
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Amphiphilic oligoamides as versatile, acid-responsive gelators†
Qiang Peiab,
Quan Tanga,
Zheng-Li Tan a,
Zhong-Lin Lu*a,
Lan He*ac and
Bing Gong*ad
a,
Zhong-Lin Lu*a,
Lan He*ac and
Bing Gong*ad
aCollege of Chemistry, Beijing Normal University, 100875, Beijing, China. E-mail: luzl@bnu.edu.cn
bCollege of Chemistry and Chemical Engineering, Xinyang Normal University, 464000, Xinyang, China
cNational Institute for Food and Drug Control, Institute of Chemical Drug Control, TianTanXiLi 2, Beijing, 100050, China. E-mail: helan1961@aliyun.com
dDepartment of Chemistry, University at Buffalo, The State University of New York, Buffalo, NY 14260, USA. E-mail: bgong@buffalo.edu
First published on 21st April 2017
Abstract
Six oligoamides with the same backbone but different side and end chains, G1–G6, were designed and synthesized. Screening the gelating abilities of these oligoamides revealed G2 as a versatile gelator capable of forming stable hydrogels as well as several organogels. From UV, fluorescence, NMR, and SEM studies, the formation of hydrogels is driven by hydrophobic forces and π–π stacking while the gelation of nonpolar organic solvents relies on hydrogen-bonding interactions. The hydrogel of G2 is able to encapsulate and release medicinally important polar substances into water with acid-responsiveness.
1. Introduction
Low-molecular-weight gelators (LMWGs) have attracted wide attention due to their potential for developing soft materials with applications in many fields.1–5 Gelators undergo self-assembly to form three-dimensional networks that entrap and immobilize solvent molecules.6–21 LMWGs that allow convenient, systematic structural modification are especially attractive since such molecules can not only lead to the discovery of new gelating properties, but also provide invaluable insights into the mechanism of gel formation.22–25 While examples of gelating organic solvents with simple amides and ureas are abundant,2 similar molecules capable of gelating distinctly different solvents are rare. Developing such gelators calls for the design of structures that incorporate hydrophilic, hydrophobic and responsive structural elements. For practical applications, the designed molecules should be readily available and modifiable.Over the years, we have developed a series of oligoamides consisting of alternating meta-substituted benzene and glycine residues.26–28 These oligoamides, which carry H-bonding sequences defined by linearly arranged amide O and H atoms, pair into H-bonded duplexes.29–37 For example, oligoamides represented by general structure 1 have a self-complementary H-bonding sequence that direct the formation of H-bonded duplexes 1·1 (Scheme 1). A study on the ability of 1·1 to template chemical reactions led to the discovery on the gelation of nonpolar, aromatic hydrocarbons by duplexes carrying appropriate alkyl side chains.25 The gelating ability of 1·1 quickly diminishes with increasing solvent polarity, suggesting that H-bonded duplexes 1·1, instead of single strand 1, were responsible for the gelation of aromatic hydrocarbons.
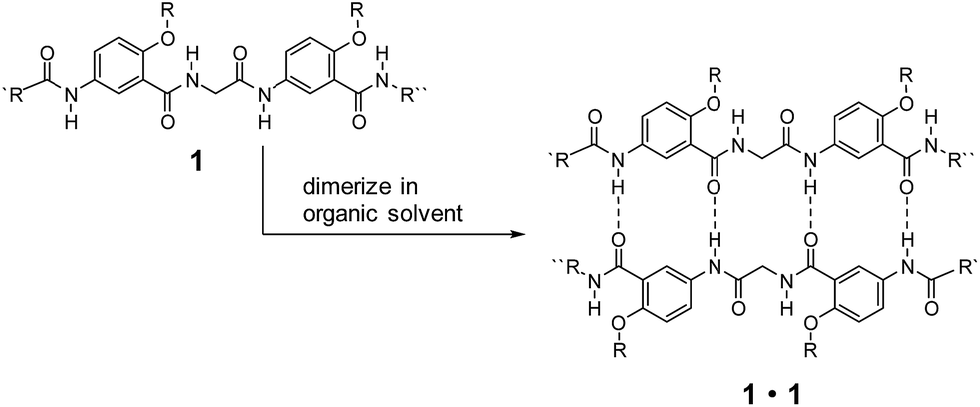 | ||
| Scheme 1 Sequence-specific formation of hydrogen-bonded duplexes 1. | ||
With their ready synthetic availability and multiple sites allowing structural modification, oligoamides 1 provide a prototype based on which analogs with wide gelation capabilities may be obtained. With the above consideration, we report herein the synthesis and characterization of six oligoamide strands with same back bone, G1–G6, (Scheme 2). Except for that of G6 which is an octyl group, the R groups of G1–G5 are derived from triethylene glycol (Tg) that is able to engage in favorable interaction with a variety of different solvents. Terminal group R1 is methyl, n-pentyl, or nonyl group. Different from 1, each of G1–G6 has a side chain consisting of a secondary amide moiety that is connected to, depending on the nature of R2 and R3 groups, a terminal primary, secondary, or tertiary amino group. In non-polar solvents, each of G1–G6 should self-dimerize into a H-bonded duplex similar to 1·1. The secondary amide moiety of each oligoamide introduces additional H-bonding between the duplexes of G1–G6, forming 3D networks that entrap solvent molecules. In polar solvents, oligoamides G1–G6 are not expected to dimerize into H-bonded duplexes. Instead, the multiple amide groups of these oligoamides should H-bond with solvent molecules while π–π stacking between the aromatic residues may lead to intermolecular aggregates, i.e., 3D networks. The terminal amino group of each oligoamide will be protonated in the presence of an acid, and may endow the potential gelator with acid-responsiveness. Screening these oligoamides against solvents of different properties revealed three strands showing gelating abilities. One of the six oligoamides, G2, serves as a versatile gelator capable of forming gels with water as well as nonpolar and polar organic solvents. The application of G2 as medicinal drug carrier was also investigated.
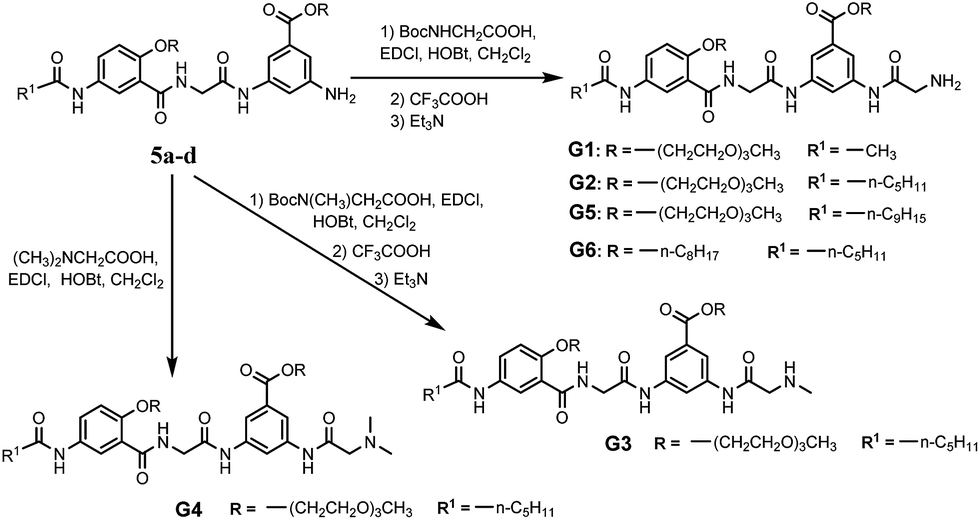 | ||
| Scheme 2 Structures of G1–G6 and their syntheses. | ||
2. Experimental
2.1 Materials and instrumentation
All compounds used were commercially available without further purification. Silica gel for analytical thin layer chromatography (TLC) and column chromatography (200–300 mesh) were purchased from Qingdao Haiyang Chemical Co., Ltd. & Special Silica Gel Factory. 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT), fetal bovine serum (FBS), Dulbecco's Modified Eagle's Medium (DMEM), and trypsin were purchased from Solarbio Science & Technology Co., Ltd. (Beijing, China). HeLa cells and HepG2 cells were obtained from Cell Resource Center (IBMS, CAMS/PUMC). Streptomycin sulfate, chloramphenicol, tetracycline, ampicillin, and methylene blue (MB) were purchased from Heowns Biochem Technologies Company (Tianjin, China).1H NMR spectra were recorded at 400 MHz and 13C NMR spectra were measured at 100 MHz on a Bruker Avance III 400 MHz spectrometer at ambient temperature, using CDCl3 or DMSO-d6 as solvents, unless otherwise noted. Chemical shifts (δ) are reported in ppm values downfield of TMS (tetramethylsilane) and coupling constants (J) are denoted in Hz. Multiplicities are denoted as follows: s = singlet, d = doublet, t = triplet, dd = double doublet, and m = multiplet. High resolution mass (HRMS) data were recorded on a Waters LCT Premier XE spectrometer. Fluorescence measurements were performed on a Varian Cary Eclipse spectrometer (USA) and UV/vis spectra measured by a Varian Cary 300 UV-vis spectrophotometer (USA). The SEM images were obtained by using a Hitachi S-4800 (Japan). The samples for SEM were prepared by dropping the formed gel on silicon wafer. The frozen specimen by liquid nitrogen was evaporated by a vacuum pump for 24 h and coated with a thin layer of Au.
2.2 Synthesis of G1–G6
Compound G1. White solid (1.6 g, 72%). IR (KBr, cm−1): 3480, 3346, 3275, 3094, 2877, 2828, 1717, 1645, 1614, 1540, 1455, 1248, 1209, 1109, 1028. 1H NMR (400 MHz, DMSO-d6): δ 10.46 (s, 1H), 10.02 (s, 1H), 8.72 (t, J = 5.4 Hz, 1H), 8.24 (s, 1H), 8.08 (d, J = 2.8 Hz, 1H), 8.01 (s, 1H), 7.97 (s, 1H), 7.80 (dd, J = 8.9, 2.7 Hz, 1H), 7.17 (d, J = 9.0 Hz, 1H), 4.40 (m, 2H), 4.27 (m, 2H), 4.17 (m, 2H), 3.85 (m, 2H), 3.74 (m, 2H), 3.61–3.33 (m, 18H), 3.19 (s, 3H), 3.17 (s, 3H), 2.02 (s, 3H); 13C NMR (100 MHz, DMSO-d6): δ 168.13, 167.91, 167.78, 165.46, 164.42, 152.43, 139.71, 139.20, 133.18, 130.59, 123.62, 121.79, 121.62, 115.08, 114.79, 114.33, 114.16, 71.27, 71.21, 69.94, 69.90, 69.80, 69.64, 69.53, 68.89, 68.65, 68.40, 64.30, 58.03, 43.57, 42.69, 23.84. HRMS (ESI, m/z) calcd for C34H49N5O13 [M + H]+: 736.3399; found: 736.3403.
Compound G2. White solid (1.9 g, 82%). IR (KBr, cm−1): 3468, 3344, 3312, 3107, 2928, 2872, 1699, 1653, 1611, 1541, 1456, 1212, 1108, 1038. 1H NMR (400 MHz, DMSO-d6): δ 10.34 (s, 1H), 9.89 (s, 1H), 8.71 (t, J = 5.3 Hz, 1H), 8.26 (s, 1H), 8.07 (d, J = 2.8 Hz, 1H), 7.99 (s, 1H), 7.74 (s, 1H), 7.82 (dd, J = 8.9, 2.8 Hz, 1H), 7.17 (d, J = 9.0 Hz, 1H), 4.46 (m, 2H), 4.27 (m, 2H), 4.19 (m, 2H), 3.86 (m, 2H), 3.74 (m, 2H), 3.59–3.35 (m, 18H), 3.20 (s, 3H), 3.17 (s, 3H), 2.28 (t, J = 7.4 Hz, 2H), 1.60 (m, 2H), 1.29 (m, 4H), 0.87 (t, J = 6.9 Hz, 3H); 13C NMR (100 MHz, DMSO-d6): δ 172.43, 171.03, 167.76, 165.48, 164.37, 152.38, 139.54, 139.51, 133.12, 130.47, 123.61, 121.84, 121.60, 114.73, 114.61, 114.30, 114.05, 71.24, 71.18, 69.89, 69.76, 69.61, 69.50, 68.87, 68.61, 68.37, 64.19, 57.97, 45.63, 43.49, 36.25, 30.90, 24.83, 21.90, 13.83. HRMS (ESI, m/z) calcd for C38H57N5O13 [M + H]+: 792.4025; found: 792.4025.
Compound G5. White solid (1.2 g, 48%). IR (KBr, cm−1): 3422, 3345, 3316, 3120, 2923, 2851, 1699, 1653, 1613, 1541, 1457, 1248, 1108. 1H NMR (400 MHz, DMSO-d6) δ 10.35 (s, 1H), 9.89 (s, 1H), 8.71 (t, J = 5.1 Hz, 1H), 8.26 (s, 1H), 8.06 (d, J = 2.7 Hz, 1H), 7.99 (s, 1H), 7.94 (s, 1H), 7.83 (dd, J = 9.0, 2.7 Hz, 1H), 7.16 (d, J = 9.0 Hz, 1H), 4.38 (m, 2H), 4.27 (m, 2H), 4.18 (m, 2H), 3.85 (m, 2H), 3.74 (m, 2H), 3.64–3.41 (m, 18H), 3.20 (s, 3H), 3.17 (s, 3H), 2.29 (t, J = 7.0 Hz, 2H), 1.60 (m, 2H), 1.29 (m, 10H), 0.85 (t, J = 6.8 Hz, 3H); 13C NMR (100 MHz, DMSO-d6) δ 172.46, 171.09, 167.79, 165.52, 164.40, 152.41, 139.56, 133.14, 130.49, 123.67, 121.88, 121.62, 114.66, 114.31, 114.12, 71.26, 71.21, 69.92, 69.79, 69.63, 69.52, 68.91, 68.65, 68.40, 64.22, 58.00, 45.63, 43.52, 36.32, 31.30, 28.93, 28.84, 28.71, 25.18, 22.12, 13.94. HRMS (ESI, m/z) calcd for C42H65N5O13 [M + H]+: 848.4651; found: 848.4652.
Compound G6. White solid (1.6 g, 72%). IR (KBr, cm−1): 3370, 3319, 3284, 3108, 2956, 2926, 2856, 1708, 1642, 1610, 1536, 1453, 1213, 1089. 1H NMR (400 MHz, DMSO-d6) δ 10.38 (s, 1H), 9.88 (s, 1H), 8.67 (d, J = 4.7 Hz, 1H), 8.18 (s, 1H), 8.07 (s, 2H), 7.94 (d, J = Hz, 1H), 7.82 (dd, J = 8.8, 2.5 Hz, 1H), 7.14 (d, J = 9.0 Hz, 1H), 4.29–4.15 (m, 6H), 3.34 (m, 2H), 2.27 (t, J = 7.3 Hz, 2H), 1.86 (m, 2H), 1.72 (m, 2H), 1.62 (m, 2H), 1.44–1.13 (m, 24H), 0.90–0.75 (m, 9H); 13C NMR (100 MHz, DMSO-d6) δ 172.01, 170.97, 165.49, 164.25, 152.55, 139.43, 132.72, 130.72, 123.63, 121.87, 121.21, 114.69, 114.58, 113.81, 113.39, 69.27, 64.71, 45.34, 43.58, 36.24, 31.21, 30.99, 28.79, 28.72, 28.60, 28.20, 25.79, 25.43, 24.83, 22.04, 21.99, 13.82. HRMS (ESI, m/z) calcd for C40H61N5O7 [M + H]+: 724.4643; found: 724.4641.
2.3 Gelation tests
The gelation tests were carried out by a inverted test tube method. Gelators and solvents were put in a septum-capped test tube and heated in water-bath (>70 °C) until the solids were completely dissolved. If a clear solution was obtained, the sample vial was then cooled to 25 °C under ambient conditions. If no flow was observed by inverting the vial, a stable gel was formed and noted as G. A gel-like, but slowly flowing substance was recorded as a partial gel (pG). Insoluble compounds (solubility < 1 mM) were noted as I. Substance that precipitated was noted as P. Those leading to clear solutions (>50 mM) were marked as soluble (S). The thermo-reversibility of a gelation process was confirmed by repeated heating and cooling.To determine the critical gelation concentration (CGC), gels were made with different gelators (10 mg) in solvents (0.5 mL) in 5 mL vials with a diameter of 1.5 cm, firstly. Subsequently, the gels were diluted, heated and then cooled to room temperature in an iterative process, until gels no longer formed or the gels became too weak to withstand gravity. The gel–sol transition temperature (Tg) was measured by the “falling drop” method. Typically, a sealed vial containing the gel was immersed in a thermostated water bath. Then the temperature of the bath was raised at a rate of 2 °C min−1 until the gel began to flow down due to the gravity, and this temperature was denoted as the gel–sol transition temperature (Tg).
2.4 Cell toxicity
In vitro cytotoxicity was measured by performing a methyl thiazolyl tetrazolium (MTT) assay on HeLa cells and HepG2 cells. Cells were cultured in DMEM medium supplemented with 10% FBS (Fetal Bovine Serum) in a humid atmosphere with 5% CO2 at 37 °C. After routinely passaged by trypsinization when nearly confluent, the cells were seeded in 96-well plates at 5000 cells and 100 μL medium per well and cultured for another 24 h for 70–80% cell confluence. Then different concentrations of G2 (100, 200, 400, 600 and 800 μmol L−1, diluted in H2O/DMEM) were then added to the wells. The cells were subsequently incubated for 24 h at 37 °C under 5% CO2. Thereafter, 20 μL of 5 mg mL−1 MTT solution in DMSO was added to each well and for additional 4 h incubation at 37 °C under 5% CO2. Finally, the medium was replaced with 150 μL of DMSO, and the plates were oscillated for 10 minutes to fully dissolve the formazan crystals formed by living cells in the wells. The UV absorbance was detected at 490 nm using the microplate reader. The following formula was used to calculate the relative viability of the cells: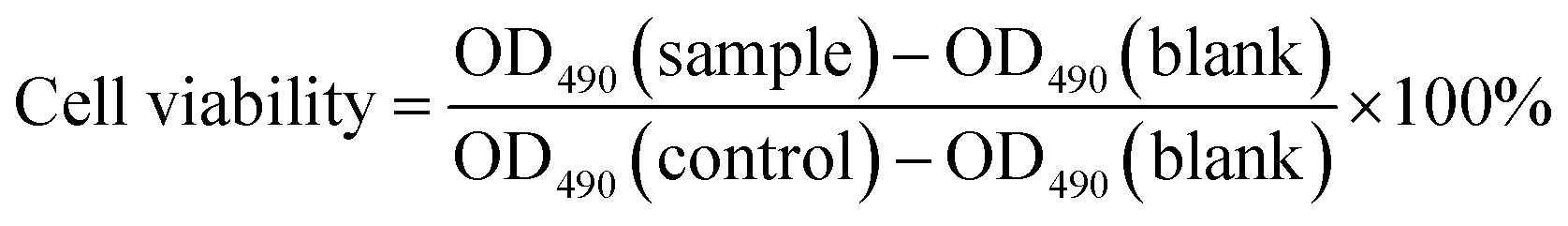
OD490 (sample) represents the optical density of the wells treated with various concentrations of the compounds, OD490 (control) represents that of the wells treated with DMEM 10% FBS and OD490 (blank) represents that of blank wells treated with pure DMSO. The finally reported percent cell survival values are relative to the untreated control cells.
2.5 Loading and releasing experiments
Loading experiments: gelator G2 (5.0 mg) and the aqueous solutions with precise concentrations of different drugs (1.0 mM, 500 μL) were taken together into a vial. The mixture was heated until complete dissolution, the solution was gradually allowed to cool to room temperature. The sample was subjected to a tube-inversion test to confirm that the compounds can be loaded.Releasing experiment: we selected methylene blue (MB) as a drug model to investigate the release behavior of hydrogel in vitro. First, the aqueous solutions of MB (0.3 mM, 500 μL) was added into a vial that contained gelator G2 (5.0 mg), and formed a gel with the method as described before. Then, 2.5 mL deionized water (or 2.5 mL deionized water containing 1‰ glacial acetic acid) was added on the above of the hydrogel matrix and kept at room temperature. The water layer was taken out at predetermined time interval. The amounts of MB released to water were monitored by an ultraviolet spectrophotometer at 665 nm at different time. Then, the liquid was added into the vial again, to maintain the total volume of the solution above the gel matrix.
3. Results and discussion
3.1 Synthesis of oligoamides G1–G6
The synthetic routes and details of compound 5a–d are depicted in Scheme S1.† The preparations were started from the acylation of m-aminobenzoic acid derivatives 1a, b with the straight aliphatic acid chlorides of 2, 6, 10 carbon atoms. The resulted compounds 2a–d were hydrolyzed in basic MeOH solution and acidified to afford the free acids 3a–d, which were then treated with compound 4a, b to give compounds 5a–d through amidation reactions. Further amidation with Boc-protected glycine and deprotection with trifluoroacetic acid resulted in oligoamides G1, G2, G5 and G6. Oligoamide G3 and G4 were obtained by using Boc-sarcosine or N,N-dimethylglycine instead of Boc-glycine (Scheme 2). All new compounds were fully characterized by 1H-NMR, 13C-NMR, HR-MS and IR. The detail of the syntheses and characterization of all new compounds except the targeted compounds G1–G6 were provided in the ESI.†3.2 Gelation capability of the compounds
Oligoamides G1–G6 were subjected to an initial screening on their ability to gelate different solvents. As shown in Table 1, G1 fails to gelate any solvent since it is either insoluble or soluble in these solvents. Replacing the terminal methyl group of G1 with a pentyl group results in G2, which is able to gelate benzene, toluene, ethyl acetate and water, four distinctly different solvents. To achieve gelation in water, there must be a balance between the hydrophilicity and hydrophobicity.5–11 In the amphiphilic gelator G2, the two triethylene glycol (Tg) and amino are hydrophilic and provide a certain solubility in water. While the hydrophobic terminal group R1 (n-pentyl) provides hydrophobic forces and facilitates the aggregation. Attaching a methyl to the terminal amino group of G2 leads to G3 which can only gelate xylene and partially gelate water. Attaching another methyl to the terminal amino group of G3 completely abolishes the gelating ability of the resultant G4 which is soluble in all solvents except for hexane and water. Replacing the methyl group of G1 or pentyl group of G2 with a nonyl group affords G5 which regains the ability of G2 to gelate benzene, toluene and ethyl acetate, and also partially gelate xylene. However, unlike G2, G5 cannot form hydrogel. Oligoamide G6, which differs from G2 by having octyl, instead of Tg chains as the R groups, becomes soluble in all solvents except for hexane and water. These results demonstrate the effectiveness of our strategy based on screening. The observed gelation behavior would otherwise be difficult to predict given the small structural difference that results in drastically different gelation behavior.| Solvent | G1 | G2 | G3 | G4 | G5 | G6 |
|---|---|---|---|---|---|---|
| a G = gel, S = soluble, I = insoluble, pG = partial gel, P = precipitate. All gels are opaque. Values denote the critical gel concentration (CGS, mg mL−1). | ||||||
| Hexane | I | I | I | I | I | P |
| Benzene | I | G10.3 | S | S | G15.3 | S |
| Toluene | I | G15.2 | S | S | G23.5 | S |
| Xylene | I | I | G21.6 | S | pG | S |
| Mesitylene | I | I | P | S | I | S |
| EtOAc | I | G14.5 | S | S | G16.5 | S |
| CH2Cl2 | S | S | S | S | S | S |
| CHCl3 | S | S | S | S | S | S |
| EtOH | S | S | S | S | S | S |
| MeOH | S | S | S | S | S | S |
| H2O | S | G6.3 | pG | I | I | I |
Among the six oligoamides with different gelating ability, G2 stands out as the most versatile by not only gelating organic solvents, but also as the only one capable of forming stable hydrogel. The gelation of both water and organic solvents with G2 was further investigated. In water, after being subjected to a heating–cooling (25–70 °C) process, G2 formed a stable hydrogel, with a critical gelation concentration (CGC) of 6.3 mg mL−1 and a gel–sol transition temperature (Tg) of 59 °C. Interestingly, gels of organic solvents had to be prepared at room temperature by sonicating the corresponding solutions of G2 for two minutes, followed by standing for another 30 minutes.38
Upon adding 5 μL of HCl (1 M) or glacial AcOH, the hydrogel of G2 quickly became a solution, which then turned back to gels when the solution was made neutral or slightly basic (pH 7–8) with Et3N (Fig. S1†). The observed acid-responsiveness is expected. The terminal NH2 group of G2, upon being protonated by an acid, introduces electrostatic repulsion, leading to the disintegration of the self-assembling networks that entrap water molecules. Reversing the protonation process allows the restoration of the 3D networks of G2, leading to gelation. The gelating ability of G2 (10 mg mL−1) in water was also examined from pH 1 to 7, which revealed that G2 formed stable hydrogels from pH 3 to 7 (Fig. S2†).
3.3 Morphologies of the gel
Additional insights into the self-assembly of G2 in its various gels were provided by examining xerogels prepared from the corresponding gels with scanning electron microscopy (SEM). As shown in Fig. 1A, a three-dimensional network composed of thin fibers with diameter of about 50 nm and several microns in length was observed in the xerogel of G2 obtained by freeze-drying method. The sizes of the meshes range from several to more than ten microns. The presence of 3D network is consistent with the observed gelation of water by G2.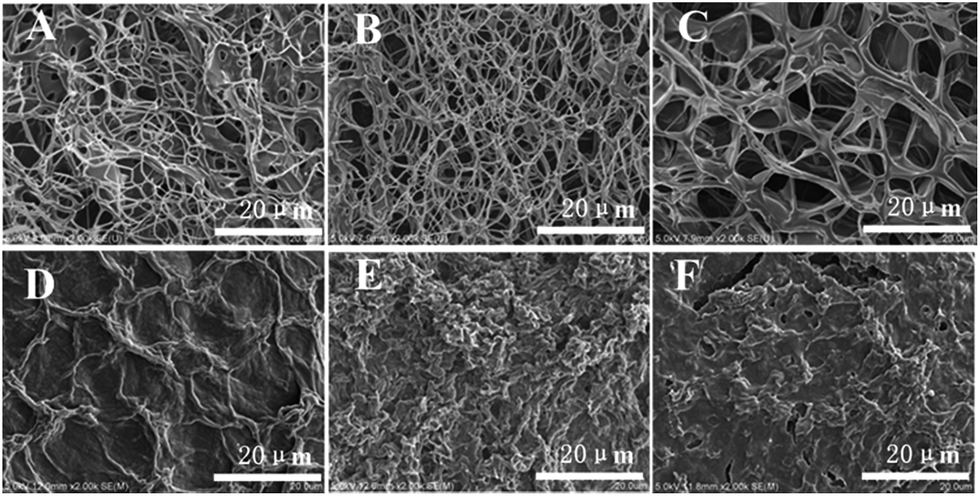 | ||
| Fig. 1 SEM images of xerogel prepared from the gels of G2 (25 °C) with: (A) water (10 mg mL−1); (B) water (10 mg mL−1) with 5 μL of added HCl (1 M), followed by regenerating gel after adding 5 μL of Et3N; (C) water (10 mg mL−1) with 5 μL of added CH3COOH, followed by regenerating gel after adding 5 μL of Et3N; (D) benzene (15 mg mL−1); (E) toluene (15 mg mL−1); (F) ethyl acetate (15 mg mL−1). | ||
The morphology of xerogels obtained from G2 and organic solvents does not show similar fibrillar network observed for that prepared from the hydrogel. Instead, irregular lamellar structures with small cavities are observed (Fig. 1D–F), which could be due to the fact that these organogels had to be prepared by sonicating, instead of heating and then cooling, the solutions of G2 in these solvents.
The morphology of xerogels (Fig. 1B and C) prepared from hydrogels regenerated by adding Et3N to the acidic solution of G2 remains essentially the same as that of the initial hydrogel (Fig. 1A). Apparently, the presence of salts, i.e., triethylammonium hydrochloride or triethylammonium acetate, in the regenerated hydrogels did not influence the regeneration and morphology of the hydrogel. This feature could be very useful for developing systems for drug delivery and release.
3.4 UV-vis and fluorescence spectroscopy
The UV-vis spectra of G2 in water and in hydrogel are shown in Fig. 2A. Compound G2 (0.1 mM) dissolved in H2O gives two absorption bands at 243 nm and 306 nm. In the gel state, the absorption band of G2 at 306 nm undergoes bathochromic shift and appears at 318 nm. The fluorescence emission (373 nm) of G2 in its hydrogel shows a large hypochromatic shift of 19 nm relative to that (392 nm) in solution (0.1 mM) (Fig. 2B). These results are consistent with the presence of π–π stacking in the gel state. In contrast, the UV-vis and fluorescence spectra of G2 in both the solution and gel with benzene do not exhibit similar shifts (Fig. S3A and S3B†), suggesting that π–π stacking is not the driving force responsible for the gelation of non-polar solvents.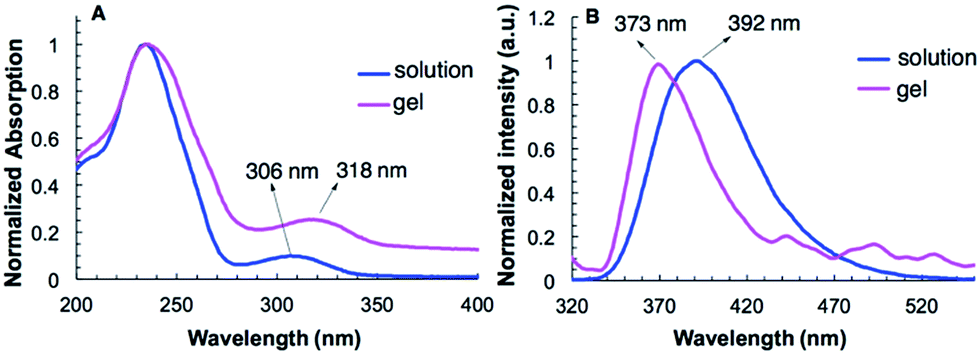 | ||
| Fig. 2 The (A) absorption (1 mm path length) and, (B) fluorescence emission spectra (λex = 307 nm) of G2 in water (0.1 mM) and in hydrogel (10 mg mL−1). | ||
The influence of temperature to the aggregation G2 in its hydrogel was further investigated with fluorescence spectroscopy. The emission intensity of G2 increased as temperature rose from 25 °C to 50 °C, followed by a sudden drop and then remained unchanged beyond 60 °C (Fig. S4†). The effect of temperature on the fluorescence intensity of G2 is consistent with the observed transition from the gel state to solution: the gel state, which showed no noticeable change from 25 °C to 50 °C, underwent an abrupt breakdown into a turbid solution when temperature rose to 60 °C.
3.5 1D and 2D NMR spectroscopy
The variable-temperature 1H NMR spectra of G2 recorded in C6D6 indicate that, as temperature changes from 10 °C to 60 °C, the broad signals of G2 in its gel state at low temperature gradually change to sharp, well resolved ones (Fig. S5†). Such a change in 1H NMR signals can be attributed to the weakening and eventual disintegration of the aggregate form of G2 with rising temperatures. The 1H NMR spectra of G2 (20 mM) recorded at 25 °C in C6D6 containing 0% to 30% DMSO-d6 (Fig. S6†) indicate that, with increasing ratio of DMSO-d6, the NMR signals of G2 become increasingly better dispersed and show no additional change with 20% or more DMSO-d6. Apparently, the presence of DMSO-d6 weakens and interrupts inter-molecular H-bonding, leading to the eventual decomposition of the organogel. These results indicate that, in benzene, intermolecular H-bonding is the driving force for the aggregation of G2, which leads to the gelation of benzene and similarly other nonpolar solvents.In nonpolar solvents, oligoamide G2, with its self-complementary H-bonding sequence, is expected to dimerize into a H-bonded duplex. Two-dimensional (NOESY) NMR (20 mM, 40 °C, C6D6, Fig. S7†) revealed strong, multiple inter-strand NOEs between non-adjacent protons of G2, which provide diagnostic evidence supporting the formation of the H-bonded duplex of this oligoamide. Such a duplex, like duplex 1·1, further assembles and aggregates, leading to 3D networks that entrap non-polar solvents.
3.6 Cytotoxicity of gelator G2
To verify the biocompatibility of the gelator G2, the cytotoxicity of it against HeLa cells and HepG2 cells over 24 h period was assessed by means of a standard MTT assay. As depicted in Fig. 3, gelator G2 could ensure that no less than 70% of the HeLa cells and HepG2 cells survived at the tested concentrations, demonstrating its low toxicity and excellent biocompatibility.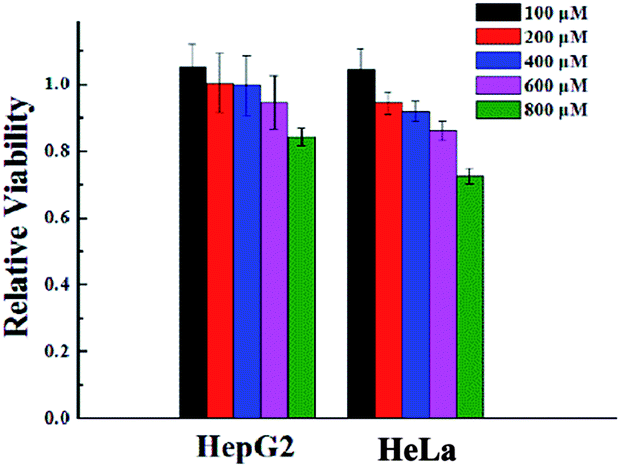 | ||
| Fig. 3 Cytotoxicity of gelator G2 at different concentrations toward different cell lines after incubation at 37 °C for 24 h. | ||
3.7 Loading and releasing experiments
To examine the encapsulation and release of hydrophilic substances from the hydrogel formed with G2, several medicinally important, water-soluble salts and compounds, including zinc acetate, streptomycin sulfate, chloramphenicol, tetracycline, ampicillin, and the cationic aromatic dye methylene blue (MB), were loaded to the hydrogels formed by G2. Zinc ion is an essential mineral in cellular metabolism and at sufficiently high concentrations, shows antibacterial properties.39 Streptomycin sulfate, chloramphenicol, tetracycline and ampicillin are common antibiotics. As shown in Fig. S8,† gelator G2 could form stable gel in the aqueous solution of each (1.0 mM) of these substances.The in vitro release behavior of MB from the hydrogel of G2 was also investigated. Hydrogel was prepared with G2 (5.0 mg) and MB (0.3 mM) in water (0.5 mL), followed by immersing the hydrogel in 2.5 mL of deionized water (or deionized water containing 1‰ acetic acid) at room temperature. A portion (2.5 mL) of the water layer was taken at predetermined time interval and was then measured at 665 nm to determine the amount of MB released to water (Fig. S9†). The 2.5 mL water was then added back to the original water layer to maintain total volume of water.
As shown in Fig. 4A, the release of MB from the hydrogel of G2 into water involves two stages. The first stage is relatively fast, with ∼20% of MB being released within the initial 3 h. The second stage is much slower, with ∼30% of MB being released in 33 h. By 36 h, about 50% of MB was released. For the MB-loaded hydrogel immersed in water containing 1‰ glacial acetic acid, a much faster release rate was observed. In this case over 70% of MB was released within 5 h (Fig. 4B), which was accompanied by the disintegration of the hydrogel. This observation is consistent with the acid-responsiveness of the hydrogel of G2.
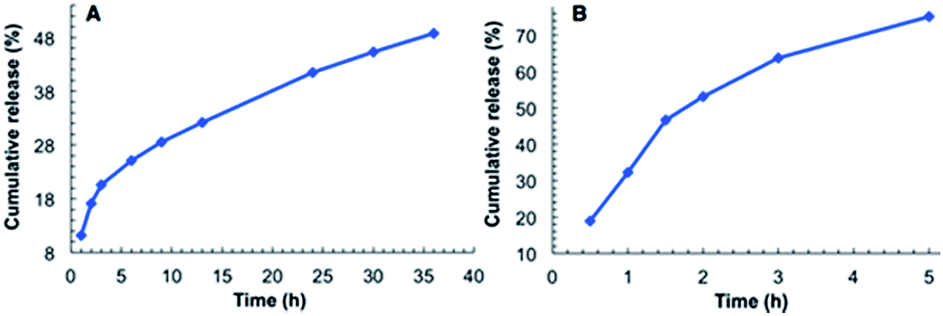 | ||
| Fig. 4 Releasing curve of MB from the hydrogel of G2: (A) into water and, (B) into water containing 1‰ glacial acetic acid. | ||
4. Conclusions
In summary, six oligoamides G1–G6, designed as potential gelators, were successfully synthesized and fully characterized. Their gelation abilities were screened against solvents of different properties. It was found that structure variation greatly affection their performance. Among three oligoamides that are found to gelate solvents, oligoamides G2 stands out as a versatile gelator capable of forming stable gels with both nonpolar and polar solvents including pure water. Through UV, fluorescent spectra and 1H-NMR study under different conditions, it indicated that the formation of hydrogel with G2 was mainly driving by hydrophobic forces and π–π stacking interaction while the gelation of non-polar solvents such as benzene is mediated by H-bonding. SEM images confirm that molecules of G2 aggregate into fibrous networks in the corresponding gels with water and organic solvents. The hydrogel formed by gelator G2 was thermally reversible and acid responsive, and can be decomposed and regenerated by adjusting pH. Finally, the hydrogel of G2 shows favorable loading and releasing ability in water, based on which applications in drug delivery and target release can be envisioned.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21372032, 21272274, and 91227109) and the US National Science Foundation (CBET-1512164). The authors also gratefully thank the Fundamental Research Funds for the Central Universities, Beijing Municipal Commission of Education, the Program for Changjiang Scholars and Innovative Research Team in University.Notes and references
- D. J. Abdallah and R. G. Weiss, Organogels and low molecular mass organic gelators, Adv. Mater., 2000, 12, 1237–1247 CrossRef CAS.
- C. Tomasini and N. Castellucci, Peptides and peptidomimetics that behave as low molecular weight gelators, Chem. Soc. Rev., 2013, 42, 156–172 RSC.
- X. W. Du, J. Zhou, J. F. Shi and B. Xu, Supramolecular Hydrogelators and Hydrogels: From Soft Matter to Molecular Biomaterials, Chem. Rev., 2015, 115, 13165–13307 CrossRef CAS PubMed.
- L. Voorhaar and R. Hoogenboom, Supramolecular polymer networks: hydrogels and bulk materials, Chem. Soc. Rev., 2016, 45, 4013–4031 RSC.
- L. A. Estroff and A. D. Hamilton, Water Gelation by Small Organic Molecules, Chem. Rev., 2004, 104, 1201–1217 CrossRef CAS PubMed.
- Y. Li, F. Zhou, Y. Wen, K. Liu, L. Chen, Y. Mao, S. Yang and T. Yi, (−)-Menthol based thixotropic hydrogel and its application as a universal antibacterial carrier, Soft Matter, 2014, 10, 3077–3085 RSC.
- S. Nandi, H. J. Altenbach, B. Jakob, K. Lange, R. Ihizane, M. P. Schneider, Ü. Gün and A. Mayer, Amphiphiles Based on D-Glucose: Efficient Low Molecular Weight Gelators, Org. Lett., 2012, 14, 3826–3829 CrossRef CAS PubMed.
- F. Xu, H. Wang, J. Zhao, X. Liu, D. Li, C. Chen and J. Ji, Chiral Packing of Cholesteryl Group as an Effective Strategy To Get Low Molecular Weight Supramolecular Hydrogels in the Absence of Intermolecular Hydrogen Bond, Macromolecules, 2013, 46, 4235–4246 CrossRef CAS.
- Z. Yuan, W. Lu, W. Liu and J. Hao, Gel phase originating from molecular quasi-crystallization and nanofiber growth of sodium laurate–water system, Soft Matter, 2008, 4, 1639–1644 RSC.
- M. Ornatska, S. Peleshanko, K. L. Genson, B. Rybak, K. N. Bergman and V. V. Tsukruk, Assembling of Amphiphilic Highly Branched Molecules in Supramolecular Nanofibers, J. Am. Chem. Soc., 2004, 126, 9675–9684 CrossRef CAS PubMed.
- M. Yamanaka, Y. Miyake, S. Akita and K. Nakano, Sol–Gel Transcription of Semi-Fluorinated Organogel Fiber into Fluorocarbon-Functionalized Silica Nanotubes, Chem. Mater., 2008, 20, 2072–2074 CrossRef CAS.
- D. Görl, B. Soberats, S. Herbst, V. Stepanenko and F. Würthner, Perylene bisimide hydrogels and lyotropic liquid crystals with temperature-responsive color change, Chem. Sci., 2016, 7, 6786–6790 RSC.
- S. Ogi, V. Stepanenko, K. Sugiyasu, M. Takeuchi and F. Würthner, Mechanism of Self-Assembly Process and Seeded Supramolecular Polymerization of Perylene Bisimide Organogelator, J. Am. Chem. Soc., 2015, 137, 3300–3307 CrossRef CAS PubMed.
- F. K. Zhan, S. M. Hsu, H. Cheng and H. C. Lin, Remarkable influence of alkyl chain lengths on supramolecular hydrogelation of naphthalene diimide-capped dipeptides, RSC Adv., 2015, 5, 48961–48964 RSC.
- J. Y. Fan, X. M. Chang, M. X. He, C. D. Shang, G. Wang, S. W. Yin, H. N. Peng and Y. Fang, Functionality-Oriented Derivatization of Naphthalene Diimide: A Molecular Gel Strategy-Based Fluorescent Film for Aniline Vapor Detection, ACS Appl. Mater. Interfaces, 2016, 8, 18584–18592 CAS.
- T. Koga, M. Matsuoka and N. Higashi, Structural Control of Self-Assembled Nanofibers by Artificial β-Sheet Peptides Composed of D- or L-Isomer, J. Am. Chem. Soc., 2005, 127, 17596–17597 CrossRef CAS PubMed.
- K. Y. Liu, L. Y. Meng, S. L. Mo, M. M. Zhang, Y. Y. Mao, X. H. Cao, C. H. Huang and T. Yi, Colour change and luminescence enhancement in a cholesterol-based terpyridyl platinum metallogel via sonication, J. Mater. Chem. C, 2013, 1, 1753–1762 RSC.
- J. R. Moffat and D. K. Smith, Controlled self-sorting in the assembly of ‘multi-gelator’ gels, Chem. Commun., 2009, 316–318 RSC.
- X. H. Cao, J. Zhou, Y. Zou, M. M. Zhang, X. D. Yu, S. Zhang, T. Yi and C. H. Huang, Fluorescence and Morphology Modulation in a Photochromic Diarylethene Self-Assembly System, Langmuir, 2011, 27, 5090–5097 CrossRef CAS PubMed.
- M. Xue, D. Gao, X. L. Chen, K. Q. Liu and Y. Fang, New dimeric cholesteryl-based A(LS)2 gelators with remarkable gelling abilities: Organogel formation at room temperature, J. Colloid Interface Sci., 2011, 361, 556–564 CrossRef CAS PubMed.
- D. K. Smith, Lost in translation? Chirality effects in the self-assembly of nanostructured gel-phase materials, Chem. Soc. Rev., 2009, 38, 684–694 RSC.
- P. F. Duan and M. H. Liu, Design and Self-Assembly of L-Glutamate-Based Aromatic Dendrons as Ambidextrous Gelators of Water and Organic Solvents, Langmuir, 2009, 25, 8706–8713 CrossRef CAS PubMed.
- N. Zweep, A. Hopkinson, A. Meetsma, W. R. Browne, B. L. Feringa and J. H. van Esch, Balancing Hydrogen Bonding and van der Waals Interactions in Cyclohexane-Based Bisamide and Bisurea Organogelators, Langmuir, 2009, 25, 8802–8809 CrossRef CAS PubMed.
- J. H. van Esch, We Can Design Molecular Gelators, But Do We Understand Them?, Langmuir, 2009, 25, 8392–8394 CrossRef CAS PubMed.
- R. K. Cao, J. J. Zhou, W. Wang, W. Feng, X. H. Li, P. H. Zhang, P. C. Deng, L. H. Yuan and B. Gong, Oligoamide Duplexes as Organogelators, Org. Lett., 2010, 12, 2958–2961 CrossRef CAS PubMed.
- B. Gong, Specifying Non-Covalent Interactions: Sequence-Specific Assembly of Hydrogen-Bonded Molecular Duplexes, Synlett, 2001, 5, 582–589 Search PubMed.
- B. Gong, Molecular Duplexes with Encoded Sequences and Stabilities, Acc. Chem. Res., 2012, 45, 2077–2087 CrossRef CAS PubMed.
- B. Gong, Mini Review Engineering hydrogen-bonded duplexes, Polym. Int., 2007, 56, 436–443 CrossRef CAS.
- B. Gong, Y. F. Yan, H. Q. Zeng, E. Skrzypczak-Jankunn, Y. W. Kim, J. Zhu and H. Ickes, A New Approach for the Design of Supramolecular Recognition Units: Hydrogen-Bonded Molecular Duplexes, J. Am. Chem. Soc., 1999, 121, 5607–5608 CrossRef CAS.
- H. Q. Zeng, R. S. Miller, R. A. Flowers and B. Gong, A Highly Stable, Six-Hydrogen-Bonded Molecular Duplex, J. Am. Chem. Soc., 2000, 122, 2635–2644 CrossRef CAS.
- H. Q. Zeng, H. Ickes, R. A. Flowers and B. Gong, Sequence Specificity of Hydrogen-Bonded Molecular Duplexes, J. Org. Chem., 2001, 66, 3574–3583 CrossRef CAS PubMed.
- H. Q. Zeng, X. W. Yang, R. A. Flowers and B. Gong, A Noncovalent Approach to Antiparallel β-Sheet Formation, J. Am. Chem. Soc., 2002, 124, 2903–2910 CrossRef CAS PubMed.
- X. W. Yang, F. J. Hua, K. Yamato, E. Ruckenstein, B. Gong, W. Kim and C. Y. Ryu, Supromolecular AB Diblock Copolymers, Angew. Chem., Int. Ed., 2004, 43, 6471–6474 CrossRef CAS PubMed.
- X. W. Yang and B. Gong, Template-Assisted Cross Olefin Metathesis, Angew. Chem., Int. Ed., 2005, 44, 1352–1356 CrossRef CAS PubMed.
- M. F. Li, K. Yamato, J. S. Ferguson and B. Gong, Sequence-Specific Association in Aqueous Media by Integrating Hydrogen Bonding and Dynamic Covalent Interactions, J. Am. Chem. Soc., 2006, 128, 12628–12629 CrossRef CAS PubMed.
- M. F. Li, K. Yamato, J. S. Ferguson and B. Gong, Sequence-Specific, Dynamic Covalent Crosslinking in Aqueous Media, J. Am. Chem. Soc., 2008, 130, 491–500 CrossRef CAS PubMed.
- P. H. Zhang, H. Z. Chu, X. H. Li, W. Feng, P. C. Deng, L. H. Yuan and B. Gong, Alternative Strategy for Adjusting the Association Specificity of Hydrogen-Bonded Duplexes, Org. Lett., 2011, 13, 54–57 CrossRef CAS PubMed.
- G. Cravotto and P. Cintas, Molecular self-assembly and patterning induced by sound waves. The case of gelation, Chem. Soc. Rev., 2009, 38, 2684–2697 RSC.
- S. Samani, S. M. Hossainalipour, M. Tamizifar and H. R. Rezaie, In vitro Antibacterial evaluation of sol–gel-derived Zn–, Ag–, and (Zn + Ag)-doped hydroxyapatite coatings against methicillin-resistant Staphylococcus aureus, J. Biomed. Mater. Res., Part A, 2013, 101, 222–230 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra03074f |
| This journal is © The Royal Society of Chemistry 2017 |