
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hybrid flavan–flavanones from Friesodielsia desmoides and their inhibitory activities against nitric oxide production†
Pornphimol Meesakula,
Khanitha Pudhomb,
Stephen G. Pynec and
Surat Laphookhieo *a
*a
aNatural Products Research Laboratory, School of Science, Mae Fah Luang University, Chiang Rai, 57100, Thailand. E-mail: surat.lap@mfu.ac.th
bDepartment of Chemistry, Faculty of Science, Chulalongkorn University, Bangkok, 10330 Thailand
cSchool of Chemistry, University of Wollongong, Wollongong, New South Wales, 2522 Australia
First published on 20th March 2017
Abstract
The first phytochemical investigation of Friesodielsia desmoides leaves and twigs led to the isolation and identification of three new hybrid flavan–flavanones, friesodielsones A–C (1–3), together with 18 known compounds (4–21). The structures of compounds 1–3 were elucidated through intensive analysis of spectroscopic data and their absolute configurations at C-2 and C-4 were determined by a combination of 1H NMR and CD spectroscopy. The configuration at C-2′′ is tentatively assigned as 2′′S based on biosynthesis considerations. Compounds 2 and 15 significantly inhibited NO production with IC50 values of 10.21 ± 0.074 and 7.56 ± 0.087 μM, respectively, whereas compounds 11 (IC50 = 28.14 ± 0.024 μM) and 12 (IC50 = 37.21 ± 0.017 μM) were moderate inhibitors.
Introduction
Friesodielsia is a small genus belonging to the Annonaceae family which is distributed in Africa and Asia. Five species, including F. desmoides, F. fornicata, F. discolor, F. filipes, and F. kingii, are found in Thailand.1 Many types of secondary metabolites are produced from this genus including flavonoids,1,2 chalcones,1,3 alkaloids,3 benzyl benzoate derivatives2 and sesquiterpenes.2 Some of these compounds show interesting biological properties, such as antiplasmodial1,3 and cytotoxicity activities.1,3 F. desmoides (Craib) Steenis (Fig. 1) is a small tree or shrub that is grown as an ornamental plant in Thailand. This plant has two synonymous names, Goniothalamus desmoides Craib and Oxymitra desmoides. To the best of our knowledge, this is the first report of phytochemical investigations of this plant. Three new hybrid flavan–flavanones (1–3) along with 18 known compounds (4–21) (Fig. 2) were isolated and identified from F. desmoides leaves and twigs that were collected from Mae Fah Luang University Health Park, Chiang Rai Province, Thailand. Most of the isolated compounds were evaluated for their NO inhibitory activities. | ||
| Fig. 1 Friesodielsia desmoides (these photos were taken by Surat Laphookhieo). | ||
 | ||
| Fig. 2 Compounds isolated from F. desmoides leaves and twigs. | ||
Results and discussion
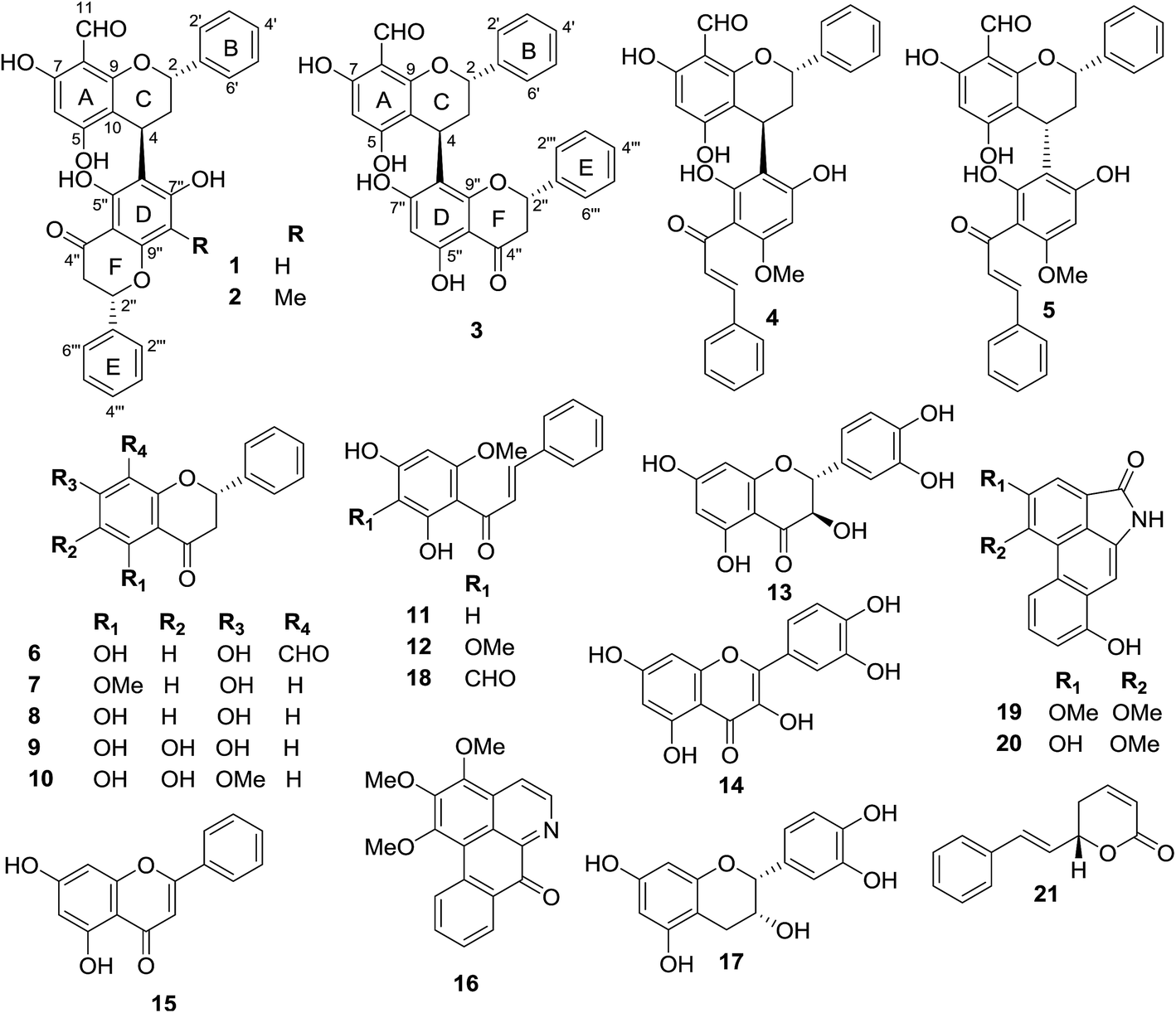
The crude extracts of F. desmoides leaves and twigs were separated by column chromatography using various stationary phases to yield three new hybrid flavan–flavanones, friesodielsones A–C (1–3), along with 18 known compounds (4–21). The known compounds were identified as desmosflavan A (4),4 desmosflavan B (5),4 (2S)-8-formyl-5,7-dihydroxyflavanone (6),1 alpinetin (7),5 pinocembrin (8),5 5,6,7-trihydroxy-flavanone (9),6 5,6-dihydroxy-7-methoxy-flavanone (10),7 cardamonin (11),5 2′,4′-dihydroxy-3′,6′-dimethoxychalcone (12),8 trans-dihydroquercetin (13),9 quercetin (14),10 chrysin (15),11 O-methylmoschatoline (16),12 (−)-epicatechin (17),13 3′-formyl-2′,4′-dihydroxy-6′-methoxychalcone (18),1 O-aristololactam BII (19),14 aristololactam AIa (20)14 and goniothalamin (21).15Friesodielsone A (1) was obtained as a yellow solid. It showed a pseudomolecular ion peak at m/z [M − H]− 523.1393 (calcd 523.1393) in the HRESIMS corresponding to a molecular formula of C31H24O8. The 1H, 13C, DEPT and 2D NMR spectroscopic data of 1 (Table 1) suggested this compound contained two moieties, a flavan unit and a flavanone unit.16 The flavan unit displayed 1H and 13C NMR signals for a hydrogen-bonded hydroxyl proton [δH 12.35 (1H, s, 7-OH)], a formyl group [δH 10.10 (1H, s, 8-CHO)/δC 192.1], a monosubstituted aromatic ring [δH 7.44–7.47 (2H, m, H-2′, H-6′)/δC 126.8, 7.40–7.42 (2H, m, H-3′, H-5′)/δC 129.4, and 7.30–7.34 (1H, m, H-4′)/δC 128.6], an isolated aromatic proton [δH 5.90 (1H, s, H-6)/δC 94.9], and an AB2C proton spin system (deduced from COSY spectrum) [δH 5.53 (1H, dd, J = 3.5, 10.0, H-2)/δC 76.7, 2.24–2.33 (2H, m, H-3)/δC 37.9, and 4.67 (1H, dd, J = 2.6, 5.4 Hz, H-4)/δC 26.1]. The formyl group was located at C-8 (δC 105.9) from the HMBC correlations (Fig. 3) of C-8 (δC 105.9) to the hydrogen-bonded hydroxyl proton (δH 12.35), the formyl proton (δH 10.10) and the isolated aromatic proton H-6 (δH 5.90).
| Position | Friesodielsone A (1) | Friesodielsone B (2) | Friesodielsone C (3) | ||||
|---|---|---|---|---|---|---|---|
| δCa | δHa (J in Hz) | δHb (J in Hz) | δCa | δHa (J in Hz) | δCa | δHa (J in Hz) | |
| a Spectrum measured in acetone-d6.b Spectrum measured in methanol-d4. | |||||||
| 2 | 76.7 | 5.53 (dd, 3.5, 10.0) | 5.40 (dd, 2.6, 11.0) | 76.7 | 5.52 (dd, 3.0, 10.8) | 76.7 | 5.53 (dd, 3.2, 10.7) |
| 3 | 37.9 | 2.24–2.33 (m) | 2.19 (ddd, 5.4, 11.0, 14.3) | 37.9 | 2.25–2.30 (m) | 37.9 | 2.25–2.32 (m) |
| 2.13 (dt, 2.6, 14.3) | |||||||
| 4 | 26.1 | 4.67 (dd, 2.6, 5.4) | 4.55 (dd, 2.6, 5.4) | 26.5 | 4.70 (dd, 2.4, 5.6) | 26.1 | 4.67 (dd, 2.6, 5.5) |
| 5 | 162.1 | — | — | 159.1 | — | 162.8 | — |
| 6 | 94.9 | 5.90 (s) | 5.80 (s) | 94.9 | 5.91 (s) | 94.8 | 5.90 (s) |
| 7 | 164.8 | — | — | 164.9 | — | 165.2 | — |
| 8 | 105.9 | — | — | 106.0 | — | 105.8 | — |
| 9 | 160.6 | — | — | 160.8 | — | 164.7 | — |
| 10 | 104.7 | — | — | 105.9 | — | 104.7 | — |
| 11 | 192.1 | 10.10 (s) | 10.05 (s) | 192.1 | 10.16 (s) | 192.1 | 10.15 (s) |
| 1′ | 142.5 | — | — | 142.3 | — | 142.4 | — |
| 2′,6′ | 126.8 | 7.44–7.47 (m) | 7.34–7.37 (m) | 126.8 | 7.45–7.47 (m) | 126.8 | 7.44–7.48 (m) |
| 3′,5′ | 129.4 | 7.40–7.42 (m) | 7.37–7.41 (m) | 129.4 | 7.38–7.42 (m) | 129.4 | 7.39–7.42 (m) |
| 4′ | 128.6 | 7.30–7.34 (m) | 7.26–7.30 (m) | 128.6 | 7.32–7.34 (m) | 128.6 | 7.33–7.35 (m) |
| 2′′ | 79.9 | 5.57 (dd, 3.0, 13.5) | 5.49 (dd, 3.0, 13.5) | 79.6 | 5.61 (dd, 3.0, 13.0) | 79.9 | 5.59 (dd, 3.0, 13.0) |
| 3′′ | 43.7 | 2.80 (dd, 3.0, 17.1) | 2.73 (dd, 3.0, 17.1) | 43.6 | 2.82 (dd, 3.0, 17.0) | 43.7 | 2.81 (dd, 3.0, 17.0) |
| 3.16 (dd, 13.5, 17.1) | 3.06 (dd, 13.5, 17.1) | 3.16 (dd, 13.0, 17.0) | 3.22 (dd, 13.0, 17.0) | ||||
| 4′′ | 197.0 | — | — | 197.7 | — | 197.1 | — |
| 5′′ | 163.1 | — | — | 160.6 | — | 162.0 | — |
| 6′′ | 111.9 | — | — | 111.8 | — | 95.8 | 6.08 (s) |
| 7′′ | 165.2 | — | — | 160.6 | — | 162.8 | — |
| 8′′ | 95.9 | 6.08 (s) | 5.98 (s) | 159.1 | — | 111.8 | — |
| 9′′ | 162.1 | — | — | 164.5 | — | 160.6 | — |
| 10′′ | 103.1 | — | — | 103.8 | — | 103.5 | — |
| 11′′ | 8.32 | 2.09 (s) | |||||
| 1′′′ | 140.2 | — | — | 140.4 | — | 140.1 | — |
| 2′′′,6′′′ | 127.8 | 7.56–7.58 (m) | 7.46–7.48 (m) | 127.2 | 7.60–7.61 (m) | 129.4 | 7.44–7.46 (m) |
| 3′′′,5′′′ | 129.5 | 7.43–7.47 (m) | 7.34–7.37 (m) | 129.5 | 7.45–7.47 (m) | 129.4 | 7.57–7.58 (m) |
| 4′′′ | 129.4 | 7.40–7.43 (m) | 7.37–7.41 (m) | 129.3 | 7.38–7.42 (m) | 128.6 | 7.39–7.42 (m) |
| 7-OH | — | 12.35 (s) | — | — | 12.35 (s) | — | 12.35 (s) |
| 5′′-OH | — | 12.73 (s) | — | — | 12.72 (s) | ||
 | ||
| Fig. 3 Selected HMBC correlations of compounds 1–3. | ||
The second fragment, a flavanone unit, showed 1H and 13C NMR resonances for a hydrogen-bonded hydroxyl [δH 12.73 (1H, s, 5′′-OH)], a monosubstituted aromatic ring [δH 7.56–7.58 (2H, m, H-2′′′, H-6′′′)/δC 127.8, 7.43–7.47 (2H, m, H-3′′′, H-5′′′)/δC 129.5, and 7.40–7.43 (1H, m, H-4′′′)/δC 129.4], an isolated aromatic proton [δH 6.08 (1H, s, H-8′′)/δC 95.9] and an AB2 proton spin system (deduced from COSY spectrum) [δH 5.57 (1H, dd, J = 3.0, 13.5 Hz, H-2′′)/δC 79.9, 2.80 (1H, dd, J = 3.0, 17.1 Hz, H-3′′)/δC 43.7 and 3.16 (1H, dd, J = 13.5, 17.1 Hz, H-3′′)/δC 43.7].
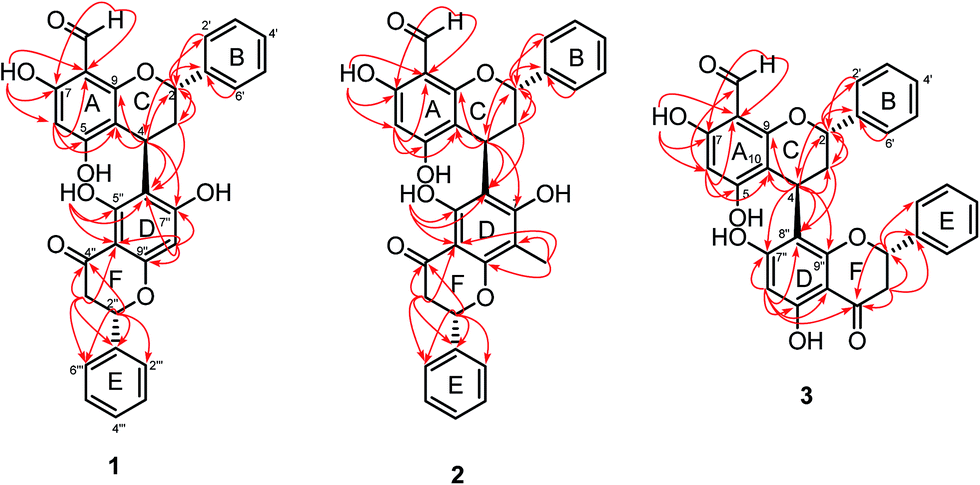
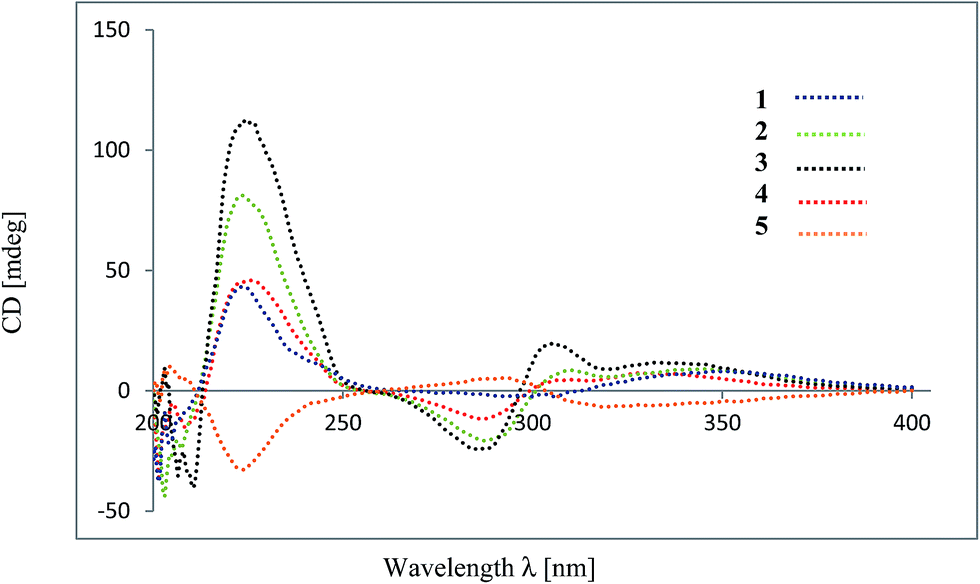
The flavan and flavanone units of 1 had a C–C bond linkage between C-4 of ring C and C-6′′ of ring D which was deduced from the following HMBC correlations: δH 4.67 (H-4) with C-5′′ (δC 163.1), C-6′′ (δC 111.9) and C-7′′ (δC 165.2); and δH 2.24–2.33 (H-3) with C-6′′ (δC 111.9). The assignments of the NMR spectroscopic data of 1 are summarized in Table 1. Therefore, friesodielsone A was identified as structure 1. The relative configuration of the C-ring of 1 was deduced to be the same as that of desmosflavan A (4),4 from the similarity of their 1H NMR coupling constants for the protons H-2, H-3 and H-4. The absolute 4S configuration of 1 was evident from the positive Cotton effect (Fig. 4) at λmax (Δε) 225.5 (2.92 × 104) nm.17–19 This allowed the assignment of the 2S configuration of 1 based on the aforementioned 1H NMR comparisons. The configuration at C-2′′ could not be unequivocally determined but is based on the biosynthetic consideration outline in Scheme 1.
 | ||
| Fig. 4 CD spectra (MeOH) of compounds 1–5. | ||
 | ||
| Scheme 1 Plausible biosynthetic pathway of compounds 1–5. | ||
Friesodielsone B (2) was obtained as a yellow solid. Its molecular formula, C32H26O8, was obtained from HRESIMS analysis which showed a [M − H]− at m/z 537.1549 (calcd for C32H25O8, 537.1549). The 1H and 13C NMR spectroscopic data of 2 (Table 1) were similar to those of 1. The main differences were the ring D resonances of the flavanone unit. Compound 2 displayed an additional resonance for methyl protons at δH 2.09 (3H, s) and the absence of the C-8′′ aromatic proton resonance at δH 6.08 (1H, s, H-8′′) as was observed in 1. The HMBC correlation (Fig. 3) between these methyl protons (δH 2.09) and C-8′′ (δC 159.1) further supported the position of this methyl group at C-8′′. The assignments of the 1H and 13C spectroscopic data of 2 are summarized in Table 1. The CD spectrum of 2 (Fig. 4) and 1H NMR coupling constants of H-2, H-3 and H-4 were similar to that of 1 indicating that the absolute configuration at C-2 and C-4 of 2 and 1 were the same. This was further supported by their similar and negative specific optical rotations, [α]25D −57.6 (c 0.03, MeOH) for 1 and [α]26D −45.1 (c 0.006, MeOH) for 2. Thus, friesodielsone B was assigned the structure 2.
Friesodielsone C (3) was obtained as a yellow solid. Its HRESIMS spectrum showed a [M − H]− at m/z 523.1393 (calcd 523.1393) corresponding to the molecular formula of C31H24O8. The 1H and 13C NMR spectroscopic data of 3 (Table 1) were similar to those of 1 and 2. The significant difference in the structure of 3 was the position of flavan–flavanone linkage. Compounds 1 and 2 were linked at C-4/C-6′′ whereas compound 3 was linked at C-4/C-8′′. The HMBC correlations of δH 4.67 (H-4) to C-7′′ (δC 162.0), C-8′′ (δC 111.8) and C-9′′ (δC 162.8) supported this C-4/C-8′′ linkage (Fig. 3). The absolute configuration at C-4 of 3 was determined by comparison of its CD spectrum with that of compounds 1 and 2. The absolute configuration at C-4 of 3 was the same as that of 1 and 2 from the positive Cotton effect seen in its CD spectrum (Fig. 4) at λ 224 nm, which was similar to those of compounds 1 and 2. Therefore, friesodielsone C was assigned the structure 3.
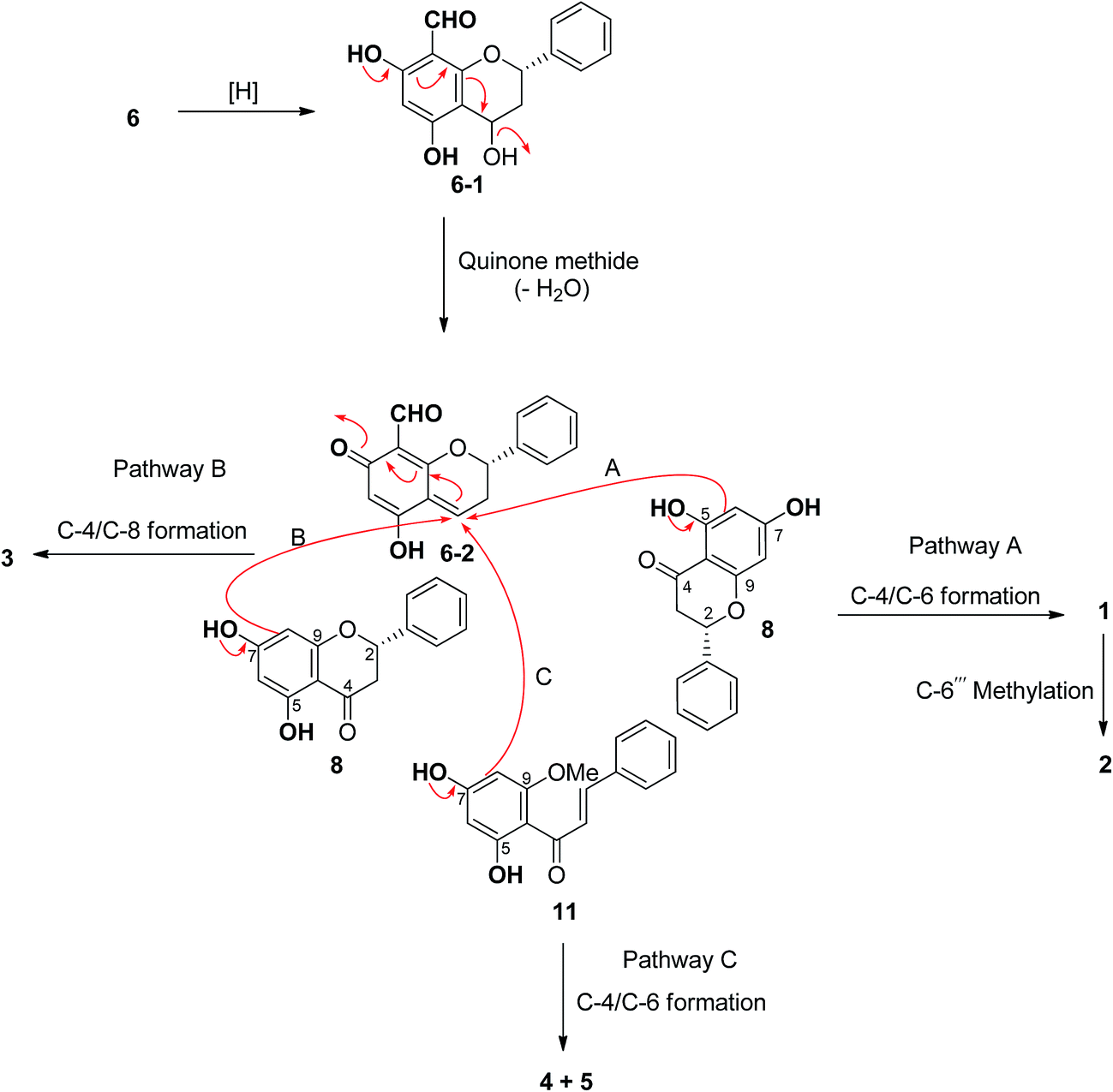
Hybrid biflavonoids comprising a linked flavan and flavanone units are rarely found as natural products.16,20–23 Biosynthetically, compounds 1–3 could be derived from compound 6 (Scheme 1) via reduction of the C-4 carbonyl group and then formation of the para-quinone methide 6–2 by dehydration.24 Intermediate 6–2 could couple with compound 8 at C-4/C-6 to provide compound 3 (via pathway B) and at C-4/C-6 to produce compounds 1 and 2 (via pathway A). The latter compound obtained from 1 via methylation at C-6′′′. Similarly, the biosynthetic pathway to compounds 4 and 5 could be derived from the coupling of compounds 6–2 and 11 (Scheme 1). The isolation of compounds 6, 8 and 11 in this study supports this biosynthetic hypothesis. The absolute configuration tentatively assigned as 2′′S at C-2′′ in compounds 1–3 is based on this biosynthetic hypothesis.
Most of the isolated compounds were evaluated for their NO inhibitory activities in J774.A1 macrophage cells. Compounds 2 and 15 significantly exhibited NO production with IC50 values of 10.21 ± 0.074 and 7.56 ± 0.087 μM, respectively, whereas compounds 11 (IC50 = 28.14 ± 0.024 μM) and 12 (IC50 = 37.21 ± 0.017 μM) were more moderate inhibitors (Table 2). Significantly, the active compounds, 2, 11, 12 and 15, did not show cytotoxicity at 10 μM against J774.A1 cells (Table 2).
| Compounds | NO production IC50, μM | MTT% cell viability at 10 μM |
|---|---|---|
| 1 | Inactive | 116.2 |
| 2 | 10.21 ± 0.074 | 126.6 |
| 5 | Inactive | 102.5 |
| 6 | Inactive | 72.80 |
| 7 | Inactive | 73.00 |
| 8 | Inactive | 90.90 |
| 9 | Inactive | 97.40 |
| 10 | Inactive | 78.10 |
| 11 | 28.14 ± 0.024 | 98.30 |
| 12 | 37.21 ± 0.017 | 97.80 |
| 13 | Inactive | 82.20 |
| 15 | 7.56 ± 0.087 | 84.30 |
| 16 | Inactive | 76.60 |
| 17 | Inactive | 82.90 |
| 21 | Inactive | 64.40 |
| Indomethacin | 28.4 ± 3.5 | — |
Conclusion
Friesodielsia is a small genus in the Annonaceae family. To the best of our knowledge, only three species have been investigated phytochemically and the major compounds are flavonoids.1–3 However, a few chalcones,1,3 alkaloids,3 benzyl benzoate derivatives2 and sesquiterpenes2 were also been found. In this study, 21 compounds were isolated and identified including three new unique hybrid flavan–flavones (1–3), two hybrid flavan–chalcones (4 and 5), nine flavonoids (6–10, 13–15, 17), three chalcones (11–12, 18), three alkaloids and one styryl lactone (21). A hypothesis for the biosynthesis of compounds 1–5 from a para-quinone methide intermediate derived from compound 6 is proposed. This is the first reported isolation of compounds 4–10, 13–17, and 19–21 from this genus. Compounds 2, 11, 12 and 15 inhibited NO production indicating that they might be potential lead compounds for further study and development as anti-inflammatory agents.Experimental
General experimental procedures
Melting points were determined on a Stuart SMP3 Melting Point Apparatus. The [α]D values were measured with a Bellingham and Stanley ADP400 polarimeter. UV-vis spectra were recorded with a BMG LABTECH/SPECTROstar Nano spectrometer. The circular dichroism (CD) spectra were measured on a JASCO J-815 apparatus. The IR spectra were recorded using a PerkinElmer FTS FT-IR spectrometer. The NMR spectra were recorded using a 400 MHz Bruker AM 400 spectrometer in acetone-d6 with TMS as an internal standard. The HRESIMS were obtained on a Bruker microTOF mass spectrometer. Silica gel C60 (0–20 μm, SiliCycle® Inc.) and silica gel G60 (60–200 μm, SiliCycle® Inc.) were used to perform quick column chromatography (QCC) and column chromatography (CC), respectively. Analytical thin-layer chromatography (TLC) was performed with the precoated plates of silica gel 60 F254. The macrophage J774.A1 cells were purchased from CLS (Cell Line Service, Germany).Plant material
The twigs and leaves of F. desmoides were collected in August 2015 from an authentically identified plant growing at Mae Fah Luang University Health Park, Chiang Rai Province, Thailand. The plant specimen (no. MFU-NPR0102) was deposited at the Natural Products Research Laboratory, School of Science, Mae Fah Luang University.Extraction and isolation
Air-dried leaves of F. desmoides (564.3 g) were extracted with EtOAc (5 L) over a period of 3 days at room temperature. Removal of the solvent provided the crude EtOAc extract (49.42 g), which was subjected to QCC over silica gel, eluting with a gradient of hexanes–EtOAc (100% hexanes to 100% EtOAc) to give compound 6 (5.2 mg), 14 (6.5 mg) and nine fractions (A–I). Fractions C (1.59 g) was further separated by CC (100% DCM) to give compounds 9 (17.1 mg) and 10 (6.3 mg). Fraction D (2.38 g) was subjected to CC using reverse phase silica gel (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 MeOH/H2O) to afford four subfractions (DA–DD). Compound 7 (14.7 mg) was obtained from subfraction DA (31.4 mg) by Sephadex LH-20 (100% MeOH). Subfraction DC (173.4 mg) was further separated by CC (100% DCM) to yield compounds 11 (13.6 mg) and 12 (14.2 mg). Fraction F (1.30 g) was subjected to CC using reverse phase silica gel (4:1 MeOH/H2O) to afford three subfractions (FA–FC). Purification of subfraction FC (526.9 mg) by CC (1:4 acetone/hexanes) gave compounds 1 (16.8 mg) and 3 (5.3 mg). Fraction G (1.09 g) was further separated by CC using reverse phase silica gel (4:1 MeOH/H2O) to obtained seven subfractions (GA–GG). Subfractions GC (376.5 mg) was further purified by CC (100% DCM), yielding compounds 2 (7.0 mg), 4 (3.1 mg) and 5 (4.5 mg). Compounds 6 (7.4 mg) and 14 (5.5 mg) were obtained from fraction H (1.21 g) by repeated CC (3:7 acetone/hexanes). Fraction I (1.84 g) was further separated by CC using reverse phase silica gel (4:1 MeOH/H2O) to obtained four subfractions (IA–ID). Subfraction IC (451.8 mg) was further purified by CC (2:3 acetone/hexanes) to afford compounds 13 (12.4 mg) and 16 (4.3 mg).
:1 MeOH/H2O) to afford four subfractions (DA–DD). Compound 7 (14.7 mg) was obtained from subfraction DA (31.4 mg) by Sephadex LH-20 (100% MeOH). Subfraction DC (173.4 mg) was further separated by CC (100% DCM) to yield compounds 11 (13.6 mg) and 12 (14.2 mg). Fraction F (1.30 g) was subjected to CC using reverse phase silica gel (4:1 MeOH/H2O) to afford three subfractions (FA–FC). Purification of subfraction FC (526.9 mg) by CC (1:4 acetone/hexanes) gave compounds 1 (16.8 mg) and 3 (5.3 mg). Fraction G (1.09 g) was further separated by CC using reverse phase silica gel (4:1 MeOH/H2O) to obtained seven subfractions (GA–GG). Subfractions GC (376.5 mg) was further purified by CC (100% DCM), yielding compounds 2 (7.0 mg), 4 (3.1 mg) and 5 (4.5 mg). Compounds 6 (7.4 mg) and 14 (5.5 mg) were obtained from fraction H (1.21 g) by repeated CC (3:7 acetone/hexanes). Fraction I (1.84 g) was further separated by CC using reverse phase silica gel (4:1 MeOH/H2O) to obtained four subfractions (IA–ID). Subfraction IC (451.8 mg) was further purified by CC (2:3 acetone/hexanes) to afford compounds 13 (12.4 mg) and 16 (4.3 mg).
Air-dried twigs of F. desmoides (1.26 kg) were extracted with EtOAc (5 L) over a period of 3 days at room temperature. Removal of the solvent provided the crude EtOAc extract (52.02 g), which was subjected to QCC over silica gel, eluting with a gradient of hexanes–EtOAc (100% hexanes to 100% EtOAc) to give five fractions (A–E). Fraction B (396.1 mg) was separated on Sephadex LH-20 (100% MeOH) to obtained four subfractions (BA–BD). Subfractions BC (165.9 mg) was further purified by CC (100% DCM), yielding compound 21 (2.8 mg). Compounds 11 (10.1 mg) and 15 (1.4 mg) were obtained from subfraction BD (93.7 mg) by CC (100% DCM). Purification of fraction C (745.9 mg) by CC (1:4 acetone/hexanes) yielded compounds 17 (6.8 mg) and 18 (6.0 mg). Fractions D (325.0 mg) was further purified by CC (1:9 EtOAc/DCM) to give compound 16 (5.3 mg). Compounds 19 (1.3 mg) and 20 (1.4 mg) were obtained from fraction E (212.5 mg) by CC (0.5:9.5 MeOH/DCM).
ε) 245 (3.51), 313 (3.40) nm; CD (MeOH) λmax (Δε) 328 (4.72 × 103), 311 (2.99 × 103), 286 (−7.41 × 103) and 225.5 (2.92 × 104) nm; IR (neat) νmax 3087, 2924, 2851, 1652, 1614, 1580, 1501, 1449, 1167, 766 cm−1; see Table 1 for 1H NMR (acetone-d6, 400 MHz) and 13C NMR (acetone-d6, 100 MHz); HRESIMS m/z 523.1393 [M − H]− (calcd for C31H23O8, 523.1393).ε) 235 (3.61), 298 (3.64), 340 (3.33) nm; CD (MeOH) λmax (Δε) 322 (3.52 × 104), 311 (4.92 × 104), 289 (−1.19 × 105), and 225 (4.70 × 105) nm; IR (neat) νmax 3434, 2920, 2851, 1634, 1441, 1373, 1275, 1111, 618 cm−1; see Table 1 for 1H NMR (acetone-d6, 400 MHz) and 13C NMR (acetone-d6, 100 MHz); HRESIMS m/z 537.1549 [M − H]− (calcd for C32H25O8, 537.1549).ε) 240 (4.27), 298 (4.33), 340 (3.75) nm; CD (MeOH) λmax (Δε) 344 (7.00 × 103), 305 (1.24 × 104), 286 (−1.56 × 104), and 224 (7.15 × 104); IR (neat) νmax 3200, 2928, 2854, 1634, 1443, 1371, 1275, 1161, 1167, 766 cm−1; see Table 1 for 1H NMR (acetone-d6, 400 MHz) and 13C NMR (acetone-d6, 100 MHz); HRESIMS m/z 523.1393 [M − H]− (calcd for C31H23O8, 523.1393).Nitric oxide production inhibitory assay
The effects of the isolated compounds on nitric oxide production in murine macrophage J774.A1 cells (Cell Line Service, Germany) in supernatant were determined using the previously reported method.22 In brief, J774.A1 cells were added in 96-well plate with 5 × 105 cells per well and incubated for 1 h at 37 °C and 5% CO2. After that, cells were treated with various concentrations of sample or vehicle (DMSO) for 2 h, followed by LPS 10 μg mL−1. After 18 h incubation, NO production in the culture medium was determined using the Griess reagent (1% sulfanilamide in 5% phosphoric acid and 0.1% naphthylethylenediamine dihydrochloride in distilled water) for 10 min and the absorbance was measured at 540 nm. Indomethacin was used as a positive control.Cell viability assay
The measurement of cell viability of the tested compounds was performed using the 3-[4,5-dimethylthiazol-2-yl-2,5-diphenyltetrazolium] bromide (MTT) assay against unstimulated J774.A1 cells.25 Briefly, 10 μL of fresh MTT solution (5 mg mL−1 in saline) was added to each well, incubated at 37 °C in CO2 for 3 h. The media was discarded and DMSO was added to dissolve the formazan crystals and the absorbance value at 540 nm was measured. The percentages of cell survival were calculated from the absorbance value of the tested compounds and control (LPS) using the equation below.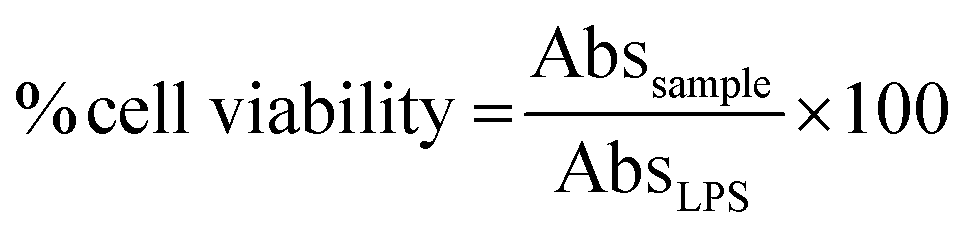
Acknowledgements
This research was financially supported by the Thailand Research Fund and Mae Fah Luang University through the Basic Research Grant (BRG5980012), the Thailand Research Fund through the Royal Golden Jubilee PhD Program (PHD/0010/2558) and Direct Basic Research Grant (DBG5980001) and the Mae Fah Luang University Graduate Student Research Grant. S. L. thanks the Australian Government via the Endeavour Award 2016 for a research fellowship. Mae Fah Luang University and the University of Wollongong are also acknowledged for laboratory facilities.Notes and references
- U. Prawat, D. Phupornprasert, A. Butsuri, A.-W. Salae, S. Boonsri and P. Tuntiwachwuttikul, Phytochem. Lett., 2012, 5, 809–813 CrossRef CAS.
- T. C. Fleischer, R. D. Waigh and P. G. Waterman, Phytochemistry, 1997, 44, 315–318 CrossRef CAS.
- C. C. Joseph, J. J. Magadula and M. H. H. Nkunya, Nat. Prod. Res., 2007, 21, 1009–1015 CrossRef CAS PubMed.
- S. P. Bajgai, V. Prachyawarakorn, C. Mahidol, S. Ruchirawat and P. Kittakoop, Phytochemistry, 2011, 72, 2062–2067 CrossRef CAS PubMed.
- M. Kuroyanagi, T. Noro, S. Fukushima, R. Aiyama, A. Ikuta, H. Itokawa and M. Morita, Chem. Pharm. Bull., 1983, 31, 1544–1550 CrossRef CAS.
- N. Narasimhachari, V. D. N. Sastri and T. R. Seshadri, Proc. Natl. Acad. Sci., India, Sect. A, 1949, 29, 404–412 Search PubMed.
- P. V. Kiem, C. V. Minh, H. T. Huong, J. J. Lee, I. S. Lee and Y. H. Kim, Arch. Pharmacal Res., 2005, 28, 1345–1349 CrossRef CAS.
- K. Ichino, H. Tanaka, K. Ito, T. Tanaka and M. Mizuno, J. Nat. Prod., 1988, 51, 906–914 CrossRef CAS PubMed.
- E. Kiehlmann and E. P. M. Li, J. Nat. Prod., 1995, 58, 450–455 CrossRef CAS.
- M. A. Aderogba, A. R. Ndhlala, K. R. Rengasamy and J. Van Staden, Molecules, 2013, 18, 12633–12644 CrossRef CAS PubMed.
- H. Shimura, M. Matsuura, N. Takada and Y. Koda, Phytochemistry, 2007, 68, 1442–1447 CrossRef CAS PubMed.
- K. Mahmood, K. C. Chan, M. H. Park, Y. N. Han and B. H. Han, Phytochemistry, 1986, 25, 1509–1510 CrossRef CAS.
- G. Wang, X. Huang, D. Pei, W. Duan, K. Quan, X. Li and D. Di, New J. Chem., 2016, 40, 3885–3891 RSC.
- H. A. Priestap, Phytochemistry, 1985, 24, 849–852 CrossRef CAS.
- K. Jewers, J. B. Davis, J. Dougan, A. H. Manchanda, G. Blunden, A. Kyi and S. Wetchapinan, Phytochemistry, 1972, 11, 2025–2030 CrossRef CAS.
- C.-M. Cao, L.-J. Xu, Y. Peng, Q.-W. Shi and P.-G. Xiao, Chem. Pharm. Bull., 2010, 58, 1395–1398 CrossRef CAS PubMed.
- Y. Ding, X.-C. Li and D. Ferreira, J. Nat. Prod., 2010, 73, 435–440 CrossRef CAS PubMed.
- M. S. Pesca, F. D. Piaz, R. Sanogo, A. Vassallo, M. B. de Abreu, A. Rapisarda, M. P. Germanó, G. Certo, S. D. Falco, N. De Tommasi and A. Braca, J. Nat. Prod., 2013, 76, 29–35 CrossRef CAS PubMed.
- J. Coetzee, J. P. Steynberg, P. J. Steynberg, E. V. Brandt and D. Ferreira, Tetrahedron, 1995, 51, 2339–2352 CrossRef CAS.
- A. J. Birch and C. J. Dahl, Aust. J. Chem., 1974, 27, 331–344 CrossRef CAS.
- A. J. Birch, C. J. Dahl and A. Pelter, Tetrahedron Lett., 1967, 481–487 CrossRef CAS.
- J. C. S. Malan, D. A. Young, J. A. Steenkamp and D. Ferreira, J. Chem. Soc., Perkin Trans. 1, 1988, 2567–2572 RSC.
- J. Coetzee, J. P. Steynberg, J. P. Steynberg, V. E. Brandt and D. Ferreira, Tetrahedron, 1995, 51, 2339–2352 CrossRef CAS.
- M. R. Attwood, B. R. Brown, S. G. Lisseter, C. L. Torrero and P. M. Weaver, J. Chem. Soc., Chem. Commun., 1984, 177–179 RSC.
- K. Pudhom and T. Teerawatananond, J. Nat. Prod., 2014, 77, 1962–1966 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra02528a |
| This journal is © The Royal Society of Chemistry 2017 |