm−1 under various pressures. The vibrations that are associated with external modes are expected in this region. The evolution of external modes can serve as an index of structural change. As shown in Fig. 2, the spectrum at 0.1 GPa consists of 11 external modes (56, 57, 67, 69, 77, 86, 93, 101, 115, 130, and 142 cm−1). At 1.4 GPa, the mode at 86 cm−1 merges into the left band and the mode at 93 cm−1 merges into the right band. In the plot of 2.1 GPa, the shape of the spectrum abruptly changes, which indicates the transition from ambient phase I to high-pressure phase II. With further compression beyond 7.0 GPa, the two modes that are marked by downward-facing arrows gradually lose their intensities and eventually vanish into the scattering background. No discontinuity is detected in external modes beyond 2.1 GPa, suggesting that phase II is stable and does not undergo further changes at up to 10.4 GPa, the highest pressure employed in this experiment. When pressure is released, the Raman pattern of phase II returns to its initial state, implying the phase transition is reversible (Fig. S1, ESI†).
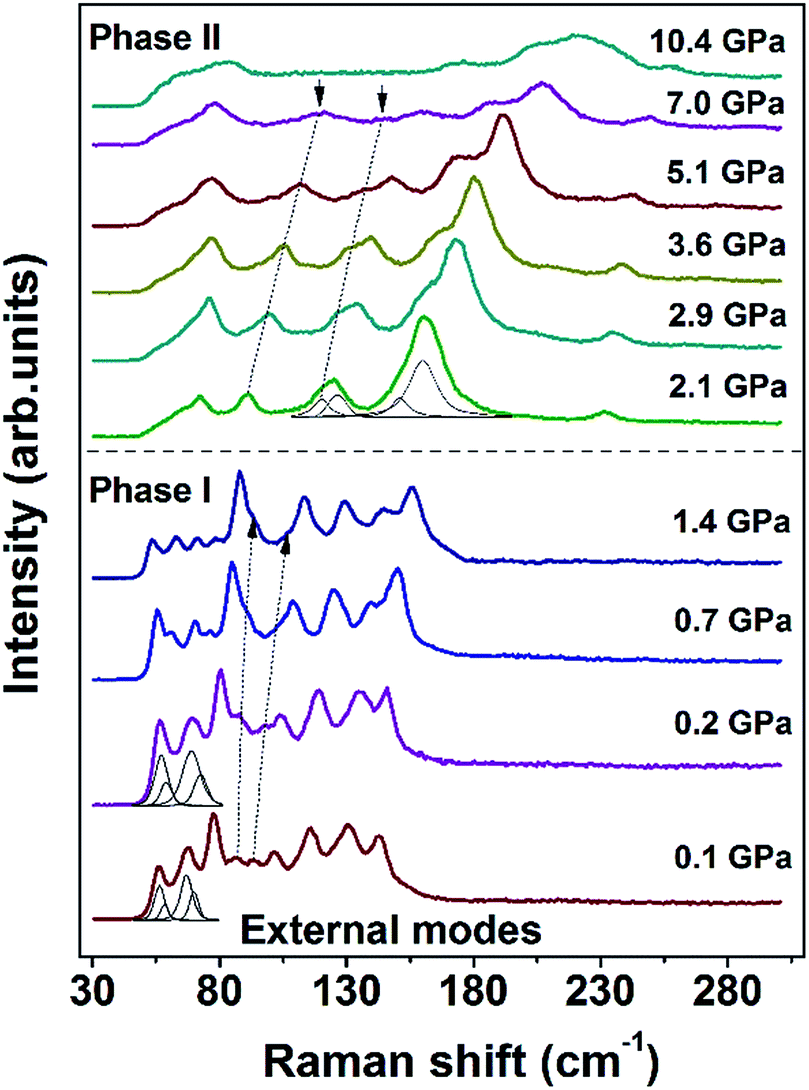 | ||
| Fig. 2 Raman spectra of malonamide at selected pressures in the external modes region in the range of 30–305 cm−1. Peak fitting and decomposition are obtained using a combination of Gaussian and Lorentzian functions. | ||
Fig. 3 shows the pressure-induced frequency shifts of the external modes. There is an obvious discontinuity at approximately 2 GPa. This discontinuity is consistent with the proposed phase transition. The mode at 56 cm−1 shifts to a low frequency as pressure increases until phase transition occurs. Based on the lattice dynamical viewpoint, the stability limit of a crystal lattice is reached as the frequency of any external mode decreases.35–38 Thus, the red shifts indicate that the crystal structure gradually becomes unstable and that molecules adopt new orientations. Apart from this mode, other modes display blue shifts in the phase I zone. After phase transition, all external modes exhibit substantial blue shifts at different pressures of up to 10.4 GPa. The observed blue shifts of the external modes are ascribed to the stronger interactions between adjacent molecules as intermolecular distances decreased.39,40
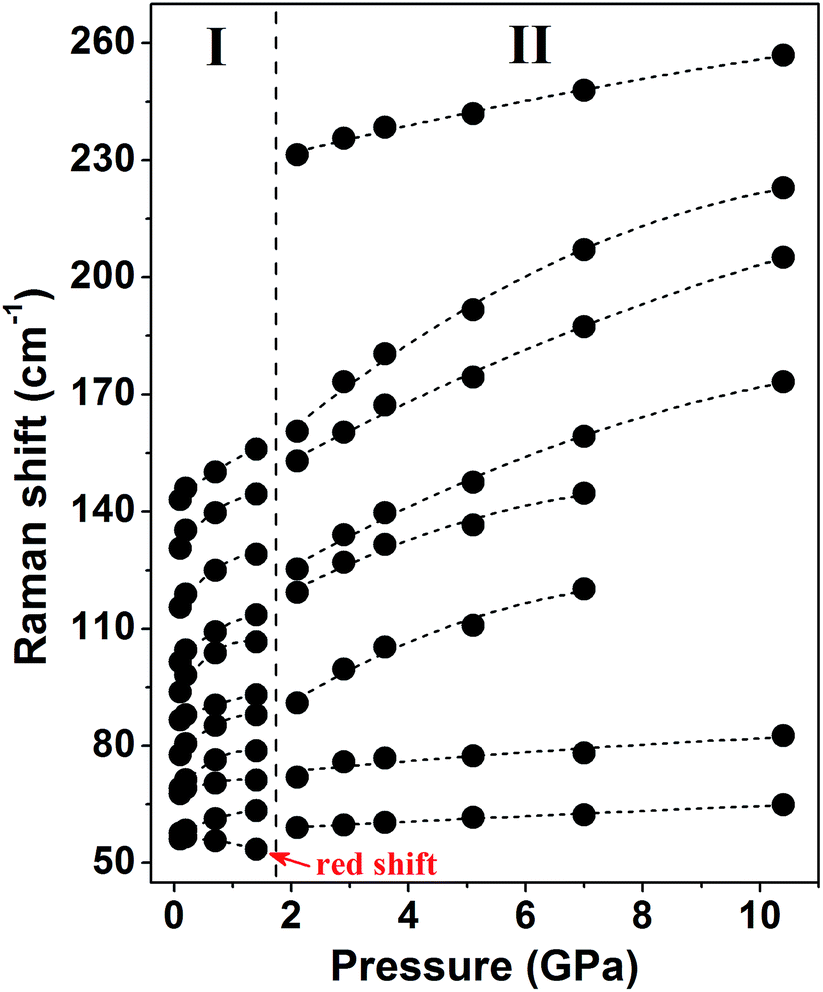 | ||
| Fig. 3 Frequency shifts of the external modes as a function of pressure. Fitting curves are performed for clarity. The vertical dotted line represents the boundary of ambient phase I and high-pressure phase II. | ||
The Raman spectra of the malonamide internal modes in 350–1750 cm−1 are depicted in Fig. 4. Internal modes are sensitive to high pressure and provide fundamental information on the chemical environments that surround a specific group. The simultaneous variations in internal modes reveal the existence of phase transition, as shown in Fig. 4. At 2.1 GPa, the modes marked by downward-facing arrows completely lose their intensities. Moreover, numerous new modes, which are marked with asterisks, are observed. Above 5.1 GPa, the mode marked with an arrow at 1644 cm−1 weakens to the detectable limit. Fig. 5 summarizes the corresponding peak positions versus pressure of the internal modes. An obvious discontinuity between the two regions is observed, which is attributed to the proposed phase transition. Most of the internal modes shift gradually toward higher frequencies throughout the entire pressure region, including the NH2 bending (amide II) and C–N stretching (amide III) vibrations. As the crystal is compressed, the higher frequencies are caused by the shortened covalent bond and the increased effective force constants.40 Meanwhile, the blue shifts of the NH2 bending and NH2 rocking modes imply the existence of weak- or moderate-strength hydrogen bonds in malonamide. The C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching mode (amide I) red-shifts up to 1.4 GPa, as shown in Fig. 4c. An increase in pressure should decrease the distance between O and C atoms. However, the weak- or moderate-strength N–H⋯OC hydrogen bonds lengthen this distance, thus counteracting pressure effects.41–43 Moreover, the new mode located at 1608 cm−1, which is identified as the CO stretching vibration, also red-shifts up to the highest experimental pressure. Based on internal mode behavior, phase transition involves a large change in the chemical environments around molecules. Changes in NH2 twisting, NH2 bending, NH2 rocking, and NH2 scissoring modes, suggest that increasing pressure causes the local distortion of amino groups. Furthermore, changes in CCC wagging, CCC stretching, CCC deformation, and NCO deformation modes demonstrate that the molecular skeleton changes during phase transition.
O stretching mode (amide I) red-shifts up to 1.4 GPa, as shown in Fig. 4c. An increase in pressure should decrease the distance between O and C atoms. However, the weak- or moderate-strength N–H⋯OC hydrogen bonds lengthen this distance, thus counteracting pressure effects.41–43 Moreover, the new mode located at 1608 cm−1, which is identified as the CO stretching vibration, also red-shifts up to the highest experimental pressure. Based on internal mode behavior, phase transition involves a large change in the chemical environments around molecules. Changes in NH2 twisting, NH2 bending, NH2 rocking, and NH2 scissoring modes, suggest that increasing pressure causes the local distortion of amino groups. Furthermore, changes in CCC wagging, CCC stretching, CCC deformation, and NCO deformation modes demonstrate that the molecular skeleton changes during phase transition.
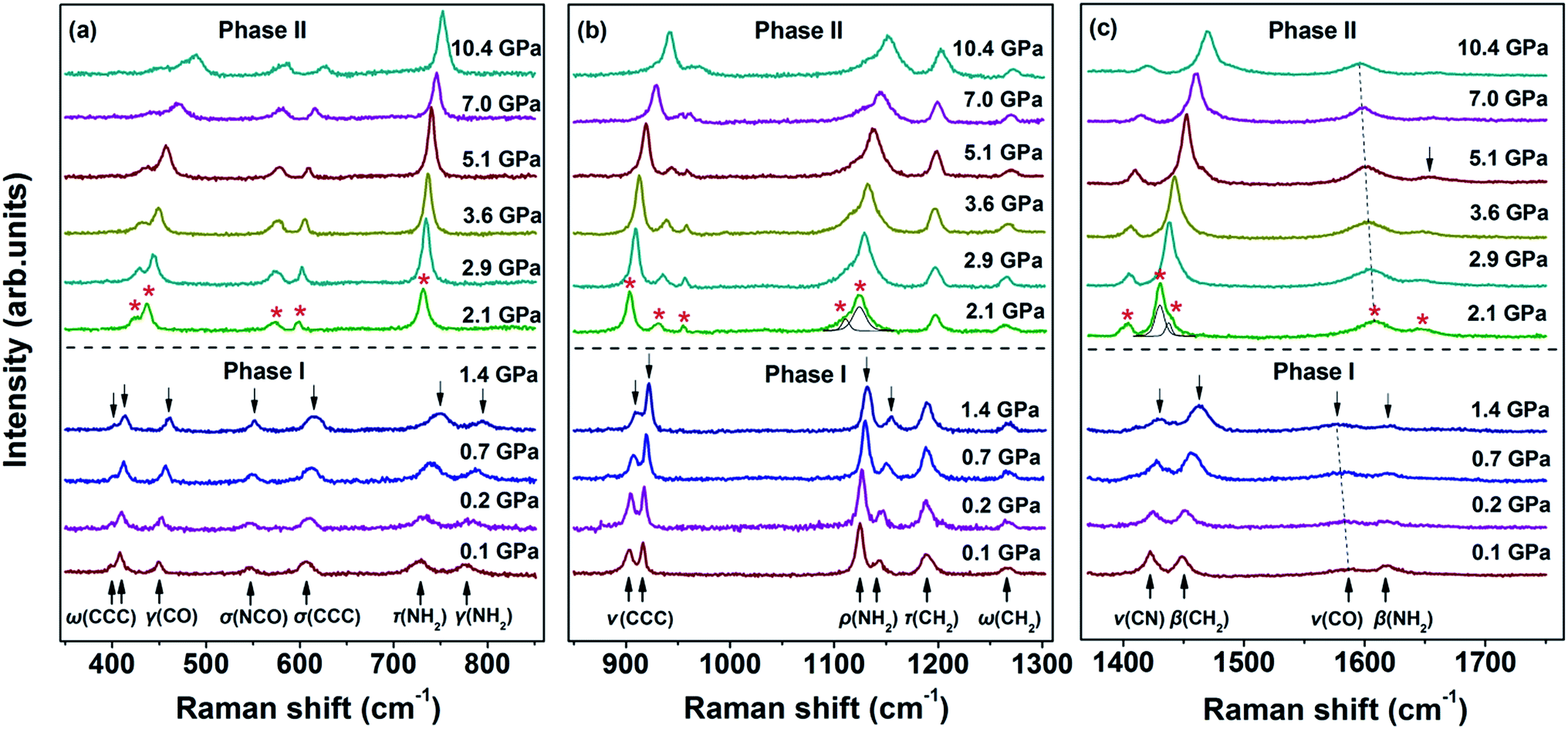 | ||
| Fig. 4 Raman spectra of the internal modes of malonamide in the ranges of (a) 350–850 cm−1, (b) 850–1300 cm−1, and (c) 1370–1750 cm−1. ω, wagging; γ, bending; σ, deformation; τ, twisting; ν, stretching; ρ, rocking; β, scissoring; s, symmetric; as, asymmetric. | ||
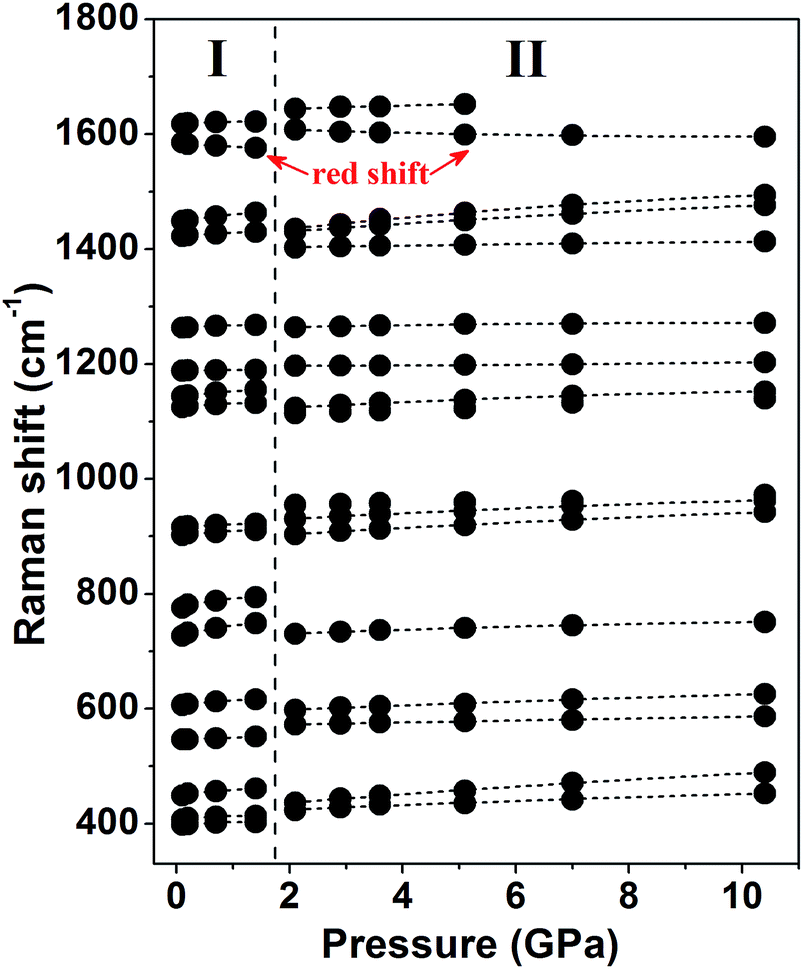 | ||
| Fig. 5 Pressure dependence of Raman peak positions of internal modes in the range of 350–1750 cm−1. The vertical dotted line denotes the proposed phase boundary. | ||
The evolution of the Raman spectra in the CH2 and NH2 stretching region under selected pressures and the pressure dependence of the corresponding modes are illustrated in Fig. 6. As shown in Fig. 6a, the spectrum comprises three bands at 0.1 GPa. The band at 2944 cm−1 is assigned to CH2 symmetric stretching vibration, whereas the band at 2981 cm−1 is identified as CH2 asymmetric stretching vibration. The other band at 3158 cm−1 corresponds to NH2 symmetric stretching vibration. When the pressure reaches 2.1 GPa, a new peak emerges at 3395 cm−1, which is identified as the NH2 asymmetric stretching mode. This pattern remains completely unchanged under pressures of up to 10.4 GPa. As seen in Fig. 6b, the NH2 symmetric stretching and NH2 asymmetric stretching modes red-shift as pressure increases. The red shifts are in accordance with the general rules that increasing pressure decreases the N–H stretching frequencies of weak- or moderate-strength N–H⋯O hydrogen bonds.41–43 Pressure can shorten the distance between H and O atoms, thus enhancing electrostatic attraction between the two atoms. This process can lengthen N–H distance, eventually causing red shifts. Since all N–H bonds participate in hydrogen bonds, the considerable changes in NH2 stretching modes indicate that the proposed phase transition is correlated with the rearrangements of the N–H⋯O hydrogen-bonded networks.
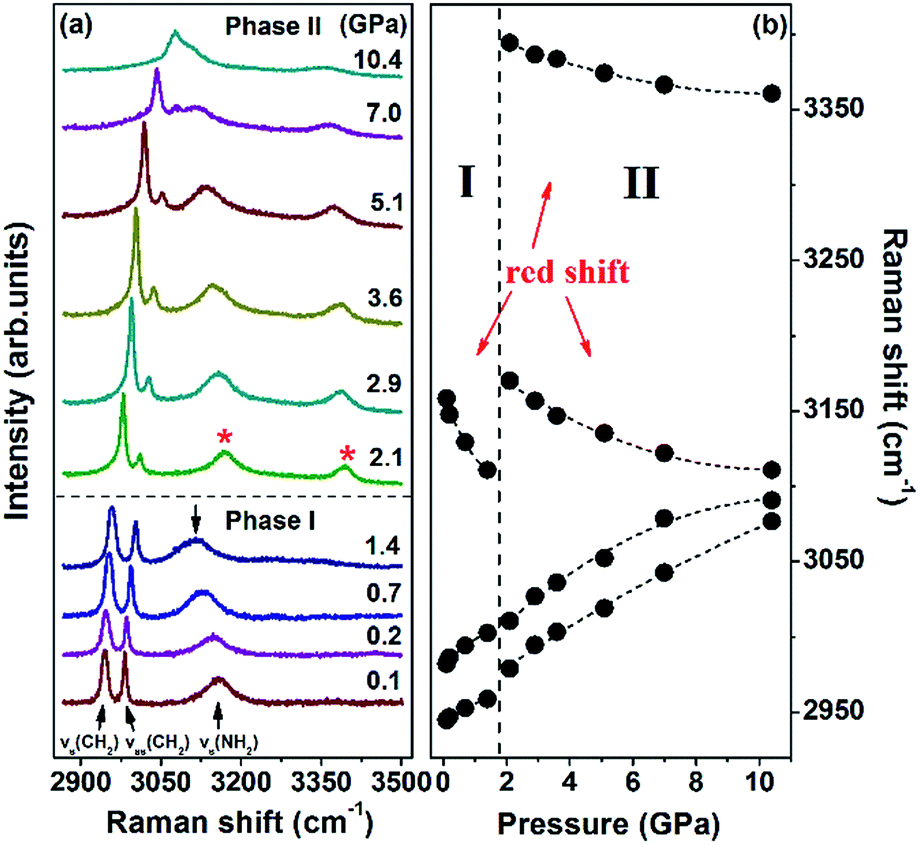 | ||
| Fig. 6 (a) Evolution of the CH2 and NH2 stretching vibrations at different pressures in the region of 2870–3500 cm−1; (b) frequency shifts of these modes. The dotted line represents the boundary of the two phases. | ||
To understand what changes occur in the local structure and the hydrogen-bonded networks of malonamide under high pressures, we performed ab initio calculations using pseudopotential plane-wave methods that are based on the density functional theory.44 Fig. 7 depicts the Hirshfeld surfaces and fingerprint plots for malonamide at ambient pressure and 10 GPa. Compression of the crystal structure on increasing pressure leads to the changes of intermolecular interactions. Hirshfeld surfaces and fingerprint plots are employed to directly compare the changes in packing patterns and intermolecular interactions.45,46 In this manner, hydrogen bonding and van der Waals radius are readily identifiable. In the Hirshfeld surfaces, the blue regions correspond to long contacts, whereas the red regions denote short contacts, as shown in Fig. 7a1 and a2. Upon increasing pressure, the blue regions decrease and the red regions increase. This trend is consistent with the general shortening of contacts at high pressure. The voids close to decrease the distances of hydrogen bonding and van der Waals interactions. In Fig. 7b1 and b2, the fingerprint plots show pseudo-symmetry about the diagonal shape and approach the origin as the contacts shorten. The decreased maximum values of de between ambient pressure (2.399 Å) and 10 GPa (1.921 Å) are attributed to the overall shortening of the long contacts. In addition, the two “spikes” in both plots represent N–H⋯O hydrogen bonds. The upper spike corresponds to the hydrogen bond donor (where de > di), whereas the lower spike corresponds to the hydrogen bond acceptor (where di > de). The single N–H⋯O hydrogen bond extends down to (di, de) = (0.8, 1.15), corresponding to a H-bond distance of 1.95 Å. This finding is clearly represented in the fingerprint plot: the N–H⋯O spikes become less pronounced as the rest of the plot approaches the origin. The contribution of the H⋯O contacts undergoes small changes from 23.9% at ambient pressure and 21.1% at 10 GPa. Furthermore, the “skirt” points (de ≈ di at around the van der Waals radius of the H-atom of 1.3 Å) in between the spikes, which are marked by a red arrow, arise from very close C–H⋯H–C contacts across the cyclic hydrogen-bonded dimer. This phenomenon corresponds to the crystal structure of malonamide, i.e., three molecules (labeled from 1 to 3) that form cyclic hydrogen-bonded dimers, as shown in Fig. 8a. The C–H⋯H–C contacts are compressed from 2.6 Å at ambient pressure to 2.2 Å at 10 GPa. Moreover, the contribution of the C–H⋯H–C interaction changes from 32.3% at ambient pressure to 30.2% at 10 GPa. In addition, the “wings” area marked with a red circle is related to C–H⋯π interaction, implying the packing patterns and intermolecular interactions in high-pressure structure have been changed significantly. Moreover, the contribution of other contacts are changed simultaneously with increasing pressure, such as H⋯N contacts from 3.1% to 3.8%, H⋯C contacts from 2.6% to 2.7%, N⋯O contacts from 0.6% to 0.7%, and C⋯O contacts from 0.6% to 1.5%, respectively.
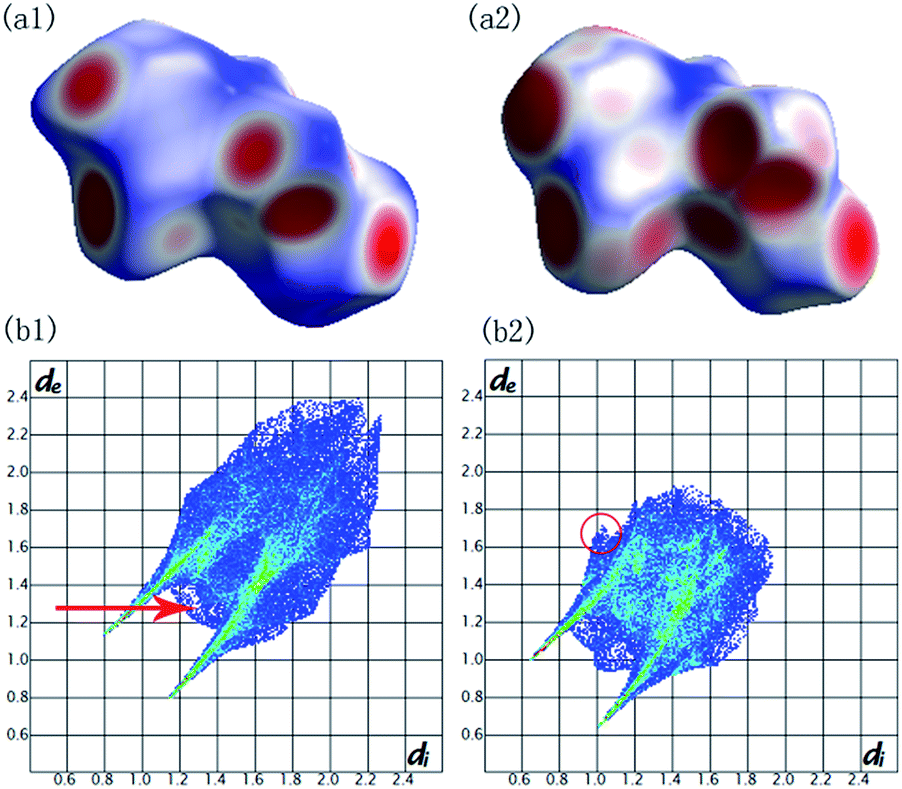 | ||
| Fig. 7 Hirshfeld surfaces mapped with dnorm and fingerprint plots for malonamide at ambient pressure (a1) (b1) and at 10 GPa (a2) (b2). | ||
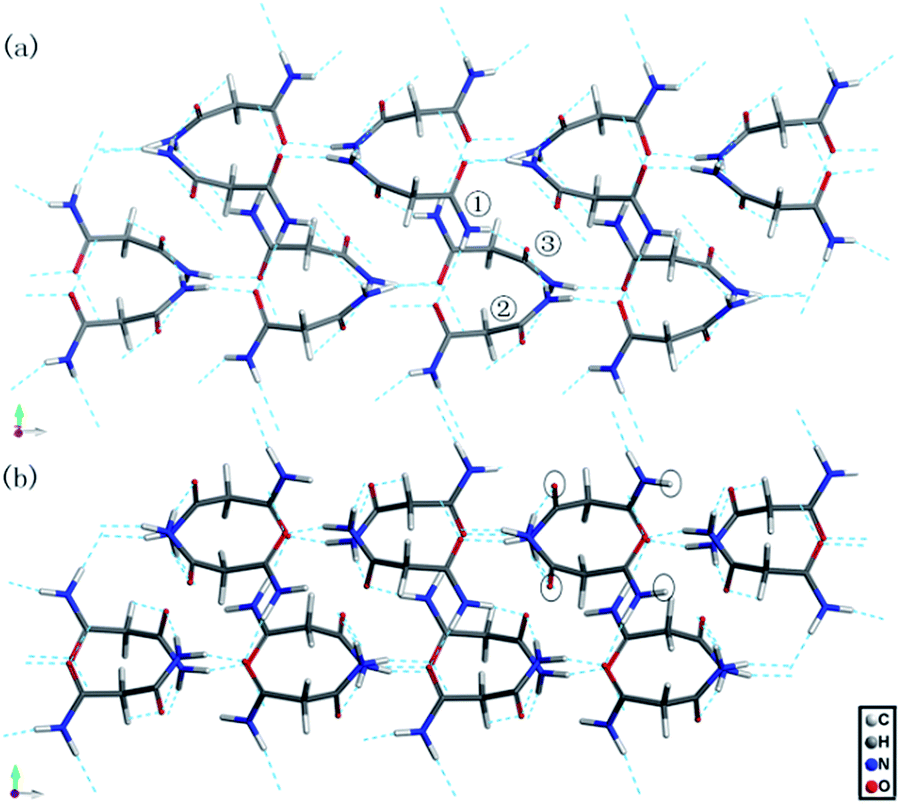 | ||
| Fig. 8 The calculated hydrogen-bonded networks of malonamide (a) at ambient pressure and (b) at 10 GPa. | ||
Fig. 8 shows the calculated hydrogen-bonded networks in malonamide at ambient pressure and 10 GPa. According to the calculated results, molecules undergo distortions to a certain extent in response to pressure. Thus, the original N–H⋯O hydrogen bonds are distorted. In addition, the hydrogen-bonded patterns considerably change. In the ambient structure, all amine groups participate in hydrogen bonds. Each molecule forms eight hydrogen bonds with four donors and four acceptors. However, at 10 GPa, one kind of molecule in the asymmetric unit participates in the formation of six hydrogen bonds with three donors and three acceptors, as indicated by the black circles in Fig. 8b. These results agree well with the variations in the NH2 stretching modes in Raman spectra. The emergence of the NH2 asymmetric stretching mode indicates that the hydrogen-bonded networks are reconstructed. Meanwhile, the modes that are related to N–H⋯O hydrogen bonds, including τ(NH2), ρ(NH2), β(NH2), γ(NH2), γ(CO), and ν(CO) modes, all vary remarkably during phase transition. These variations indicate that the hydrogen bond donors and acceptors adopt new orientations. Furthermore, in view of the essential structure, malonamide has one degree of freedom only, that is, the ability of the two amide units to rotate around the C–C single bonds of the single sp3-carbon atom. Thus, changes of the molecular conformations by a rotation of the amide units significantly modify the N–H⋯O hydrogen-bonded networks.
On the basis of the experimental and calculated results, a mechanism for the phase transition is proposed. Hydrogen bonding and van der Waals interactions are the predominant cohesive factors for crystal packing of malonamide at ambient pressure. Compared with the strength of covalent bonds (150–400 kJ mol−1), hydrogen bonds (8–50 kJ mol−1) and van der Waals interactions (0.4–4.0 kJ mol−1) are far weaker, although van der Waals interactions have the additive property. The application of low pressure continuously shortens molecular distances. Molecules in the crystals become closer to each other inevitably to achieve closer packing. Thus, hydrogen bonding and van der Waals interactions are enhanced. As shown in the Raman patterns, below 2.1 GPa, as the enhanced hydrogen bonding interaction, modes related to N–H⋯O hydrogen bonds, including NH2 twisting, NH2 rocking, NH2 scissoring, NH2 bending, and CO bending modes exhibit blue shifts, and CO stretching mode displays red shift. Meanwhile, Gibbs free energy in the system is increased from −15.8253 keV at ambient pressure to −15.8241 keV at 2 GPa, due to the strengthened hydrogen bonding and van der Waals interactions. With further compression at 2.1 GPa, the balance between hydrogen bonding and van der Waals interactions is broken. The hydrogen-bonded networks can no longer support the increased Gibbs free energy. The bond geometry parameters of hydrogen bonds (bond length, bond angle, and bond strength) are changed. Thus, the hydrogen-bonded networks are rearranged to reduce the free energy. As can be seen from the spectra obtained, various variations of the Raman modes related to N–H⋯O hydrogen bonds confirm the rearrangement of hydrogen-bonded networks in malonamide during phase transition. Changes in the hydrogen bonds may break through the energy barriers, inducing large changes in the crystal structures. Overall, rearrangement of the hydrogen-bonded networks causes molecular rotation and translation, eventually resulting in phase transition. After releasing pressure, these deformed molecules and distorted hydrogen bonds are recovered, so the high-pressure structure returns to the ambient state. The proposed mechanism can be well-supported by the Raman results. The external modes involve the collective motions of all atoms in the unit cell and exert an important function in monitoring structural changes under high pressures. Drastic changes in these external modes, such as the disappearance of numerous peaks and appearance of new peaks, suggest that great changes in intermolecular interactions take place. The results suggest that hydrogen bonds have an important function in the structural transition of malonamide. High-pressure X-ray and neutron diffraction, however, are needed to reliably determine atomic positions in malonamide.
Conclusions
The pressure-induced phase transition in hydrogen-bonded crystalline malonamide was investigated by in situ Raman scattering spectroscopy. The remarkable changes in the Raman spectra at 2.1 GPa strongly suggested the emergence of a high-pressure phase, which was stable at up to 10.4 GPa of pressure. The observed phase transition was reversible following decompression to 0.1 GPa. Ab initio calculations were conducted to explore the changes in the local structure and hydrogen-bonded networks of malonamide under high pressures. Hirshfeld surfaces and fingerprint plots were employed to compare the variations in intermolecular interaction. Raman spectra and ab initio calculations revealed that the phase transition mechanism resulted from the rearrangements of hydrogen-bonded networks and the deformation of the molecular framework. This work furthers the current understanding of hydrogen bonds and the structural stability of hydrogen-bonded molecular crystals under high pressures.Acknowledgements
This work is supported by NSFC (Grant No. 11604224 and 11604308), project of the Education Department of Liaoning Province (LJZ2016031 and LJZ2016030), and project of Shenyang Jianzhu University Discipline Content Education (XKHY2-105 and XKHY2-101).Notes and references
- T. Steiner, Angew. Chem., Int. Ed., 2002, 41, 48–76 CrossRef CAS.
- C. B. Aakeröy and K. R. Seddon, Chem. Soc. Rev., 1993, 22, 397–407 RSC.
- J. D. Dunitz and A. Gavezzotti, Angew. Chem., Int. Ed., 2005, 44, 1766–1787 CrossRef CAS PubMed.
- F. Fillaux, Int. Rev. Phys. Chem., 2000, 19, 553–564 CrossRef CAS.
- G. R. Desiraju, Acc. Chem. Res., 2002, 35, 565–573 CrossRef CAS PubMed.
- S. K. Sikka and S. M. Sharma, Phase Transitions, 2008, 81, 907–934 CrossRef CAS.
- E. V. Boldyreva, J. Mol. Struct., 2004, 700, 151–155 CrossRef CAS.
- S. V. Goryainov, E. V. Boldyreva and E. Kolesnik, Chem. Phys. Lett., 2006, 419, 496–500 CrossRef CAS.
- A. F. Goncharov, M. R. Manaa, J. M. Zaug, R. H. Gee, L. E. Fried and W. B. Montgomery, Phys. Rev. Lett., 2005, 94, 065505 CrossRef PubMed.
- M. Mookherjee and L. Stixrude, Am. Mineral., 2006, 91, 127–134 CrossRef CAS.
- J. Joseph and E. D. Jemmis, J. Am. Chem. Soc., 2007, 129, 4620–4632 CrossRef CAS PubMed.
- V. S. Minkov, A. S. Krylov, E. V. Boldyreva, S. V. Goryainov, S. N. Bizyaev and A. N. Vtyurin, J. Phys. Chem. B, 2008, 112, 8851–8854 CrossRef CAS PubMed.
- M. Bernasconi, P. Silvestrelli and M. Parrinello, Phys. Rev. Lett., 1998, 81, 1235–1238 CrossRef CAS.
- A. Luzar and D. Chandler, Nature, 1996, 379, 55–57 CrossRef CAS.
- A. F. Goncharov, V. V. Struzhkin, H.-k. Mao and R. J. Hemley, Phys. Rev. Lett., 1999, 83, 1998–2001 CrossRef CAS.
- T. T. Yan, K. Wang, X. Tan, K. Yang, B. B. Liu and B. Zou, J. Phys. Chem. C, 2014, 118, 15162–15168 CAS.
- R. S. Chellappa, D. M. Dattelbaum, N. Velisavljevic and S. Sheffield, J. Chem. Phys., 2012, 137, 064504 CrossRef PubMed.
- T. T. Yan, K. Wang, X. Tan, J. Liu, B. B. Liu and B. Zou, J. Phys. Chem. C, 2014, 118, 22960–22967 CAS.
- T. T. Yan, K. Wang, D. F. Duan, X. Tan, B. B. Liu and B. Zou, RSC Adv., 2014, 4, 15534–15541 RSC.
- A. Katrusiak, Crystallogr. Rev., 1996, 5, 133–175 CrossRef CAS.
- P. F. McMillan, Chem. Soc. Rev., 2006, 35, 855–857 RSC.
- S. Moggach and S. Parsons, CrystEngComm, 2010, 12, 2515 RSC.
- C. Murli, N. Lu, Z. Dong and Y. Song, J. Phys. Chem. B, 2012, 116, 12574–12580 CrossRef CAS PubMed.
- G. A. Jeffrey and W. Saenger, Hydrogen Bonding in Biological Structures, Springer-Verlag, Berlin, 1991 Search PubMed.
- R. D. L. Johnstone, A. R. Lennie, S. F. Parker, S. Parsons, E. Pidcock, P. R. Richardson, J. E. Warren and P. A. Wood, CrystEngComm, 2010, 12, 1065–1078 RSC.
- H. Shimizu, K. Nagata and S. Sasaki, J. Chem. Phys., 1988, 89, 2743–2747 CrossRef CAS.
- R. Gajda and A. Katrusiak, Cryst. Growth Des., 2011, 11, 4768–4774 CAS.
- T. T. Yan, S. R. Li, K. Wang, X. Tan, Z. M. Jiang, K. Yang, B. B. Liu, G. T. Zou and B. Zou, J. Phys. Chem. B, 2012, 116, 9796–9802 CrossRef CAS PubMed.
- A. K. Mishra, C. Murli and S. M. Sharma, J. Phys. Chem. B, 2008, 112, 15867–15874 CrossRef CAS PubMed.
- G. S. Nichol and W. Clegg, Acta Crystallogr., 2005, E61, o3424–o3426 Search PubMed.
- G. S. Nichol and W. Clegg, Acta Crystallogr., 2005, E61, o3427–o3429 Search PubMed.
- M. M. Schiavoni, H.-G. Mack, S. E. Ulic and C. O. D. Védova, Spectrochim. Acta, Part A, 2000, 56, 1533–1541 CrossRef.
- P. C. Chieh, E. Subramanian and J. Trotter, J. Chem. Soc., 1970, 92, 179–184 RSC.
- H. K. Mao, P. M. Bell, J. W. Shaner and D. J. Steinberg, J. Appl. Phys., 1978, 49, 3276–3283 CrossRef CAS.
- N. Binggeli, N. R. Keskar and J. R. Chelikowsky, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 3075–3081 CrossRef CAS.
- G. Samara and P. Peercy, Solid State Phys., 1982, 36, 1–118 Search PubMed.
- J. F. Scott, Rev. Mod. Phys., 1974, 46, 83–128 CrossRef CAS.
- H. Chihara, N. Nakamura and M. Tachiki, J. Chem. Phys., 1973, 59, 5387–5391 CrossRef CAS.
- T. R. Park, Z. A. Dreger and Y. M. Gupta, J. Phys. Chem. B, 2004, 108, 3174–3184 CrossRef CAS.
- J. A. Ciezak, T. A. Jenkins, Z. Liu and R. J. Hemley, J. Phys. Chem. A, 2007, 111, 59–63 CrossRef CAS PubMed.
- S. D. Hamann and M. Linton, Aust. J. Chem., 1976, 29, 1641–1647 CrossRef CAS.
- Y. Lin, H. W. Ma, C. W. Matthews, B. Kolb, S. Sinogeikin, T. Thonhauser and W. L. Mao, J. Phys. Chem. C, 2012, 116, 2172–2178 CAS.
- B. B. Sharma, C. Murli and S. M. Sharma, J. Raman Spectrosc., 2013, 44, 785–790 CrossRef CAS.
- M. D. Segall, P. J. D. Lindan, M. J. Probert, C. J. Pickard, P. J. Hasnip, S. J. Clark and M. C. Payne, J. Phys.: Condens. Matter, 2002, 14, 2717 CrossRef CAS.
- P. A. Wood, J. J. McKinnon, S. Parsons, E. Pidcock and M. A. Spackman, CrystEngComm, 2008, 10, 368–376 CAS.
- M. A. Spackman and J. J. McKinnon, CrystEngComm, 2002, 46, 378–392 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Raman spectra on compression at 0.1 GPa and decompression at 0.1 GPa, ranging from 50–3500 cm−1, respectively. See DOI: 10.1039/c7ra02205k |
| This journal is © The Royal Society of Chemistry 2017 |