
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A facile synthetic route to novel thermotropic liquid crystalline polymers and characterization of their mesophases†
Kyoung Hwan Ohab,
Hoyun Kima and
Yongsok Seo *a
*a
aRIAM, School of Materials Science and Engineering, College of Engineering, Seoul National University, Kwanakro-1, Kwanakgu, Seoul, 151-744, Republic of Korea. E-mail: ysseo@snu.ac.kr
bResearch & Development Division, Materials Development Center, 150, Hyundaiyeonguso-ro, Namyang-eup, Hwaseong-si, Gyeonggi-do 18280, Korea
First published on 7th June 2017
Abstract
A novel method for the synthesis of a new class of thermotropic liquid crystalline polymers has been developed: it proceeds via a polyaddition reaction involving diepoxy-containing mesogene groups and a monoamine (aniline). The reaction between the epoxy ring and the primary amine produces an intermediate containing a secondary amine which undergoes further reaction with an epoxy ring of another molecule in the melt to generate a thermotropic liquid crystalline polymer. This polymerization process is one step. The two new thermotropic liquid crystalline polymers prepared with this method have high molecular weights. Both polymers were found to exhibit nematic mesophase characteristics. The prepared polymers exhibit high thermal stabilities (degradation temperatures above 350 °C) but low melting temperatures (lower than 250 °C). When examined with polarized optical microscopy in the melt state, the nematic phases of the polymers produce characteristic textures. The liquid crystalline structures of the two polymers were confirmed by performing 2D X-ray diffraction measurements. Scanning electron microscopy images of drawn polymer fibers reveal a strong orientation on the surface along the flow direction and many microfibrillar structures extruding out of the fractured surfaces normal to the direction of flow. These results demonstrate the potential for the facile mass production of novel thermotropic liquid crystalline polymers for high performance composites or fibers of high mechanical strength.
Introduction
Thermotropic liquid crystalline polymers (TLCPs) are high performance engineering materials because of their outstanding physical properties, particularly their high stiffness, strength, excellent dimensional and thermal stability, high chemical resistance, and remarkable barrier properties.1–5 These superior properties arise from the unique structures of their mesogenic units which induce liquid crystalline phase ordering in the melt. Compared to lyotropic liquid crystalline polymers that form liquid crystalline structures in solution, thermotropic liquid crystalline polymers form liquid crystalline structures upon heating, which means they can be utilized in the fabrication of general thermoplastics with three-dimensional structures by using conventional processing equipment. However, the inclusion of a rigid mesogenic unit in the main chains of TLCPs provides high thermal stability, but also gives rise to poor melt processibility because of their high melting temperatures. The melting points of some TLCPs even exceed their decomposition temperatures, which can complicate their processing and their use in special structure applications. The high processing temperatures of such TLCPs makes it difficult to compound them with other thermoplastic polymers to prepare so-called in situ composites.6–13 In situ composite preparation requires thermoplastic matrices that are capable of withstanding the high transition temperatures of TLCPs so that a TLCP phase with fibril shapes can be produced that reinforces the thermoplastic matrix.14,15 TLCPs also have low molar masses in general because they are mostly synthesized via condensation polymerizations.Herein we report a facile method for generating a new kind of thermotropic liquid crystalline polymers with a polyaddition process: the reaction between a diepoxy compound and a primary monoamine (aniline) links two epoxy groups to form linear chains (Scheme 1). Diepoxy units have mainly been used to prepare liquid crystalline polymer networks (LCPNs) rather than TLCPs.16 Our approach is notable for enabling the preparation of high molar mass TLCPs with low processing temperatures because it does not produce any byproducts that must be removed. By varying the structure of the diepoxy compound, we can control the processing temperature of the resulting TLCP. To the best of our knowledge, this is the first report of the synthesis of TLCPs via a polyaddition process, which enables the production of the high molar mass TLCPs with low processing temperatures that are required for compounding with other thermoplastics.6–13
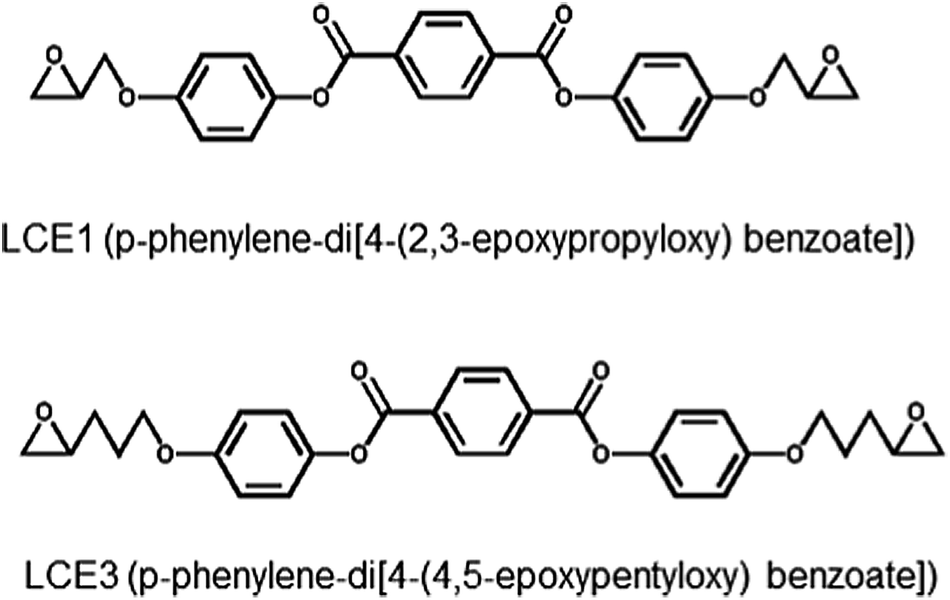 | ||
| Scheme 1 Chemical structures of LCE1 and LCE3. | ||
Experimental
The two diepoxides, LCE1 (p-phenylene-di-4-(2,3-epoxypropyloxy)benzoate) and LCE3 (p-phenylene-di-4-(4,5-epoxypentyloxy)benzoate) (Scheme 1) were synthesized. A mixture of ethyl-4-hydroxybenzoate, 3-bromopropene, K2CO3, and acetone was refluxed for 24 h. After filtering off the solid and performing solvent evaporation, diethylether and water were added. The diethylether layer was separated and washed three times with a 10% NaOH solution. After evaporating the diethylether, the residue was boiled in an ethanol/water (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) solution containing KOH until the solution became clear. 2 N HCl solution was added to lower the pH to 2. After filtration, the solids were washed with excess water and recrystallized in ethanol to yield (4-(2-propyloxy)benzoic acid) as a white powder. A mixture of 4-(2-propyloxy)benzoic acid and SOCl2 was refluxed for 2 h. After evaporating excess SOCl2, pyridine and hydroquinone were added. The mixture was reacted in an acidified aqueous solution, then filtered and washed with a 5% Na2CO3 aqueous solution and water. After drying, the solid product (LCE1) was recrystallized in ethyl acetate. LCE1 was purified with column chromatography by using CH2Cl2 as the eluent. After evaporating the solvent, the LCE1 product was recrystallized in ethyl acetate with a yield of 55%. LCE3 was synthesized by using 5-bromopentene instead of 3-bromopropene. The yield of LCE3 was 69%. More details of the synthetic procedure is described in the ESI.†
:2) solution containing KOH until the solution became clear. 2 N HCl solution was added to lower the pH to 2. After filtration, the solids were washed with excess water and recrystallized in ethanol to yield (4-(2-propyloxy)benzoic acid) as a white powder. A mixture of 4-(2-propyloxy)benzoic acid and SOCl2 was refluxed for 2 h. After evaporating excess SOCl2, pyridine and hydroquinone were added. The mixture was reacted in an acidified aqueous solution, then filtered and washed with a 5% Na2CO3 aqueous solution and water. After drying, the solid product (LCE1) was recrystallized in ethyl acetate. LCE1 was purified with column chromatography by using CH2Cl2 as the eluent. After evaporating the solvent, the LCE1 product was recrystallized in ethyl acetate with a yield of 55%. LCE3 was synthesized by using 5-bromopentene instead of 3-bromopropene. The yield of LCE3 was 69%. More details of the synthetic procedure is described in the ESI.†
The melt polymerization of the synthesized monomers (LCE1 and LCE3 having propyl epoxy and pentyl epoxy groups, respectively) with a primary monoamine (aniline) were performed in a flask to produce poly(diepoxy aniline)s (PDEAs, Scheme 2). The resultant polymers (PDEA1 and PDEA3) were characterized by using Fourier transform infrared spectrometry (FT-IR, Nicolet 6700, Thermo Scientific (USA)) with ATR accessory. The spectra were recorded with 32 scans at 8 cm−1 resolution. 1H NMR and 13C NMR. 1H-NMR and 13C-NMR spectra were obtained by Bruker spectrometer (300 MHz). Samples were prepared by dissolving in THF-d8 solvent. H-NMR spectra were referenced to residual TMS (0 ppm) except D2O (solvent reference, 4.79 ppm) and DMF-d7 (solvent reference, 8.03 ppm). Chemical shifts of the C-NMR spectra were measured relative to DMF-d7 (67.21 and 25.31 ppm).
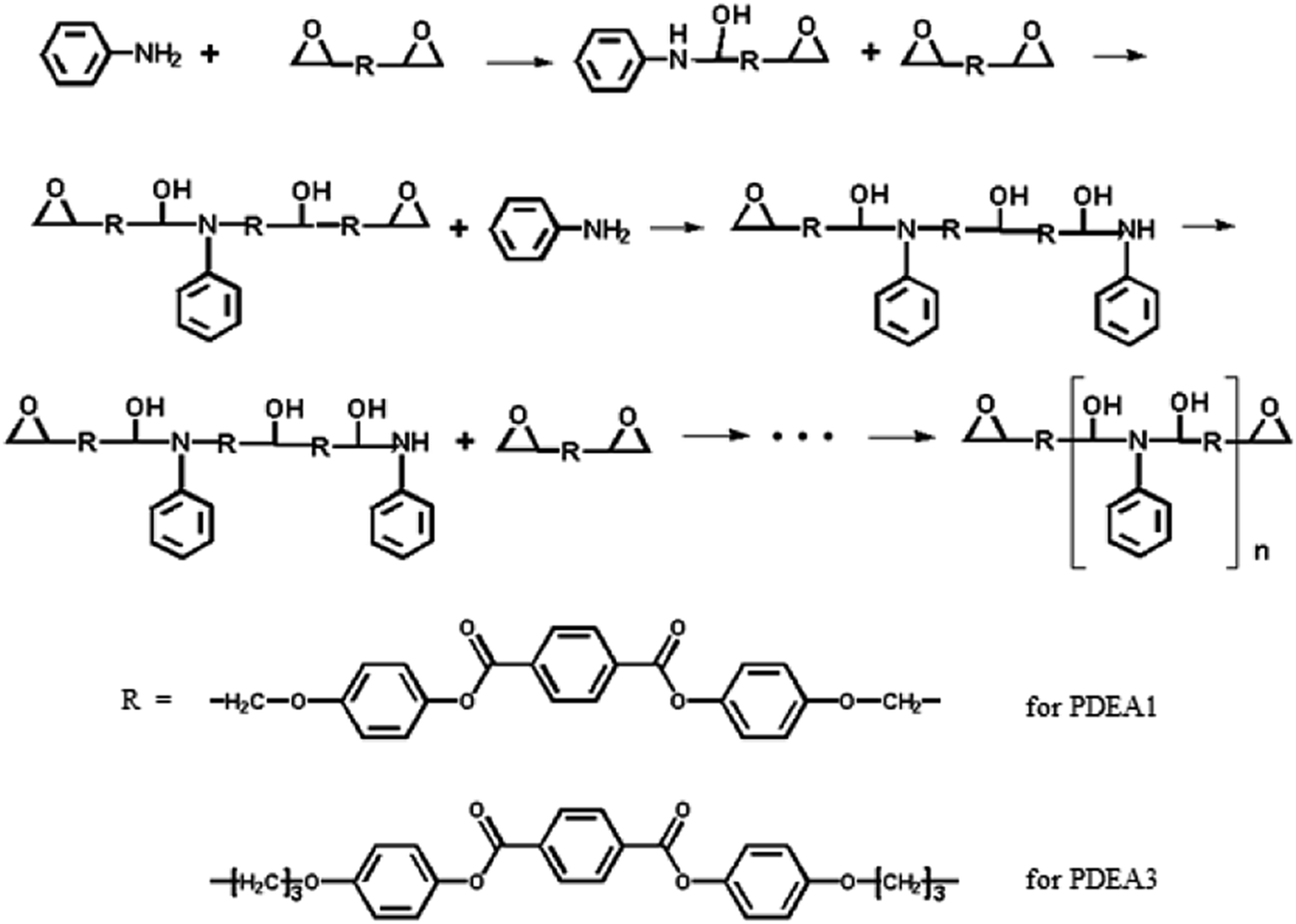 | ||
| Scheme 2 Synthesis of PDEA1 and PDEA3 via a polyaddition reaction. | ||
Thermal gravimetric analysis (TGA, TGA/DSC1, Mettler Toledo) and differential scanning calorimetry (DSC, DSC 823e, Mettler Toledo) were used to analyze the thermal properties, degradation temperature and transition behaviors of LCE/amine LCPs. Under a nitrogen atmosphere, samples were heated from 25 °C to 450 °C at 10 °C min−1 for TGA. For DSC, samples were first heated from 25 °C to 320 °C at 10 °C min−1, cooled to 25 °C and heated to 320 °C again at same heating rate, under a nitrogen atmosphere. The Fourier transform infrared spectrometer (FT-IR) spectra analysis was also carried out. Before measurement, LCE/amine samples were hot pressed at 250 °C and cooled to room temperature to obtain films.
Gel Permeation Chromatography (GPC, Viscotek model 250) was carried out with a RI750F refractive index detector. Waters Styragel HP 4E and Styragel HR 5E columns were used with THF as eluent as flow rate of 1 mL min−1 at 40 °C. Approximate calibration of the column was accomplished by polystyrene standards.
Thermal transition behaviors of polymers were observed by polarized light optical microscopy (POM, BX51, Olympus) using polarization mode. Samples were prepared by hot pressing the samples between two slide glasses at 250 °C. The measurement temperature range was from 25 °C to 320 °C with Mettler Toledo FP82HT hotstage. The polymer melts were pinched small parts off to make fibrils which were used to observe the morphology by a scanning electron microscopy (SEM, Hitach S 2200C). Fractured sample surface in the liquid nitrogen was coated Pt/Pd before SEM observation.
2-D high temperature X-ray diffraction (XRD) experimental was carried out with D8 Discover (Bruker) with GADDS (General Area Detector Diffraction System) program. XRD scans were carried out from 25 °C to 230 °C with CuKα radiation.
Results and discussion
The FT-IR spectra of the polymers are shown in Fig. 1. The sharp absorption peak at 914 cm−1 is due to the vibration of the oxirane ring in LCE1. The intensity of this peak is significantly lower for the TLCPs, whereas broad shoulder absorption peaks are evident at 3450 cm−1 for the TLCPs; these peaks are due to the stretching vibrations of the OH groups produced by the reaction between the oxirane rings and the amine group of aniline (Scheme 2). These results confirm that the epoxide groups on the LCEs have reacted with the amine group and polymerized to form the TLCPs (PDEA1 and PDEA3).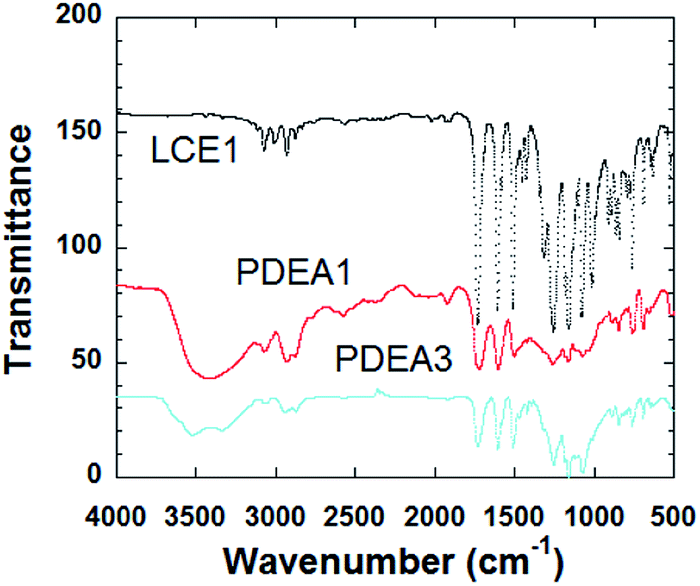 | ||
| Fig. 1 FTIR of the LCE1, PDEA1 and PDEA3. | ||
Analysis of the 1H NMR spectra of a representative monomer and polymer (PDEA1) were performed with Chemsketch software; by assigning the signals to the corresponding protons and carbon atoms, the polymerizations were confirmed (Fig. 2(a); 1H-NMR spectrum (CDCl3): δ (ppm) 8.13 (4H, d), 7.23 (4H, s), 6.99 (4H, d), 4.33 (2H, dd, CH2 of glycidyl), 4.02 (2H, dd, CH2 of glycidyl), 3.38 (2H, s, CH of epoxy), 2.92 (2H, dd, CH of epoxy), 2.78 (2H, dd, CH of epoxy). See the ESI† for the details of the 13C NMR spectrum of PDEA3). The mesogenic monomer LCE1 contains five protons on its phenyl rings and its two aliphatic groups, including the methyl group of the spacer unit and the methyl group on the oxirane ring. The signals of the aromatic protons appeared at 6.5–8.2 ppm whereas the methyl group signals of the TLCPs are at 3.2–5.3 ppm: the corresponding protons of the LCEs appeared at 2.5 and 4.1 ppm respectively. The reaction between the primary amine group and the epoxide ring (a ring opening reaction via nucleophilic attack on the amine) is accompanied by the appearance of new peaks at 4.7 and 5.3 ppm. The 13C NMR spectrum of PDEA1 is presented in Fig. 2(b). The characteristic peaks are in agreement with those from the polymer structures. The doublet at 3.5 ppm is due to the hydrogen atoms in aniline and disappears as the reaction progresses. The appearance of the aniline aromatic peak at 149.2 ppm confirms the formation of the polymer. These spectral results also clearly reveal the structures of the polymers. The epoxy group reacts vigorously with the amine group to produce a compound containing a secondary amine that reacted further with other epoxy groups to form a linear polymer chains (Scheme 1). The reaction between the epoxy groups and the secondary amine (that formed after the reaction of the first epoxy group with the amine group) is known to be less favorable than that with the primary amine;17,18 however, this reaction was found to proceed rapidly to yield the polymers.
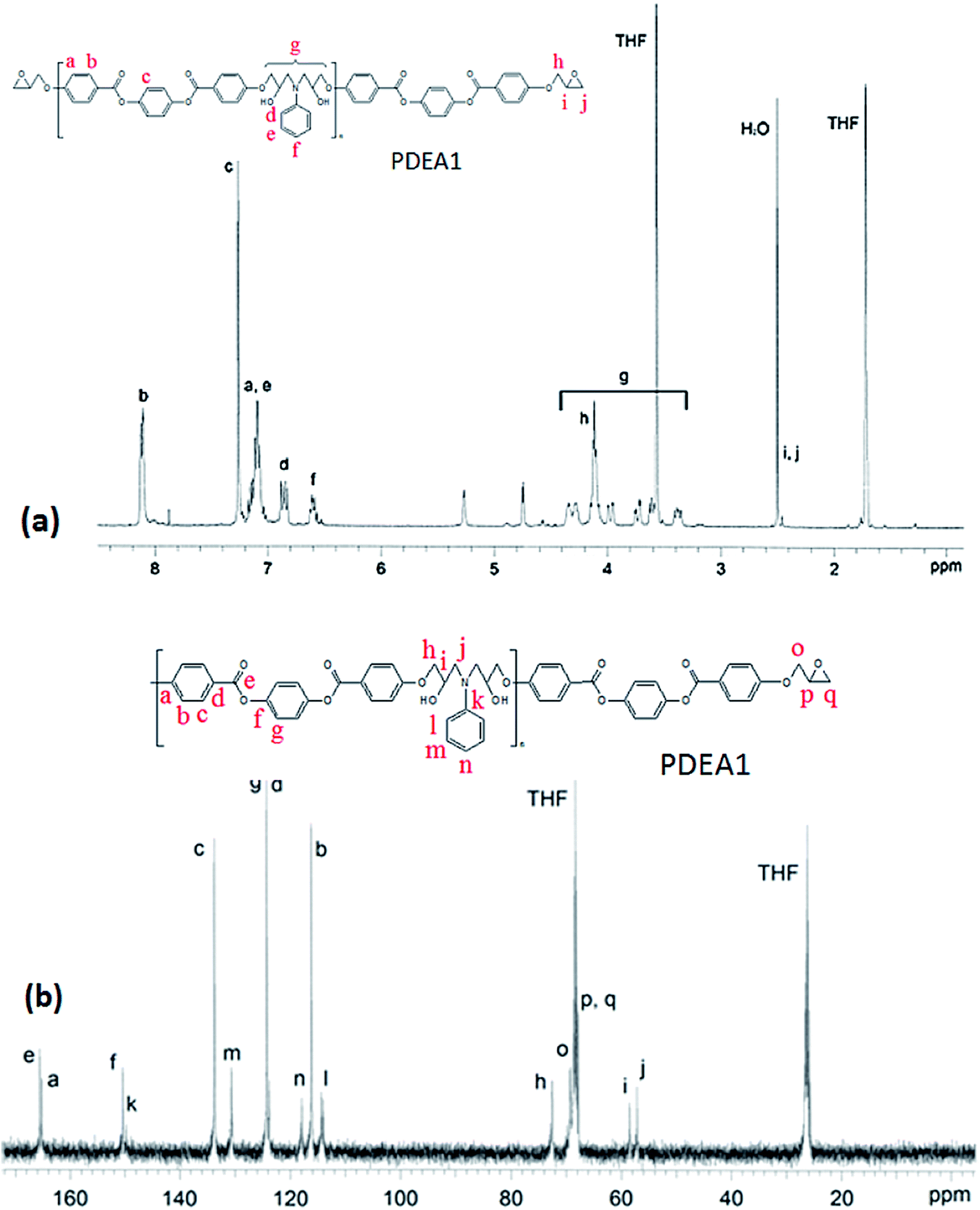 | ||
| Fig. 2 (a) 1H NMR and (b) 13C NMR of PDEA1. | ||
The 2-D wide angle X-ray diffraction patterns of the synthesized polymers are presented in Fig. 3, which shows that there are inner layer reflections on the meridional scan at a temperature below Tm. Sharp inner peaks are observed at a diffraction angle of 2θ = 7.6°, which corresponds to 11.7 Å according to Bragg's law. Broader outer peaks were observed at a diffraction angle of 2θ = 19–20°, which corresponds to 4.4–4.6 Å on the equatorial scan. This diffraction patter is typical of LC chains with a lateral-packing 3–6 Å.2,19,20 The broad peak in the wide angle region indicates that the ordering of the TLCP chains is liquid-like. As the temperature increases to 230 °C, the small angle reflections disappear whereas the broad peak at wide angles remains intact, which suggests that at this temperature there is a breakdown of chain regularity and a nematic phase outcome.21
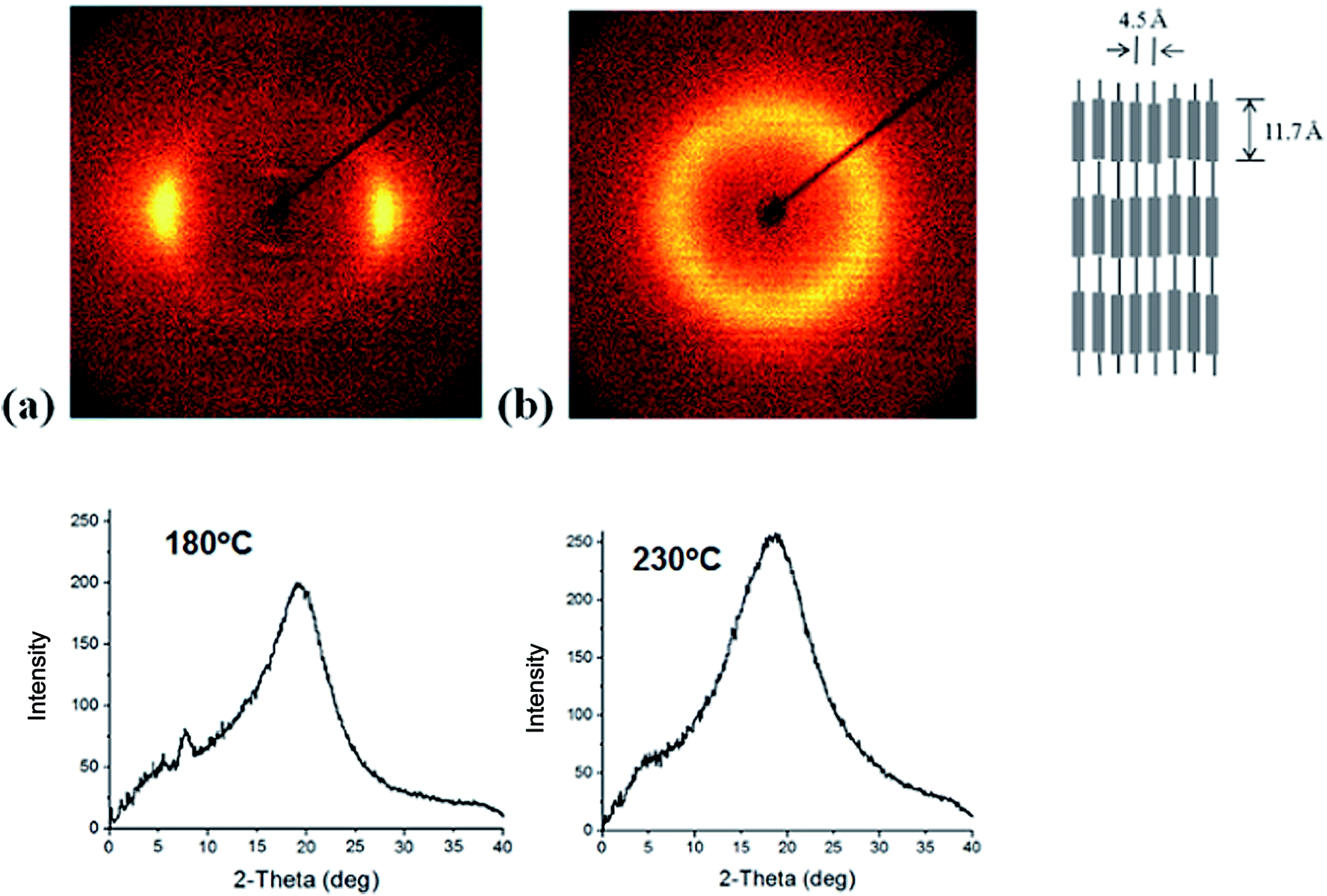 | ||
| Fig. 3 XRD patterns of (a) PDEA1 at 180 °C and (b) PDEA1 at 230 °C. | ||
The thermal behaviors of the polymers were investigated by using differential scanning calorimetry (DSC) and thermogravimetric analysis (TGA). The thermograms of both synthesized polymers exhibited a single endothermic peak during the heating scans attributed to the melting point (Fig. 4(a)). The complex thermal history of the samples was removed by recording the second heating thermograms to determine their glass transition temperatures (Tg), melting temperature (Tm), and nematic liquid crystalline-to-isotropic phase transition temperatures (TNI). The DSC scans revealed the Tg and Tm values, listed in Table 1. Tg is related to the flexibility of the chains and corresponds to the ease of chain relaxation. Increasing the length of the flexible spacer in the main chain decreases the rigidity of the polymer molecules by reducing the mesogene proportion. Thus PDEA3 has a lower Tg than PDEA1. The polymer with a longer alkyl group, PDEA3, has a higher Tm values than the polymer with the short alkyl group (LCE1), PDEA1. Table 1 also provides the degradation temperatures (Tdg of 5 wt% loss). Both the polymers have a high thermal stability (Fig. 4(b)): degradation does not begin until 350 °C. Degradation proceeds in a single stage, which implies that the presence of the flexible spacer groups does not affect the high thermal stability off the lengthy cores with ester connecting units. Both polymers have a low melting temperature (189 °C for PDEA1 and 249 °C for PDEA3), so they can be easily processed in an extruder or injection molder.
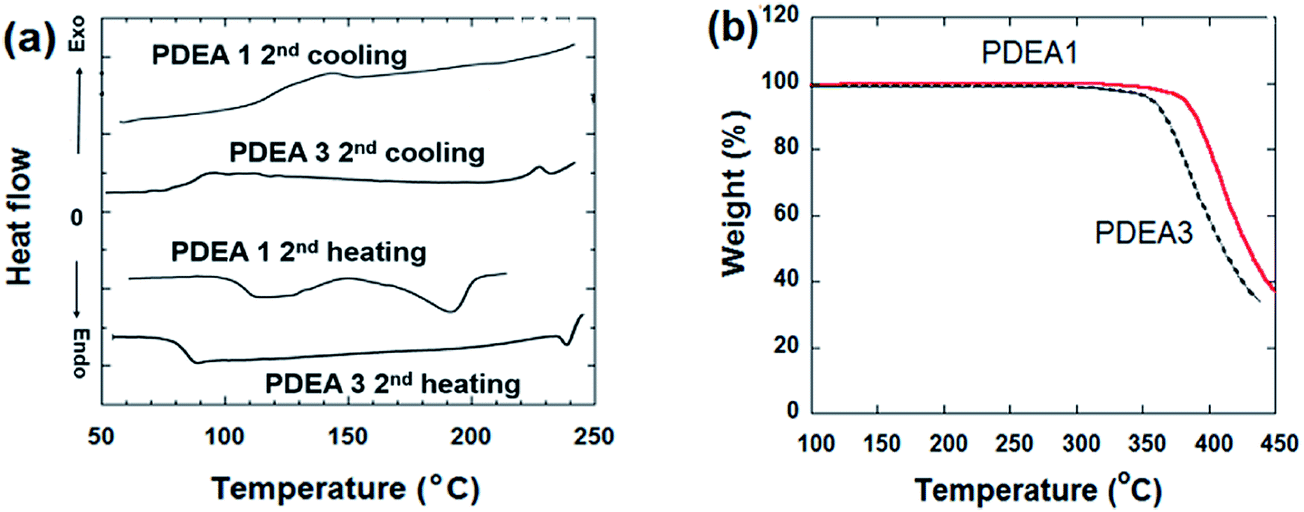 | ||
| Fig. 4 (a) DSC thermograms and (b) TGA degradation curves. | ||
| Samples | Mn | Mw | PDI | Tg (°C) | Tm (°C) | Tdg (5 wt% °C) |
|---|---|---|---|---|---|---|
| a Tg: glass transition temperature, Tm: melt temperature, Tdg: 5 wt% degradation temperature. | ||||||
| PDEA1 | 24100 |
56100 |
2.3 | 95 | 189 | 358 |
| PDEA3 | 10100 |
31400 |
3.1 | 74 | 239 | 355 |
The optical textures of the polymers near the transition temperature are presented in Fig. 5. During the heating, both polymers exhibited Schlieren-texture characteristic to the nematic mesophase near the transition temperature. These images were collected during the second heating process, so all samples exhibit the characteristic birefringence texture even at lower temperatures. When the samples were cooled to room temperature after the first heating, the colorful textures remained almost unchanged, which implies that the mesophase of the polymers formed at high temperatures are stable. The surface morphologies of the fibers drawn from the melt were examined with SEM (Fig. 6). All the samples contain highly oriented TLCP fibrils. The surfaces of the fibers after cryogenic fracturing in liquid nitrogen contain protruding fibrils. This appearance of protruding fibrils on the fractured surfaces is consistent with a continuous fibril morphology on the TLCP surface that is oriented parallel to the fibril direction.10,22
 | ||
| Fig. 5 POM images of (a) PDEA1 at 190 °C and (b) PDEA3 at 250 °C. | ||
 | ||
| Fig. 6 SEM micrographs of polymers (fractured surface normal to fibril direction (left); parallel to fibril direction (middle); slant view of the normal surface (right)) (×2000 magnification). | ||
Conclusion
The novel liquid crystalline polymers with low melting temperatures have been successfully synthesized by performing melt reactions between diepoxy molecules and a primary monoamine (aniline). This is the first report of the synthesis of TLCPs via a polyaddition process: this approach has significant commercial-importance because of its potential for the mass production. The synthesized polymers have good thermal stabilities and high decomposition temperatures exceeding 350 °C, as well as low melting temperatures that make their processing facile. Longer spacer groups in PDEA3 enhanced the transition temperatures, but the thermal stabilities of both polymers remained comparable due to the not-so-long spacer groups (methyl and propyl). Both polymers exhibit a nematic liquid crystal mesophase in the melt state. The polymers form well-ordered structures. Once elongated, the drawn polymer fibers have a strong orientation on the surface along the flow direction and many microfibrillar structures extrude out of the fractured surface normal to the direction of flow which indicates these nematic polymers can be applied for in situ composites preparation.Acknowledgements
This work was supported by NRF (MSE BK21 Program of SNU) and MIKE (No. I-AC24-16(0417-20160111), Core Original Materials Development Program).References
- F. Chen, Y. Cong and B. Zhang, Liq. Cryst., 2016, 43, 1100 CrossRef CAS.
- P. K. Bhowmik, S. T. Killarney, J. Koh, O. Tanthmanatham, H. Han, T. Harris, M. R. Fisch, D. M. Agra-Kooijman, L. Sharpnack and S. Kumar, Liq. Cryst., 2016, 43, 1560 CrossRef CAS.
- A. Muthusamy, K. Balaji and S. C. Murugavel, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 1707 CrossRef CAS.
- Y. Kimura, K. Kuboyama and T. Ougizawa, Liq. Cryst., 2016, 43, 587 CrossRef CAS.
- A. B. Samui, S. Pandey and S. P. Mishra, RSC Adv., 2015, 5, 68351 RSC.
- Y. Seo, S. M. Hong and K. U. Kim, Macromolecules, 1997, 30, 2978–2988 CrossRef CAS.
- Y. Seo, T. H. Ninh, S. M. Hong, S. Kim, T. J. Kang, H. S. Kim and J. Kim, Langmuir, 2006, 8, 3062–3067 CrossRef PubMed.
- Y. Seo, B. Kim, S. Kwak and K. U. Kim, Polymer, 1999, 40, 4483–4492 CrossRef CAS.
- Y. Seo, B. Kim and K. U. Kim, Polymer, 1999, 40, 4441–4450 CrossRef CAS.
- E. K. Fleischmann and R. Zentel, Angew. Chem., Int. Ed., 2013, 52, 8810–8827 CrossRef CAS PubMed.
- Y. Seo, S. M. Hong, S. S. Hwang, T. S. Park, K. U. Kim, S. M. Lee and J. Lee, Polymer, 1995, 36, 515–524 CrossRef CAS.
- Y. Seo, S. M. Hong, S. S. Hwang, T. S. Park, K. U. Kim, S. M. Lee and J. Lee, Polymer, 1995, 36, 525–534 CrossRef CAS.
- S. M. Lee, S. M. Hong, Y. Seo, T. S. Park, S. S. Hwang, K. U. Kim and J. Lee, Polymer, 1994, 35, 519–531 CrossRef CAS.
- Thermotropic Liquid Crystal Polymer Blends, ed. F. R. La Mantia, Technomic Publishing, Lancaster, PA, 1993 Search PubMed.
- R. K. Krishwaswamy and D. G. Baird, Polymer, 1999, 40, 701–716 CrossRef.
- K. I. Aly and M. M. Sayed, Liq. Cryst., 2014, 41, 67 CrossRef CAS; K. I. Aly and M. M. Sayed, Liq. Cryst., 2017 DOI:10.1080/02678292.2017.1309699.
- N. S. Enikolopiyan, Pure Appl. Chem., 1976, 48, 317–328 CrossRef CAS.
- M. Blanco, M. A. Corcuera, C. C. Riccard and I. Mandryar, Polymer, 2005, 46, 7989–8000 CrossRef CAS.
- J. Watanabe and M. Hayashi, Macromolecules, 1989, 22, 4083–4088 CrossRef CAS.
- S. W. Kantor, T. C. Sung and E. D. T. Atkins, Macromolecules, 1992, 25, 2789–2795 CrossRef CAS.
- Z. Zhu, J. Zhi, A. Liu, H. Tang, W. Qiao, X. Wan and Q. Zhou, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 800–847 Search PubMed.
- B. Chena, S. Tsaya and J. Chen, Polymer, 2005, 46, 8624–8633 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis details of the mesogenic epoxides, the polymerization process, and NMR spectra of PDEA3 are available. See DOI: 10.1039/c7ra02809a |
| This journal is © The Royal Society of Chemistry 2017 |