
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Experimental and computational study of the inclusion complexes of β-cyclodextrin with the chemical warfare agent soman (GD) and commonly used simulants†
Mark R. Sambrook *a,
Jack C. Vincenta,
Jayne A. Edea,
Ian A. Gassb and
Peter J. Cragg*b
*a,
Jack C. Vincenta,
Jayne A. Edea,
Ian A. Gassb and
Peter J. Cragg*b
aCBR Division, Dstl Porton Down, Salisbury, SP4 0JQ, UK. E-mail: msambrook@dstl.gov.uk; Tel: +44 (0)1980 952077
bSchool of Pharmacy and Biomolecular Sciences, University of Brighton, Brighton, BN2 4GJ, UK. E-mail: P.J.Cragg@brighton.ac.uk; Tel: +44 (0)1273 642037
First published on 2nd August 2017
Abstract
The hydrophobically driven inclusion complexation of the Chemical Warfare Agent (CWA) pinacolyl methylphosphonofluoridate (soman, or GD) by β-cyclodextrin (β-CD) is studied both experimentally and computationally. Semiempirical modelling (PM6) adds further insight to the understanding of the CD–GD complex and the preferential binding of P(+) isomers over P(−). Comparison of the CD–GD complex to those formed by β-CD with commonly used trialkyl phosphate and dialkyl methylphosphonate simulants furnishes preliminary supramolecular agent–simulant correlation data. Such comparison studies will have utility in the design of new CWA-responsive materials.
Introduction
Supramolecular approaches to the mitigation of the hazard posed by chemical warfare agents (CWAs) have attracted recent interest,1 with chemical sensors,2,3 catalysts4,5 and CWA-responsive materials being proposed,6–8 amongst other applications. In the pursuit of recognition strategies for the complexation of chemical warfare agents, and the subsequent transduction of this event to furnish functional materials, there is a need for academic and industrial research groups to use appropriate chemical simulants (also referred to as surrogates or analogues). This is mandated by both the health and safety impact of CWAs and also by their regulation under the Chemical Weapons Convention. The challenge arises in determining the most appropriate chemical simulant; in the case of chemical reactivity and vapour pressure, model compounds have been described and evaluated and their limitations understood.9,10 In the case of supramolecular binding events the choice of CWA simulant for many studies is not yet clear.Commonly used simulants include the dialkyl methylphosphonates and the trialkyl phosphates (Fig. 1), with examples of inclusion in supramolecular ‘baskets’ and coordination to metal centres being reported in recent years.11,12 Studies of CWA recognition behaviour are not common, and comparisons of agent behaviour to simulants even less so. Some rare examples of CWA recognition and also of comparative CWA-simulant behaviour include hydrogen bond-mediated recognition of the nerve agent O-pinacolyl methylphosphonofluoridate or soman (hereafter referred to as GD),13 studies of the coordination of both V- and G-series CWAs and simulants to lanthanide complexes,14 and the performance of responsive supramolecular gels to the presence of GD, dimethyl methylphosphonate (DMMP) and diethyl chlorophosphate (DCP).15,16
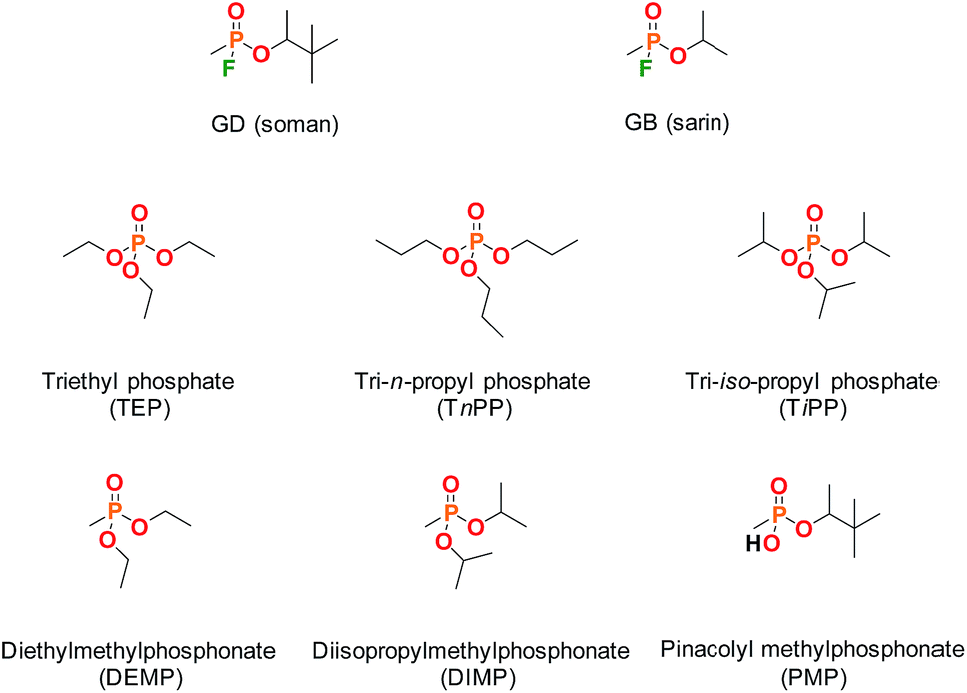 | ||
| Fig. 1 Molecular structures of the CWAs soman (GD) and sarin (GB) and some commonly used organophosphorus simulants. | ||
With regard to the use of the hydrophobic effect to drive inclusion of organophosphorus (OP) CWAs such as GB (sarin) and GD, cyclodextrins (CDs) provide an ideal test case for establishing supramolecular agent–simulant correlations. The recognition of OP CWAs by CDs has been demonstrated previously by a number of research groups;17,18 GB is bound by α- and GD by β-CD. Furthermore, functionalised-CDs have been proposed for use in CWA decontamination and medical countermeasures, particularly with regard to cyclosarin (GF).19,20 A recent review by Estour et al. summarises work in the interactions of both OP nerve agents and other toxic OP compounds, such as pesticides, with CDs.21 Studies of the complexes formed between OP CWAs and CDs by NMR methods and appropriate comparison to the inclusion behaviour of simulants has not, to the best of our knowledge, been widely investigated.
Herein, we describe experimental and computational methods for the study of inclusion complexes of GD by β-CD and make comparison to the binding behaviour of commonly used simulants. Such data will find utility in the development of new supramolecular approaches to hazard mitigation in which simulants are likely to be used, and, in addition, furnishes new understanding of the hydrophobic inclusion of GD by β-CD.
Results and discussion
The interaction of GD with β-cyclodextrin
Initial evidence that indicates the formation of complexes in solution is obtained from 1H NMR spectra of equimolar solutions of β-CD and GD in D2O and referenced against an internal MeOH standard.22,23 As can be seen in Fig. 2, significant upfield chemical shift perturbations of the β-CD internal cavity proton environments H-3 and H-5 (conventional proton numbering, see ESI†) are clearly seen and are highly indicative of inclusion of the GD guest species.24 It might be expected that, due to the aliphatic nature of the guests, that these perturbations in chemical shift would be small. In agreement with this proposed inclusion complex, the proton environments external to the cavity (e.g. H-2, H-4; Fig. 2) are not perturbed by the presence of the guest species.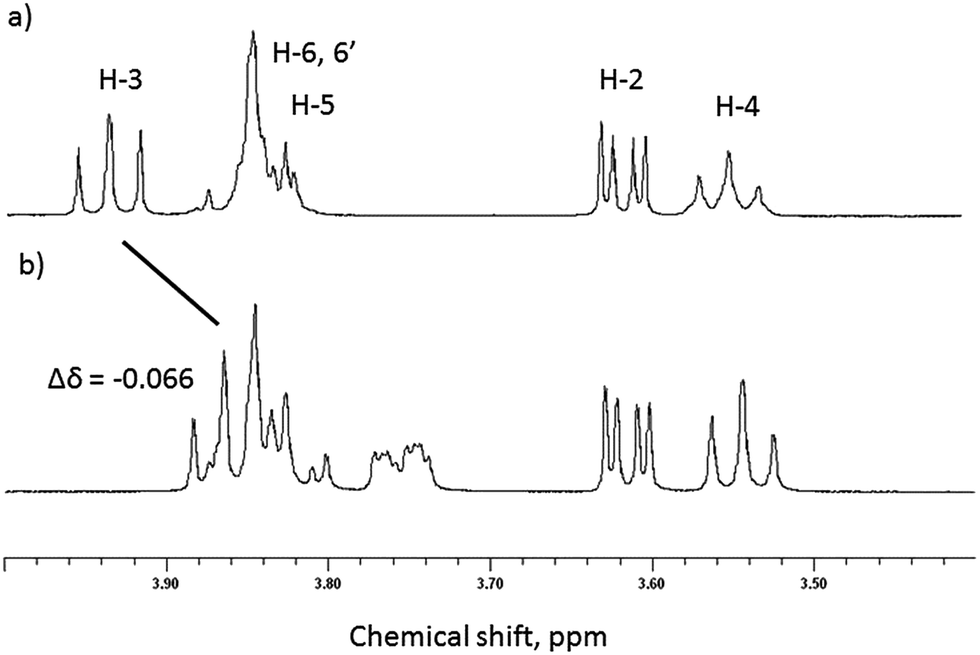 | ||
| Fig. 2 Selected region of the 1H NMR spectra of (a) β-CD and (b) equimolar β-CD and GD in D2O (293 K). | ||
Proton environments in the GD molecule were also observed to undergo significant chemical shift changes. The proton resonances of the tert-butyl group of the pinacolyl side-chain were observed to move downfield from 0.89 to 0.99 ppm, and the side-chain methyl group from 1.30 to 1.41 ppm. Conversely, the P–Me group underwent very small magnitude perturbations of <0.01 ppm, indicating that the inclusion of the pinacolyl side-chain in the CD cavity dominates the complexation event.
The presence of both phosphorus and fluorine nuclei on the GD guest molecule also provides additional information for the study of these complexes. As can be seen in Fig. 3 the presence of β-CD results in the resolution of all four stereoisomers (P(±), C(±)) of GD, indicating preferential binding of some isomers over others, in agreement with previous reports25 and as will be shown computationally. In addition, upfield chemical shift perturbations (≈0.3 ppm) are also observed for the phosphorus nuclei, again indicative of complex formation. Similarly significant perturbations are observed in the 19F NMR spectra of GD–β-CD mixtures, indicative once more of both inclusion complex formation and chiral resolution (see ESI†).
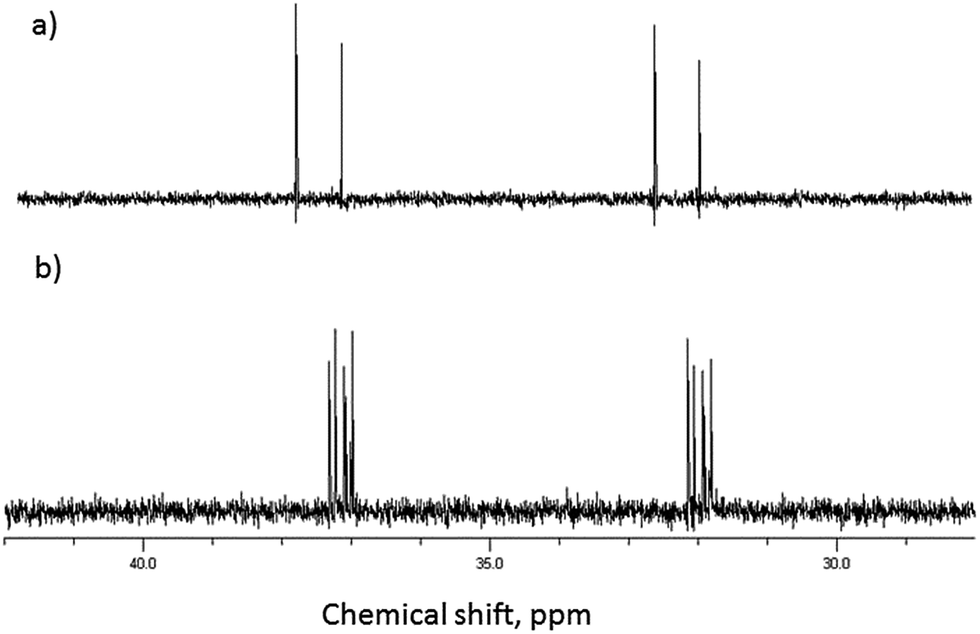 | ||
| Fig. 3 31P NMR of (a) GD and (b) equimolar solution of GD and β-CD in D2O (293 K). | ||
Clearly, the presence of multiple stereoisomers of the guest GD species results in complication of the binding phenomena. However, it should also be noted that the presence of isomerically pure samples of GD is unlikely, and their synthesis challenging. To that end, racemic mixtures of GD were studied with regard to determining complex stoichiometry and complex affinity. Standard methods for Job plot analysis were conducted, and the solutions checked for degradation of GD to furnish PMP as would be evidenced by the appearance of a singlet in the 31P NMR at 28.6 ppm (see ESI;† lit.26 25.99 ppm) over the course of the experiment. This was estimated to be <5% degradation, and as such unlikely to significantly influence the complex stoichiometry determination. Analysis of the data by monitoring the H-3 and H-5 environments of the β-CD cavity furnished a Job plot with maxima at ≈0.5 mol fraction, indicative of the formation of a complex of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometry (Fig. 4).
:1 stoichiometry (Fig. 4).
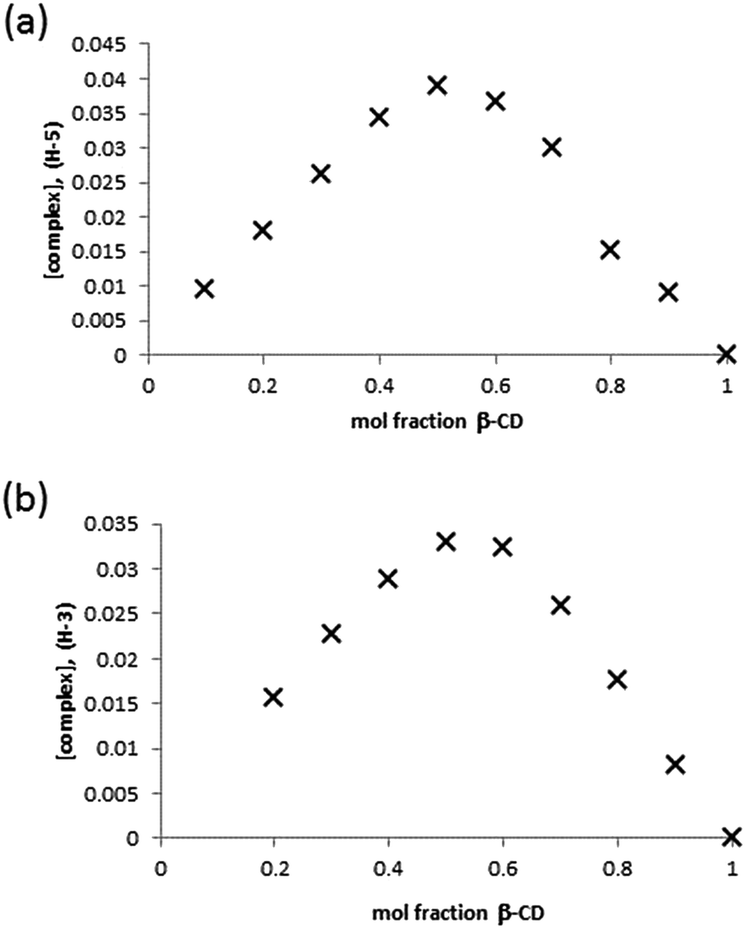 | ||
| Fig. 4 Job plot analysis of the complex formed between β-CD and GD in D2O by monitoring of the β-CD (a) H-3 and (b) H-5 internal cavity proton environments. | ||
Quantitative NMR titrations were then conducted in order to determine the association constant for the racemic mixture of GD with β-CD. Experiments were conducted using seventeen separate NMR samples of the β-CD solution with the addition of aliquots of a stock GD solution to furnish solutions containing appropriate molar equivalents of GD to construct a binding curve (0, 0.2, 0.4…2.0, 2.5, 3.0, 4.0, 5.0, 7.0 and 10.0 molar equivalents). Due to solubility limitations of GD in an aqueous environment, aliquot volumes of 8 μL per 0.2 mol equivalents were used. As with the Job plot analysis, titrations could potentially be further complicated by the presence of GD hydrolysis products that may interact with the β-CD cavity, namely PMP (vide infra), and a fresh GD stock solution was prepared at the start and at two intermediate points of the titration. Analysis of the 31P NMR data (see ESI† for spectra) for GD and GD–β-CD mixtures indicates minimal PMP present in the mixtures (typically <5%). Furthermore, 1H NMR spectra are in agreement with less than 5% PMP present in all samples. In addition to this, examination of the 1H NMR environments of the β-CD host indicates no reaction between host and guest over the time period of the experiments. The binding curves obtained by monitoring the changes in the chemical shift of both the H-3 and H-5 β-CD proton environments are shown in Fig. 5 and were fit satisfactorily to a 1:1 binding model using the software package EQNMR.27 Association constants of 2075 ± 66 and 2060 ± 79 M−1 for the H-3 and H-5 binding curves, respectively, were obtained (Table 1).
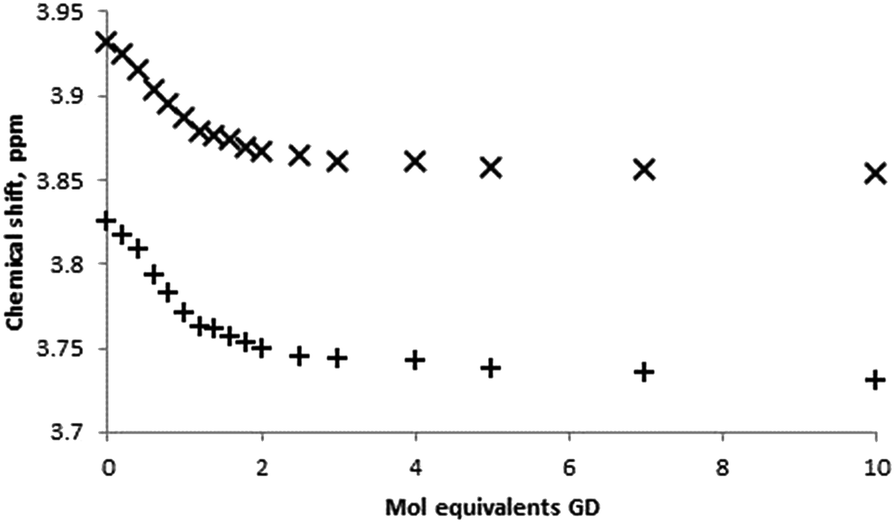 | ||
| Fig. 5 Binding curves obtained from 1H NMR titrations of β-CD with GD in D2O monitoring the β-CD H-3 (×) and H-5 (+) proton environments. | ||
:1 models and association constants determined by EQNMR27
| Guest species | Association constant M−1 |
|---|---|
| β-GD | 2075 ± 75; 2060 ± 79 |
| TEP | 40 ± 9 |
| TnPP | 115 ± 6 |
| TiPP | 56 ± 8 |
| DEMP | ≈0 |
| DIMP | 22 ± 4 |
| PMP | 230 ± 26 |
The interaction of trialkylphosphates and dialkyl methylphophonates with β-cyclodextrin
A series of commonly used trialkyl phosphates and dialkylmethyl phosphonate species were chosen as non-reactive simulants for the G-series agents. As with the GD experiments, initial screening of the guests with β-CD for complex formation was conducted using equimolar aqueous solutions of approximately 5 mmol concentration. Consideration of the internal (H-3 and H-5) protons once more indicates inclusion behaviour, although unlike the example with GD, the upfield perturbations are small: 0.012 for triethyl phosphate (TEP), 0.013 for diisopropyl methylphosphonate (DIMP), and <0.01 ppm for (diethyl methylphosphonate) DEMP, for the H-3 environment. Again, external protons (H-1, H-2, H-4, H-6) were not affected by the presence of the guest species.Determination of the stoichiometry of the complex formed between the simulant guest molecule and the β-CD host was once again determined by Job plot analysis. Fig. 6a is a typical Job plot obtained for DIMP and β-CD, indicating a 1:1 complex stoichiometry; the same complex stoichiometry was determined for all simulants studied. In the case of DEMP it should be noted that the perturbations in chemical shift were very small, indicating very low affinity complexes.
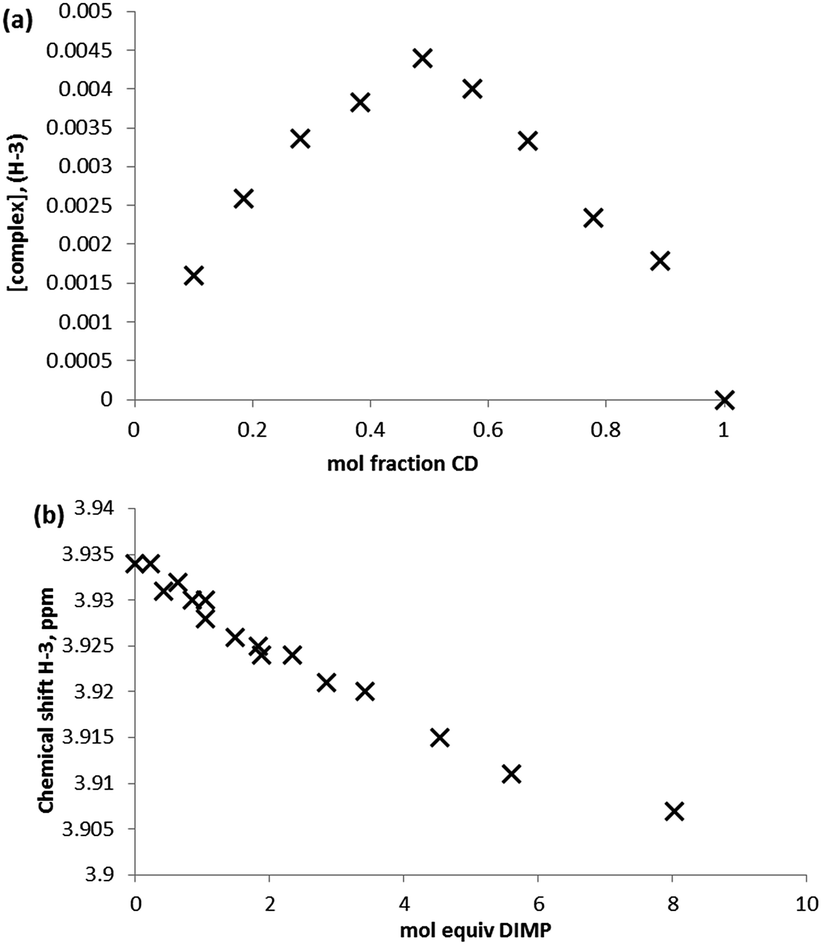 | ||
| Fig. 6 1H NMR studies of the complexation behaviour of DIMP with β-CD, where (a) Job plot analysis constructed form monitoring the β-CD H-3 proton environment, and (b) binding curve obtained by titration of β-CD with DIMP and monitoring of the β-CD H-3 proton environment (298 K, D2O). | ||
Determination of the association constants for the formation of the simulant-CD complexes was conducted by quantitative 1H NMR titration experiments. In all cases, the titration was performed by addition of minimum volume aliquots of the guest species into the β-CD solution in order to hold the concentration of the latter near-constant and to generate molar ratio solutions of up to 10:1 guest:host. Binding isotherms were constructed by monitoring the chemical shift perturbations in the β-CD internal proton environments H-3 and H-5 with changing guest concentration (Fig. 6b), although the small chemical shift perturbations led to some overlap of neighbouring data points. The determination of association constants for the complexation of simulants with β-CD were determined by fitting of the 1H NMR titration data using EQNMR.27 In the case of DEMP a binding curve could not be obtained, with shift perturbations of <0.01 ppm observed at a DEMP:CD molar ratio of 10:1. As such any DEMP–CD complex formation is extremely weak, and the binding constant reported as approximately zero. All CD-simulant association constants are reported in Table 1.
The interaction of pinacolyl methylphosphonate with β-cyclodextrin
Also of interest as a supramolecular G-agent simulant is pinacolylmethyl phosphonate (PMP), the hydrolysis product of the nerve agent GD. Structurally this bears closest resemblance to GD, but again the choice of this as a G-agent simulant is still not ideal. The pKa of PMP is estimated at ≈2.5, thus under the current conditions (and most conditions under which such binding events would be considered) the molecule will be dissociated and present as an anionic species.As with the previous simulants, 1H NMR revealed perturbations in the β-CD internal proton environments H-3 and H-5 and these were of a slightly greater magnitude (Δδ H-3 = 0.03 ppm at equimolar concentrations) to those observed with the trialkylphosphate and dialkyl methyl phosphonate simulants. Interestingly, external proton environments were also perturbed, albeit to a lesser extent, with environments H-1 and H-2 and H-4 perturbed upfield by approximately 0.01 ppm. Furthermore, it was found that the PMP guest protons were also affected significantly by the complexation, an effect which could not be measured easily on the previous simulants studied but was apparent with GD. In this case, the changes to the guest proton environments were larger than those observed for β-CD protons, with the side chain methyl group in particular perturbed by 0.12 ppm. Alongside significant chemical shift changes for the t-butyl group (approx. 0.088 ppm at 1:1 CD:PMP molar ratio) this is highly indicative of the inclusion of the aliphatic side-chain in the CD cavity (see ESI† for tabulated chemical shift observations). Analysis of 31P NMR spectra of PMP and equimolar PMP–β-CD mixtures revealed small upfield perturbations (≈0.03 ppm) supportive of complexation, and also resolution of stereoisomers (see ESI†).
Job plot analysis and NMR titration binding studies (Fig. 7) were undertaken for comparison to both the neutral simulant and GD studies. Fitting of the H-3 internal proton data yielded an association constant of 230 M−1.
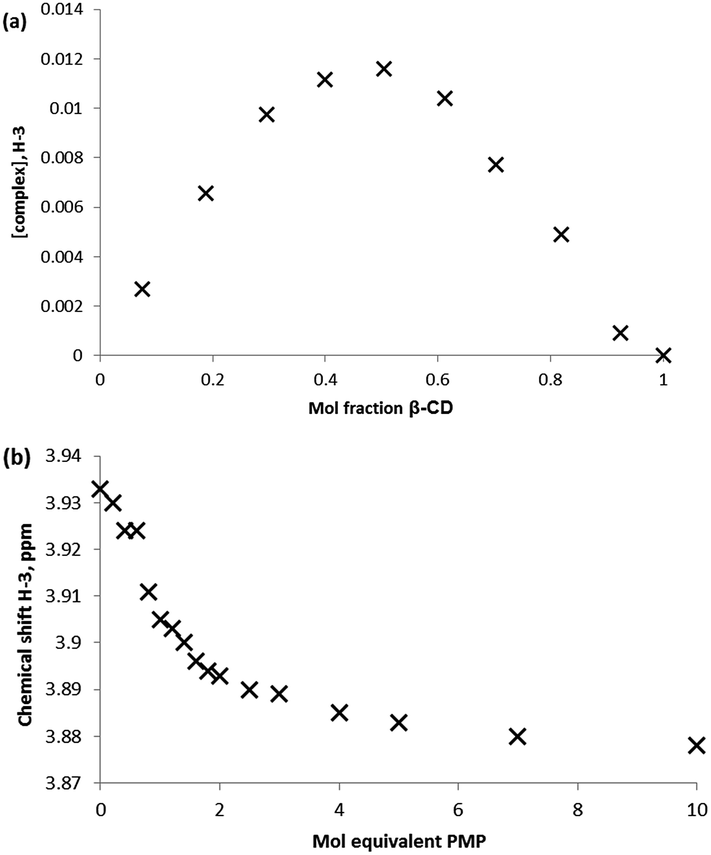 | ||
| Fig. 7 1H NMR studies of the complexation behaviour of PMP with β-CD, where (a) Job plot analysis constructed form monitoring the β-CD H-3 proton environment, and (b) binding curve obtained by titration of β-CD with PMP and monitoring of the β-CD H-3 proton environment (298 K, D2O). | ||
Computational modelling
As stated earlier, GD exists in four stereoisomers that possess different affinities for inclusion by β-CD and also possess different toxicities. According to Benschop and De Jong, the P(−) isomers possess greater affinity for human acetylcholine esterase (HuAChE), and therefore greater toxicity (LD50 (mouse) 99 μg kg−1 for P(−)C(+) and 38 μg kg−1 for P(−)C(−) following subcutaneous administration).28 In contrast, they state the analogous LD50 value for the P(+) isomers in the range 2000–5000 μg kg−1, and refer to these compounds as ‘non-toxic’. In agreement with Benschop and De Jong,28 van Dongen et al.29 also state that the C(±)P(+) isomers are non-toxic and furthermore, that they are hydrolysed more rapidly.More recent work by Ordenlich et al. measured the bimolecular rate constants of phosphorylation of HuAChE and showed a selectivity for the P(−)C(+) as 7.5 × 104-fold over the P(+)C(+) isomer.30 They also demonstrated negligible effects of the chiral Cα centre on the rate of phosphorylation. The authors explain the greater toxicity of the P(−) isomers through the combination of the orientation of the P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group within the active site and the accommodation of the alkoxy substituent. This complementarity between the P(−) isomers and the active centre allows for polarisation of the PO bond by the oxyanion hole and the close proximity of this group to the nucleophilic residues. In contrast, the P(+) diastereoisomers exhibit low reactivity that results from steric constraints preventing accommodation of the alkoxy side-chain within the enzyme acyl pocket.
O group within the active site and the accommodation of the alkoxy substituent. This complementarity between the P(−) isomers and the active centre allows for polarisation of the PO bond by the oxyanion hole and the close proximity of this group to the nucleophilic residues. In contrast, the P(+) diastereoisomers exhibit low reactivity that results from steric constraints preventing accommodation of the alkoxy side-chain within the enzyme acyl pocket.
As discrimination between these four isomers has been reported28,29 for β-CD it was decided to determine if computational methods could reproduce the experimental data. As a test of this approach all four isomers of GD were modelled as their β-CD complexes. Binding in water was presumed to occur through dehydration of both β-CD host and GD guest followed by complexation and ejection of water:
| β-CD·mH2O + GD·nH2O → β-CD·GD + (H2O)m+n |
Inspection of an X-ray structure of β-CD hydrate shows that it has ten water molecules inside the central cavity.31 These interact strongly with each other and the surrounding hydroxyl groups. Consequently the atomic coordinates from that structure were used as a basis for a model of β-CD·10H2O to simulate the solvated host. Initial calculations were undertaken with the semi-empirical PM6 method which is rapid compared to ab initio methods. Although the magnitudes of binding energies determined by this method are often unrealistic, they generally predict the correct rank order of complex formation.32,33 To introduce a further level of complexity it was assumed that GD would be solvated so a simulation of GD surrounded by 55 water molecules (to approximate a molar solution) was initiated to determine if there were any significant hydrogen bonds formed between the CWA and surrounding solvent. All non-hydrogen bonding water molecules were removed prior to a second geometry optimisation iteration by PM6. Free energies were calculated for the resulting PM6 optimised geometries for each chemical entity. The simulation indicated four interactions so the free energy of GD·4H2O was calculated. The process of host and guest dehydration involved the removal of 14 water molecules so the free energy of a (H2O)14 cluster was also calculated. The β-CD·GD complex was created by introducing GD into the cavity of solvent-free β-CD. Starting geometries were created using the Spartan′14 Minimizer. As β-CD has a slight funnel shape, GD was introduced in two orientations and the lower energy complex from each pair of simulations retained. The geometries of all entities were calculated from molecular mechanics coordinates (Merck Molecular Force Field) followed by semi-empirical calculations using PM6. The process can be formalised as:
| β-CD·10H2O + GD·4H2O → β-CD·GD + (H2O)14 |
The free energy for the complexation process is given by:
| ΔG(binding) = [β-CD·GD + (H2O)14] − [β-CD·10H2O + GD·4H2O] |
All four isomers are predicted to bind with the C(−)P(+) and C(+)P(+) isomers held more strongly than the C(−)P(−) and C(+)P(−) isomers. From these calculated values (Table 2) the average association constant from the simulations is 13742 at 298.15 K. Thermodynamic properties derived from semi-empirical models are generally far less accurate than those calculated by more sophisticated ab initio approaches, however, for supramolecular complexation involving relatively large host molecules the latter can be highly CPU intensive and time consuming. Using the PM6 data and fitting the experimental data from Table 1 for H-3, assuming that the value of K is likely to be an average of all four isomers, allows the derivation of a normalisation coefficient, n, of 1.27 from:
| ΔG(calcd) = −nRTlnK(expt) |
| G (kJ mol−1) | ΔG (kJ mol−1) | |
|---|---|---|
| GD·4H2O | 606.29 | — |
| β-CD·10H2O | 3057.00 | — |
| (H2O)14 | 518.55 | — |
| β-CD·GD[C(−)P(+)] | 3120.03 | −24.71 |
| β-CD·GD[C(+)P(+)] | 3120.38 | −24.36 |
| β-CD·GD[C(+)P(−)] | 3121.05 | −23.69 |
| β-CD·GD[C(−)P(−)] | 3121.35 | −23.36 |
Applying this to the individual calculated ΔG(binding) values gives K for the individual isomers as shown in Table 3.
| Association constants K11, M−1 | |||
|---|---|---|---|
| C(−)P(+), [(S)-C, (R)-P] | C(+)P(+), [(R)-C, (R)-P] | C(+)P(−), [(R)-C, (S)-P] | C(−)P(−), [(S)-C, (S)-P] |
| 2554 | 2285 | 1848 | 1664 |
These results indicate that the P(−) stereoisomers that possess greater complementarity for the active site of HuAChE, and exhibit greater toxicity, are in fact bound with lower affinity by β-CD than the lower toxicity P(+). The value of 1848 and 1664 M−1 for the two P(−) isomers are in close agreement with the value reported by Desire and Saint-Andre of 1887 M−1 (Kd = 5.3 × 10−4 M) at pH 7.40, determined from enzyme inhibition assays and fluoride release.25 While the difference between the toxic and non-toxic isomers determined computationally in our study is not as great as would be expected based on hydrolysis data, it has been possible to differentiate between the two classes using a low level, semi-empirical approach. Analysis of the resulting structures using Olex234 indicates that hydrogen bonds form between GD and the cyclodextrin for the C(±)P(−) isomers but not for the C(±)P(+) isomers, as shown in Fig. 8 and Table 4.32 Furthermore, in every case the t-butyl and methyl substituents of the pinacolyl group were drawn into the central cavity of the CD whereas the P–Me group remained at the annulus of the CD, in agreement with the chemical shifts observed in the proton NMR spectra. The chirality of the carbon centre is therefore of secondary importance to that of the phosphorus. In their 1.95 Å resolution crystal structure of GD with Torpedo californica acetylcholine esterase (TcAChE), Sanson et al. note that the terminal GD oxygen interacts weakly with hydrogen bond donors at distances of 2.6, 2.7 and 3.0 Å and that the bridging oxygen interacts with a histidine nitrogen atom at 3.2 Å.35 A similar effect is seen for the CD–GD complexes with C(±)P(−) chirality, where hydrogen bonds of 2.7 Å are found in the model systems. In all cases C–H⋯O interactions are predicted and it is presumably these weak arrays, coupled to hydrophobic effects, which stabilise complex formation leading to slightly more favourable ΔG values for the β-CD complexes if the C(±)P(+) isomers. It appears that in this case the less stable complexes happen to allow for some hydrogen bonding despite it often being associated with increased stability in supramolecular chemistry.
 | ||
| Fig. 8 β-CD·GD complexes: (from top) C(−)P(+), C(+)P(+), C(+)P(−), C(−)P(−). Dashed lines indicate intermolecular interactions. | ||
| Compound | X–H⋯O | ||
|---|---|---|---|
| X⋯O (Å) | H⋯O (Å) | X–H⋯O (°) | |
| GD[C(−)P(+)]–CD | |||
| C–H⋯O | 3.19 | 2.15 | 156.2 |
|
|||
| GD[C(+)P(+)]–CD | |||
| C–H⋯O | 3.21 | 2.17 | 154.2 |
| C–H⋯O | 3.23 | 2.13 | 175.9 |
|
|||
| GD[C(+)P(−)]–CD | |||
| O–H⋯O | 2.76 | 1.74 | 176.7 |
| C–H⋯O | 3.17 | 2.14 | 151.2 |
| C–H⋯O | 3.23 | 2.13 | 168.5 |
|
|||
| GD[C(−)P(−)]–CD | |||
| O–H⋯O | 2.74 | 1.71 | 172.6 |
| C–H⋯O | 3.14 | 2.11 | 151.6 |
| C–H⋯O | 3.22 | 2.12 | 169.9 |
| C–H⋯O | 3.24 | 2.17 | 159.5 |
Agent–simulant comparisons and implications for simulant selection
The use of simulants to test the efficacy of CWA disclosure and decontamination systems is necessary due to the prohibited use and safety impacts of the CWAs themselves. Indeed, there are many examples of responsive materials that claim functional properties towards CWAS based on extrapolation of simulant, rather than agent, data, however, the reason for selecting a specific simulant is not always clear and may be dependent on material availability or literature precedent.36–38 Whilst some agent–simulant correlations such as vapour pressure10 or material permeability39 are understood (or at least partially so), those involving host–guest systems are not to the same extent.In this work many of the commonly used simulants do not mirror the inclusion behaviour of GD in the hydrophobic cavity of β-CD. Almost all of the dialkyl methylphosphonates, such as DIMP, exhibit affinities two orders of magnitude lower than the GD·β-CD complex. Concerns could be raised with regard to pursuing synthetic host systems for CWA recognition based on simulant binding data, or conversely, the dismissal of hosts systems based upon the lack of simulant binding. The former could result in development of functional systems optimised towards binding simulant molecules and not CWAs, whereas the latter could result in missed opportunities where CWA binding could potentially be exploitable.
The work here presents a further case, alongside other supramolecular studies,1,14 that simulant selection must be considered carefully, and extrapolation of simulant data towards implied performance against CWAs must be done cautiously. It also demonstrates that computational methods can furnish additional useful information with regard to complex formation and, importantly, stereoisomer-specific complexation.
Conclusions
A combined experimental and computational study of the complexation of the CWA GD with β-CD has been conducted, with further insights provided by semiempirical modelling of the four isomers of GD bound within a hydrophobic cavity. Comparison to commonly used simulants provides agent–simulant correlation data, albeit for a specific rather than general case; further studies will seek to establish how generic some of these comparative behaviours are for a wider range of host systems. The significant discrepancy in complex affinity between the CWA and all the studied simulants highlights the importance of appropriate simulant choice in the study of supramolecular system.Acknowledgements
M. R. S. thanks Dr James Jones (Dstl) for assistance with NMR. P. J. C. thanks the US ARO for financial support (Contract W911NF-15-1-0624). Content includes material subject to © Crown Copyright (2015), Dstl. This material is licensed under the terms of the Open Government Licence except where otherwise stated. To view this licence, visit http://www.nationalarchives.gov.uk/doc/open-government-licence/version/3 or write to the Information Policy Team, The National Archives, Kew, London TW9 4DU, or email: E-mail: psi@nationalarchives.gsi.gov.uk.Notes and references
- M. R. Sambrook and S. Notman, Chem. Soc. Rev., 2013, 42, 9251 RSC.
- M. Burnworth, S. J. Rowan and C. Weder, Chem.–Eur. J., 2007, 13, 7828 CrossRef CAS PubMed.
- S. Royo, R. Martinez-Máñez, F. Sancenón, A. M. Costero, M. Parra and S. Gil, Chem. Commun., 2007, 4839 RSC.
- A. Barbra-Bon, A. M. Costero, M. Parra, S. Gil, R. Martinez-Máñez, F. Sancenón, P. A. Gale and J. R. Hiscock, Chem.–Eur. J., 2013, 19, 1586 CrossRef PubMed.
- J. R. Hiscock, M. R. Sambrook, P. B. Cranwell, P. Watts, J. C. Vincent, D. J. Xuereb, N. J. Wells, R. Raja and P. A. Gale, Chem. Commun., 2014, 50, 6217 RSC.
- S. Chen, Y. Ruan, J. D. Brown, J. Gallucci, V. Maslak, C. M. Hadad and J. D. Badjić, J. Am. Chem. Soc., 2013, 135, 14964 CrossRef CAS PubMed.
- C. Wilson, M. J. Marcus, N. J. Cooper, M. E. Briggs, A. I. Cooper and D. J. Adams, Polym. Chem., 2017, 8, 1914–1922 RSC.
- B. O. Okesola and D. K. Smith, Chem. Soc. Rev., 2016, 45, 4226 RSC.
- S. L. Bartelt-Hunt, D. R. U. Knappe and M. A. Barlaz, Crit. Rev. Environ. Sci. Technol., 2008, 38, 112 CrossRef CAS.
- Y. C. Yang, J. A. Baker and J. R. Ward, Chem. Rev., 1992, 92, 1729 CrossRef CAS.
- Y. Ruan, H. A. Taha, R. J. Yoder, V. Maslak, C. M. Hadad and J. D. Badjić, J. Phys. Chem. B, 2013, 117, 3240 CrossRef CAS PubMed.
- D. Knapton, M. Burnworth, S. J. Rowan and C. Weder, Angew. Chem., Int. Ed., 2006, 45, 5825 CrossRef CAS PubMed.
- M. R. Sambrook, J. R. Hiscock, A. Cook, A. C. Green, I. Holden, J. C. Vincent and P. A. Gale, Chem. Commun., 2012, 48, 5605–5607 RSC.
- G. H. Dennison, C. G. Bochet, C. Curty, J. Ducry, D. J. Nielsen, M. R. Sambrook, A. Zaugg and M. R. Johnston, Eur. J. Inorg. Chem., 2016, 1348 CrossRef CAS.
- J. R. Hiscock, F. Piana, M. R. Sambrook, N. J. Wells, A. J. Clark, J. C. Vincent, N. Busschaert, R. C. D. Brown and P. A. Gale, Chem. Commun., 2013, 6217–6220 Search PubMed.
- J. R. Hiscock, M. R. Sambrook, J. A. Ede, N. J. Wells and P. A. Gale, J. Mater. Chem. A, 2015, 3, 1230 CAS.
- C. Van Hooidonk and J. C. A. E. Breebart-Hansen, Recl. Trav. Chim. Pays-Bas, 1971, 90, 680 CAS.
- C. Van Hooidonk and J. C. A. E. Breebart-Hansen, Recl. Trav. Chim. Pays-Bas, 1972, 92, 958 Search PubMed.
- F. Worek, T. Seeger, M. Zengerle, S. Kubik, H. Thiermann and T. Wille, Toxicol. Lett., 2014, 226, 222 CrossRef CAS PubMed.
- A. Kranawetvogl, S. Müller, S. Kubik, H. Spruit, H. Thiermann, F. Worek, D. Noort and G. Reiter, Toxicol. Lett., 2015, 239, 41 CrossRef CAS PubMed.
- S. Letort, S. Balieu, W. Erb, G. Gouhier and F. Estour, Beilstein J. Org. Chem., 2016, 12, 204 CrossRef CAS PubMed.
- Y. Matsui and S. Tokunaga, Bull. Chem. Soc. Jpn., 1996, 69, 2477 CrossRef CAS.
- N. Funasaki, M. Nomura, S. Ishikawa and S. Neya, J. Phys. Chem. B, 2001, 105, 7361 CrossRef CAS.
- H.-J. Schneider, F. Hacket and V. Rüdiger, Chem. Rev., 1998, 98, 1755 CrossRef CAS PubMed.
- B. Desire and S. Saint-Andre, Fundam. Appl. Toxicol., 1986, 7, 646 CrossRef CAS PubMed.
- H. Kaskela, Anal. Chim. Acta, 2012, 751, 105–111 CrossRef PubMed.
- M. J. Hynes, J. Chem. Soc., Dalton Trans., 1993, 311 RSC.
- H. P. Benschop and L. P. A. De Jong, Acc. Chem. Res., 1988, 21, 368–374 CrossRef CAS.
- C. J. van Dongen, J. de Lange and J. van Genderen, Biochem. Pharmacol., 1989, 38, 2263 CrossRef CAS PubMed.
- A. Ordentlich, D. Barak, C. Kronman, H. P. Benschop, L. P. A. De Jong, N. Ariel, R. Barak, Y. Segall, B. Velan and A. Shafferman, Biochemistry, 1999, 38, 3055 CrossRef CAS PubMed.
- W. Saenger and T. Steiner, Acta Crystallogr., Sect. A: Found. Crystallogr., 1998, 54, 798 CrossRef.
- F. Fucassi, A. Heikal, L. I. Mikhalovska, G. Standen, I. U. Allan, S. V. Mikhalovsky and P. J. Cragg, J. Inclusion Phenom. Macrocyclic Chem., 2014, 80, 345 CrossRef CAS.
- L. X. Song, H. M. Wang, X. Q. Guo and L. Bai, J. Inorg. Chem., 2008, 73, 8305–8316 CAS.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339 CrossRef CAS.
- B. Sanson, F. Nachon, J.-P. Colletier, M.-T. Froment, L. Toker, H. M. Greenblatt, J. L. Sussman, Y. Ashani, P. Masson, I. Silman and M. Weik, J. Med. Chem., 2009, 52, 7593 CrossRef CAS PubMed.
- L. Pascual, S. El Sayed, R. Martinez-Manez, A. M. Costero, S. Gil, P. Gavina and F. Sanceon, Org. Lett., 2016, 18, 5548 CrossRef CAS PubMed.
- C. Belger, J. G. Weis, E. Egap and T. M. Swager, Macromolecules, 2015, 48, 7990 CrossRef CAS.
- S. Chen, Y. Ruan, J. D. Brown, J. Gallucci, V. Maslak, C. M. Hadad and J. D. Badjić, J. Am. Chem. Soc., 2013, 135, 14964 CrossRef CAS PubMed.
- K. H. Jung, B. Pourdeyhimi and X. W. Zhang, J. Membr. Sci., 2010, 362, 137 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Methods for Job plot and titration experiments, data fitting. See DOI: 10.1039/c7ra03328a |
| This journal is © The Royal Society of Chemistry 2017 |