
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Recent progress in ergot alkaloid research†
Jing-Jing Chen,
Meng-Yao Han,
Ting Gong,
Jin-Ling Yang and
Ping Zhu *
*
State Key Laboratory of Bioactive Substance and Function of Natural Medicines, Key Laboratory of Biosynthesis of Natural Products of National Health and Family Planning Commission, Institute of Materia Medica, Peking Union Medical College & Chinese Academy of Medical Sciences, 1 Xian Nong Tan Street, Beijing 100050, China. E-mail: zhuping@imm.ac.cn; Fax: +86-10-63017757; Tel: +86-10-63165197
First published on 23rd May 2017
Abstract
Ergot alkaloids are a class of indole derivatives produced by the genera of Ascomycota including Claviceps, Aspergillus, Penicillium, and Epichloë. Many natural and semi-synthetic ergot alkaloids exhibit valuable pharmacological activities and have been widely used in the therapy of human CNS disorders. Owing to the development of genome sequencing technology, the gene clusters involved in the biosynthesis of ergot alkaloids have been identified from these fungi. In this review, we briefly introduce the pharmacological activities and possible mechanisms of action of some ergot alkaloids. Then we summarize the recent progress in the functional characterization of the key genes and gene clusters involved in the biosynthetic pathways of ergot alkaloids from different genera. Particularly, we summarize and discuss the constructions of ergot alkaloid biosynthetic pathways in different heterologous hosts and the optimization strategies performed on the recombinant strains, which provide references for producing ergot alkaloids and the derivatives in cell factories by synthetic biology in the future.
1. Introduction
Ergot alkaloids are a class of indole derivatives having biological and medicinal activities. Over the past decades, the pharmacological activities of these compounds have been extensively studied.1 In the Middle Ages, ergot alkaloids were thought to be natural toxins with an impact on human health.2 It is well known that the main producers of ergot alkaloids are members of the genus Claviceps, which belongs to Ascomycotina in taxonomy. As a parasitic fungus, Claviceps can grow on grass or grain and the ingestion of the infected grass or grain by humans or animals led to the breakout of severe epidemics.3,4 In 1850, a clearer understanding of ergot poisoning was provided when the French scientist Tulasne comprehensively elucidated the life cycle of Claviceps. The Swiss biochemist Stoll first isolated ergotamine in 1918, and the Swiss psychiatrist Maier found it to be effective for the treatment of migraines in 1926, marking the beginning of the modern study of ergot alkaloids.5,6 In recent years, other genera of Ascomycota including Aspergillus7 and Penicillium,8 even Epichloë have also been proven to produce ergot alkaloids.All ergot alkaloids have an indole-derived tetracyclic ring structure (ergoline) in common, wherein the A and B rings are from L-tryptophan, and the C and D rings are from the cyclization of dimethylallyl pyrophosphate (DMAPP) and L-tryptophan. As shown in Fig. 1, based on their structures, ergot alkaloids are usually divided into three classes.1,9,10 Clavine-type alkaloids (ergoclavines), in which chanoclavine-I aldehyde is the common structural scaffold, are mainly produced by Aspergillus and Penicillium. Some ergoclavines such as chanoclavine-I, agroclavine and elymoclavine are also the intermediates of ergoamides and ergopeptines. Ergoamides, including D-lysergic acid amide, D-lysergic acid α-hydroxyethylamide and ergometrine, are the simpler derivatives of D-lysergic acid. Ergopeptines, the complicated derivatives of D-lysergic acid, are the largest and most diverse class in ergot alkaloids. D-Lysergic acid derivatives are mainly produced by the genus Claviceps, some of them are produced by Epichloë.8,11–13 Most of the D-lysergic acid derivatives are bioactive, and have been widely used in the therapy of human CNS disorders. The detailed classification and structures of ergot alkaloids have been recently summarized elsewhere.1 The NMR spectrums of some bioactive ergot alkaloids have been identified (Tables S1–S3†). Here, we briefly introduce the pharmacological activities and clinical applications of some ergot alkaloids and highlights the recent progress in the identification of the catalytic enzymes involved in the biosynthetic pathways of ergot alkaloids. The future perspective of the ergot alkaloids production in cell factories is also discussed.
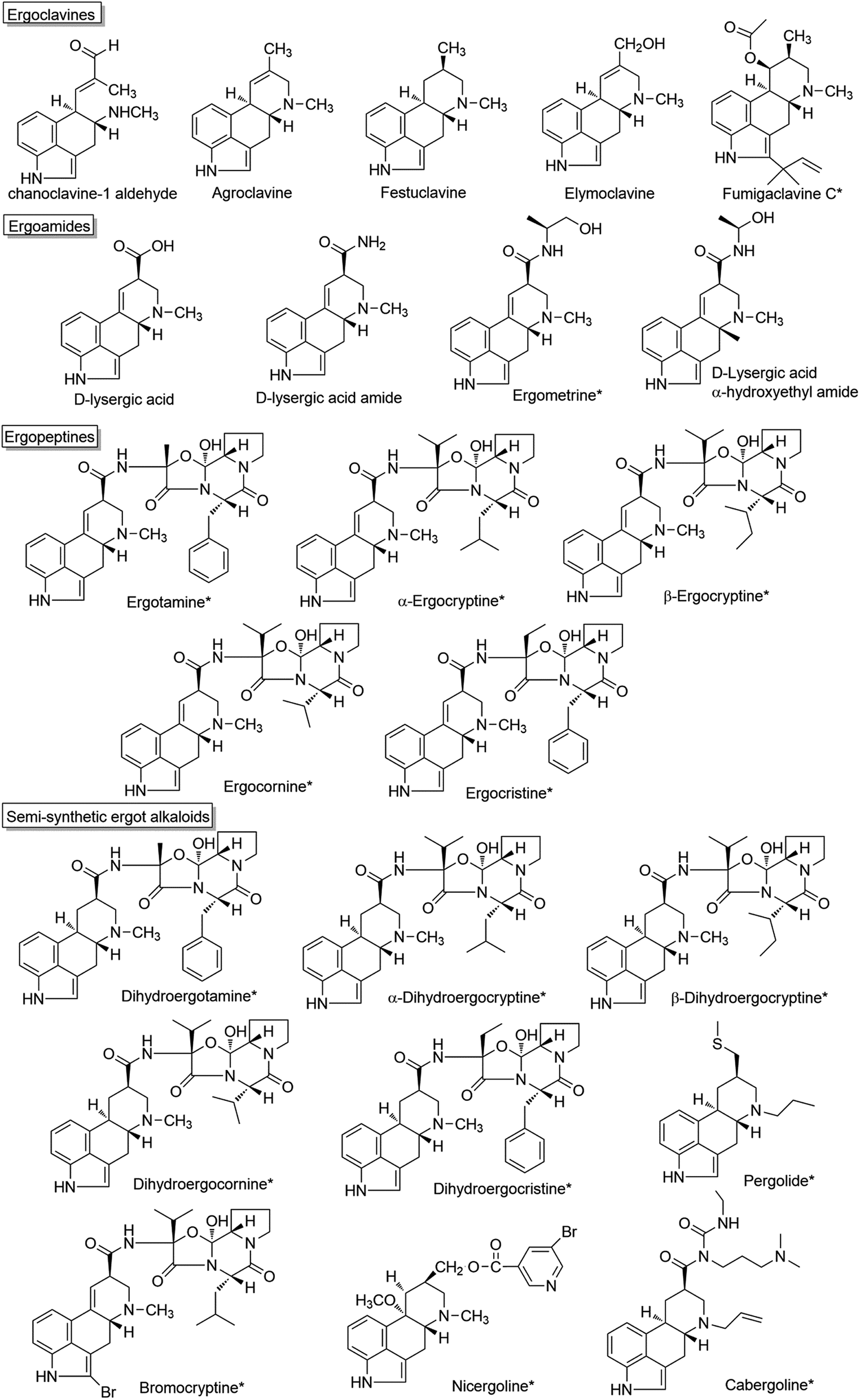 | ||
| Fig. 1 Structures of some ergot alkaloids in which the compounds with clinical applications are marked with *. | ||
2. Pharmaceutic activities and clinical applications of some ergot alkaloids
Fumigaclavine C, a clavine-type alkaloid, is a promising molecule for the treatment of inflammatory bowel diseases and atherosclerosis.14–19 Ergometrine (also known as ergonovine, an amide derivative of D-lysergic acid) has the function of causing contractions of the uterus and can be used to treat bleeding after childbirth.2 Many natural ergopeptines and semi-synthetic ergot alkaloids exhibit remarkable pharmaceutical activities due to their interactions with receptors in the central nervous system, and are of value in the treatment of neurological disorders (Fig. 1). For example, ergotamine and its derivative dihydroergotamine are used for treatment of acute migraine attacks.20–23 Hydergine (trade name), a mixture of the methanesulfonate salts of four dihydrogenated ergot alkaloids (dihydroergocristine, dihydroergocornine, α-dihydroergocryptine and β-dihydroergocryptine), has been used to treat dementia and age-related cognitive impairment (such as Alzheimer's disease).24–26 α-Dihydroergocryptine (the trade name: Vasobral) is effective as monotherapy in the early stages of Parkinson's disease and for the treatment of low blood pressure.27,28 Bromocryptine synthesized by the bromination of α-ergocryptin using N-bromosuccinimideis is used for the treatment of Parkinson's disease and hyperprolactinaemia.29,30 Nicergoline is a derivative of D-lysergic acid and is used in the treatment of dementia and vascular disorders such as cerebral thrombosis.31–33 Another D-lysergic acid derivative, Cabergoline, is frequently used as a first-line agent in the treatment of prolactinomas, since it has a more convenient dosing schedule than Bromocryptine.34–38 Pergolide is also a dopamine receptor agonist used in some countries for the treatment of Parkinson's disease, although it was withdrawn from the U.S. market for human use in March 2007 due to serious valvular damage.39,40 These clinical applications are summarized in Table 1.| Name | Pharmaceutic activity | Indications | Reference |
|---|---|---|---|
| a Mixture of dihydroergocristine, dihydroergocornine, α-dihydroergocryptine, β-dihydroergocryptine. | |||
| Fumigaclavine C | Inhibiting NLRP3 inflammasome activation; attenuating TNFα via the TLR4-NFκB signaling transduction pathway | Inflammatory bowel diseases and atherosclerosis | 14–19 |
| Ergometrine | Promoting contraction of the uterus | Bleeding after childbirth | 2 |
| Ergotamine | 5-HT receptor agonist | Acute migraine attack | 22 and 23 |
| Dihydroergotamine | 5-HT receptor agonist | Acute migraine attack | 20–23 |
| α-Dihydroergocryptine | α-Adrenergic antagonist and dopamine agonist | Parkinson's disease andlow blood pressure | 27 and 28 |
| Hyderginea | Interacting with D1- and D2-subtypes of dopamine receptors | Dementia and Alzheimer disease | 24–26 |
| Bromocryptine | Dopamine agonist | Parkinson's disease andhyperprolactinaemia | 29 and 30 |
| Nicergoline | α-1A adrenergic receptor antagonist | Vascular disorders and dementia | 31–33 |
| Cabergoline | Inhibiting the secretion of prolactin from the pituitary; dopamine agonist | Prolactinomas and Parkinson's disease | 34–38 |
| Pergolide | Dopamine agonist | Parkinson's disease | 39 and 40 |
3. Biosynthetic pathways of ergot alkaloids
3.1 Common biosynthetic pathway towards chanoclavine-I-aldehyde
The primary biosynthetic pathway of ergot alkaloids is shown in Fig. 2, which led to the formation of chanoclavine-I aldehyde.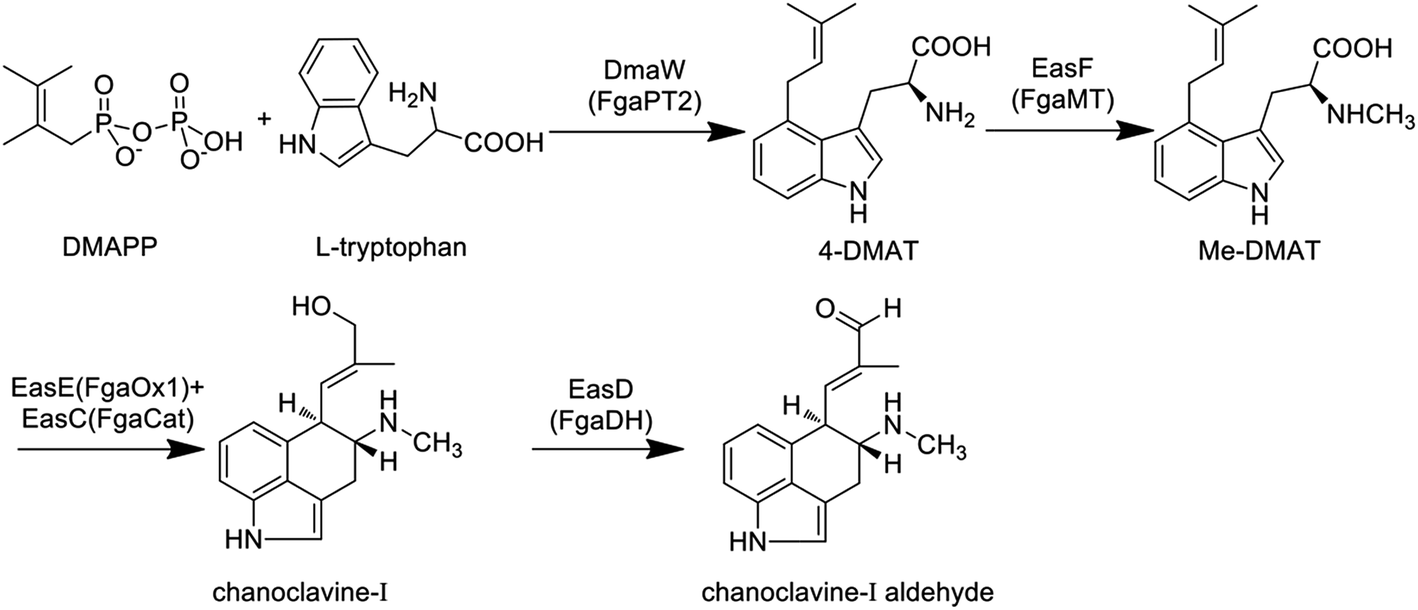 | ||
| Fig. 2 The common biosynthetic pathway of ergot alkaloids in different fungal species. (DmaW, EasF, EasE, EasC and EasD are the enzymes responsible for the synthesis of chanoclavine-I-aldehyde in C. purpurea; the homologous enzymes in A. fumigatus are FgaPT2, FgaMT, FgaOx1, FagCat and FgaDH, respectively). | ||
The first step of the whole pathway is the condensation reaction of L-tryptophan and DMAPP, in which DMAPP as the product of mevalonate pathway provides the isoprene group for this reaction. The enzyme responsible for this step is dimethylallytryptophan (DMAT) synthase (DMATS), which is the rate-limiting enzyme in the synthesis of ergoline and is regulated positively by tryptophan and negatively by the intermediates agroclavine and elymoclavine.41 The coding genes of DMATS in C. purpurea and C. fusiformis have been cloned and named cpd1 and dmaW, respectively, but the corresponding sequences of encoded proteins are quite different.42–44 DMATS is a homodimer with a molecular weight of 105 kDa.42,44 It was reported that fgaPT2 in Aspergillus fumigatus is the homologous gene of dmaW, which was supported by the result that no ergot alkaloids could be detected in the fgaPT2 knockout strain while the production ability of the mutant could be restored when fgaPT2 was re-transformed.45 Similar phenomena were confirmed in C. fusiformis and Epichloë species.46 Unsöld et al. cloned fgaPT2 from A. fumigatus and expressed it in Saccharomyces cerevisiae, and the results showed that fgaPT2 is a soluble dimer with a molecular weight of 104 kDa.47 Sequence analysis showed that there was no assumed binding site of the enzyme for isoprene pyrophosphate, however, it could still use L-tryptophan and DMAPP as substrates to form DMAT. In this reaction, metal ions such as magnesium and calcium ions can increase the reaction rate.48 Sixteen new members of the DMATS family from other species have also been cloned, expressed and identified in recent years. Most of these enzymes only accept DMAPP as the donor of the isoprene group, and the prenylation often occurs at the C4 position of the substrates.49 The existence of a dimethylallyl cation intermediate in the aromatic prenyltransferase reaction could explain the mechanism of DMATS, which involves a discrete dimethylallyl cation intermediate or an associative mechanism in which the indole ring directly displaces diphosphate.50–52 The phylogenetic analysis indicated that dmaW of A. fumigatus has the same origin as those from clavicipitaceous fungi.53 The relationships of authentic dmaW genes also suggest that they originated from multiple gene duplications with subsequent losses of original or duplicate versions in some lineages.
The second reaction is the methylation of 4-DMAT, while the methyl group of S-adenosine methionine (SAM) is transferred to the amino nitrogen of DMAT under the catalysis of N-methyl transferase, generating N-methyl-dimethylallyltryptophan (Me-DMAT). The enzymes responsible for this reaction in C. purpurea and A. fumigatus are encoded by easF and fgaMT, respectively, and both of them are SAM-dependent methyltransferases. In addition, Rigbers et al. successfully expressed fgaMT in Escherichia coli, and the results suggest that recombinant FgaMT with a molecular weight of 38.1 kDa transfers DMAT into Me-DMAT in the presence of SAM in vitro without the participation of metal ions.54
The third reaction is the transformation of Me-DMAT to chanoclavine-I. Tudzynski et al. speculated that this process comprised at least three reactions, including cyclization, decarboxylation and hydroxylation.55 First, the bond linking C8 and C9 in Me-DMAT is desaturated, while the proton of C17 is lost, resulting in the formation of a diene. Subsequently, the rotation around the C8–C9 bond and the oxidation of C8–C7 occur to give an epoxide intermediate. Following this, the decarboxylation reaction of this intermediate occurs, accompanied by the formation of a new bond between C5 and C10. Ring opening of the epoxide occurs by proton attack, with the shift of the double bond between C9 and C10 to C8 and C9. In recent years, some laboratories have conducted detailed research on the enzyme responsible for the formation of chanoclavine-I in C. purpurea and A. fumigatus by gene knockout experiments. Lorenz et al. found that chanoclavine-I and other downstream products could not be detected in the easE (previously referred to as ccsA) deleted strain of C. purpurea P1, while the amount of 4-DMAT was increased. Meanwhile, the complementation of the ΔeasE mutant with its respective gene fragments resulted in the mutant indistinguishable from the wild-type strain in its capacity for ergot alkaloid biosynthesis. All of the above results suggest that EasE plays a distinct role in the formation of chanoclavine-I.55 The easE gene contains two exons plus one intron and is 1503 bp in length, and the encoded protein is 483 amino acids. EasE has a high similarity with redox enzymes from other fungi that produce ergot alkaloids, such as the homologous protein EasE in C. fusiformis (E values for e−160), EasE in Neotyphodium lolii, the genus being merged into Epichloë in 2014 56 (E values for e−118) and FgaOx1 in A. fumigatus (E values for e−96). The domain located in the 14–161 amino acids of the protein is determined to be the flavin adenine dinucleotide (FAD)-binding domain by sequence analysis, suggesting that EasE might work in a FAD-dependent manner.55 A similar conclusion was revealed by Goetz et al., who found that no chanoclavine-I could be detected in the fgaOx1 (the homologous gene of easE in C. purpurea) mutant of A. fumigatus.57 More importantly, the gene fgaCat in A. fumigatus (the homologous gene of easC in C. purpurea) is essential for the production of ergot alkaloids. The fgaCat deletion strain could not produce chanoclavine-I and other products such as festuclavine and fumigaclavine A, B and C, instead it accumulated the intermediate Me-DMAT.57 Thus, the results suggest that FgaCat and FgaOx1 are both required for the transformation of Me-DMAT to chanoclavine-I, and function as catalase and oxidoreductase, respectively. FgaCat might decompose the hydrogen peroxide produced by FgaOx1 in the oxidation reaction.57
The next reaction is the generation of chanoclavine-I-aldehyde by the redox of chanoclavine-I under the action of oxidoreductase. Wallwey et al. determined that the enzyme (designated as FgaDH) for A. fumigatus is composed of 261 amino acids with a molecular weight of approximately 27.8 kDa and contains conserved motifs of short-chain dehydrogenase/reductases (SDRs). The oxidation and dehydrogenation of chanoclavine-I were catalyzed by FgaDH in vitro in the presence of NAD+.58 The homologous enzyme (designated as EasD) of FgaDH has been discovered in Claviceps.1 In addition, the genome sequence analysis of Arthrodermataceae revealed the presence of a gene cluster consisting of five genes with high similarity to those involved in the biosynthesis of chanoclavine-I aldehyde in A. fumigatus and C. purpurea. The gene ARB_04646 in Arthroderma benhamiae was expressed in E. coli, and the results showed that the encoded enzyme catalyzed the oxidation of chanoclavine-I in the presence of NAD+, resulting in the formation of chanoclavine-I aldehyde.59 Thus, this gene as the homologous gene of easD and fgaDH also functioned as the oxidoreductase. At the present time, the four reactions described above constitute the common biosynthetic pathway of ergot alkaloids in different fungal species (Fig. 2).
3.2 Biosynthetic pathway of ergoclavines
Chanoclavine-I-aldehyde is proposed as a branch point via festuclavine or pyroclavine to clavine-type alkaloids in Aspergillus and Penicillium, and via agroclavine to ergoamides and ergopeptines in Claviceps.60 The disruption of fgaOx3 in A. fumigatus, a gene predicted to encode a flavin-dependent oxidoreductase of the old yellow enzyme class, led to the accumulation of the upstream product chanoclavine-I-aldehyde. The complementation of the fgaOx3 mutant with a wild-type allele restored the original profile of ergot alkaloids. The results suggest that FgaOx3 is required for the incorporation of chanoclavine-I-aldehyde into more complex ergot alkaloids.61 In addition, FgaOx3 reduces the double band between C8 and C9 of chanoclavine-I aldehyde, and the reduction facilitates an intramolecular reaction between the aldehyde and the amine moieties to allow the formation of festuclavine, which was recently confirmed by the crystal structure resolution of the FMN-loaded FgaOx3.60,62,63 Wallwey et al. reported that the oxidoreductase FgaFS in A. fumigatus reduced the cyclic iminium product formed by FgaOx3 to produce festuclavine.64 In an in vitro reaction system, festuclavine was formed when chanoclavine-I aldehyde was incubated with the recombinant FgaOx3 and FgaFS simultaneously or as a tandem-reaction with FgaOx3 before FgaFS in the presence of NADH (Fig. 3).64 In Penicillium, chanoclavine-I-aldehyde is usually converted to pyroclavine. It was reported that FgaOx3 and FgaFS from P. commune catalyzed the formation of pyroclavine and a small amount of festuclavine (Fig. 3). More importantly, a detailed inspection of the HPLC chromatogram of the incubation mixture with FgaOx3 and FgaFS from A. fumigatus also revealed the presence of a minor product pyroclavine.65 The two products both can be produced by using different combinations of FgaOx3 and FgaFS from A. fumigatus and P. commune,65 and the ratio of pyroclavine to festuclavine was controlled by the reduction of an imine intermediate catalyzed by FgaFS (Fig. 3).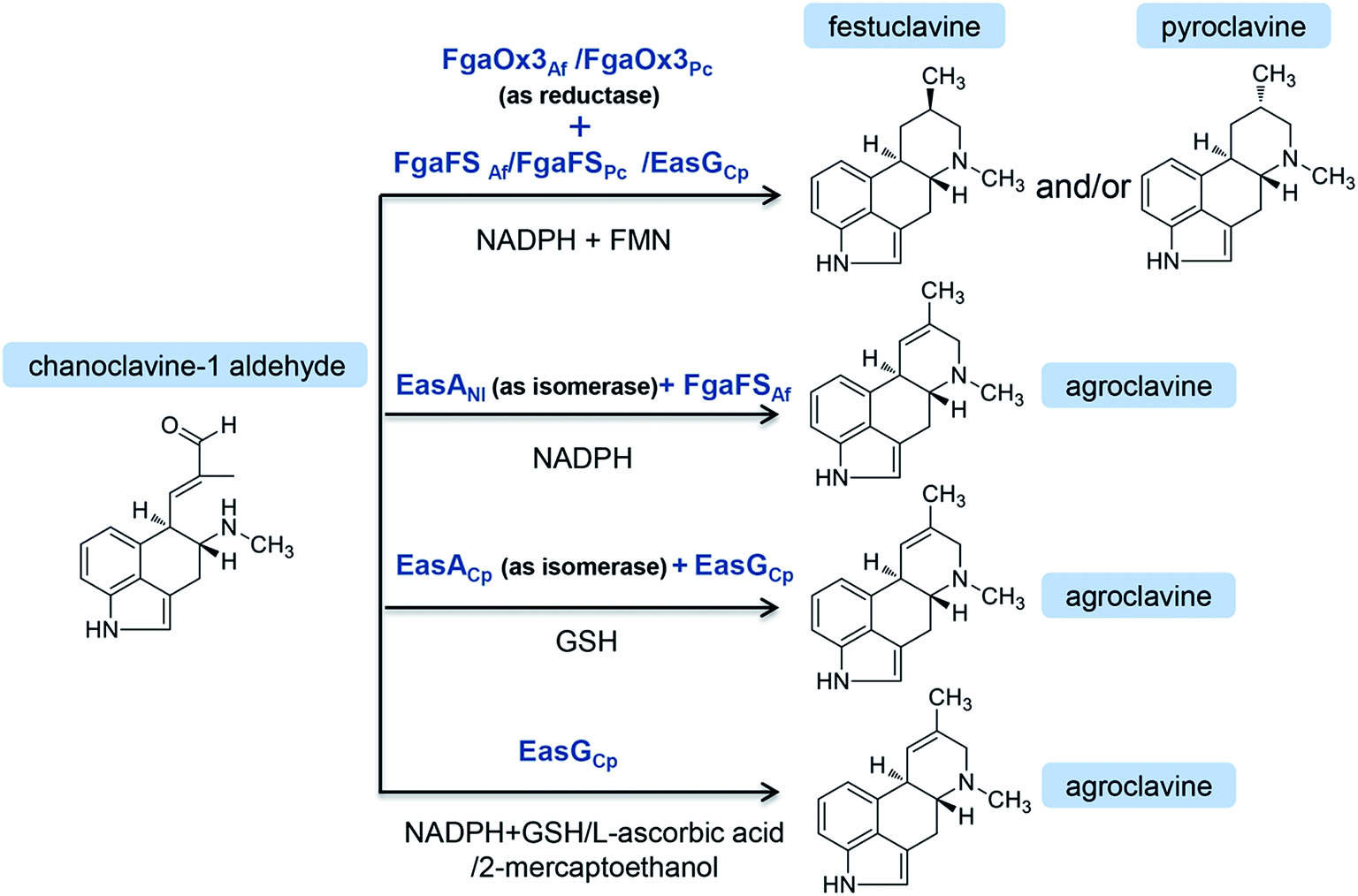 | ||
| Fig. 3 In vitro reaction from chanoclavine-I-aldehyde to festuclavine and/or pyroclavine and agroclavine catalyzed by the recombinant EasA and EasG from different strains in varied combinations. | ||
On the other hand, chanoclavine-I-aldehyde is usually converted to agroclavine in C. purpurea (Fig. 4). The structural difference between festuclavine and agroclavine is whether there is a double bond between the C8 and C9 positions. The homologs of FgaOx3 (designated as EasA) and FgaFS (designated as EasG) in C. purpurea comprise 290 and 369 amino acids, respectively, with molecular weights of 31.9 and 41.5 kDa.66 Cheng proposed that EasA controlled the conversion of chanoclavine-I aldehyde to festuclavine or agroclavine. In the presence of NADPH, festuclavine was observed when FgaOx3Af and FgaFSAf were incubated with chanoclavine-I aldehyde, which is consistent with Wallywey's results mentioned above.64,67 In contrast, agroclavine was also yielded when chanoclavine-I aldehyde was incubated with EasANl from Epichloë lolii (the homolog of FgaOx3 in A. fumigatus) and FgaFSAf (Fig. 3) simultaneously. The results indicated that FgaOx3Af acted as a reductase and EasANl acted as an isomerase. The mutational analysis suggests that Tyr to Phe mutation of EasA is responsible for this switch in activity from a reductase (as found in A. fumigatus) to an isomerase (as found in N. lolii).67 In addition, it was reported that in the presence of reduced glutathione (GSH), chanoclavine-I aldehyde was converted to agroclavine by the recombinant EasA and EasG from C. purpurea. Most surprisingly, they found that only EasG itself could catalyze the conversion of chanoclavine-I aldehyde to agroclavine in the presence of reduced GSH and NADPH in vitro. GSH and NADPH could be replaced by NADH and other thiol compounds, such as 2-mercaptoethanol and dithiothreitol, indicating that EasG is sufficient for the conversion of chanoclavine-I aldehyde to agroclavine without EasA in vitro (Fig. 3).66 In this reaction, EasG possibly serves as an oxidoreductase to reduce the Schiff base.
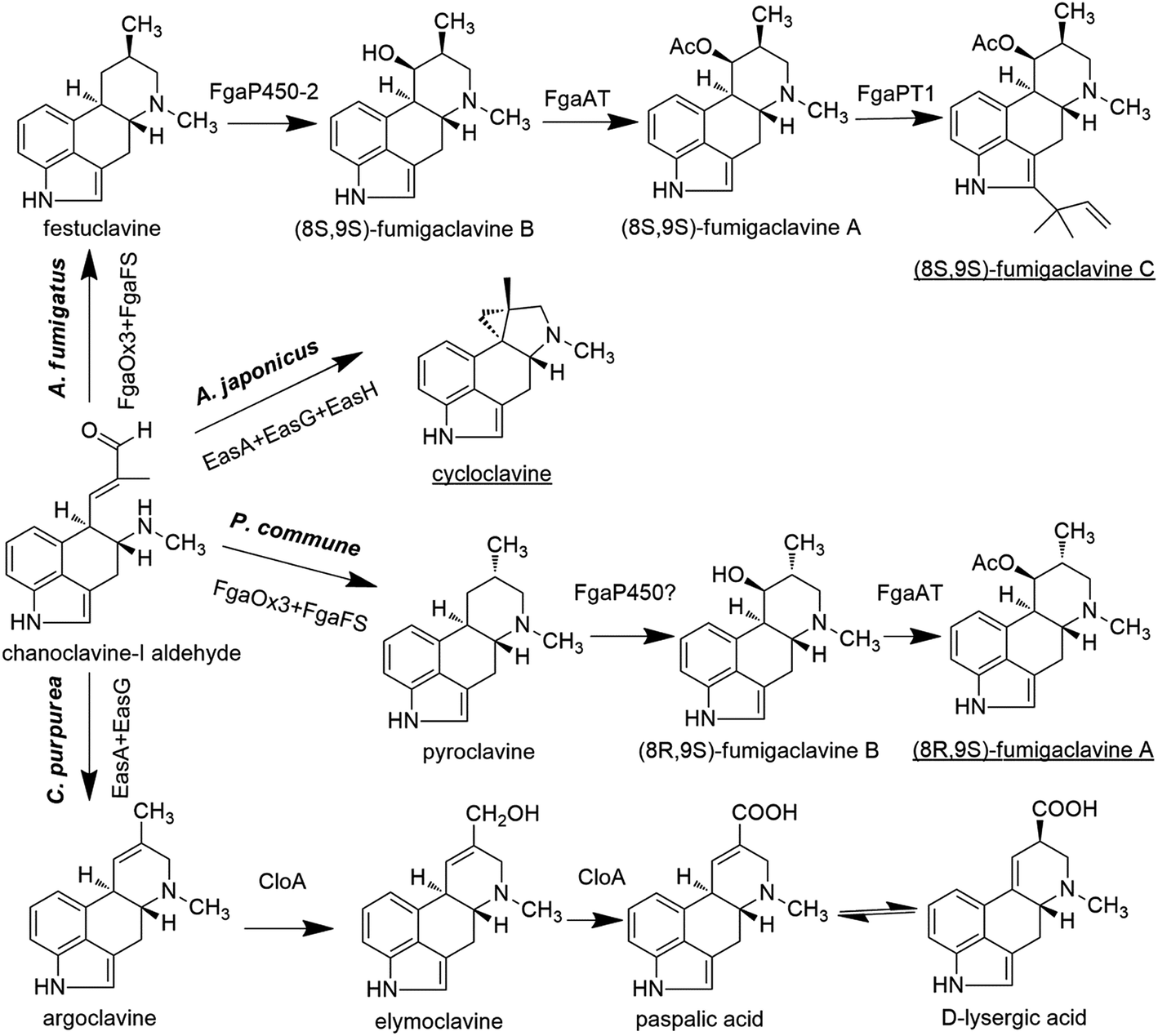 | ||
| Fig. 4 The biosynthetic pathways of clavine-type alkaloids or D-lysergic acid from the branch point chanoclavine-I aldehyde in different species. The end products in different species are underlined. | ||
In A. fumigatus, festuclavine was transformed to the final product fumigaclavine C by some enzymes.68 Compared with festuclavine, a prenyl group and acetyl groups are present at the C2 and C9 of fumigaclavine C, respectively. Thus, the formation of fumigaclavine C is proposed to occur through hydroxylation, acetylation and prenylation. It is reported that only festuclavine was produced in the fgaP450-2 (also refered as easM) deletion mutant. Complementation with fgaP450-2 gene restored the ability of the fungus to produce the downstream compounds.69 The results suggest that festuclavine is converted to (8S,9S)-fumigaclavine B by the hydroxylase FgaP450-2 (EasM). The acetyl group provided by acetyl coenzyme A is further transferred to the C8 of (8S,9S)-fumigaclavine B by acetyltransferase FgaAT, generating (8S,9S)-fumigaclavine A, and then (8S,9S)-fumigaclavine A is prenylated by prenyltransferase with DMAPP as the prenyl group donor, to form the final product (8S,9S)-fumigaclavine C.70 The prenyltransferase gene fgaPT1 has been identified in A. fumigatus AF293. FgaPT1 was demonstrated to cause the formation of a C–C bond between the C-3 of the prenyl group from DMAPP and the C-2 of the indole nucleus and therefore to function as a reverse prenyltransferase.70,71 The enzyme shares high sequence similarity with DMATS which functions as a prenyltransferase in the first pathway-specific step of the ergot alkaloid biosynthesis in different fungi, such as C. purpurea, C. fusiformis, Neothyphodium coenophialum, Balansia obtecta, and A. fumigatus AF293. Thus, it can be concluded that FgaPT1 is evolved from a regular prenyltransferase in fungi.
Cycloclavine contains an unusual cyclopropyl moiety, and this alkaloid has recently been found in A. japonicus (Fig. 4).72 The analysis of the A. japonicus genome revealed an EAS gene cluster, in which most of the genes are homologous to those involved in the biosynthesis of festuclavine or agroclavine in other filamentous fungi. Upon the incubation of chanoclavine-I aldehyde with cofactors and enzymes heterologously expressed from E. coli (EasAAj, EasGAj) and yeast (EasHAj), the formation of cycloclavine and festuclavine was detected. EasH acted as a hydroxylase in this reaction. Most interestingly, the manipulation of the EasAAj/EasGAj/EasHAj ratio controlled the contents of cycloclavine and festuclavine.72
In Penicillium, the final product is (8R,9S)-fumigaclavine A, resulted from a subsequent reaction via the intermediates 8R-pyroclavine and (8R,9S)-fumigaclavine B (Fig. 4).8 Compared with the structures of the clavine-type final products in Aspergillus and Penicillium, the differences are the prenyl group present or absent at the C2 position and the stereochemistry of the C8. Fumigaclavine A is the final product in Penicillium due to the lack of a specific prenyltransferase FgaPT1 that exists in A. fumigatus.71
In summary, the early stages of the ergot alkaloid pathway (up to chanoclavine-I aldehyde) are shared by different fungi, including Claviceps, Aspergillus and Penicillium, whereas the later stages of the pathway are different (Fig. 4).
3.3 Biosynthetic pathway of D-lysergic acid derivatives
Four lps genes have been identified from the C. purpurea P1 strain, which has been supported by the genome sequencing of C. purpurea 20.1.12,13,75,78 Correia et al. identified the structure and function of LPSB (formerly known as LPS2) by transcriptional analysis, gene disruption and heterologous expression. LPSB is a mono-modular NRPS containing three domains in the order of A-T-C, with a molecular weight of 140 kDa, and it is encoded by lpsB. LPSB is responsible for the D-lysergic acid activation and incorporation into the D-lysergic acid amide or ergopeptine backbone. Ergopeptines were undetectable in lpsB gene deletion strain, whereas the accumulation of D-lysergic acid was clearly detected.12 LPSA is a triple-modular NRPS catalyzing ergopeptide formation. Two LPSA variants were found, namely, LPSA1 (370 kDa, formerly known as LPS1) and LPSA2 (370 kDa, formerly known as LPS4), encoded by lpsA1 and lpsA2, respectively.79 The phylogenetic analysis revealed that the first and third AT-didomains in the module 1 and module 3 of the LPSA-type enzymes were likely evolved from a common ancestor, or that the module 3 was derived from the module 1 by gene duplication. In contrast, the AT-didomain of module 2 was phylogenetically distant from those of the modules 1 and 3.69 Most interestingly, the phylogenetic tree also exhibited that both the C domains of the modules 1 and 3 were clustered into the same clade, whereas the C domain from the module 2 was attributed to another clade. All of the above results suggest that the modules of LPSs in the Claviceps species are organized as A–T–C units but not canonical C–A–T units (Fig. 5).75 The ergopeptine biosynthesis is initiated by the binding of D-lysergic acid to the T domain of LPSB as a thioester followed by the transfer of the thioester to the triple-modular LPSA1 or LPSA2, where three successive condensations into the D-lysergyl mono-, di-, and tripeptide thioester intermediates occur, with the final release of the end product D-lysergyl tripeptide lactam (ergopeptam).12,75 In this process, the LPSB and LPSA are probably in the form of an LPSB/LPSA complex. Haarmann et al. reported that the mutant lacking the lpsA1 gene was incapable of producing ergotamine but still able to produce α-ergocryptine, suggesting that LPSA1 is involved in the synthesis of ergotamine in the C. purpurea P1 strain and that the three A domains of LPSA1 are responsible for the activation of alanine, phenylalanine, and proline, respectively.78 This result also implies that LPSA2 is responsible for the biosynthesis of α-ergocryptine.78,79 However, this speculation is questionable, because it cannot explain how one strain can sometimes produce more than two ergopeptides. Besides the variation in the modules of the LPSs, the promiscuity of the substrate amino acids for LPSs should be considered. Therefore, the accurate relationships between the different ergopeptines and LPSA1 or LPSA2, i.e., the product specificities of LPSA1 and LPSA2, need to be further investigated.
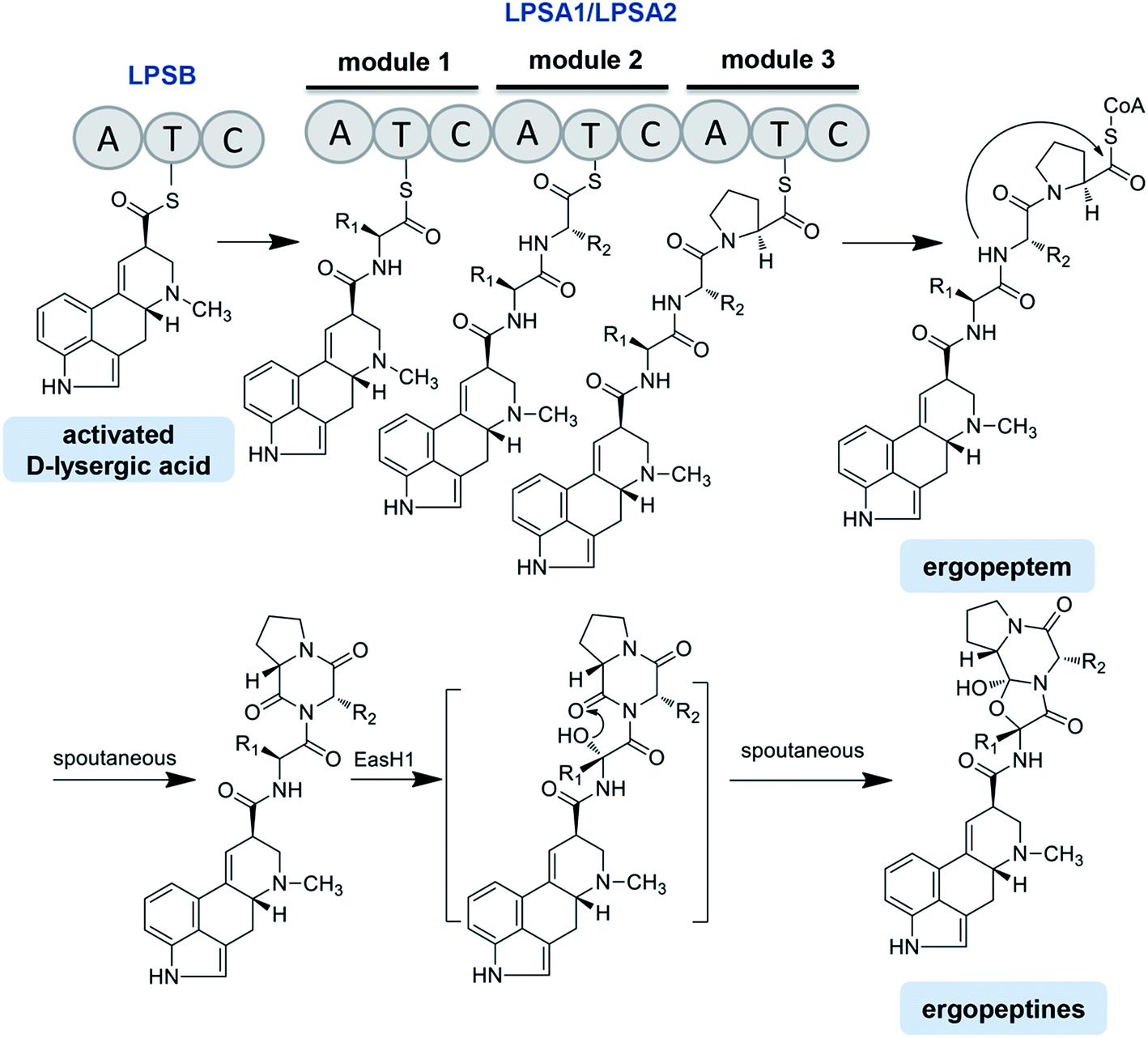 | ||
| Fig. 5 Ergopeptine biosynthesis in Claviveps. Note: R1 and R2 represent different groups. | ||
After releasing from the corresponding LPS, ergopeptam is oxidized to form ergopeptine under the action of a P450 monooxygenase named EasH1, with a molecular weight of 35 kDa. The only difference between the ergopeptam and the corresponding ergopeptine is that the former lacks a cyclol group between the α-C of the first amino acid and the carbonyl group of the third amino acid proline. Havemann and Keller carried out a detailed study of this process.80 The results showed that EasH1 acted as an oxygenase or a hydroxylase dependent on the presence of Fe2+/2-ketoglutarate (Fig. 5). EasH1 introduced a hydroxyl group into the D-lysergyl tripeptide lactam at the α-C of the aminoacyl residue followed by the spontaneous condensation with the terminal lactam carbonyl group. The sequence analysis revealed that EasH1 belonged to the wide and diverse family of phytanoyl coenzyme A hydroxylases.80
The C. purpurea Ecc93 strain, an ergocristine producer, can also produce a smaller amount of ergometrine. Ortel and Keller discovered the enzyme (designated as LPSC or LPS3) responsible for ergometrine synthesis.75 LPSC is a monomodular NRPS with a molecular weight of 180 kDa and contains not only an A domain, T domain and C domain but also a C-terminal reductase domain (Red/R domain). The R domain acts as a reductase with homology to the C-terminal Red-domains of other NRPS members. This R domain is capable of catalyzing the electron reduction of the respective carrier protein-bound thioester, thereby releasing the former intermediate as an alcohol-type product.75,81 As shown in Fig. 6, D-lysergic acid is attached to alanine under the functions of LPSB and LPSC, which are responsible for the D-lysergic acid and alanine activation, respectively. Finally, ergometrine is released in the presence of NADPH. It was speculated that the C domain of LPSB is responsible for the amide bond formation between the T domain-bound D-lysergic acid and alanine. However, according to the blasting result LPSC also harbors a C domain whose function is uncertain. Crocacins are natural antifungal compounds produced by myxobacteria. These compounds are most likely derived from an NRPS/PKS hybrid biosynthetic pathway. Its biosynthesis involves an unusual hydrolytic release domain similar to the C domain of LPSC.81 Consequently, the C domain of LPSC together with its R domain may catalyze the release of the final product but not catalyze the amide bond formation.
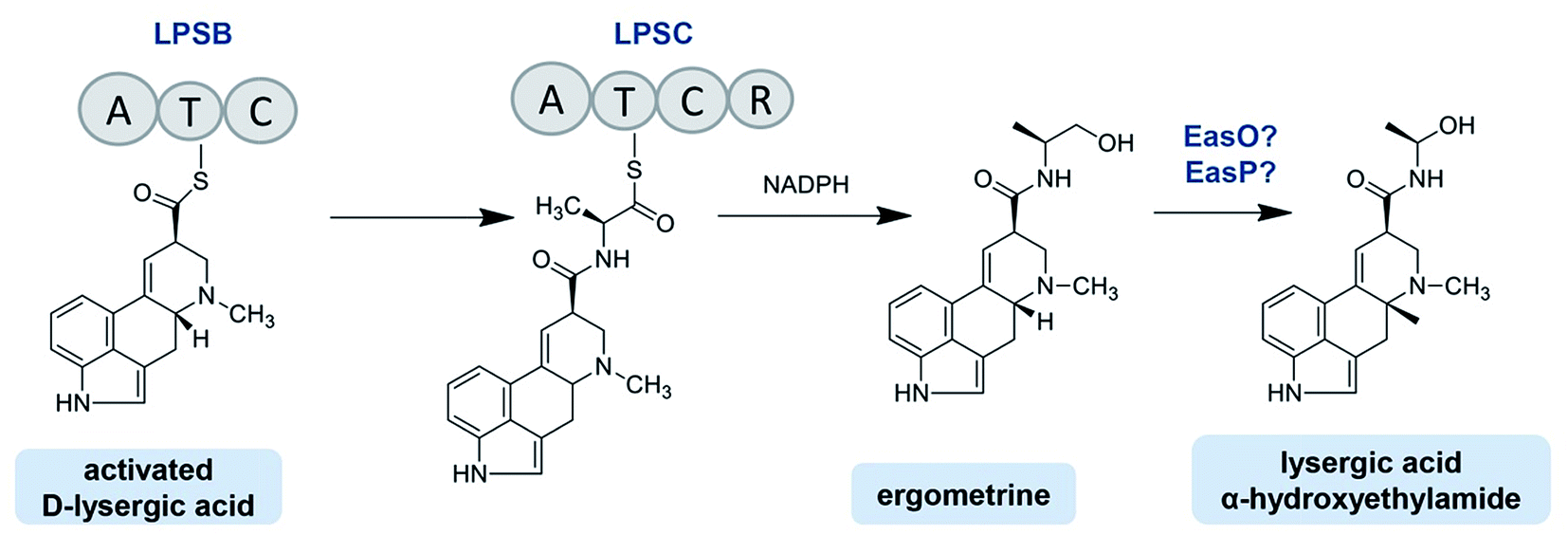 | ||
| Fig. 6 Ergoamide biosynthesis in Claviveps. | ||
The lpsC genes were also cloned from the C. purpurea D1 and P1 strains, and the two nucleotide sequences share up to 90% identities with that of the Ecc93 strain. However, the D1 and P1 strains mainly produced ergopeptines and only minimal ergometrine. The RT-PCR analysis showed that the lpsC expressions occurred at the transcriptional level in all strains, but the LPSC proteins in the D1 and P1 strains were not detected by the antibody prepared with the partial LPSC fragment from the C. purpurea Ecc93 strain, which may be the reason why ergometrine was not detected in the two strains.75
C. paspali mainly produces simple lysergic acid derivatives such as lysergic acid α-hydroxyethylamide and ergometrine. The enzymes EasO and EasP in C. paspali are speculated to be involved in the biosynthesis of lysergic acid α-hydroxyethylamide (Fig. 6). The sequence analysis indicated that easO encodes a flavin-binding monooxygenase (EasO) that may oxidize the α-carbon of the alanine-derived residue in the ergometrine precursor attached to the LpsB/LpsC complex, while easP encodes an α/β hydrolase-fold enzyme (EasP), which could be involved in the subsequent hydrolysis to release lysergic acid α-hydroxyethylamide.13,82
4. Gene cluster for ergot alkaloid biosynthesis
In fungi and bacteria, the genes involved in the biosynthesis of the corresponding secondary metabolites are generally located on the chromosome with a continuous and clustered manner. This facilitates the identification of the genes responsible for the biosynthesis of certain compounds. Tudzynski et al. explored the adjacent gene sequences of dmaW using chromosome walking and found an ergot alkaloid synthesis (EAS) gene cluster located at the 3′ end of dmaW in the C. purpurea P1 strain.83 Then, a total of 11 genes were determined in which easA and lpsA1 were located on the left and right flanks of dmaW, respectively. Subsequently, Harrmann et al. discovered another two new NPRS members (lpsC and lpsA2). The length of the gene cluster was eventually confirmed to be 68.5 kb, containing a total of 14 genes (Fig. 7),79 whose functions have been characterized. The core genes involved in the two clusters of C. purpurea P1 (ergotamine producer) and C. purpurea ECC93 (ergocristine producer) are highly conserved. Yet the NRPS genes are various, which may contribute the various ergot alkaloid spectra among the two strains except for the promiscuity of the substrate amino acids for the LPSs. Lorenz et al. screened the positive clone containing fragment contigs from the C. fusiformis SD58 plasmid library by using the cDNA of dmaW as a probe43. After sequencing these contigs, they found a sequence with a size of 19.6 kb containing 9 homologous genes of the C. purpurea EAS gene cluster (Fig. 7).84 This gene cluster in C. fusiformis does not contain the homologous genes of lpsC, lpsA1 and lpsA2 of C. purpurea. The CloA and LPSB in C. fusiformis have lost their functions although the coding genes (cloA and lpsB) were expressed normally. Consequently, only ergoclavines such as agroclavine and elymoclavine are produced by C. fusiformis84. The gene cluster of C. hirtella has also been investigated. This strain mainly produces ergoclavines together with a small amount of ergometrine (3–8% of total ergot alkaloids).85 The analysis of its EAS gene cluster indicated that no homologous genes of lpsA1 and lpsA2 but only genes of lpsB and lpsC existed in it.86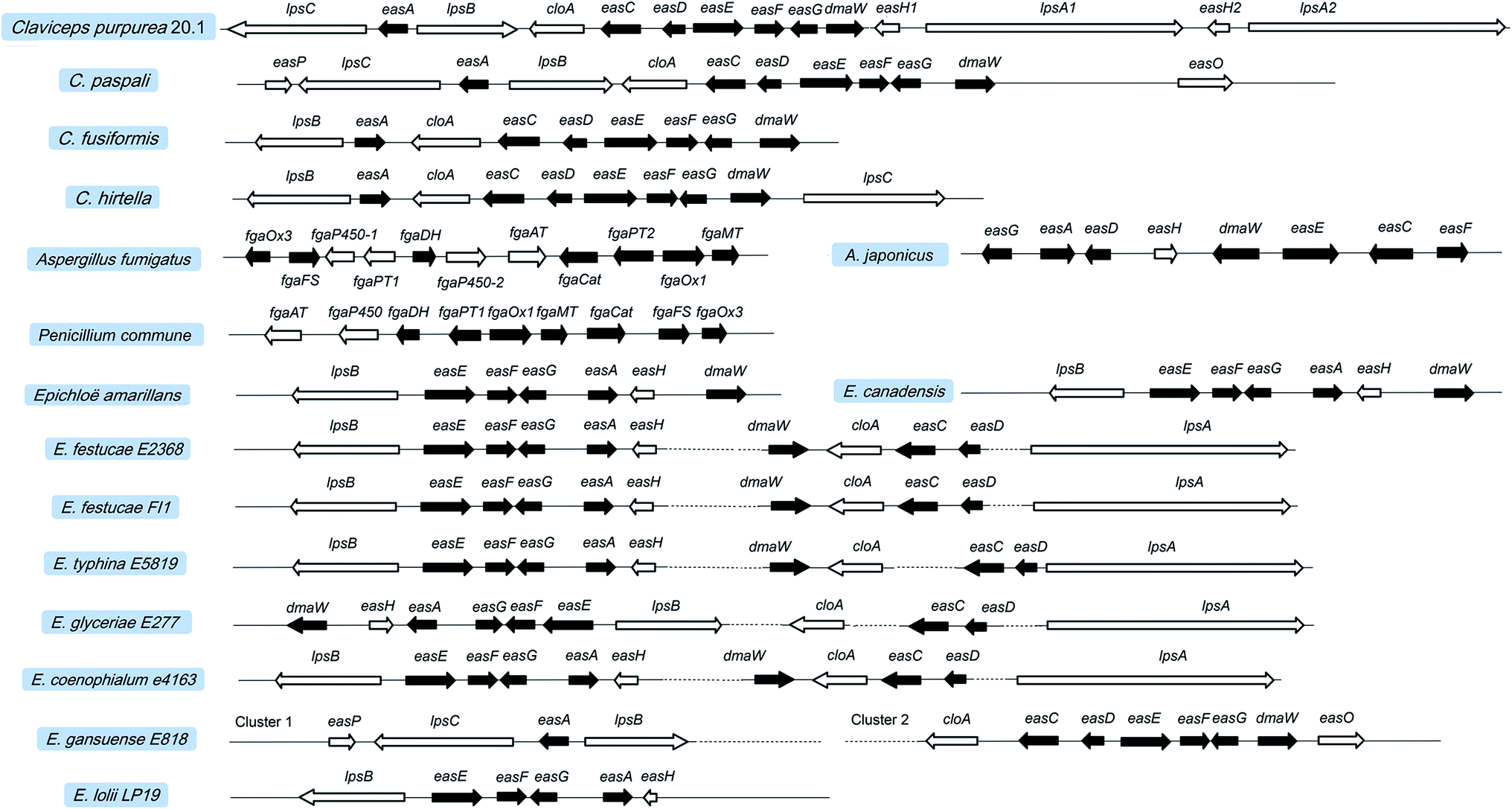 | ||
| Fig. 7 Ergot alkaloid synthesis (EAS) gene clusters in Claviceps, Aspergillus, Penicillium, and Epichloë. Black boxes represent the homologous genes between different species. | ||
Recently, the EAS gene cluster with a length of 27 kb from A. fumigatus Af293 has also been identified by the DNA blast with the gene sequence of dmaW from C. purpurea. This cluster is located on chromosome 2 and contains 11 genes 45 in which seven homologous genes with 46–66% identities were found in both A. fumigatus Af293 and C. purpurea. As shown in Fig. 7, the homologous genes of A. fumigatus, P. commune and C. purpurea are involved in the early stage of the ergot alkaloid biosynthetic pathway (up to agroclavine). Other than ergopeptines being produced in Claviceps, fumigaclavines are the final products of A. fumigatus7 and P. commune.8 The specific gene fgaPT1 located 10 kb away from fgaPT2 in the genome of A. fumigatus Af293 was found to catalyze the last reaction step in fumigaclavine C biosynthesis.71 In contrast, no fgaPT1 gene was found in the EAS gene cluster of P. commune, leading to the production of fumigaclavine A which lacks the prenyl group at the C2 position. In addition, the EAS cluster of A. japonicus has been found to be 16.8 kb in length and contain 8 genes (Fig. 7), among which seven (dmaW, easF, easE, easC, easD, easA, easG) are homologous to those previously found in other filamentous fungi, leading to the formation of festuclavine. The easH in A. japonicus is highly identical to the easH1 in C. purpurea, leading to the formation of cycloclavine which contains a cyclopropyl moiety.72
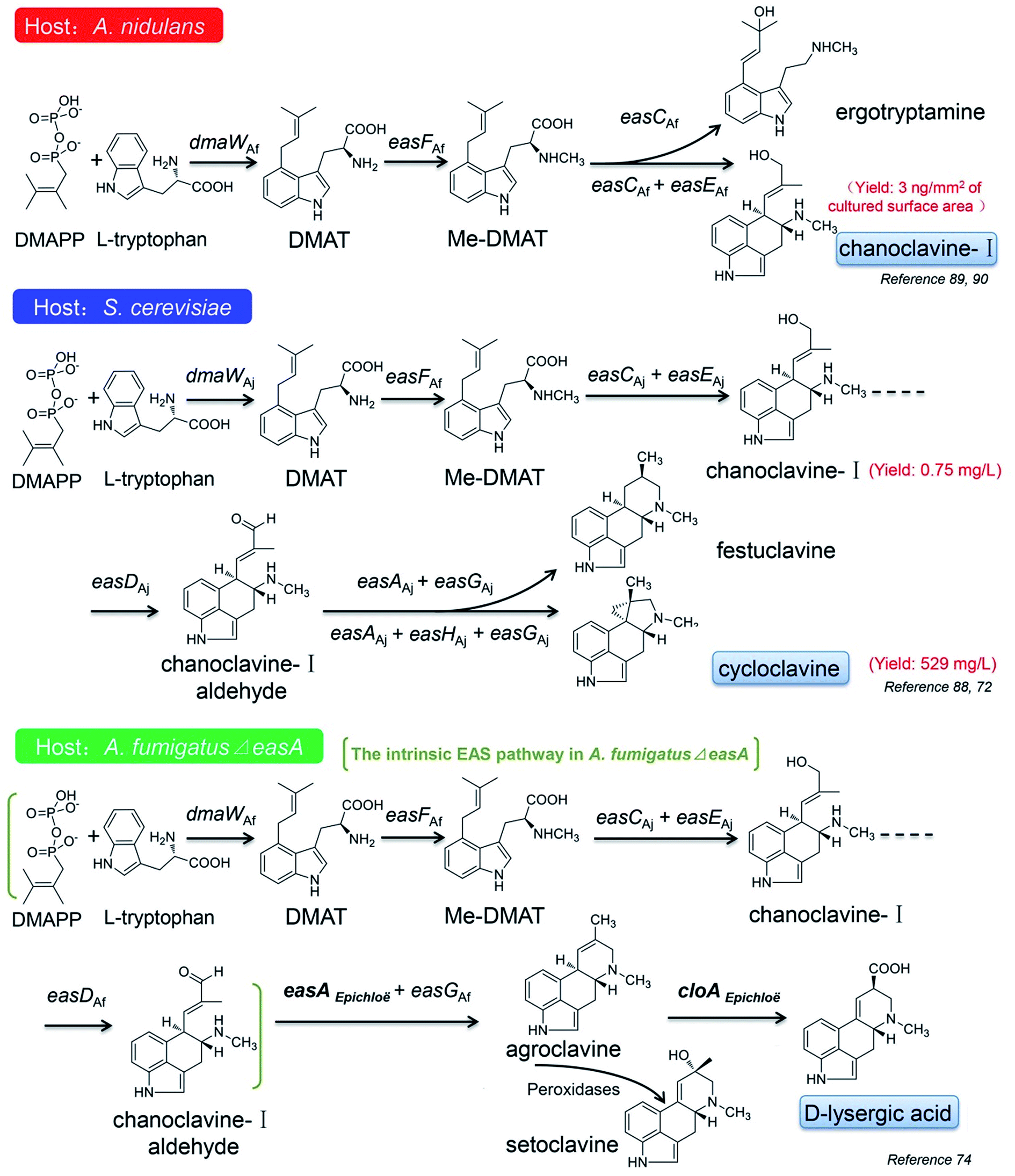 | ||
| Fig. 8 The constructed ergot alkaloid biosynthetic pathways in different hosts. | ||
The Epichloë EAS clusters are also available in the NCBI database, such as those of Epichloë festucae Fl1, E. glyceriae E277, E. typhina E5819 and E. festucae E2368. However, the distances between some genes in the EAS cluster are quite long. For example, in E. festucae E2368 easH is almost 25 kb far away from dmaW, and easD is almost 15 kb far away from lpsA. Due to the lack of lpsC, the aforementioned strains mainly produce ergovaline but not ergometrine or lysergic acid α-hydroxyethylamide. Another advance is the discovery of the EAS clusters in Neotyphodium (Epichloë) species. The principal alkaloids produced by E. gansuense E818 are simpler lysergyl amides, including high levels of ergometrine and low levels of lysergic acid α-hydroxyethylamide.13 Its EAS cluster is arranged similarly to that of Claviceps, although it is separated into two parts. The lack of lpsA1 and lpsA2 makes the strain as a producer of ergometrine. Like C. paspali, the easO and easP present in the EAS cluster of E. gansuense E818 make it produce lysergic acid α-hydroxyethylamide. The EAS gene cluster of E. coenophialum E4163 contains 11 genes. The arrangement order and physical distance of genes are extremely similar to those of Epichloë. Therefore, ergovaline is speculated to be its product, and its pharmacological activity needs to be evaluated. In addition, E. lolii Lp19 mainly produces ergovaline. However, the reported EAS cluster of E. lolii Lp19 in NCBI database only contains 6 genes.87 Compared with the EAS clusters of other Epichloë spieces, it is possible that the other genes (dmaW, cloA, easC, easD and lpsA) also exist in the EAS cluster of N. lolii Lp19 but have not been found due to the long distance between dmaW and easH. Overall, the biosynthetic pathways of ergot alkaloids in different species are summarized in Fig. S1.†
5. Production of ergot alkaloids by synthetic biology or metabolic engineering
In recent years, the applications of synthetic biological techniques provide probabilities for mass production of natural pharmaceutical products or their precursors in cell factories, which leads to a new era of biotechnology. Some progress have also been made in the construction of the EAS pathways in different heterologous hosts, such as Aspergillus nidulans, A. fumigatus and S. cerevisiae, to produce some intermediates or novel ergot alkaloids derivatives.72,74,88–90The first attempt for the construction of the ergot alkaloid biosynthetic pathway was taken in the A. nidulans host. A. nidulans as a model fungus has a very close genetic relationship with A. fumigatus, but it does not produce ergot alkaloids due to the absence of the EAS gene cluster, which makes it a suitable host for observing the effectiveness of the engineered EAS pathway. The partial EAS gene cluster of A. fumigatus, which harbored dmaW, easF, easC and easE, was introduced into the A. nidulans host, and with the intrinsic DMAPP and L-tryptophan as the substrates, the engineered mold acquired the ability to produce chanoclavine-I, although the yield was only 3 ng mm−2 of the culture surface area (Fig. 8).90 If the gene cluster only harbored dmaW, easF and easC, the recombinant strain produced ergotryptamine instead of chanoclavine-I. The role of ergotryptamine as a pathway intermediate or byproduct was tested by feeding ergotryptamine to A. fumigatus easC knockout (ko) and A. fumigatus easE ko strains. Although both the strains contain downstream pathway enzymes, they failed to turn over ergotryptamine, indicating that ergotryptamine is not the precursor of chanoclavine-I.89
The similar work has also been done in the S. cerevisiae host (Fig. 8).72,88 The four EAS genes of dmaW (A. japonicas origin), easF (A. fumigatus origin), easC (A. japonicas origin) and easE (A. japonicas origin) were combined in the host and the engineered yeast produced little amount of chanoclavine-I.88 The chanoclavine-I amount was increased to 0.75 mg L−1 when the recombinant strain overexpressed the molecular chaperones PDI1 and ERO1, which may improve the folding of EasE.88 The pathway was further stretched to cycloclavine after easD (A. japonicas origin), easA (A. japonicas origin), easH (A. japonicas origin) and easG (A. japonicas origin) were introduced into the engineered yeast. By increasing the copy number of dmaW, easC, easD, easH, easG, pdi1 and fad1, the cycloclavine yield was further increased to 529 mg L−1.72 However, when easH was not involved in the aforementioned pathway, the recombinant strain produced another product festuclavine (Fig. 8). It is worth mentioning that cycloclavine is the first relatively more complicated ergot alkaloid successfully produced by the engineered yeast (Fig. 8).
Robinson and Panaccione74 even introduced easA (functioned as an isomerase) from Epichloë species into A. fumigatus ΔeasA (also referred as fgaOx3) mutant, which harbored the intrinsic EAS pathway from DMAPP to chanoclavine-I aldehyde, and instead of the intermediate product festuclavine, agroclavine together with its oxidation product setoclavine was yielded, and the former is also one of the key EAS intermediates found in Claviceps species (Fig. 4). Then the cloA gene from Epichloë species was added to the pathway, and the engineered mold acquired the ability to produce D-lysergic acid (Fig. 8).74
6. Prospective
Studies on the biosynthetic pathway of ergot alkaloids have achieved great progress. So far, most of the related enzymes involved in the biosynthetic pathway of ergot alkaloids are clearly elucidated. These breakthroughs provide a better understanding of the biosynthesis of ergot alkaloids, and make it possible to improve the yields of ergot alkaloids through the enzyme regulation. At present, the production of ergot alkaloids with various pharmacological activities is mainly based on the liquid fermentation of the selected Claviceps strains. However, these selected Claviceps strains, especially high producers, are generally unstable and apt to degeneration during the cultivation and preservation process. Moreover, the components of the alkaloids are usually complex, which leads to the increase in the cost of isolation and purification. Therefore, the alternative approaches need to be developed. The booming synthetic biology may solve these problems, which is based on the reconstruction of the EAS pathway in an engineered microbial cell factory. Currently, the partial biosynthetic pathways of ergot alkaloids have been constructed in the cell factories and the corresponding intermediates are produced. However, the yields of these intermediates are quite low. Furthermore, these intermediates are much simple and far from the final products on the biosynthetic pathway. More efforts are required to improve the productivity, which include further optimizing the EAS pathway, genome editing, improving the enzyme activities and fitness in the engineered host, and increasing the precursor supply. There is still a long way to realize the efficient and large-scale productions of ergot alkaloids or their novel derivatives in the cell factories by the metabolic engineering.Acknowledgements
This work was supported by the Fundamental Research Funds for the Institute of Materia Medica (Grant No. 2015CX07), Fundamental Research Funds for the Central Universities (Grant No. 2016ZX350018), National Natural Science Foundation of China (Grant No. 81603002) and CAMS Innovation Fund for Medical Sciences (CIFMS) (Grant No. 2016-I2M-2-002).References
- C. Wallwey and S.-M. Li, Nat. Prod. Rep., 2011, 28, 496–510 RSC.
- C. De Costa, Lancet, 2002, 359, 1768–1770 CrossRef.
- A. N. de Groot, P. W. van Dongen, T. B. Vree, Y. A. Hekster and J. van Roosmalen, Drugs, 1998, 56, 523–535 CrossRef CAS PubMed.
- T. Haarmann, Y. Rolke, S. Giesbert and P. Tudzynski, Mol. Plant Pathol., 2009, 10, 563–577 CrossRef CAS PubMed.
- M. Lee, J. Roy. Coll. Phys. Edinb., 2009, 39, 365–369 CrossRef CAS.
- M. Lee, J. Roy. Coll. Phys. Edinb., 2009, 39, 179–184 CAS.
- H. M. Ge, Z. G. Yu, J. Zhang, J. H. Wu and R. X. Tan, J. Nat. Prod., 2009, 72, 753–755 CrossRef CAS PubMed.
- N. Vinokurova, S. Ozerskaya, B. Baskunov and M. Arinbasarov, Microbiology, 2003, 72, 149–151 CAS.
- M. Flieger, M. Wurst and R. Shelby, Folia Microbiol., 1997, 42, 3–30 CrossRef CAS.
- H. Hulvová, P. Galuszka, J. Frébortová and I. Frébort, Biotechnol. Adv., 2013, 31, 79–89 CrossRef PubMed.
- N. Flieger, P. Sedmera, J. Vokoun, Z. Řeháček, J. Stuchlík, Z. Malinka, L. Cvak and P. Harazim, J. Nat. Prod., 1984, 47, 970–976 CrossRef.
- T. Correia, N. Grammel, I. Ortel, U. Keller and P. Tudzynski, Chem. Biol., 2003, 10, 1281–1292 CrossRef CAS PubMed.
- C. L. Schardl, C. A. Young, U. Hesse, S. G. Amyotte, K. Andreeva, P. J. Calie, D. J. Fleetwood, D. C. Haws, N. Moore and B. Oeser, PLoS Genet., 2013, 9, e1003323 CAS.
- R. H. Du, E. G. Li, Y. Cao, Y. C. Song and R. X. Tan, Life Sci., 2011, 89, 235–240 CrossRef CAS PubMed.
- R. H. Du, S. Y. Qin, L. S. Shi, Z. Q. Zhou, X. Y. Zhu, J. Liu, R. X. Tan and W. Cao, Atherosclerosis, 2014, 234, 120–128 CrossRef CAS PubMed.
- W. Guo, S. Hu, F. Shao, R. Ren, W. Liu, W. Zhang, X. Wang, R. Tan, Q. Xu and Y. Sun, J. Pharmacol. Sci., 2015, 129, 101–106 CrossRef CAS PubMed.
- Y.-X. Li, S. Himaya, P. Dewapriya, C. Zhang and S.-K. Kim, Mar. Drugs, 2013, 11, 5063–5086 CrossRef CAS PubMed.
- X.-F. Wu, M.-J. Fei, R.-G. Shu, R.-X. Tan and Q. Xu, Int. Immunopharmacol., 2005, 5, 1543–1553 CrossRef CAS PubMed.
- Y. Zhao, J. Liu, J. Wang, L. Wang, H. Yin, R. Tan and Q. Xu, J. Pharm. Pharmacol., 2004, 56, 775–782 CrossRef CAS PubMed.
- G. Jourdan, P. Verwaerde, A. Pathak, M. A. Tran, J. L. Montastruc and J. M. Senard, Fundam. Clin. Pharmacol., 2007, 21, 45–53 CrossRef CAS PubMed.
- J. R. Saper and S. Silberstein, Headache, 2006, 46, S171–S181 CrossRef PubMed.
- S. D. Silberstein and D. C. McCrory, Headache, 2003, 43, 144–166 CrossRef PubMed.
- S. D. Silberstein, Headache, 1996, 37, S15–S25 Search PubMed.
- R. McDonald, Pharmacopsychiatry, 1979, 12, 407–422 CrossRef CAS PubMed.
- I. Setnikar, K. Schmid, L. C. Rovati, B. Vens-Cappell, D. Mazur and I. Kozak, Arzneimittelforschung, 2001, 51, 2–6 CAS.
- R. Markstein, Eur. J. Pharmacol., 1982, 86, 145–155 CrossRef CAS PubMed.
- B. Bergamasco, L. Frattola, A. Muratorio, F. Piccoli, F. Mailland and L. Parnetti, Acta Neurol. Scand., 2000, 101, 372–380 CAS.
- L. T. Williams, D. Mullikin and R. Lefkowitz, J. Biol. Chem., 1976, 251, 6915–6923 CAS.
- M. J. Bannon, A. A. Grace, B. S. Bunney and R. H. Roth, Naunyn-Schmiedeberg's Arch. Pharmacol., 1980, 312, 37–41 CrossRef CAS PubMed.
- J. McMurtry, N. Kazama and B. Wexler, Exp. Biol. Med., 1979, 161, 186–188 CrossRef CAS.
- A. Battaglia, G. Bruni, A. Ardia, G. Sacchetti, A. Baroni, R. Cisbani, P. Bovi, G. Roncolato, M. Dotto and G. Frascella, J. Am. Geriatr. Soc., 1989, 37, 295–302 CAS.
- B. Saletu, E. Paulus, L. Linzmayer, P. Anderer, H. V. Semlitsch, J. Grünberger, L. Wicke, A. Neuhold and I. Podreka, Psychopharmacology, 1995, 117, 385–395 CrossRef CAS PubMed.
- B. Winblad, M. Fioravanti, T. Dolezal, I. Logina, I. G. Milanov, D. C. Popescu and A. Solomon, Clin. Drug Invest., 2008, 28, 533–552 CrossRef CAS PubMed.
- F. Bracco, A. Battaglia, C. Chouza, E. Dupont, O. Gershanik, J. F. M. Masso and J.-L. Montastruc, CNS Drugs, 2004, 18, 733–746 CrossRef CAS PubMed.
- M. P. Curran and C. M. Perry, Drugs, 2004, 64, 2125–2141 CrossRef CAS PubMed.
- U. Rinne, F. Bracco, C. Chouza, E. Dupont, O. Gershanik, J. M. Masso, J. Montastruc and C. Marsden, Drugs, 1998, 55, 23–30 CrossRef CAS PubMed.
- J. Verhelst, R. Abs, D. Maiter, A. van den Bruel, M. Vandeweghe, B. Velkeniers, J. Mockel, G. Lamberigts, P. Petrossians and P. Coremans, J. Clin. Endocrinol. Metab., 1999, 84, 2518–2522 CrossRef CAS PubMed.
- A. Colao, A. Di Sarno, M. L. Landi, F. Scavuzzo, P. Cappabianca, R. Pivonello, R. Volpe, F. Di Salle, S. Cirillo and L. Annunziato, J. Clin. Endocrinol. Metab., 2000, 85, 2247–2252 CAS.
- G. Van Camp, A. Flamez, B. Cosyns, C. Weytjens, L. Muyldermans, M. Van Zandijcke, J. De Sutter, P. Santens, P. Decoodt and C. Moerman, Lancet, 2004, 363, 1179–1183 CrossRef CAS.
- M. Goldstein, A. Lieberman, J. Y. Lew, T. Asano, M. R. Rosenfeld and M. H. Makman, Proc. Natl. Acad. Sci. U. S. A., 1980, 77, 3725–3728 CrossRef CAS.
- L.-J. Cheng, J. E. Robbers and H. G. Floss, J. Nat. Prod., 1980, 43, 329–339 CrossRef CAS.
- J. C. Gebler and C. D. Poulter, Arch. Biochem. Biophys., 1992, 296, 308–313 CrossRef CAS PubMed.
- H.-F. Tsai, H. Wang, J. C. Gebler, C. D. Poulter and C. L. Schardl, Biochem. Biophys. Res. Commun., 1995, 216, 119–125 CrossRef CAS PubMed.
- C. Arntz and P. Tudzynski, Curr. Genet., 1997, 31, 357–360 CrossRef CAS PubMed.
- C. M. Coyle and D. G. Panaccione, Appl. Environ. Microbiol., 2005, 71, 3112–3118 CrossRef CAS PubMed.
- J. Wang, C. Machado, D. G. Panaccione, H.-F. Tsai and C. L. Schardl, Fungal Genet. Biol., 2004, 41, 189–198 CrossRef CAS PubMed.
- I. A. Unsöld and S.-M. Li, Microbiology, 2005, 151, 1499–1505 CrossRef PubMed.
- E. Kranen, N. Steffan, R. Maas, S. M. Li and J. Jose, ChemCatChem, 2011, 3, 1200–1207 CrossRef CAS.
- N. Steffan, I. A. Unsöld and S. M. Li, ChemBioChem, 2007, 8, 1298–1307 CrossRef CAS PubMed.
- L. Y. Luk and M. E. Tanner, J. Am. Chem. Soc., 2009, 131, 13932–13933 CrossRef CAS PubMed.
- U. Metzger, C. Schall, G. Zocher, I. Unsöld, E. Stec, S.-M. Li, L. Heide and T. Stehle, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 14309–14314 CrossRef CAS PubMed.
- N. Steffan and S.-M. Li, Archives of microbiology, 2009, 191, 461–466 CrossRef CAS PubMed.
- M. Liu, D. G. Panaccione and C. L. Schardl, Evol. Bioinf. Online, 2009, 5, 15 CAS.
- O. Rigbers and S.-M. Li, J. Biol. Chem., 2008, 283, 26859–26868 CrossRef CAS PubMed.
- N. Lorenz, J. Olšovská, M. Šulc and P. Tudzynski, Appl. Environ. Microbiol., 2010, 76, 1822–1830 CrossRef CAS PubMed.
- A. Leuchtmann, C. W. Bacon, C. L. Schardl, J. F. White Jr and M. Tadych, Mycologia, 2014, 106, 202–215 CAS.
- K. E. Goetz, C. M. Coyle, J. Z. Cheng, S. E. O'Connor and D. G. Panaccione, Curr. Genet., 2011, 57, 201–211 CrossRef CAS PubMed.
- C. Wallwey, M. Matuschek and S.-M. Li, Arch. Microbiol., 2010, 192, 127–134 CrossRef CAS PubMed.
- C. Wallwey, C. Heddergott, X. Xie, A. A. Brakhage and S.-M. Li, Microbiology, 2012, 158, 1634–1644 CrossRef CAS PubMed.
- J. Z. Cheng, C. M. Coyle, D. G. Panaccione and S. E. O'Connor, J. Am. Chem. Soc., 2010, 132, 1776–1777 CrossRef CAS PubMed.
- C. M. Coyle, J. Z. Cheng, S. E. O'Connor and D. G. Panaccione, Appl. Environ. Microbiol., 2010, 76, 3898–3903 CrossRef CAS PubMed.
- X. Xie, C. Wallwey, M. Matuschek, K. Steinbach and S. M. Li, Magn. Reson. Chem., 2011, 49, 678–681 CrossRef CAS PubMed.
- A. S. Chilton, A. L. Ellis and A. L. Lamb, Acta Crystallogr., Sect. F: Struct. Biol. Commun., 2014, 70, 1328–1332 CrossRef CAS PubMed.
- C. Wallwey, M. Matuschek, X.-L. Xie and S.-M. Li, Org. Biomol. Chem., 2010, 8, 3500–3508 CAS.
- M. Matuschek, C. Wallwey, B. Wollinsky, X. Xie and S.-M. Li, RSC Adv., 2012, 2, 3662–3669 RSC.
- M. Matuschek, C. Wallwey, X. Xie and S.-M. Li, Org. Biomol. Chem., 2011, 9, 4328–4335 CAS.
- J. Z. Cheng, C. M. Coyle, D. G. Panaccione and S. E. O'Connor, J. Am. Chem. Soc., 2010, 132, 12835–12837 CrossRef CAS PubMed.
- D. G. Panaccione and C. M. Coyle, Appl. Environ. Microbiol., 2005, 71, 3106–3111 CrossRef CAS PubMed.
- Y. Bilovol and D. G. Panaccione, Curr. Genet., 2016, 62, 853–860 CrossRef CAS PubMed.
- S.-M. Li and I. A. Unsöld, Planta Med., 2006, 72, 1117–1120 CrossRef CAS PubMed.
- I. A. Unsöld and S. M. Li, ChemBioChem, 2006, 7, 158–164 CrossRef PubMed.
- D. Jakubczyk, L. Caputi, A. Hatsch, C. A. Nielsen, M. Diefenbacher, J. Klein, A. Molt, H. Schroder, J. Z. Cheng, M. Naesby and S. E. O'Connor, Angew. Chem., 2015, 54, 5117–5121 CrossRef CAS PubMed.
- T. Haarmann, I. Ortel, P. Tudzynski and U. Keller, ChemBioChem, 2006, 7, 645–652 CrossRef CAS PubMed.
- S. L. Robinson and D. G. Panaccione, Appl. Environ. Microbiol., 2014, 80, 6465–6472 CrossRef PubMed.
- I. Ortel and U. Keller, J. Biol. Chem., 2009, 284, 6650–6660 CrossRef CAS PubMed.
- M. A. Marahiel, Nat. Prod. Rep., 2016, 33, 136–140 RSC.
- M. Z. Ansari, G. Yadav, R. S. Gokhale and D. Mohanty, Nucleic Acids Res., 2004, 32, W405–W413 CrossRef CAS PubMed.
- T. Haarmann, N. Lorenz and P. Tudzynski, Fungal Genet. Biol., 2008, 45, 35–44 CrossRef CAS PubMed.
- T. Haarmann, C. Machado, Y. Lübbe, T. Correia, C. L. Schardl, D. G. Panaccione and P. Tudzynski, Phytochemistry, 2005, 66, 1312–1320 CrossRef CAS PubMed.
- J. Havemann, D. Vogel, B. Loll and U. Keller, Chem. Biol., 2014, 21, 146–155 CrossRef CAS PubMed.
- S. Müller, S. Rachid, T. Hoffmann, F. Surup, C. Volz, N. Zaburannyi and R. Müller, Chem. Biol., 2014, 21, 855–865 CrossRef PubMed.
- N. Castagnoli, K. Corbett, E. Chain and R. Thomas, Biochem. J., 1970, 117, 451–455 CrossRef CAS PubMed.
- P. Tudzynski, K. Hölter, T. Correia, C. Arntz, N. Grammel and U. Keller, Mol. Gen. Genet., 1999, 261, 133–141 CrossRef CAS PubMed.
- N. Lorenz, E. V. Wilson, C. Machado, C. L. Schardl and P. Tudzynski, Appl. Environ. Microbiol., 2007, 73, 7185–7191 CrossRef CAS PubMed.
- S. Pazoutová, M. Kolařík, G. Odvody, D. Frederickson, J. Olšovská and P. Man, Fungal. Divers., 2008, 95–111 Search PubMed.
- N. Lorenz, T. Haarmann, S. Pažoutová, M. Jung and P. Tudzynski, Phytochemistry, 2009, 70, 1822–1832 CrossRef CAS PubMed.
- D. J. Fleetwood, B. Scott, G. A. Lane, A. Tanaka and R. D. Johnson, Appl. Environ. Microbiol., 2007, 73, 2571–2579 CrossRef CAS PubMed.
- C. A. Nielsen, C. Folly, A. Hatsch, A. Molt, H. Schröder, S. E. O'Connor and M. Naesby, Microb. Cell Fact., 2014, 13, 95 CrossRef PubMed.
- K. L. Ryan, N. G. Akhmedov and D. G. Panaccione, J. Agric. Food Chem., 2014, 63, 61–67 CrossRef PubMed.
- K. L. Ryan, C. T. Moore and D. G. Panaccione, Toxins, 2013, 5, 445–455 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra03152a |
| This journal is © The Royal Society of Chemistry 2017 |