
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licencep-Hydroxybenzenesulfonic acid–formaldehyde solid acid resin for the conversion of fructose and glucose to 5-hydroxymethylfurfural†
Wenzhi Li a,
Tingwei Zhang*a,
Haosheng Xinb,
Mingxue Suc,
Longlong Ma*d,
Hason Jameele,
Hou-min Change and
Gang Peia
a,
Tingwei Zhang*a,
Haosheng Xinb,
Mingxue Suc,
Longlong Ma*d,
Hason Jameele,
Hou-min Change and
Gang Peia
aDepartment of Thermal Science and Energy Engineering, University of Science and Technology of China, Hefei 230026, PR China. E-mail: ztwei@mail.ustc.edu.cn
bInstitute of Materials and Chemical Engineering, Anhui Jianzhu Univerisity, Hefei 230022, PR China
cDepartment of Chemistry, University of Science and Technology of China, Hefei 230026, PR China
dCAS Key Laboratory of Renewable Energy, Guangzhou Institute of Energy Conversion, Chinese Academy of Sciences, Guangzhou 510640, PR China
eDepartment of Forest Biomaterials, North Carolina State University, Raleigh, NC 27695-8005, USA
First published on 24th May 2017
Abstract
A novel solid p-hydroxybenzenesulfonic acid–formaldehyde resin (SPFR) was prepared via a straightforward hydrothermal method. The catalytic properties of SPFR solid acids were evaluated in the dehydration reaction of fructose and glucose to 5-hydroxymethylfurfural (HMF). SEM, TEM, N2 adsorption–desorption, elemental analysis (EA), thermogravimetric analysis (TGA), and FT-IR were used to explore the effects of catalyst structure and composition on the HMF preparation from fructose. The effects of reaction time and temperature on the dehydration of fructose and glucose were also investigated. An HMF yield as high as 82.6% was achieved from fructose at 140 °C after 30 min, and 33.0% was achieved from glucose at 190 °C in 30 min. Furthermore, the recyclability of SPFR for the HMF production from fructose in 5 cycles was good.
Introduction
In recent years, the utilization of renewable biomass resources as feedstocks for the production of sustainable biofuels and valuable chemicals has been regarded as a promising strategy to deal with limited petroleum reserves and environmental problems.1,2 In this context, 5-hydroxymethylfurfural (HMF), a pivotal and versatile biomass-derived platform compound, plays a crucial role in the synthesis of many fine chemicals, biofuels, liquid alkanes, polymer monomers and solvents.3–6 Although the conversion of fructose to HMF is rather complicated due to the occurrence of a number of side reactions, HMF can be readily obtained via acid-catalyzed dehydration of fructose because of its fructofuranose structure.7,8 However, the conversion of glucose to HMF is more difficult, and involves an initial isomerization of glucose to fructose, followed by dehydration of fructose to HMF.9–11Both homogeneous and heterogeneous catalytic systems were studied for HMF production from hexoses. Homogeneous catalysts, including mineral acids (H2SO4, H3PO4, and HCl), metal chlorides (CrCl3 and AlCl3), and ionic liquids have been applied for the production of HMF.12–14 Li et al. found that 1-butyl-3-methylimidazolium bromide, without using any other additive or catalyst, unexpectedly showed catalytic activity in the fructose to HMF conversion at mild temperatures, and the yield of HMF was 95%.12 In addition, homogeneous catalysts used together with a solid catalyst that contained Brønsted acid sites and/or Lewis acid sites were effective in the conversion of carbohydrates to HMF.15–20 Zhang et al. developed a hierarchically carbonaceous catalyst with Brønsted–Lewis acid sites, which was used to prepare HMF from cellulose in the ionic liquid 1-butyl-3-methylimidazolium chloride, and an HMF yield of 43.1% was achieved.19 However, reactor corrosion, difficult downstream separation of the catalyst and the high cost of ionic liquids limited the application of the abovementioned methods for HMF production.
Prompted by the disadvantages of homogeneous catalyst, tremendous efforts have been made for developing heterogeneous catalysts for the synthesis of HMF in organic solvents and/or water. The generated HMF in a heterogeneous catalytic system remains more stable. The dehydration of fructose over a variety of solid catalysts such as functionalized silica,1,3,21 carbon-based solid acids,22,23 zeolites,24 ion-exchange resins,25 metal phosphate,26 and heteropoly acids27 has been investigated. Alamillo et al. synthesized an ordered mesoporous acid-functionalized silica catalyst by the introduction of poly(vinylpyrrolidone) (PVP). PVP improved the HMF selectivity from fructose, and a HMF yield of 87% was achieved at a relatively low temperature (130 °C) in a mixture of tetrahydrofuran and water.28 Mazzotta et al. prepared a porous sulfonated carbonaceous TiO2 catalyst (Glu-TsOH-Ti) containing Brønsted and Lewis acid sites by a one-pot hydrothermal method. A good catalytic activity of Glu-TsOH-Ti in the conversion of fructose to HMF was evidenced by the high HMF yield of 59% and 99% in a methyltetrahydrofuran/H2O biphasic solvent system and dimethyl sulfoxide (DMSO), respectively.29 Previously, our group synthesized a strong solid Brønsted acid PTSA–POM resin via copolymerization of paraformaldehyde (POM) and p-toluenesulfonic acid (PTSA), which was used for fructose dehydration, and a HMF yield of 78.1% was obtained in γ-valerolactone (GVL)/H2O.7 Ordomskyet et al. studied the effect of organic solvents on fructose dehydration catalyzed by a series of zeolites (MOR, ZSM-5, BEA, amorphous aluminosilicate) in a bisphasic system, and found that the addition of methyl isobutyl ketone significantly improved HMF selectivity. Especifically, the improvement in selectivity decreased in the following order: MOR > ZSM-5 > BEA > amorphous aluminosilicate.24 In summary, fructose dehydration over solid acids is an efficient and convenient way to synthesize HMF.
Glucose, the most abundant hexose in nature, which can even be obtained from non-edible agricultural and forestry waste, has promising prospects in a number of applications. A considerable body of research has been conducted on the transformation of glucose into HMF over heterogeneous catalysts.4–6,30–34 Jiménez-Morales et al. prepared the Al-MCM-550 catalyst, containing Brønsted and Lewis acid sites, for the dehydration of glucose in a MIBK/H2O system. A HMF yield of 36% and a glucose conversion of 87% were achieved after 150 min at 195 °C.35 Gallo et al. exploited the combination of Amberlyst-70 with Sn-based catalysts (Sn-SBA-15, Sn-beta) for the production of HMF from glucose in different mixed solvent systems (THF/H2O, γ-hexalactone/H2O and GVL/H2O).36 The combination of Amberlyst-70 with Sn-based catalysts led to a remarkable enhancement of the HMF selectivity. Sn-beta, with higher Lewis acidity, showed a higher HMF selectivity than Sn-SBA-15, and the highest HMF yield, of 63%, was achieved in a THF/H2O system. In general, it is difficult to develop an efficient heterogeneous catalyst for producing HMF from glucose, and the synthesis for most of these catalysts is complicated and expensive.4–6 Therefore, the development of a simple, economical and efficient catalyst for glucose dehydration is of great importance.
Yu's group developed an energy and time efficient hydrothermal method to synthesize a phenol–formaldehyde resin (PFR), which had interesting mechanical properties.37 PFR was obtained via polymerization of phenol and formaldehyde in acetic acid aqueous solution using chitosan as a soft template. In our work, according to Yu's method, a novel and straightforward route for the synthesis of a solid acid resin catalyst was reported, where phenol was substituted with p-hydroxybenzenesulfonic acid (PHSA). The as-prepared solid acid resins were named SPFR-x, where x is the molar ratio of PHSA to formaldehyde. Since the raw materials used for the preparation of SPFR are relatively inexpensive and their synthesis is straightforward, their large-scale production is feasible and thus, SPFR may have great potential applications in acid catalysis. The catalytic activity of SPFR-x catalysts for the dehydration of fructose and glucose to HMF were studied in a mixture of GVL/H2O. GVL, a nontoxic and eco-friendly solvent, may favour the conversion of fructose to HMF.9 In addition, GVL can be separated from the reaction media by distillation. The structure and elemental composition of the SPFR catalysts were characterized by SEM, TEM, N2 adsorption–desorption isotherms, elemental analysis (EA), thermogravimetric analysis (TGA) and FT-IR, and the effect of structure and composition on HMF production from fructose were studied. Furthermore, the effect of reaction time and temperature on fructose and glucose dehydration were also studied, along with the reusability of SPFR.
Experimental
Formaldehyde solution (AR, 37–40%), acetic acid (GR), acetone (AR), and sodium chloride (NaCl, AR) were supplied by Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China). D-(+)-glucose (GC, 99.5%), D-fructose (99%), chitosan (degree of deacetylation ≥ 95%), HMF (99%), p-hydroxybenzenesulfonic acid hydrate (85%), and sodium hydroxide standard solution (0.02 M) were purchased from Aladdin Industrial, Inc. (Shanghai, China). GVL (95%) was obtained from LangFang Hawk Technology and Development Co., Ltd. All chemicals were used without further purification.Two kinds of SPFR catalysts with different amounts of PHSA were synthesized: SPFR-0.38 and SPFR-0.26. In a typical synthetic procedure for SPFR-0.38, 0.45 g of chitosan were added to 30 mL of distilled water (DIW) under constant magnetic stirring, and 0.6 mL of acetic acid were subsequently added to completely dissolve chitosan. After the addition of 3.1 g of p-hydroxybenzenesulfonic acid hydrate, mixture was stirred for ten minutes. Then, 3 mL of formaldehyde solution were added quickly to the mixture, and stirring was continued for further 5 minutes. The mixture was sealed in a 50 mL Teflon-lined autoclave, and heated in an oven for 10 h at 120 °C (SPFR gels were also successfully synthesized at a larger scale without changing the concentrations of reactants or reaction time). After cooling down, the product of SPFR filled the reaction autoclave, because the autoclave is cylindrical, so finally columned SPFR gels formed. The gels were mashed to a paste, washed with acetone/DIW once, then washed with plenty of DIW several times until the filtrate was neutral, and finally dried at 100 °C overnight. The gels shrank significantly during the drying process (shrinkage for SPFR-0.38 was more pronounced than for SPFR-0.26). Moreover, the gels developed a brownish colour during the drying process, so polymerization of the SPFR gels by condensation must have occurred during drying. The as-obtained brownish solids were ground to a powder, and dried at 100 °C to a constant weight.
The microstructure and morphology of SPFR-x was analyzed by SEM (FEI Sirion scanning electron microscope) and TEM (JEM 2010 electron microscope). The BET specific surface areas, BJH desorption pore diameters and total pore volumes of SPFR-x were obtained based on the N2 adsorption–desorption isotherms, which were recorded using an Autosorb iQ (Quantachrome) device. Thermogravimetric analysis (TGA) was performed on a TGA Q5000 analyzer. FTIR spectra were recorded on a Nicolet 8700 instrument (with KBr disks). The elemental composition of the samples was determined with an Elementar model Vario EL III. The Brønsted acidity of SPFR-0.38 and SPFR-0.26 was measured by acid base titration.38 The strong acid site concentration for fresh SPFR-0.38 and SPFR-0.26 were 0.7 mmol g−1 and 0.23 mmol g−1, respectively.
The experimental parameters, including the dose of substrate and catalyst, the amount of solvent, the reaction time, and the temperature, employed in this work were determined according to previous studies published by our group.7,9 In a typical dehydration experiment, 0.4 g of substrate (glucose or fructose), 0.2 g of SPFR-0.38 and a mixture of GVL/DIW (1.5 mL/15 mL) were added to a 25 mL stainless steel reactor. Then, the reactor was sealed and heated to the target temperature within 30 minutes under magnetic stirring at 500 rpm. After reaction completion, the reactor was immersed in cool water to quench the reaction immediately. The mixture was filtrated, and the filtrate was dissolved in DIW.
The dissolved samples were analyzed by HPLC (Waters 515 pump, equipped with a Waters 2414 refractive index detector using a Waters XBridge Amide column; and a Waters 2489 ultraviolet detector using a Waters Symmetry C18 column) to determine the yield of HMF, as well as fructose and glucose conversion.7,9,38 Standard calibration curves were used for the quantification of glucose, fructose and HMF in the diluted samples.
The HMF yield and selectivity, furfural yield, and glucose and fructose conversion were defined as follows:
| HMF yield = (moles of HMF produced/moles of starting glucose or fructose) × 100% |
| HMF selectivity = (HMF yield/glucose or fructose conversion) × 100% |
| Furfural yield = (moles of furfural produced/moles of starting fructose) × 100% |
| Glucose or fructose conversion = 1 − (moles of glucose or fructose in products/moles of starting glucose or fructose) × 100% |
Results and discussion
Chitosan can dissolve well in an acetic acid solution and form a viscous and transparent gel. It can absorb p-hydroxybenzenesulfonic acid (PHSA) and can be cross-linked by formaldehyde. Moreover, PHSA and formaldehyde can polymerize in an acetic acid solution. This way, chitosan was used as a template in which formaldehyde reacted with PHSA and chitosan concurrently. The rapidly generated SPFR micromolecules gradually deposited on the chitosan template to form the SPFR gels. During the hydrothermal process, chitosan gels were gradually dissolved away, and only SPFR gels were retained. The SPFR gels were washed with distilled water (DIW), and finally dried in an oven at 100 °C to afford a brownish solid, the SPFR catalyst. Since PHSA was used to prepare the phenolic resin, a certain content of –SO3H groups should be found in the resin (ESI, Scheme S1†).37,39Fig. S1† shows the SEM and TEM images of the SPFR catalysts (SPFR-0.38 and SPFR-0.26). The majority of particles in SPFR-0.38 exhibited an irregular bulk-like structure with a relatively uniform size. On the other hand, SPFR-0.26 exhibited a clustered irregular structure with a much larger particle size. No net-like framework or nanoporous structures were observed in SPFR-0.38 or SPFR-0.26, which was attributed to the thermal drying process, in agreement with the results obtained by Yu et al.37 Compared with PTSA–POM, a resin catalyst synthesized without a template, the particle size of SPFR-0.38 was much smaller and therefore, the dispersibility of SPFR was expected to be better. The textural properties of SPFR-0.38, SPF-0.26 and PTSA–POM are summarized in Table 1. SPFR catalysts had a small surface area, total pore volume and BJH desorption pore diameter. The surface area and total pore volume of SPFR-0.38 were slightly larger than those of SPFR-0.26, and the two catalysts exhibited a similar BJH desorption pore diameter of 1.42 nm. Also, the surface area and total pore volume of both SPFR catalysts were higher than those of PTSA–POM.9,38
| Sample | SBETa (m2 g−1) | VPb (cm3 g−1) | rpc (nm) | rPd (nm) | Refs. |
|---|---|---|---|---|---|
| a Multipoint BET.b Total pore volume.c BJH desorption pore diameter.d Average pore diameter. | |||||
| SPFR-0.26 | 21.13 | 0.06 | 1.42 | 11.74 | This work |
| SPFR-0.38 | 27.98 | 0.11 | 1.42 | 15.48 | This work |
| PTSA–POM | 0.4503 | 0.001 | — | — | 38 |
| First used SPFR-0.38 | 23.96 | 0.06 | — | 9.70 | This work |
| 5th used SPFR-0.38 | 21.65 | 0.05 | — | 9.28 | This work |
The FTIR spectra of SPFR-0.38 and SPFR-0.26 were similar (Fig. 1). The bands at 1040 cm−1 (O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) SO stretching vibration) and 1167 cm−1 (SO3–H stretching vibration) in the FTIR spectra clearly revealed the presence of sulfonic acid groups in SPFR. The broad band at around 3436 cm−1 was ascribed to the N–H and O–H stretching vibrations, which were attributed to the amino groups and phenolic hydroxils in SPFR, respectively.37
SO stretching vibration) and 1167 cm−1 (SO3–H stretching vibration) in the FTIR spectra clearly revealed the presence of sulfonic acid groups in SPFR. The broad band at around 3436 cm−1 was ascribed to the N–H and O–H stretching vibrations, which were attributed to the amino groups and phenolic hydroxils in SPFR, respectively.37
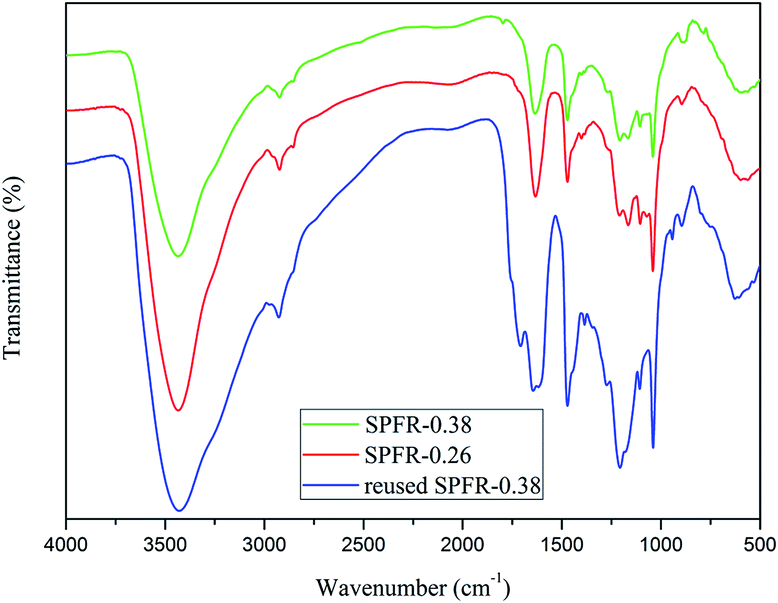 | ||
| Fig. 1 FT-IR spectra of SPFR-0.26 and SPFR-0.38. | ||
Table 2 summarizes the elemental composition of the SPFR catalysts. The H, O and S content in SPFR-0.26 were higher than in SPFR-0.38. The presence of N in the SPFR catalysts suggested that a certain amount of chitosan was incorporated. According to Yu's work, a reaction between chitosan and SPFR occurred.37 The O/S ratio in the SPFR catalysts as calculated from the elemental analysis was greater than 3, indicating that SPFR catalysts contained other oxygen functional groups.
| Sample | Atomic ratio (%) | S content mmol g−1 | Strong acid acidity mmol H+/g | ||||
|---|---|---|---|---|---|---|---|
| C | H | O | N | S | |||
| SPFR-0.26 | 29.04 | 55.33 | 13.87 | 0.76 | 1.00 | 1.48 | 0.23 |
| SPFR-0.38 | 31.99 | 53.12 | 13.38 | 0.53 | 0.98 | 1.42 | 0.7 |
Fig. 2 shows the weight loss curves of the SPFR catalysts as obtained by TGA. SPFR-0.38 (Fig. 2A) and SPFR-0.26 (Fig. 2B) displayed similar weight loss trends, which could be divided into 3 stages during the decomposition process. In stage 1 (30 to 140 °C), the weight loss was more pronounced for SPFR-0.38. The weight loss in this stage corresponded to the evaporation of water, mainly in the form of physically adsorbed water and water weakly bonded to SPFR via hydrogen bonds. In stage 2 (160 to 410 °C), the weight loss was higher for SPFR-0.26, and probably corresponded to the elimination of strongly bonded water via hydrogen bonds, sulfonic acid groups and thermal decomposition of chitosan.40 In stage 3 (410 to 760 °C), the weight loss was again higher for SPFR-0.26, and could be attributed to the decomposition and carbonization of the phenolic–formaldehyde resin. Throughout the whole process, more carbonization products formed from the thermal decomposition of SPFR-0.38. This was ascribed to the higher carbon content and the more rigid structure of SPFR-0.38, which is in good agreement with the results obtained by EA, SEM and TEM. However, SPFR-0.26 was more stable in the low temperature stage. This may be ascribed to a further condensation polymerization in SPFR-0.38, which would lead to a more pronounced weight loss at low temperatures.
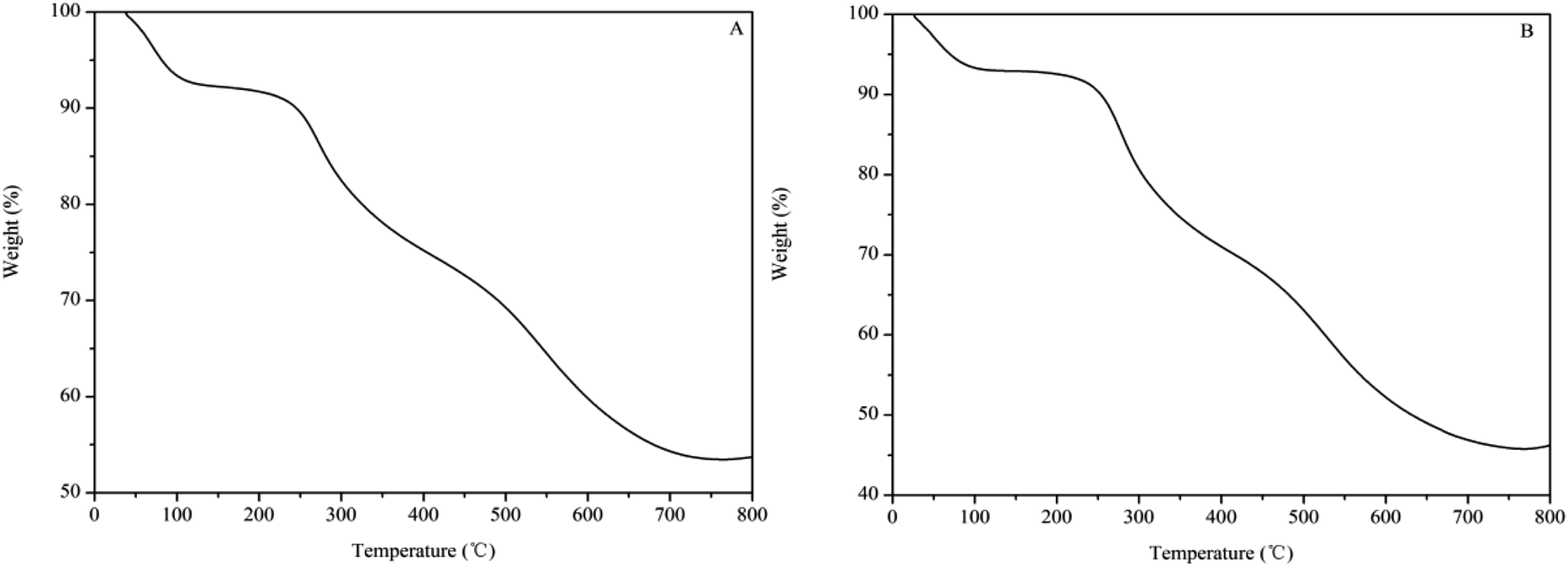 | ||
| Fig. 2 Weight loss curves of SPFR-0.38 (A) and SPFR-0.26 (B). | ||
The catalytic activity of the SPFR catalysts was evaluated in terms of the dehydration rate of fructose and glucose to form HMF in GVL/H2O. No insoluble polymers or clogging in the reactor were observed during the experiments.
The effect of temperature and reaction time on HMF production from fructose using SPFR-0.38 as the catalyst was investigated by performing the reaction at 120–150 °C for 10–50 min. The results, shown in Fig. 3, indicate that SPFR-0.38 is an effective catalyst for fructose dehydration. In addition, the reaction temperature and time play a crucial role in the conversion of fructose to HMF. At a temperature of 120 °C for 10 min, the HMF yield was of only 32.0%, with a fructose conversion of 60.2%. At 120 and 130 °C, the HMF yield increased with reaction time. Upon further increasing the temperature to 140 °C, the HMF yield initially increased (for a reaction time of 10–30 min), and then decreased as reaction time increased. At 150 °C, the HMF yield decreased with reaction time. A higher temperature promoted the formation of HMF, but also favoured undesired side reactions. Fructose conversion increased with time and/or temperature. A HMF yield as high as 82.6% with a fructose conversion of 99.3% was achieved at 140 °C in 30 min, and complete conversion was reached under harsher reaction conditions. Although the variation trend in the HMF selectivity is complicated, it was concluded that a high HMF selectivity of 87.3% and yield of 67.2% can be achieved at 130 °C in 20 min (Fig. S2†). Compared with the much stronger pure Brønsted acid catalyst PTSA–POM (2.4 mmol g−1), the HMF yield with SPFR-0.38 was slightly higher. This was attributed to the higher dispersibility of SPFR-0.38, which may favour HMF production.7 As shown in Fig. 4, a small amount of furfural was generated in this reaction system, which is in good agreement with Dumesic et al. studies.41 Furfural yield increased with temperature and time in all experiments.
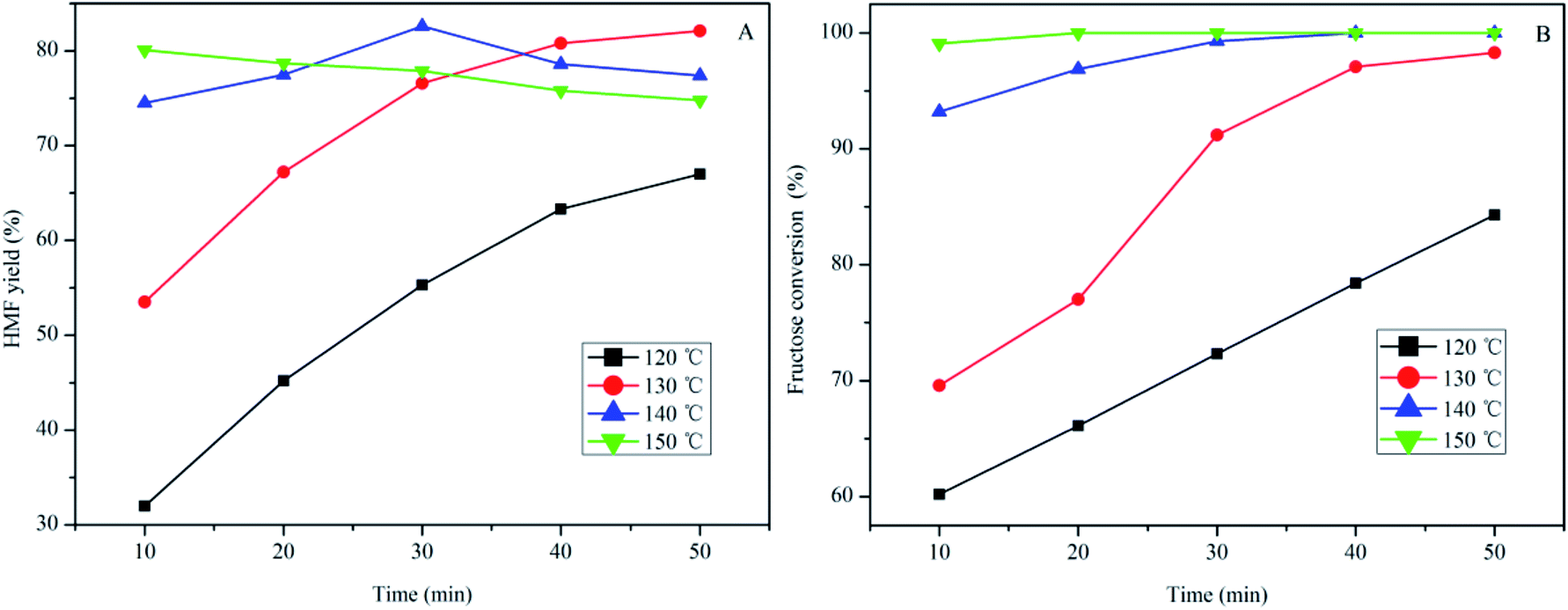 | ||
| Fig. 3 Effect of temperature and reaction time on HMF yield (A) and fructose (0.40 g) conversion to HMF (B) in GVL/DIW (10/1 v/v) catalyzed by SPFR-0.38 (0.20 g). | ||
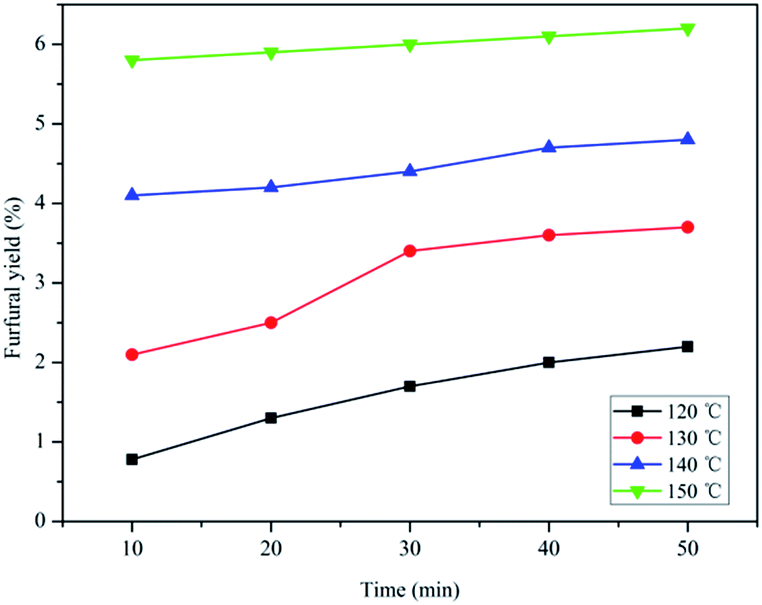 | ||
| Fig. 4 Effect of temperature and reaction time on fructose (0.40 g) conversion to furfural in GVL/DIW (10/1 v/v) catalyzed by SPFR-0.38 (0.2 g). | ||
The effect of adding different amounts of water (0–30 wt%, based on GVL) on HMF production from fructose was studied, and the results are shown in Table S1.† The addition of a certain amount of water had a significant improvement on the conversion of fructose to HMF, and higher HMF yields were obtained compared with the pure GVL solvent system. Upon adding 10 wt% water, a HMF yield as high as 82.6% was achieved. However, the conversion of fructose to HMF became slow with the addition of more water. Furthermore, it was found that the formation of furfural was hindered when water was added, thus promoting HMF formation. Actually, several secondary reactions were prevented by the addition of water, which would explain the increase in HMF yield. Thus, GVL/DIW is an ideal mixture of solvents for HMF production.
The recyclability of the SPFR catalysts is very relevant for potential industrial applications. A five-cycle experiment was conducted to assess the stability of the SPFR catalysts. After each cycle, the used solid acids were washed with DIW and acetone, and dried at 80 °C. The results of the reusability test are shown in Fig. 5. The yield of HMF decreased from 82.6% to 64.2% with reused SPFR-0.38 after five cycles. The leaching of H+ from the SO3–H groups during the 5 cycles may be responsible for the reduction in the HMF yield. This hypothesis was verified by the attenuated band at 1167 cm−1 (SO3–H stretching vibration) in the IR spectrum of the catalyst after 5 cycles. This band was so attenuated that it was almost covered by the band at 1208 cm−1 (Fig. 2). Furthermore, the surface area, total pore volume and average pore diameter of the used catalyst decreased, indicating that insoluble byproducts may form and deposit on the catalyst, blocking the pores (Table 2). This may be another reason for the reduction in the HMF yield. However, despite this, the recyclability of SPFR-0.38 was still considered to be good.
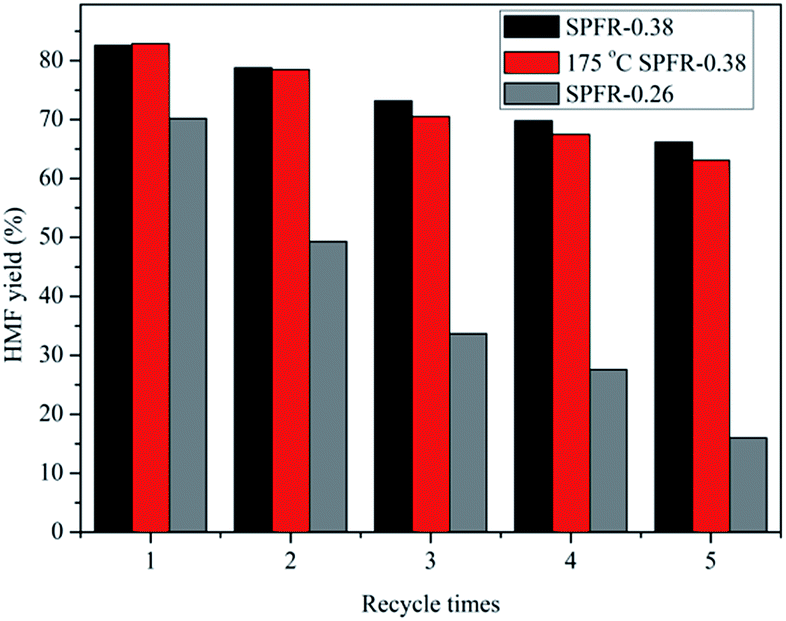 | ||
| Fig. 5 Reusability study for the SPFR catalysts. Reaction conditions: 0.40 g of fructose, catalyst (0.20 g of SPFR-0.38 or 175 °C-treated SPFR-0.38, 0.30 g of SPFR-0.26), GVL/DIW (10/1 v/v), 140 °C, 30 min reaction time. | ||
SPFR-0.26 was also used for the conversion of fructose to HMF at 140 °C for 30 min, but the catalytic activity was low, only 70.2% HMF yield was obtained even with the use of 0.3 g SPFR-0.26 (Fig. 5). In addition, the recyclability of SPFR-0.26 was poor, with a HMF yield of only 16.0% after the 5th cycle (Fig. 5). According to the characterization results, the main differences between SPFR-0.26 and SPFR-0.38 lie in the morphology, microstructure and Brønsted acidity. Thus, we inferred that the higher Brønsted acidity and relatively small and uniform particle size of SPFR-0.38 played a key role in its high catalytic activity.
Previously, we found that for PTSA–POM, a further drying treatment at high temperatures significantly improved thermal stability and water resistance. The PTSA–POM catalyst obtained by this procedure exhibited a higher catalytic activity in the furfural production from xylose.38 Therefore, the catalytic activity of SPFR-0.38 after a further treatment at a high temperature (175 °C) was examined in a reaction conducted at 140 °C for 30 min. The additional drying treatment was conducted in an oven at 175 °C for 6 h the SPFR-0.38 catalyst that had previously been dried at 100 °C. There was no obvious distinction in the HMF yield and catalyst reusability between the SPFR catalyst dried at 100 °C and the same catalyst after a further drying treatment at 175 °C (Fig. 5), indicating that the thermal stability and water resistance of SPFR-0.38 was already optimal after the drying treatment at 100 °C, and a further thermal treatment was not necessary.
Finally, glucose was used as the substrate for HMF production, and the effect of temperature and reaction time were studied using SPFR-0.38 as the catalyst. As shown in Fig. 6, the temperature and time were also important factors for the glucose to HMF conversion. When performing the reaction at 160 and 170 °C, the HMF yield increased with reaction time. By contrast, when performing the reaction at 180 and 190 °C, the HMF yield initially increased with time (10–30 min), and thereafter decreased (40–60 min). A high HMF yield of 33.0% with glucose conversion of 97.6% was achieved at 190 °C in 30 min. Glucose conversion increased as time and temperature increased, and conversion was complete at 190 °C and 60 min. Compared with PTSA–POM, a relatively higher HMF yield was obtained using SPFR-0.38. The introduction of nitrogen functional groups in SPFR may promote glucose isomerization, thereby increasing the yield of HMF.7
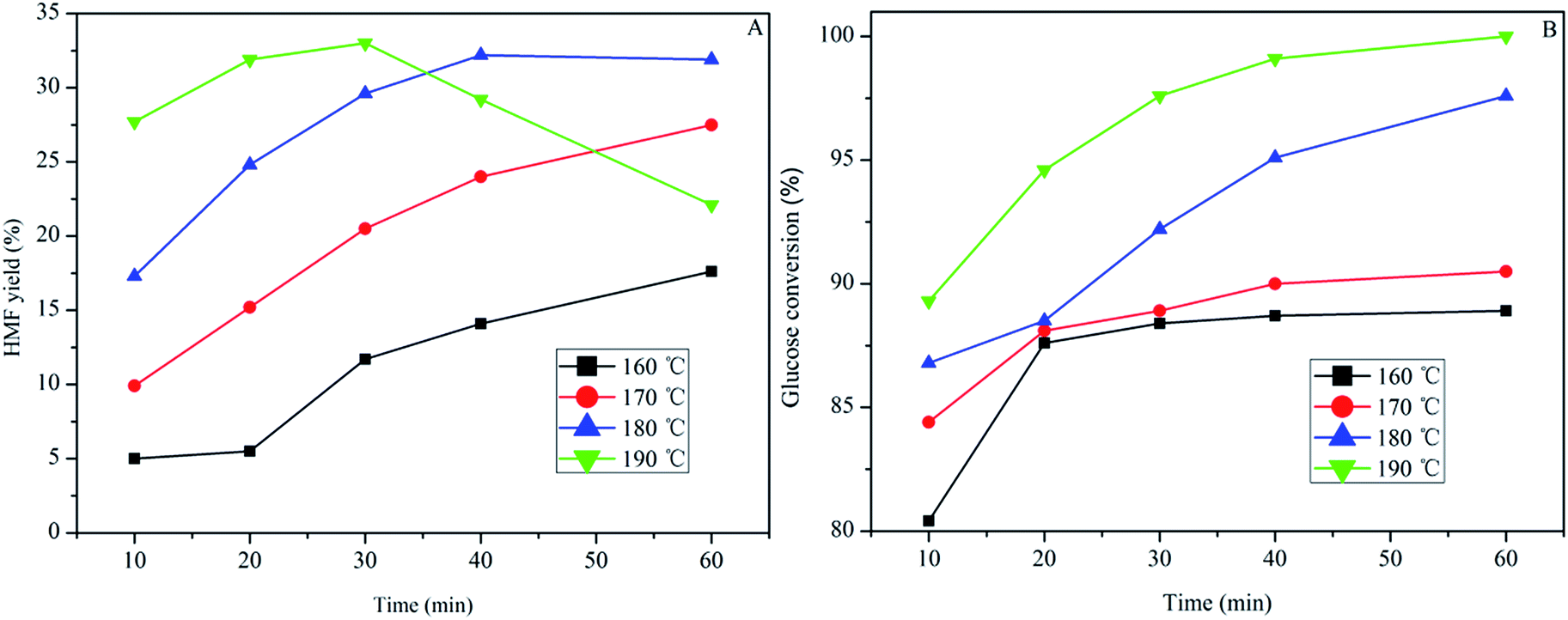 | ||
| Fig. 6 Effect of temperature and reaction time (A and B) on glucose (0.40 g) conversion to HMF in GVL/DIW (10/1 v/v) catalyzed by SPFR-0.38 (0.20 g). | ||
Conclusions
In summary, a novel and highly efficient solid acid resin catalyst, SPFR, was successfully synthesized by a straightforward hydrothermal route. The catalytic activity of SPFR was evaluated in the dehydration of fructose and glucose. Different amounts of PHSA were added for the synthesis of SPFR to obtain SPFR-0.38 and SPFR-0.26: SPFR-0.38, with a higher acidity, shows a better catalytic activity and recyclability for the HMF production from fructose than SPFR-0.26. The morphology and microstructure of the SPFR catalysts plays an important role in the catalytic activity. A HMF yield as high as 82.6% was obtained from fructose at 140 °C in 30 min, and 33.0% was achieved from glucose at 190 °C in 30 min. Compared with PTSA–POM, a relatively higher HMF yield was achieved with SPFR-0.38. This may be explained by the presence of nitrogen functional groups in SPFR-0.38, which may promote glucose isomerization. Also, quite importantly, the recyclability of SPFR-0.38 was good.Acknowledgements
This study was financially supported by the State Key Program of the National Natural Science Foundation of China (51536009), the National Key Technology R&D Program of China (No. 2015BAD15B06) and the Science and Technological Fund of the Anhui Province for Outstanding Youth (1508085J01).Notes and references
- H. Hafizi, A. N. Chermahini, M. Saraji and G. Mohammadnezhad, Chem. Eng. J., 2016, 294, 380–388 CrossRef CAS.
- G. Tsilomelekis, M. J. Orella, Z. Lin, Z. Cheng, W. Zheng, V. Nikolakis and D. G. Vlachos, Green Chem., 2016, 18(7), 1983–1993 RSC.
- A. N. Chermahini, F. Shahangi, H. A. Dabbagh and M. Saraji, RSC Adv., 2016, 6(40), 33804–33810 RSC.
- P. Zhou and Z. Zhang, Catal. Sci. Technol., 2016, 6(11), 3694–3712 CAS.
- R. J. van Putten, J. C. van der Waal, E. D. De Jong, C. B. Rasrendra, H. J. Heeres and J. G. de Vries, Chem. Rev., 2013, 113, 1499–1597 CrossRef CAS PubMed.
- A. Mukherjee, M. J. Dumont and V. Raghavan, Biomass Bioenergy, 2015, 72, 143–183 CrossRef CAS.
- W. Li, Z. Xu, T. Zhang, G. Li, H. Jameel, H. M. Chang and L. Ma, BioResources, 2016, 11(3), 5839–5853 CAS.
- X. Wang, H. Zhang, J. Ma and Z. H. Ma, RSC Adv., 2016, 6(49), 43152–43158 RSC.
- T. Zhang, W. Fan, W. Li, Z. Xu, H. Xin, M. Su, Y. Lu and L. Ma, Energy Technol., 2017, 5, 747–755 CrossRef CAS.
- A. Takagaki, M. Ohara, S. Nishimura and K. Ebitani, Chem. Commun., 2009, 41, 6276–6278 RSC.
- E. Nikolla, Y. Román-Leshkov, M. Moliner and M. E. Davis, ACS Catal., 2011, 1, 408–410 CrossRef CAS.
- Y. N. Li, J. Q. Wang, L. N. He, Z. Z. Yang, A. H. Liu, B. Yu and C. R. Luan, Green Chem., 2012, 14(10), 2752–2758 RSC.
- J. N. Chheda, Y. Román-Leshkov and J. A. Dumesic, Green Chem., 2007, 9(4), 342–350 RSC.
- Y. Yang, C. W. Hu and M. M. Abu-Omar, Green Chem., 2012, 14(2), 509–513 RSC.
- V. Choudhary, S. H. Mushrif, C. Ho, A. Anderko, V. Nikolakis, N. S. Marinkovic, A. I. Frenkel, S. I. Sandler and D. G. Vlachos, J. Am. Chem. Soc., 2013, 135, 3997–4006 CrossRef CAS PubMed.
- B. Kim, J. Jeong, D. Lee, S. Kim, H. J. Yoon, Y. S. Lee and J. K. Cho, Green Chem., 2011, 13(6), 1503–1506 RSC.
- Y. Zhang, Y. Chen, Y. Shen, Y. Yan, J. Pan, W. Shi and L. Yu, ChemPlusChem, 2016, 81, 108–118 CrossRef CAS.
- Y. Li, H. Liu, C. Song, X. Gu, H. Li, W. Zhu, S. Yin and C. Han, Bioresour. Technol., 2013, 133, 347–353 CrossRef CAS PubMed.
- Y. Zhang, Y. Shen, Y. Chen, Y. Yan, J. Pan, Q. Xiong and L. Yu, Chem. Eng. J., 2016, 294, 222–235 CrossRef CAS.
- L. Hu, Z. Wu, J. Xu, Y. Sun, L. Lin and S. Liu, Chem. Eng. J., 2014, 244, 137–144 CrossRef CAS.
- Z. Yang, W. Qi, R. Huang, J. Fang, R. Su and Z. He, Chem. Eng. J., 2016, 296, 209–216 CrossRef CAS.
- J. M. R. Gallo, R. Alamillo and J. A. Dumesic, J. Mol. Catal. A: Chem., 2016, 422, 13–17 CrossRef CAS.
- J. Zhao, C. M. Zhou, C. He, Y. H. Dai, X. L. Jia and Y. H. Yang, Catal. Today, 2016, 264, 123–130 CrossRef CAS.
- V. V. Ordomsky, J. Van der Schaaf, J. C. Schouten and T. A. Nijhuis, J. Catal., 2012, 287, 68–75 CrossRef CAS.
- G. Sampath and S. Kannan, Catal. Commun., 2013, 37, 41–44 CrossRef CAS.
- L. Yang, X. Yan, S. Xu, H. Chen, H. Xia and S. Zuo, RSC Adv., 2015, 5(26), 19900–19906 RSC.
- C. Fan, H. Guan, H. Zhang, J. Wang, S. Wang and X. Wang, Biomass Bioenergy, 2011, 35(7), 2659–2665 CrossRef CAS.
- R. Alamillo, A. J. Crisci, J. M. R. Gallo, S. L. Scott and J. A. Dumesic, Angew. Chem., 2013, 125(39), 10539–10541 CrossRef.
- M. G. Mazzotta, D. Gupta, B. Saha, A. K. Patra, A. Bhaumik and M. M. Abu-Omar, ChemSusChem, 2014, 7, 2342–2350 CrossRef CAS PubMed.
- T. D. Swift, H. Nguyen, Z. Erdman, J. S. Kruger, V. Nikolakis and D. G. Vlachos, J. Catal., 2016, 333, 149–161 CrossRef CAS.
- X. Yi, I. Delidovich, Z. Sun, S. Wang, X. Wang and R. Palkovits, Catal. Sci. Technol., 2015, 5(4), 2496–2502 CAS.
- A. Osatiashtiani, A. F. Lee, D. R. Brown, J. A. Melero, G. Morales and K. Wilson, Catal. Sci. Technol., 2014, 4(2), 333–342 CAS.
- V. V. Ordomsky, J. van der Schaaf, J. C. Schouten and T. A. Nijhuis, ChemSusChem, 2013, 6(9), 1697–1707 CrossRef CAS PubMed.
- I. Jiménez-Morales, J. Santamaría-González, A. Jiménez-López and P. Maireles-Torres, Fuel, 2014, 118, 265–271 CrossRef.
- I. Jimenez-Morales, M. Moreno-Recio, J. Santamaria-Gonzalez, P. Maireles-Torres and A. Jiménez-López, Appl. Catal., B, 2015, 164, 70–76 CrossRef CAS.
- J. M. R. Gallo, D. M. Alonso, M. A. Mellmer and J. A. Dumesic, Green Chem., 2013, 15, 85–90 RSC.
- Z. L. Yu, Z. Y. Wu, S. Xin, C. Qiao, Z. Y. Yu, H. P. Cong and S. H. Yu, Chem. Mater., 2014, 26(24), 6915–6918 CrossRef CAS.
- Z. P. Xu, W. Z. Li, Z. J. Du, H. Wu, H. Jameel, H. M. Chang and L. L. Ma, Bioresour. Technol., 2015, 198, 764–771 CrossRef CAS PubMed.
- J. M. Li, X. G. Meng, C. W. Hu and J. Du, Bioresour. Technol., 2009, 100(3), 1168–1173 CrossRef CAS PubMed.
- J. Zawadzki and H. Kaczmarek, Carbohydr. Polym., 2010, 80(2), 394–400 CrossRef CAS.
- E. I. Gürbüz, J. M. R. Gallo, D. M. Alonso, S. G. Wettstein, W. Y. Lim and J. A. Dumesic, Angew. Chem., Int. Ed., 2013, 52, 1270–1274 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra03155f |
| This journal is © The Royal Society of Chemistry 2017 |