
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
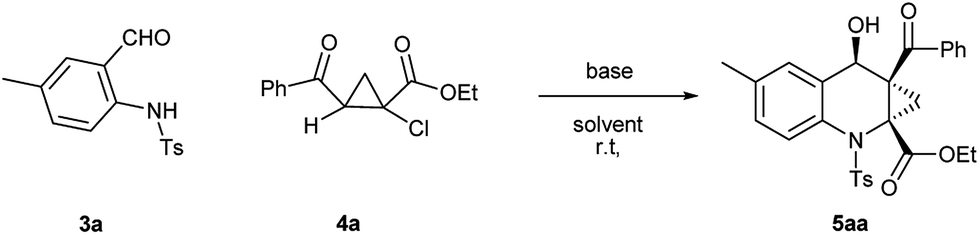
Direct stereoselective construction of cyclopropane α-amino acid with contiguous quaternary centers via [4 + 2] annulation reaction†
Jun-Hao Hu
b,
Yun-Chao Xua,
Dan-Dan Liua,
Bing Suna,
Ying Yi*a and
Fang-Lin Zhang *a
*a
aSchool of Chemistry, Chemical Engineering and Life Sciences, Wuhan University of Technology, Wuhan 430070, People's Republic of China. E-mail: fanglinzhang@whut.edu.cn; yying630@163.com
bDepartment of Chemistry and Material Science, Key Laboratory of Functional Organometallic Materials of Hunan Provincial College, Hengyang Normal University, Hengyang, Hunan 421008, People's Republic of China
First published on 2nd August 2017
Abstract
A direct diastereoselective synthetic approach to useful cyclopropane α-amino acid was established via base-promoted [4 + 2] annulations between o-aminobenzaldehydes and alkyl 2-aroyl-1-chlorocyclopropanecarboxylates. The annulation reaction proceeded quickly under mildly basic conditions, affording α-aminocyclopropanecarboxylic acid derivatives in moderate to excellent yields with high diastereoselectivities (up to 19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1).
:1).
Since α-aminocyclopropanecarboxylic acid (ACC) was first isolated from cowberries by Vähätalo and Virtanen in 1955, ACC motifs and their derivatives have attracted considerable attention.1 ACC not only exists in many natural products, bioactive compounds and pharmaceuticals,2 but also serves as valuable tool for the mechanistic study and characterization of enzymes.3 Some representative bioactive examples bearing this skeleton are given in Fig. 1. Great interest has been shown in the efficient construction of ACC frameworks, especially those with tertiary–quaternary carbon centers.4 In addition, recent medicinal research revealed that compound I and its derivatives as a combination of 1-aminocyclopropane-1-carboxylic acid and 1,2,3,4-tetrahydroquinoline-2-carboxylic acid moieties have strong affinities with the glycine binding site of the NMDA receptor,5 and also play an important role in neuronal cell death during ischaemic or hypoxic conditions such as stroke or epilepsy.6–8 Unfortunately, however, a multiple-step synthesis is essential to construct two contiguous quaternary centers.9 Therefore, the development of more efficient and general methods for the synthesis of ACC derivatives containing contiguous quaternary centers is highly desirable.
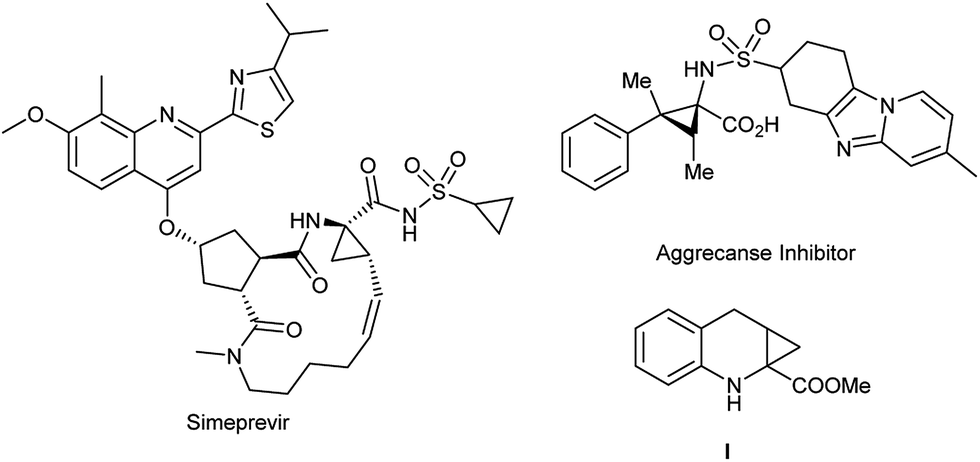 | ||
| Fig. 1 Typical bioactive molecules containing ACC motifs. | ||
The previously reported synthetic methods of ACC framework, such as insertion reaction of transition-metal-mediated α-nitroacetate carbene with disubstituted alkenes (Scheme 1A),10 [3 + 2] cycloaddition (Scheme 1B)11 and multi-step synthesis (Scheme 1C),9 are outlined in Scheme 1. The main disadvantages for these routes include low functional group tolerance and the expensive precursors prepared through multiple steps. Thus, it still remains a challenge to develop a more practical method for this core structure.
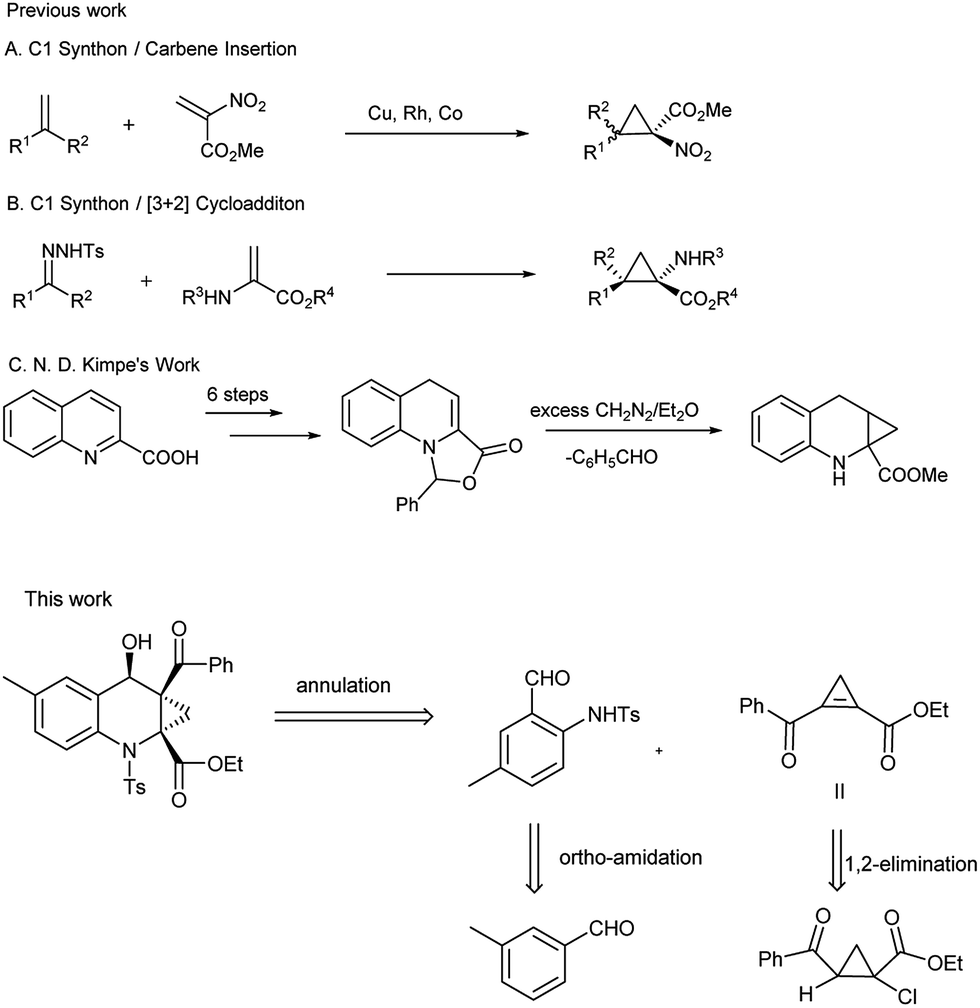 | ||
| Scheme 1 Diastereoselective synthesis of contiguous quaternary centers containing ACC. | ||
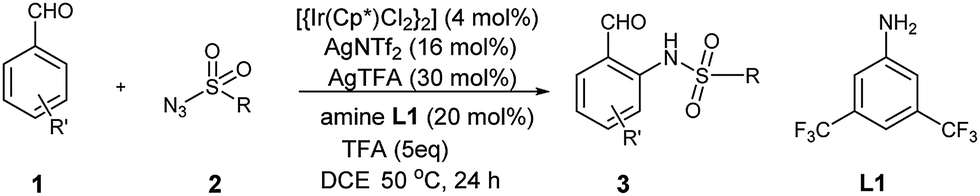
Over the last decade, the field of transition-metal-catalyzed aromatic C–H bond functionalization has gained significant development, and these strategies to form carbon–carbon and carbon–heteroatom bond have become increasingly commonplace.12 Recently, Yu et al. reported the use of glycine as a transient directing group for functionalization of C(sp3)–H bond of aldehyde.13 Then, Yu and our group have collectively demonstrated an ortho-C(sp2)–H functionalization of benzaldehyde using transient directing groups.14 As part of our ongoing work, we found that the products 3 derived from ortho-amidation of aldehydes were donor–acceptor species substantially which can be further transformed into useful core architectures. In addition, Gong et al. developed a convenient way to prepare a new type of electron-deficient cyclopropene intermediate II (marked in Scheme 1) in situ by carrying out a simple 1,2-elimination of alkyl 2-aroyl-1-chlorocyclopropanecarboxylates under basic conditions.15 This reagent has been successfully applied to construct strained ring-fused bioactive molecules through its base-promoted annulation reactions.16 The above progress encouraged us to assess the possibility of constructing the important ACC subunit I directly through the designed [4 + 2] annulation reaction between donor–acceptor reagents 3 and alkyl 2-aroyl-1-chlorocyclopropanecarboxylates. To our delight, the expected products were obtained in moderate to high yields under mildly basic conditions. The details about the Ir-catalyzed amidation of benzaldehydes with sulfonyl azides and subsequent [4 + 2] annulation reaction with alkyl 2-aroyl-1-chlorocyclopropanecarboxylates are described herein.
Firstly, the preparation of donor–acceptor reagents 3 followed our previous work.14 Under the same conditions, the yield of 3a was obtained only in 50%. Therefore, we preliminarily screened the influence of various amines, and found that amine L1 gave the desired product 3a in 75% yield (Table S1†).
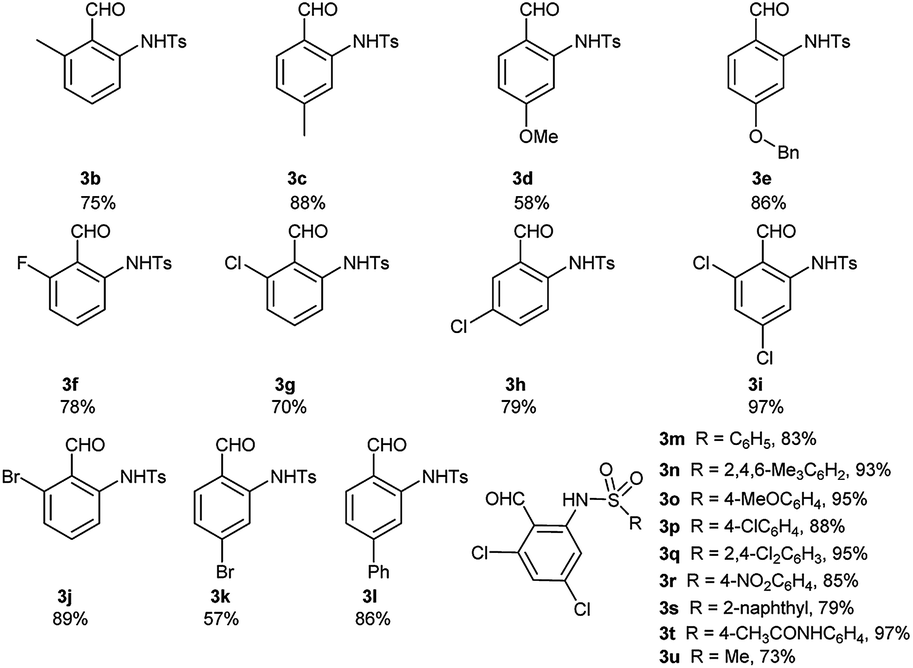
Under the optimal reaction conditions, we obtained various ortho-aminobenzaldehyde products 3, which are summarized in Table 1, in good (57%) to excellent (97%) yields.
With ortho-aminobenzaldehyde products 3 in hand, we envisioned the construction of ACC subunit though annulation reaction of 3 with ethyl 2-aroyl-1-chlorocyclopropanecarboxylates 4, the reaction of 3a with 4a was first carried out in the presence of Cs2CO3 as model. In THF, the reaction readily proceeded at room temperature, and 4a was almost completely consumed after 12 h. The product, isolated through silica gel column chromatography in 15% yield with 9:1 dr value, was identified to be the fused ACC ester 5aa by spectroscopic means (Table 2, entry 1). The diastereomeric ratio of 5aa was determined by 1H NMR spectroscopy. The stereochemistry for this process was confirmed by single crystal X-ray diffraction analysis.17 To optimize the reaction conditions, various solvents were taken into account. As shown in Table 2, in aprotic polar solvents such as N,N-dimethylformamide (DMF) and dimethyl sulfoxide (DMSO), this reaction proceeded smoothly to produce 5aa with high dr value (Table 2, entries 2 and 3), and a satisfactory yield (84%) was observed in DMF. In acetonitrile, the reaction proceeded slowly, and the yield of 5aa was relatively lower than the observed in DMF (Table 2, entry 4). In weakly polar solvent 1,4-dioxane, almost none of the desired product was detected (Table 2, entry 5).
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | Base | T (h) | Yieldb (%) | drc |
| a Reactions carried out using 0.10 mmol of 3a, 0.11 mmol of 4a and 0.20 mmol of Cs2CO3 in 1.0 mL of solvent at room temperature.b Isolated yields given.c Diastereomeric ratio (cis:trans) of the crude product determined by 1H NMR. |
|||||
| 1 | THF | Cs2CO3 | 12 | 15 | 9:1 |
| 2 | DMSO | Cs2CO3 | 2 | 84 | 13:1 |
| 3 | DMF | Cs2CO3 | 2 | 86 | 14:1 |
| 4 | CH3CN | Cs2CO3 | 12 | 70 | 14:1 |
| 5 | 1,4-Dioxane | Cs2CO3 | 12 | <5 | — |
| 6 | DMF | NaOH | 1 | 23 | 10:1 |
| 7 | DMF | K3PO4 | 12 | 56 | 9:1 |
| 8 | DMF | K2CO3 | 12 | <5 | — |
| 9 | DMF | TEA | 12 | — | — |
| 10 | DMF | DBU | 12 | — | — |
In view of the yields observed above, we chose DMF as the most promising solvent to optimize the various bases. As shown in Table 2, the yield of 5aa was remarkably dependent on the properties of the bases used. Strong inorganic base like NaOH could greatly promote this reaction, giving 5aa only in 23% yield (Table 2, entry 6). In contrast, a satisfactory result was achieved when K3PO4 was employed (Table 2, entry 7). On the other hand, the common inorganic base K2CO3, the organic base Et3N and strong organic base DBU (Table 2, entries 8, 9 and 10) were hardly able to promote this process.
With the optimized conditions of annulation reaction in hand, we subsequently examined the substrate scope of this reaction. First, the range of substrates 3 was investigated and the observed results are summarized in Table 3. Under the optimal reaction conditions, substrates 3a–3e with electron-donating groups at the benzene ring afforded the products 5aa–5ea in good yields and good dr values, respectively. Among the substrates 3f–3k with electron-withdrawing groups, the substrate 3f with 2-fluoro group afforded the highest yield and high dr value of the annulation product 5fa, whereas the substrate 3g with 2-chloro group furnished a high yield of the product 5ga with an excellent dr value (19:1) and the substrate 5i with 2,4-dichloro group gave the lowest yield of 5ia. Besides, substrate 3l with phenyl group was well tolerated in this reaction, producing the product 5la in a high yield with a good dr value.
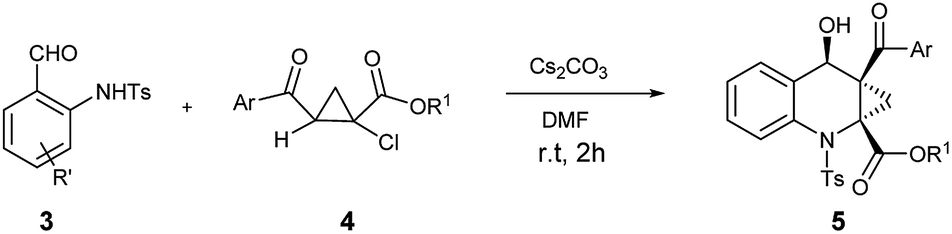
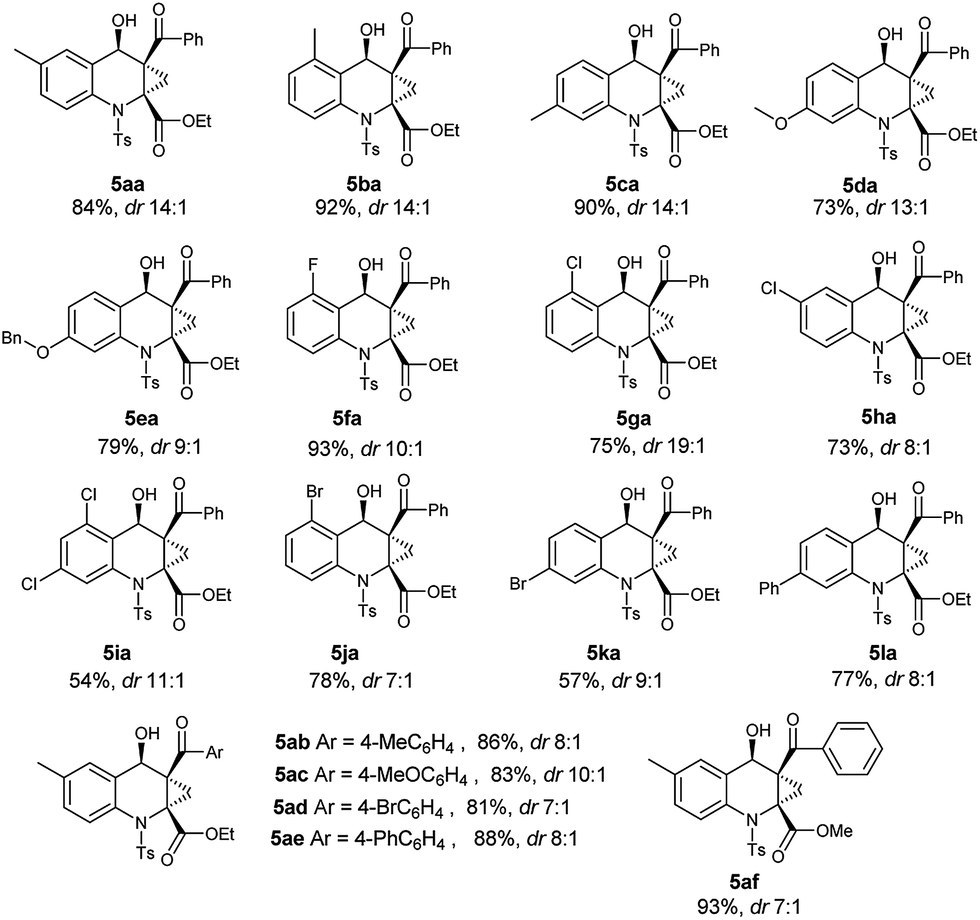
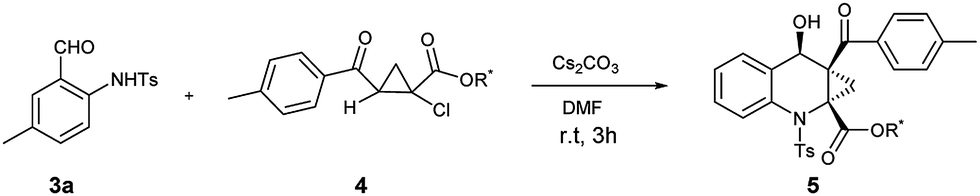
Next, we further investigated the structure effect of 2-aroyl-1-chlorocyclopropanecarboxylates 4 on the reaction. As shown in Table 3, the electronic nature of the Ar group couldn't obviously influence the product yields and the diastereomeric ratios, affording the products 5ab–5ae in high yields with good dr values, respectively. When the R1 group was replaced with a small methyl group, the corresponding product 5af could be obtained in the highest yield and good dr value.
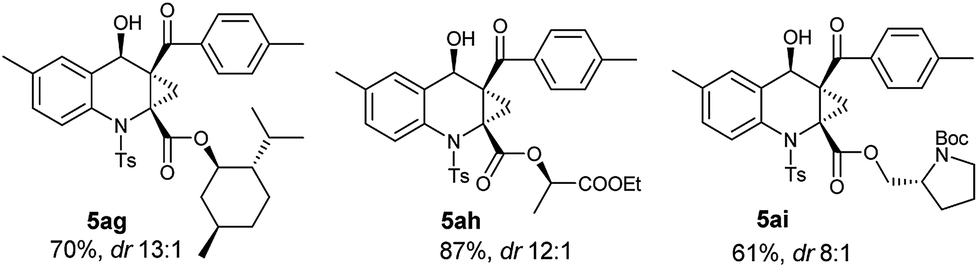
As known to all, stereoselective construction of ACC subunits is a challenging but demanding target. Encouraged by above results, we chose substrates 4 containing chiral auxiliary to expand this protocol to the synthesis of chiral ACC subunits. To our delight, the substrates 4g–4i underwent a smooth transformation to afford products 5ag–5ai in high yields and good dr values, respectively (Table 4).
Based on the above observations, we proposed a possible reaction mechanism as shown in Scheme 2. The reaction could proceed through a highly regioselective aza-Michael addition to the strained C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond of the highly reactive cyclopropene intermediate II, generated in situ in the presence of base. We realize that the diastereoselectivity of the reaction may be dominated by the coordination state of the intermediate 6. Owing to the apparent steric hindrance, the coordination state 6 was converted into fused polycyclic intermediate 7 with high dr value. Then 7 was subsequently protonated into the final product 5.
C bond of the highly reactive cyclopropene intermediate II, generated in situ in the presence of base. We realize that the diastereoselectivity of the reaction may be dominated by the coordination state of the intermediate 6. Owing to the apparent steric hindrance, the coordination state 6 was converted into fused polycyclic intermediate 7 with high dr value. Then 7 was subsequently protonated into the final product 5.
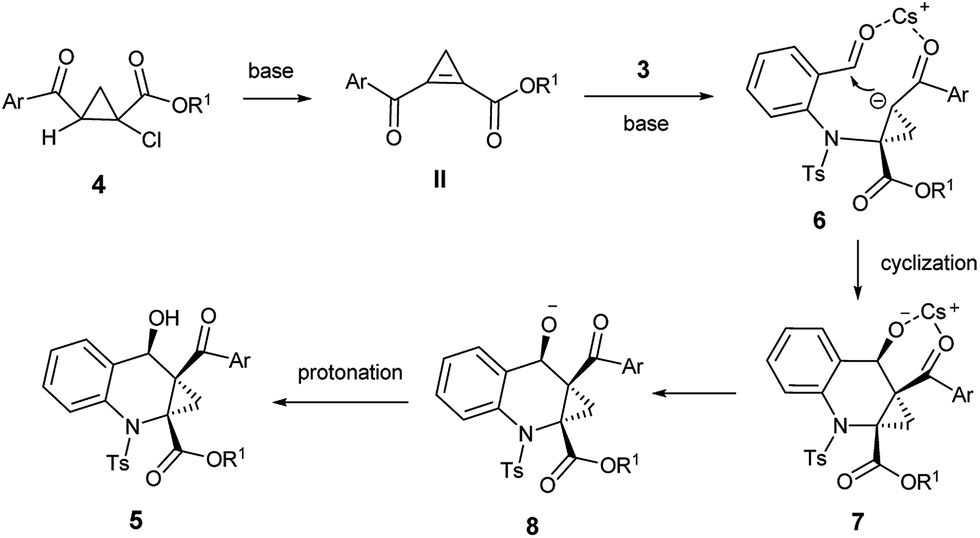 | ||
| Scheme 2 A possible mechanistic process for the [4 + 2] annulation. | ||
In summary, we have developed an efficient and practical [4 + 2] annulation reaction between alkyl 2-aroyl-1-chlorocyclopropanecarboxylates and donor–acceptor reagents derived from ortho-amidation of aldehydes in the presence of an inorganic base. This protocol is suitable for directly constructing the biologically and pharmaceutically useful cyclopropane α-amino acid bearing three continuous chiral carbon atoms and two quaternary stereogenic centers. This base-promoted cascade process does not require a transition metal catalyst, and avoids multiple steps. This reaction is tolerant to the steric hindrance and electronic properties of the reactants and can be easily performed under very mild conditions, giving ACC subunits in high yields and diastereoselectivities. Notably, ACC subunits can be stereoselectively constructed in high yields and diastereoselectivities through chiral auxiliary.
Notes and references
- C. Cativiela and M. D. Díaz-de-Villegas, Tetrahedron: Asymmetry, 2000, 11, 645 CrossRef CAS; W. A. Donaldson, Tetrahedron, 2001, 57, 8589 CrossRef; F. Brackmann and A. de Meijere, Chem. Rev., 2007, 107, 4493 CrossRef PubMed.
- H. Kakeya, H.-P. Zhang, K. Kobinata, R. Onose, C. Onozawa, T. Kudo and H. Osada, J. Antibiot., 1997, 50, 370 CrossRef CAS PubMed; M. Hara, S. Soga, M. Itoh, K. Shono, J. Eishima and T. Mizukami, J. Antibiot., 2000, 53, 720 CrossRef PubMed; M. Nishio, J. Kohno, M. Sakurai, S.-I. Suzuki, N. Okada, K. Kawano and S. Komatsubara, J. Antibiot., 2000, 53, 724 CrossRef PubMed.
- K. R. Hill, S. R. Prakash, R. Wiesendanger, W. Angst, B. Martinoni, D. Arigoni, H. W. Liu and C. T. Walsh, J. Am. Chem. Soc., 1984, 106, 795 CrossRef CAS; M. F. White, J. Vasquez, S. F. Yang and J. F. Kirsch, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 12428 CrossRef; J. Zhou, A. M. Rocklin, J. D. Lipscomb, L. Que and E. I. Solomon, J. Am. Chem. Soc., 2002, 124, 4602 CrossRef PubMed.
- P. K. Mykhailiuk, S. Afonin, A. S. Ulrich and I. V. Komarov, Synthesis, 2008, 2008, 1757 CrossRef CAS; V. N. G. Lindsay, W. Lin and A. B. Charette, J. Am. Chem. Soc., 2009, 131, 16383 CrossRef PubMed; C.-L. Zhu, L.-J. Yang, S. Li, Y. Zheng and J.-A. Ma, Org. Lett., 2015, 17, 3442 CrossRef PubMed.
- R. Nagata, N. Tanno, T. Kodo, N. Ae, H. Yamaguchi, T. Nishimura, F. Antoku, T. Tatsuno, T. Kato, Y. Tanaka and M. Nakamura, J. Med. Chem., 1994, 37, 3956 CrossRef CAS PubMed; S. Katayama, N. Ae and R. Nagata, Tetrahedron: Asymmetry, 1998, 9, 4295 CrossRef; G. Dannhardt, M. Gruchalla, K. B. Kohl and G. C. Parsons, Arch. Pharm., 2000, 333, 267 CrossRef PubMed.
- B. Meldrum and J. Garthwaite, Trends Pharmacol. Sci., 1990, 11, 379 CrossRef CAS PubMed; S. M. Rothman and J. W. Olney, Trends Neurosci., 1995, 18, 57 Search PubMed.
- A. J. Robl, S. D. Karanewsky and M. M. Assad, Tetrahedron Lett., 1995, 36, 1593 CrossRef; P. G. Zecchini and P. M. Paradisi, J. Heterocycl. Chem., 1979, 16, 1589 CrossRef.
- N. Gruenfeld, US Pat. 4,401,818, 1983Chem. Abstr. 1984, 100, 34421.
- M. Shiozaki, K. Maeda, T. Miura, M. Kotoku, T. Yamasaki, I. Matsuda, K. Aoki, K. Yasue, H. Imai, M. Ubukata, A. Suma, M. Yokota, T. Hotta, M. Tanaka, Y. Hase, J. Haas, A. M. Fryer, E. R. Laird, N. M. Littmann, S. W. Andrews, J. A. Josey, T. Mimura, Y. Shinozaki, H. Yoshiuchi and T. Inaba, J. Med. Chem., 2011, 54, 2839 CrossRef CAS PubMed; Z. Szakonyi, F. Fülöp, D. Tourwé, N. De and Kimpe, J. Org. Chem., 2002, 67, 2192 CrossRef PubMed.
- B. Moreau and A. B. Charette, J. Am. Chem. Soc., 2005, 127, 18014 CrossRef CAS PubMed; S. Zhu, J. A. Perman and X. P. Zhang, Angew. Chem., Int. Ed., 2008, 47, 8460 CrossRef PubMed; A. Pons, H. Beucher, P. Ivashkin, G. Lemonnier, T. Poisson, A. B. Charette, P. Jubault and X. Pannecoucke, Org. Lett., 2015, 17, 1790 CrossRef PubMed.
- C. Zhu, J. Li, P. Chen, W. Wu, Y. Ren and H. Jiang, Org. Lett., 2016, 18, 1470 CrossRef CAS PubMed.
- D. A. Colby, R. G. Bergman and J. A. Ellman, Chem. Rev., 2010, 110, 624 CrossRef CAS PubMed; T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147 CrossRef PubMed; G. Song, F. Wang and X. Li, Chem. Soc. Rev., 2012, 41, 3651 RSC; I. P. Beletskaya and A. V. Cheprakov, Organometallics, 2012, 31, 7753 CrossRef; P. B. Arockiam, C. Bruneau and P. H. Dixneuf, Chem. Rev., 2012, 112, 5879 CrossRef PubMed; G. Rouquet and N. Chatani, Angew. Chem, 2013, 125, 11942 (Angew. Chem. Int. Ed, 2013, 52, 11726) CrossRef.
- F. L. Zhang, K. Hong, T. J. Li, H. Park and J. Q. Yu, Science, 2016, 351, 252 CrossRef CAS PubMed.
- X. H. Liu, H. Park, J. H. Hu, Y. Hu, Q. L. Zhang, B. L. Wang, B. Sun, K. S. Yeung, F. L. Zhang and J. Q. Yu, J. Am. Chem. Soc., 2017, 139, 888 CrossRef CAS PubMed.
- M. Zhang, Y. F. Gong and W. Z. Wang, Eur. J. Org. Chem., 2013, 7372 CrossRef CAS.
- M. Zhang, J. K. Guo and Y. F. Gong, Eur. J. Org. Chem., 2014, 1942 CrossRef CAS; M. Zhang, F. Luo and Y. F. Gong, J. Org. Chem., 2014, 79, 1335 CrossRef PubMed; J. H. Hu, M. Zhang and Y. F. Gong, Eur. J. Org. Chem., 2015, 1970 CrossRef; Y. Q. Zhu, M. Zhang, H. L. Yuan and Y. F. Gong, Org. Biomol. Chem., 2014, 12, 8828 Search PubMed; Y. Q. Zhu and Y. F. Gong, J. Org. Chem., 2015, 80, 1446 CrossRef PubMed; Z. M. Huang, J. H. Hu and Y. F. Gong, Org. Biomol. Chem., 2015, 13, 8561 Search PubMed; J. H. Hu, Y. Liu and Y. F. Gong, Adv. Synth. Catal., 2015, 357, 2781 CrossRef.
- CCDC 1525861 contains the supplementary crystallographic data for this paper.†.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1525861. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra06465a |
| ‡ J. H. and Y. X. contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |