
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Dispersing hydrophilic nanoparticles in nonaqueous solvents with superior long-term stability†
Muhan Cao‡
,
Qipeng Liu‡,
Min Chen‡,
Peipei Yang,
Yong Xu,
Haihua Wu,
Jia Yu,
Le He *,
Xiao-Hong Zhang* and
Qiao Zhang*
*,
Xiao-Hong Zhang* and
Qiao Zhang*
Institute of Functional Nano & Soft Materials (FUNSOM), Jiangsu Key Laboratory for Carbon-Based Functional Materials & Devices, Soochow University, 199 Ren'ai Road, Suzhou, 215123, Jiangsu, PR China. E-mail: lehe@suda.edu.cn; xiaohong_zhang@suda.edu.cn; qiaozhang@suda.edu.cn
First published on 12th May 2017
Abstract
We report a general and robust polymerization–dissolution strategy for phase transfer of hydrophilic nanoparticles into nonaqueous solvents with a 100% transfer efficiency. This process involves the coating of hydrophilic nanoparticles with a layer of linear-chained polystyrene through seeded emulsion polymerization and a subsequent dissolution of polystyrene layer by toluene. Since one end of the linear polystyrene chain is covalently bonded to the particle surface which provides strong steric stabilization, the transferred nanoparticles exhibit superior dispersity and long-term colloidal stability in many nonpolar and polar aprotic solvents. Moreover, the present approach allows for the storage of transferred nanoparticles in a powder form which can be completely re-dispersed in solvents before the usage. Based on this strategy, we demonstrate the phase transfer of Au nanorods and nanospheres, silica, titania and resorcinol-formaldehyde spheres, which just represents a few examples of transferrable hydrophilic nanoparticles with different morphologies, sizes, compositions, functions and surface properties. This general and robust phase transfer protocol will greatly facilitate the applications of hydrophilic nanoparticle in organic catalysis, optoelectronics, energy storage and conversion, and organic light emitting diodes.
Introduction
Aqueous solvents are unique media for wet-chemical preparations of a large number of nanostructured materials that are difficult to produce in nonaqueous environments.1–5 For example, aqueous synthesis routes have been reported to obtain anisotropic nanoparticles of noble metals, including nanorods,6,7 nanospheres,8 nanocubes,9,10 nanoplates,11,12 octahedrons,13 and nanocages,14,15 mainly owing to the ease of control over the thermodynamics and kinetics of the growth reactions. Water is also required for sol–gel reactions to prepare metal oxide nanoparticles such as silica spheres.16 The advances in the preparation of hydrophilic nanoparticles have driven their applications in diverse fields, such as catalysis, optics, photonics, sensing, and biomedicine.15,17–20 For many applications such as organic catalysis and solution-processible optoelectronics, it is desirable if the nanoparticles originally prepared in aqueous solvents can be dispersed and stabilized in nonaqueous media, such as nonpolar and polar aprotic solvents, which requires the surface modification of hydrophilic nanoparticles.21Phase transfer of hydrophilic nanoparticles to hydrophobic solvents has mainly been accomplished through ligand exchange processes to modify the surface wettability.21–24 Taking metal nanoparticles as an example, many ligand-exchange techniques have been developed.25–27 Underwood and Melvaney pioneered the study on the phase transfer of gold nanoparticles from water to an organic solvent such as butyl acetate with a co-polymer based ‘comb stabilizer’.25 Many alkanethiol and alkylamine molecules have been used to hydrophobize aqueous Au nanoparticles.28–32 Despite recent advances in the phase transfer of nanoparticles, there are still several general concerns regarding ligand exchange methods.23,24,33 First, the transfer process is usually not complete as some residual particles are left in the aqueous phase. Second, in many cases, the transferred nanoparticles are not fully dispersed in nonaqueous solvents but rather agglomerate into larger clusters, which requires additional size-selection processes to remove the aggregates. Third, the long-term colloidal stability of the transferred particles in nonaqueous solvents during storage is usually questionable. Last but not the least, most ligand exchange processes are applicable to a very limited types of nanoparticles. Therefore, it is necessary to find a more general and robust phase transfer strategy for dispersion and long-term stabilization of hydrophilic nanoparticles in nonaqueous solvents.
Here we present a general and effective polymerization–dissolution strategy for phase transfer of hydrophilic nanoparticles into nonaqueous solvents by first covalently grafting the particle surfaces with a layer of linear-chained polystyrene through a seeded emulsion polymerization process, followed by the dissolution of polystyrene layer by toluene. Since one end of the linear polystyrene chain is covalently bonded to the particle surface, the transferred nanoparticles experience strong steric repulsions between each other in many nonpolar and polar aprotic solvents, resulting in superior dispersity and long-term colloidal stability. Both the polystyrene-coated and polystyrene-capped nanoparticles can be kept in a powder form which can later be dispersed in nonaqueous solvents, facilitating their storage and transportation toward practical applications. Based on this strategy, we demonstrate the phase transfer of gold nanospheres and nanorods, as well as silica, titania and resorcinol-formaldehyde spheres, to nonpolar solvents, which just represents a few examples of aqueous nanoparticles that can be transferred.
Results and discussion
Fig. 1 depicts the polymerization–dissolution strategy employed in the present work for phase transfer of hydrophilic nanoparticles into nonaqueous solvents. This process involves the surface coating of a polystyrene layer on aqueous nanoparticles (NPs) via seeded emulsion polymerization and the subsequent dissolution of the polymer layer to obtain polystyrene-capped nanoparticles. It is worth noting that seeded emulsion polymerization is a classic protocol in colloidal chemistry that has been widely used to synthesize NP@PS core–shell nanostructures.34–37 It is also well known that uncross-linked polystyrene spheres can be dissolved in toluene owing to the hydrophobic nature of polystyrene chains. Similarly, the polystyrene shell of the NP@PS particles can be dissolved by toluene to become individual polymer chains. As one end of the linear polymer chain is covalently bonded to the particle surface, the polystyrene-capped nanoparticles can be transferred to nonpolar and polar aprotic solvents that are compatible with polystyrene.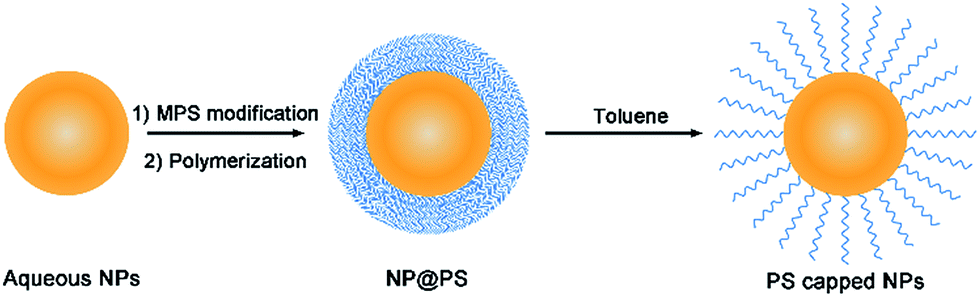 | ||
| Fig. 1 Schematic illustration of the polymerization–dissolution strategy to obtain polystyrene-capped nanoparticles (NPs) which can be dispersed in many nonaqueous solvents. | ||
Experimentally, we first demonstrate the polymerization–dissolution technique for the phase transfer of Au nanorods, which has attracted much attention due to their excellent surface plasmon resonance (SPR) property but the direct syntheses of hydrophobic Au nanorods (Au-NRs) in nonpolar solvents have rarely been reported.38 CTAB-capped Au nanorods were first synthesized through a seed-assisted growth approach (Fig. 2a).7 The CTAB-capped Au nanorods were treated with PVP molecules before the functionalization with the coupling reagent, [3-(methacryloyloxy)propyl]trimethoxysilane (MPS), to introduce surface C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds. Notably, the density of modified MPS molecules on the nanorod surface will influence the surface wettability of nanorod seeds and, thereby, the morphology of final particles. After surface modification with MPS, seeded emulsion polymerization was employed to coat the nanorods with a layer of polystyrene. A mixture solvent of water and ethanol with an optimized ratio was chosen to ensure the great dispersity of the seeds to avoid the aggregation of nanorods and the solubility of styrene monomer. The Au-NRs@PS particles exhibit an eccentric core–shell structures (Fig. 2b). The formation of such eccentric nanostructures could be ascribed to the heterogeneous polymerization process of styrene monomers on the metal surface.39 Upon the addition of initiator, potassium persulfate (KPS), styrene monomers and/or PS oligomers are anchored in the forms of some small particles on the Au nanorods via copolymerization with the CC double bonds of MPS. As the polymerization process continued, more hydrophobic styrene monomers absorbed on the existing PS particles and the heterogeneous PS nucleus grew larger and finally into PS spheres, resulting in an asymmetrical distribution of polymers and the formation of thick PS shells.
C bonds. Notably, the density of modified MPS molecules on the nanorod surface will influence the surface wettability of nanorod seeds and, thereby, the morphology of final particles. After surface modification with MPS, seeded emulsion polymerization was employed to coat the nanorods with a layer of polystyrene. A mixture solvent of water and ethanol with an optimized ratio was chosen to ensure the great dispersity of the seeds to avoid the aggregation of nanorods and the solubility of styrene monomer. The Au-NRs@PS particles exhibit an eccentric core–shell structures (Fig. 2b). The formation of such eccentric nanostructures could be ascribed to the heterogeneous polymerization process of styrene monomers on the metal surface.39 Upon the addition of initiator, potassium persulfate (KPS), styrene monomers and/or PS oligomers are anchored in the forms of some small particles on the Au nanorods via copolymerization with the CC double bonds of MPS. As the polymerization process continued, more hydrophobic styrene monomers absorbed on the existing PS particles and the heterogeneous PS nucleus grew larger and finally into PS spheres, resulting in an asymmetrical distribution of polymers and the formation of thick PS shells.
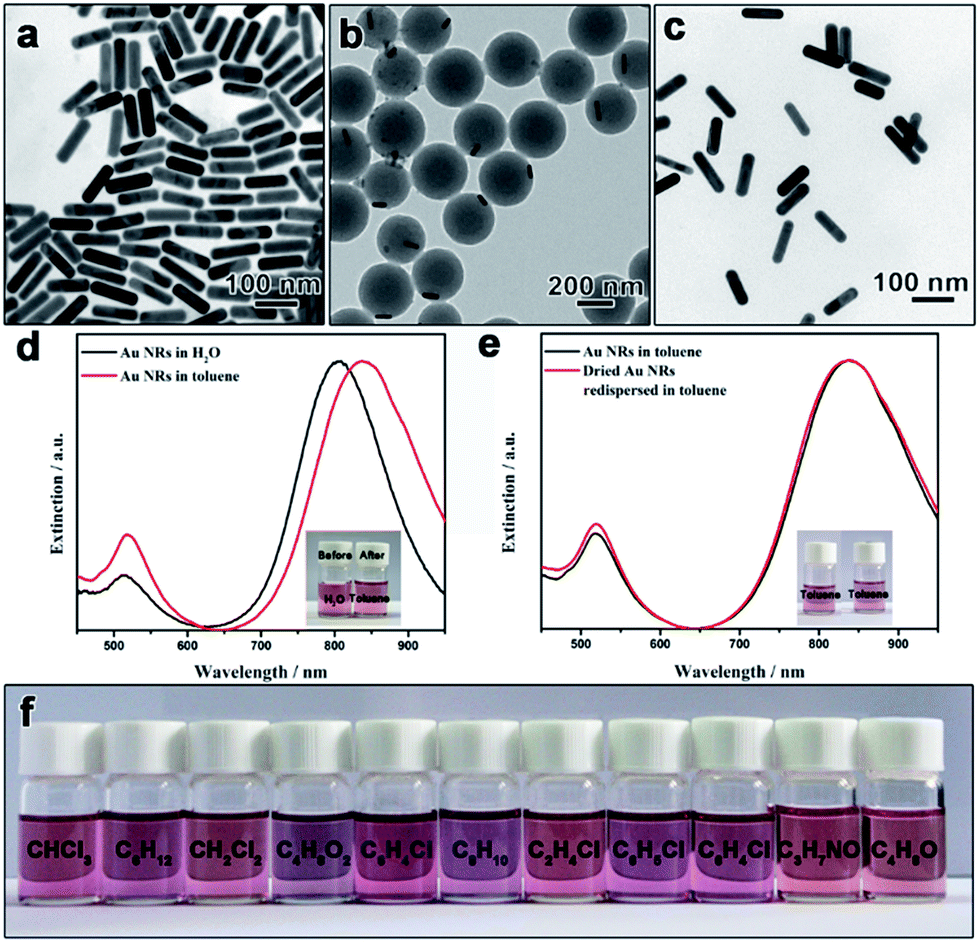 | ||
| Fig. 2 TEM images of (a) original, (b) polystyrene-coated, and (c) polystyrene-capped Au NRs. The Au-NR@PS particles in (b) were obtained by coating a layer of polystyrene on the Au NRs in (a) via seeded emulsion polymerization. The dissolution of the polystyrene layer converts the Au-NR@PS particles into polystyrene Au NRs in (c). (d) UV-vis extinction spectra of original Au NRs in water and polystyrene-capped Au NRs in toluene. Inset in (d) shows the digital photos of samples before and after the phase transfer. (e) UV-vis extinction spectra of freshly prepared polystyrene-capped Au NRs in toluene and the corresponding re-dispersed dried sample. Inset in (e) shows the digital photos of two samples. (f) Digital photos showing dispersions of polystyrene-capped Au NRs in other nonpolar solvents and polar aprotic solvents, including trichloromethane (CHCl3), cyclohexane (C6H12), dichloromethane (CH2Cl2), ethyl acetate (C4H8O2), 1,2-dichlorobenzene (C6H4Cl2), m-xylene (C8H10), 1,2-dichloroethane (C2H4Cl2), chlorobenzene (C6H5Cl), m-dichlorobenzene (C6H4Cl2), N,N-dimethylformamide (C3H7NO) and tetrahydrofuran (C4H8O) (from left to right). | ||
To dissolve the polystyrene layer, toluene was added into the dried Au-NRs@PS sample and a reddish colloidal dispersion immediately formed with slight agitation, indicating the effective transfer of nanoparticles to toluene. Notably, all Au-NRs@PS particles were dispersed in toluene without any insoluble residue, indicating a 100% transfer efficiency. TEM images of the Au nanorods in toluene revealed that the dense polystyrene layer was disappeared and the particles were separated by a few nanometers after drying on copper grids, which implies that the particle surface was capped by long polystyrene chains (Fig. 2c). It is worth noting that the existence of free polystyrene spheres in the Au-NRs@PS sample formed during the emulsion polymerization does not interfere with the phase transfer process as free spheres will be completely dissolved by toluene and the resulting free polystyrene chains can be separated from polystyrene-capped nanorods by centrifugation.
Thanks to the strong steric repulsion provided by the linear polymer chains, the polystyrene-capped Au nanorods are expected to have superior dispersity in toluene. Fig. 2d shows the UV-vis extinction spectra of original hydrophilic Au nanorods in water and the polystyrene-capped counterpart in toluene. The extinction spectrum of the aqueous dispersion shows two peaks located at 806 and 514 nm, corresponding to a strong longitudinal mode and a weak transverse mode of localized surface plasmon resonance (LSPR), respectively. Both peaks shifted to longer wavelength in the extinction spectrum of the toluene dispersion owing to the change in refractive index of the surrounding medium (nwater = 1.333 and ntoluene = 1.496).25,40 No new peak from the coupling or aggregation of nanorods was observed, confirming their good dispersity in toluene after the phase transfer. The effective phase transfer of Au nanorods was further supported by the photographs showing the samples before and after the transfer (Fig. 2d inset).
One distinguishing feature of our strategy is that it allows the storage of nanoparticles in the powder form which can be re-dispersed in solvents. Unlike most hydrophilic particles, many hydrophobic nanoparticles can be kept as a powder without sintering of the particles (which can be well re-dispersed in solvents), making it more convenient to store and transport. Owing to the unique feature of the polymerization-dissolution strategy, there are two options for storing the nanoparticles in the powder form, as either Au-NRs@PS core–shell structures or polystyrene-capped Au nanorods. For Au-NRs@PS particles, the addition of toluene to dried samples can quickly dissolve the polystyrene layer to form stable colloidal dispersions before the usage. For polystyrene-capped Au nanorods, owing to the existence of the long-range steric repulsion, they can undergo many drying–dispersing cycles without noticeable aggregation in dispersions. As shown in Fig. 2e, the extinction spectra of toluene dispersions of the original polystyrene-capped Au nanorods and re-dispersed after drying are essentially the same. The photographs of both dispersions show the same color, furthering proving the good dispersity of the transferred Au nanorods even after drying (Fig. 2e inset).
Another advantage of our strategy is that aqueous nanoparticles can be transferred to many nonpolar solvents and polar aprotic solvents which are compatible with polystyrene. As shown in Fig. 2f, after dissolving the polystyrene layer by toluene, the polystyrene-capped Au nanorods were found to exhibit excellent dispersity not only in a series of nonpolar solvents but also in polar aprotic solvents, such as N,N-dimethylformamide (DMF) and tetrahydrofuran (THF). It is expected the polystyrene-capped particles can be dispersed in more solvents not shown in this work. The superior dispersity of polystyrene-capped nanoparticles in both nonpolar and polar aprotic solvents will provide more choices of dispersion media for practical applications.
To demonstrate the universality of the polymerization–dissolution strategy in transferring different types of aqueous nanoparticles, we then study the phase transfer of hydrophilic Au nanoparticles into nonaqueous solvents. Citrate-capped Au nanospheres (Au-NSs) with an average size of ca. 10 nm were synthesized through the reduction of HAuCl4 by sodium citrate (Fig. 3a).41 The citrate-capped Au nanospheres were treated with PVP molecules before the functionalization with the coupling reagent (MPS) to introduce surface CC bonds. Core–shell structured Au-NSs@PS particles were then obtained through emulsion polymerization of styrene by using MPS-modified Au nanospheres as the seeds. Similar to the case of Au nanorods, the particles exhibit a core–shell structure with the Au-NSs cores eccentrically positioned in the PS spheres (Fig. 3b). After dissolving the polystyrene layer by toluene, Au nanoparticles were found to be well dispersed in toluene. TEM images clearly show the disappearance of the dense polystyrene layer (Fig. 3c). The presence of polystyrene on the particle surface was proved by the Fourier transform infrared (FTIR) spectrum of polystyrene-capped Au nanospheres which exhibits the characteristic peaks of polystyrene (Fig. S1†).
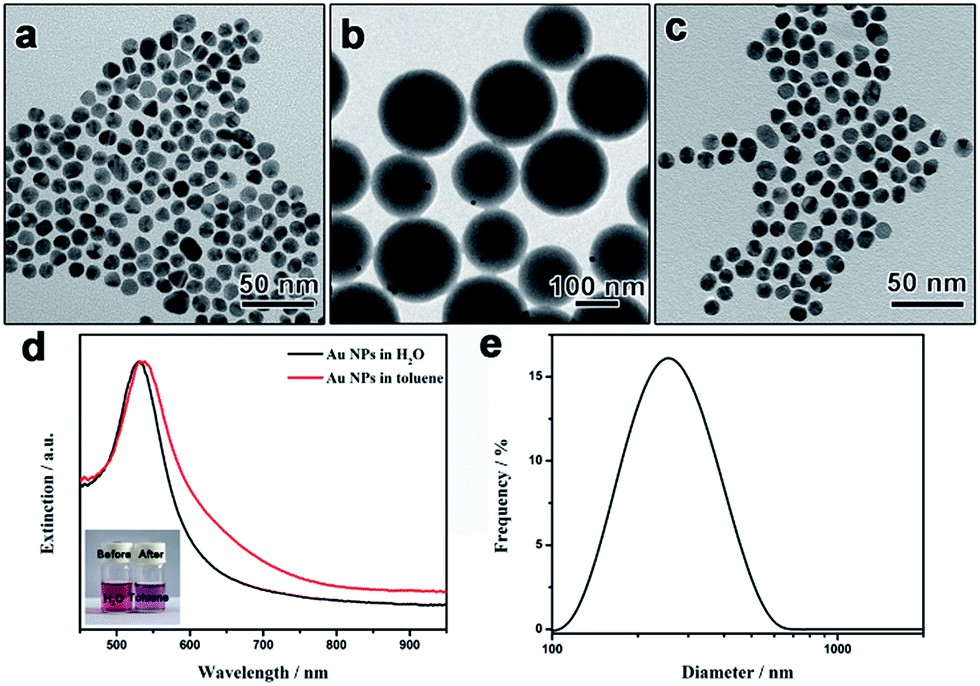 | ||
| Fig. 3 TEM images of (a) original, (b) polystyrene-coated, and (c) polystyrene-capped Au nanospheres. (d) UV-vis extinction spectra of original Au nanospheres in water and polystyrene-capped Au nanospheres in toluene. Inset in (d) shows the photographs of a typical Au nanosphere sample before and after the phase transfer. (e) Particle size distribution of polystyrene-capped Au nanospheres in toluene from dynamic light scattering (DLS) measurement. | ||
Fig. 3d shows the UV-vis extinction spectra of original hydrophilic Au nanospheres in water and the corresponding polystyrene-capped particles in toluene. The spectrum of the aqueous sample exhibited one extinction peak at ca. 530 nm, originated from the localized surface plasmon resonance of Au nanospheres. The extinction spectrum of the polystyrene-capped Au nanospheres in toluene exhibit a similar profile with the extinction peak shifted to 532 nm, indicating the successful phase transfer to toluene. The slight red-shift of the extinction peak can be explained by the fact that the refractive index of the surrounding medium near the Au particle surface was increased.25,40 More importantly, no shoulder peak in the long-wavelength range was detected, proving that there was no obvious aggregation of transferred Au nanospheres in toluene. Digital photos of the samples before and after the transfer further confirms that the surface of the nanoparticles changed from hydrophilic to hydrophobic (Fig. 3d inset). No obvious difference in the size distribution was found between the original Au NPs and the final PS capped NPs (Fig. S2†). The good dispersity of polystyrene-capped Au nanospheres in toluene is evidenced by the dynamic light scattering (DLS) measurement. Fig. 3e depicts a representative DLS curve for polystyrene-capped Au nanospheres in toluene, showing a narrow size distribution with a single peak at 260 nm. It is worth noting that the hydrodynamic diameter of 260 nm is much larger than the size of Au spheres from the TEM measurement (ca. 10 nm), which can be traced to the fact that the capping of long polystyrene chains strongly slows the Brownian movement of Au nanospheres in toluene.42
For many ligand exchange processes, the freshly transferred nanoparticles can have good dispersity in target solvents but suffer from gradual decay of the colloidal stability due to the detachment of ligands from the particle surface, which hinders the applications in areas that requires the long-term storage of nanoparticles in dispersions. In sharp contrast, the long polystyrene chains which are covalently bonded to the nanoparticle surface provides the strong steric stabilization so that the polystyrene-capped nanoparticles exhibit excellent long-term colloidal stability during storage in solvents. The extinction spectra of polystyrene-capped Au nanorods and nanospheres in toluene were monitored during a three-month period of storage. To avoid the evaporation of the solvent, the samples were kept in a closed cuvette. As shown in Fig. 4a, no obvious change was found in the profile of the extinction spectra of polystyrene-capped Au nanorods in toluene during three months, which clearly demonstrate superior long-term colloidal stability of the dispersion. For polystyrene-capped Au nanoparticles with smaller size, the extinction spectra of their toluene dispersion did not show any noticeable change during the storage for 3 months (Fig. 4b). The above results clearly demonstrate the advantages of covalently linked polymer chains to the nanoparticle surface in terms of long-term colloidal stability.
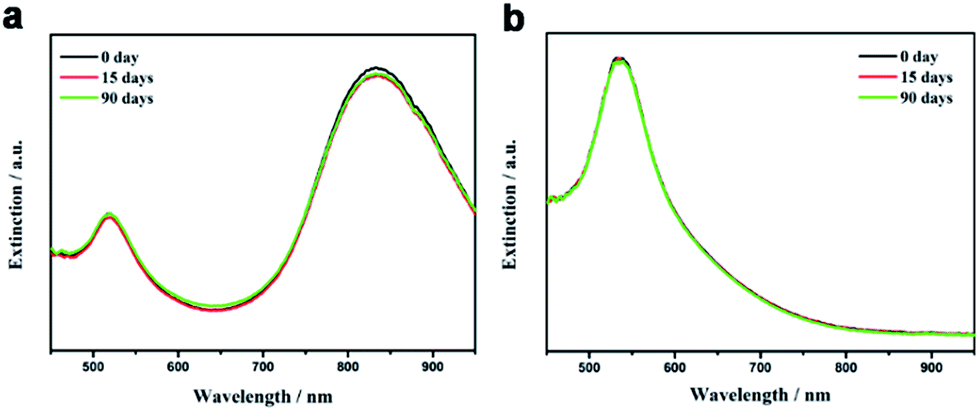 | ||
| Fig. 4 UV-vis extinction spectra of (a) polystyrene-capped Au nanorods and (b) polystyrene-capped Au nanospheres in toluene stored for different periods. The dispersions were kept in a closed cuvette to avoid the evaporation of toluene. | ||
Since seeded emulsion polymerization is a general process for coating a layer of linear polymers on the surface of various nanoparticles, our method is suitable for phase transfer of different types of aqueous nanoparticles into nonaqueous solvents. To further demonstrate the universality of the polymerization–dissolution strategy, the phase transfer of SiO2, SiO2@TiO2 and resorcinol-formaldehyde (RF) polymer spheres was also studied (Fig. 5). These well-defined nanoparticles with excellent uniformity and controlled morphologies can easily be obtained in aqueous phase while it has been extremely difficult to directly synthesize them in organic media. As shown in Fig. 5, polystyrene-capped SiO2, SiO2@TiO2 and RF spheres were obtained following the same procedures. All three particles were effectively transferred to nonaqueous solvents. The aggregate-free nature of the toluene dispersions of these nanoparticles was well supported by the DLS measurement, showing the narrow distribution of particle size with a single peak for each sample (Fig. S3†).
 | ||
| Fig. 5 TEM images of (a) SiO2, (b) SiO2@PS and (c) polystyrene-capped SiO2, (d) SiO2@TiO2, (e) SiO2@TiO2@PS, (f) polystyrene-capped SiO2@TiO2, (g) resorcinol-formaldehyde (RF), (h) RF@PS and (i) polystyrene-capped RF. | ||
Both the synthesis of core–shell structured nanoparticles via seeded emulsion polymerization and the dissolution of polystyrene by toluene have been known for decades. However, to the best of our knowledge, the combination of these two processes to transfer aqueous nanoparticles to nonaqueous solvents has been rarely reported. Our polymerization–dissolution route represents a general and robust phase transfer strategy with many unique features compared with ligand-exchange methods. First, the transferred nanoparticles can be dispersed in various nonpolar and polar aprotic solvents that are compatible with polystyrene polymers. Second, the strategy allows for the transfer of aqueous nanoparticles with a 100% efficiency. Third, the long polymer chains covalently bonded to the particle surface provides strong steric stabilization to ensure the superior dispersity and long-term colloidal stability of polystyrene-capped nanoparticles in nonaqueous solvents. Besides, both the NP@PS and polystyrene-capped nanoparticles can be stored in a power form which can be quickly re-dispersed in solvents before usage. Last but not the least, this strategy is general and robust for transferring many types of aqueous nanoparticles with different morphologies, sizes, compositions and properties. Presumably, the most striking part of our strategy is that nanostructured materials that are difficult to produce in nonaqueous environments can now become available as the stable dispersion form in nonpolar and polar aprotic solvents, which is expected to bring up new exciting applications in the near future.
Conclusions
In summary, we report a general and robust polymerization–dissolution strategy for the phase transfer of aqueously synthesized nanoparticles into nonpolar and polar aprotic solvents with a 100% efficiency. The polystyrene chains that are covalently bonded to particle surface provides strong steric stabilization, resulting in superior dispersity and long-term colloidal stability in solvents. The present strategy also allows the storage of nanoparticles in the powder form that can be re-dispersed in solvents, which is more convenient for practical applications. We demonstrate the universality of this process in the phase transfer of Au nanorods and nanospheres, SiO2, SiO2@TiO2 and RF sphere, which just represents a few examples of transferable hydrophilic nanoparticles with different morphologies, sizes, compositions, functions and surface properties. Future work will be focused on phase transfer of functional hydrophilic nanoparticles and their applications in diverse areas, such as organic catalysis, optoelectronics, organic light emitting diodes and energy conversion.Experimental section
Chemicals
Trisodium citrate (TSC), HAuCl4, PVP (M.W. = 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000), AgNO3, L-ascorbic acid, ammonium hydroxide (NH4OH, 28%), hydroxypropyl cellulose, titanium tertbutoxide (TBOT, 97%), resorcinol, formaldehyde (37%), [3-(methacryloyloxy)propyl]trimethoxysilane (MPS, 98%), sodium dodecyl sulfate (SDS), styrene (99%), potassium persulfate (KPS) were obtained from Sigma Aldrich. Sodium oleate, CTAB, tetraethylorthosilicate (TEOS, 96%) were purchased from Tokyo Chemical Industry (TCI). All chemicals were used as received.
000), AgNO3, L-ascorbic acid, ammonium hydroxide (NH4OH, 28%), hydroxypropyl cellulose, titanium tertbutoxide (TBOT, 97%), resorcinol, formaldehyde (37%), [3-(methacryloyloxy)propyl]trimethoxysilane (MPS, 98%), sodium dodecyl sulfate (SDS), styrene (99%), potassium persulfate (KPS) were obtained from Sigma Aldrich. Sodium oleate, CTAB, tetraethylorthosilicate (TEOS, 96%) were purchased from Tokyo Chemical Industry (TCI). All chemicals were used as received.
Synthesis of Au nanorods
Au nanorods were synthesized through a seed-assisted growth method in a binary surfactant system reported by Ye et al.7 The seed solution was prepared through the reduction of HAuCl4 by NaBH4. In a typical synthesis, sodium oleate (0.124 g), CTAB (0.7 g), AgNO3 (1.6 mL, 4 mM), HAuCl4 (0.984 mL, 25.4 mM) and HCl (0.3 mL, 37 wt%) were mixed in 50 mL of water. Under vigorous stirring, the L-ascorbic acid solution (0.125 mL, 64 mM) and the seed solution (0.06 mL) were then added. The reaction was continued at 30 °C for 4 h. The as-prepared Au nanorods were centrifuged at 5000 rpm for 10 min, and re-dispersed in 2 mL of water. Then 60 mg of poly(sodium-4-styrenesulfonate) (PSS) and 2 mg of NaCl were added into the Au nanorod solution. After magnetic stirring overnight, the Au nanorods were collected by centrifugation and re-dispersed in 30 mL of water. Then PVP solution (M.W. 55000, 20 mL, 12.8 g L−1) was then added under stirring. After 12 h of stirring, the PSS–PVP-modified Au nanorods were centrifuged and re-dispersed in 1 mL of water with a final concentration of 5.6 mg mL−1.
Synthesis of Au nanospheres
Spherical Au nanoparticles were prepared via the reduction of HAuCl4 with TSC at 100 °C according to a reported recipe.41 In a typical synthesis, an aqueous TSC solution (3 mL, 4 wt%) was added into a boiling HAuCl4 aqueous solution (63 mL, 1.2 mM) under vigorous stirring. The yellow solution turned colorless, then black, and finally deep red, indicating the formation of Au nanoparticles. The obtained products were centrifuged at 11000 rpm for 20 min to remove the residual reactants. To prevent the possible aggregation, an aqueous PVP solution (M.W. = 40000, 4.7 mL, 12.8 g L−1) was added and stirred overnight to ensure the adsorption of PVP on the surface of Au nanospheres. The PVP-modified Au nanospheres was collected by centrifugation and re-dispersed in 8 mL of water with a final concentration of 1.8 mg mL−1.
Synthesis of SiO2 spheres
SiO2 NPs were prepared by a modified Stöber method reported by Yin et al.43 In a mixed solution of water (24.8 mL), ethanol (61.7 mL) and ammonium hydroxide (9 mL, 28 wt%), TEOS (4.5 mL) was rapidly added under vigorous stirring. After stirring for 3 h, the white products were separated through centrifugation, cleaned with ethanol for three times, and finally re-dispersed in 10 mL of ethanol.Synthesis of SiO2@TiO2 spheres
SiO2@TiO2 spheres were obtained by coating a thick TiO2 shell on the surface of the obtained SiO2 NPs.19 Briefly, 3.8 mL of SiO2 colloid was mixed with hydroxypropyl cellulose (0.1 g), ethanol (21.2 mL) and water (0.1 mL), and stirred for 1 h to form solution A. TBOT (1 mL) was mixed with ethanol (5 mL) to form a mixing solution B, which was injected into the solution A through a syringe pump at 0.5 mL min−1, followed by refluxing for 2 h under stirring. The as-prepared SiO2@TiO2 spheres were isolated through centrifugation, cleaned with ethanol for three times, and finally re-dispersed in 10 mL of ethanol.Synthesis of resorcinol-formaldehyde resin (RF) spheres
RF spheres were prepared through a reported Stöber method.44 Typically, ammonium hydroxide (NH4OH, 0.1 mL, 28%) was mixed with ethanol (8 mL) and water (20 mL), and stirred for 1 h. Then resorcinol (0.2 g) was added, followed by the addition of formaldehyde (0.28 mL). The solution was stirred for 24 h and transferred into a Teflon-lined autoclave, followed by hydrothermal treatment at 100 °C for 48 h. The as-prepared RF spheres were collected through centrifugation, cleaned with ethanol for three times, and finally re-dispersed in 10 mL of ethanol.Surface modification
The obtained nanoparticles were functionalized with a coupling agent, [3-(methacryloyloxy)propyl]trimethoxysilane (MPS), which containing a CC double bond. In a typical procedure, 1 mL of PSS–PVP-modified Au NRs (5.6 mg mL−1) or 1 mL of PVP-functionalized Au NPs (1.8 mg mL−1) was added into a mixing solution of ethanol (20 mL) and water (1 mL). Then NH4OH (0.4 mL, 30 wt%) was added, followed by the addition of 140 μL of MPS/TEOS mixture (volume ratio = 2:5). After stirring for 2 h, the samples were collected through centrifugation, and re-dispersed in 1 mL of ethanol. For SiO2, SiO2@TiO2, and RF spheres, the particle surfaces were directly grafted with MPS. In a typical reaction, 2 mL of SiO2 (or SiO2@TiO2, RF) dispersion was mixed with isopropanol (20 mL) and MPS (100 μL), followed by refluxing at 80 °C for 2 h. The colloids were isolated by centrifugation and re-dispersed in 2 mL of ethanol as the seed solution for the subsequent seeded emulsion polymerization process.
Synthesis of NP@PS core–shell nanostructures
The MPS-grafted “seeds” (5.6 mg for Au nanorods, 1.8 mg for Au nanospheres) were firstly dispersed in 20 mL of water and ethanol mixture (Vwater/Vethanol = 5/15 for Au nanoparticles and nanorods; Vwater/Vethanol = 15/5 for SiO2, SiO2@TiO2, and RF spheres). After degassing with N2 for 30 min, 1 mL of SDS aqueous solution (3 mg mL−1) and 0.3 mL of styrene were introduced into the reaction. The reaction temperature was raised to 75 °C. An aqueous solution of KPS (0.5 mL, 20 mg mL−1) was injected to initiate the polymerization process. After reacted at 75 °C for 7 h, the products were isolated by centrifugation, cleaned with ethanol for 3 times, and dried at 60 °C.Phase transfer of NP@PS core–shell nanostructures
A certain amount of toluene was added into the dried NP@PS samples to dissolve the polystyrene layer. A colloidal dispersion was rapidly formed in a few minutes, indicating the successful transfer into toluene. Possible free polystyrene chains are removed by centrifugation. To transfer the samples into other solvents, toluene was evaporated or removed by centrifugation and the target solvent was added to form a stable colloidal dispersion.Characterization
Transmission electron microscopy (TEM) images were collected with LaB6 TEM (TECNAI G2, FEI), operating at 200 kV. The absorption spectra of Au NPs and NRs were measured by an Ocean Optics DH2000BAL-UV-NIR spectrometer. The particle size distribution was measured on a MALVERN ZEN3690 system.Acknowledgements
We acknowledge the support from the National Basic Research Program of China (973 Program, Grant No. 2013CB933500), the National Natural Science Foundation of China (Grant No. 51373188, 21401135, 21673150), the Major Research Plan of the National Natural Science Foundation of China (Grant No. 91333208), the Natural Science Foundation of Jiangsu Province (BK20140304, BK20160309), the 111 project, the Collaborative Innovation Centre of Suzhou Nano Science & Technology, and the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD) and Qing Lan Project.References
- H. Zhang, M. S. Jin, Y. J. Xiong, B. Lim and Y. N. Xia, Acc. Chem. Res., 2013, 46, 1783–1794 CrossRef CAS PubMed.
- M. Sindoro, N. Yanai, A. Y. Jee and S. Granick, Acc. Chem. Res., 2014, 47, 459–469 CrossRef CAS PubMed.
- W. S. Lu, H. F. Wang, J. P. Zhang and L. Jiang, Prog. Chem., 2015, 27, 785–793 Search PubMed.
- K. D. Gilroy, A. Ruditskiy, H.-C. Peng, D. Qin and Y. Xia, Chem. Rev., 2016, 116, 10414–10472 CrossRef CAS PubMed.
- Y. Sun and Y. Xia, Science, 2002, 298, 2176–2179 CrossRef CAS PubMed.
- N. Tian, Z.-Y. Zhou and S.-G. Sun, Chem. Commun., 2009, 1502–1504 RSC.
- X. Ye, C. Zheng, J. Chen, Y. Gao and C. B. Murray, Nano Lett., 2013, 13, 765–771 CrossRef CAS PubMed.
- N. R. Jana, L. Gearheart and C. J. Murphy, Adv. Mater., 2001, 13, 1389–1393 CrossRef CAS.
- M. Jin, H. Zhang, Z. Xie and Y. Xia, Energy Environ. Sci., 2012, 5, 6352–6357 CAS.
- S. Xie, N. Lu, Z. Xie, J. Wang, M. J. Kim and Y. Xia, Angew. Chem., Int. Ed., 2012, 51, 10266–10270 CrossRef CAS PubMed.
- Q. Zhang, N. Li, J. Goebl, Z. Lu and Y. Yin, J. Am. Chem. Soc., 2011, 133, 18931–18939 CrossRef CAS PubMed.
- L. Bi, Y. Rao, Q. Tao, J. Dong, T. Su, F. Liu and W. Qian, Biosens. Bioelectron., 2013, 43, 193–199 CrossRef CAS PubMed.
- M. Kim, Y. Kim, J. W. Hong, S. Ahn, W. Y. Kim and S. W. Han, Chem. Commun., 2014, 50, 9454–9457 RSC.
- Y. Sun, B. T. Mayers and Y. Xia, Nano Lett., 2002, 2, 481–485 CrossRef CAS.
- Y. Xia, W. Li, C. M. Cobley, J. Chen, X. Xia, Q. Zhang, M. Yang, E. C. Cho and P. K. Brown, Acc. Chem. Res., 2011, 44, 914–924 CrossRef CAS PubMed.
- W. Stöber, A. Fink and E. Bohn, J. Colloid Interface Sci., 1968, 26, 62–69 CrossRef.
- M.-C. Daniel and D. Astruc, Chem. Rev., 2004, 104, 293–346 CrossRef CAS PubMed.
- W. Li, F. Wang, S. Feng, J. Wang, Z. Sun, B. Li, Y. Li, J. Yang, A. A. Elzatahry, Y. Xia and D. Zhao, J. Am. Chem. Soc., 2013, 135, 18300–18303 CrossRef CAS PubMed.
- J. B. Joo, Q. Zhang, M. Dahl, I. Lee, J. Goebl, F. Zaera and Y. Yin, Energy Environ. Sci., 2012, 5, 6321–6327 CAS.
- Y. Xia, B. Gates, Y. Yin and Y. Lu, Adv. Mater., 2000, 12, 693–713 CrossRef CAS.
- J. Yang, J. Y. Lee and J. Y. Ying, Chem. Soc. Rev., 2011, 40, 1672–1696 RSC.
- G. H. Woehrle, L. O. Brown and J. E. Hutchison, J. Am. Chem. Soc., 2005, 127, 2172–2183 CrossRef CAS PubMed.
- A. Dong, X. Ye, J. Chen, Y. Kang, T. Gordon, J. M. Kikkawa and C. B. Murray, J. Am. Chem. Soc., 2011, 133, 998–1006 CrossRef CAS PubMed.
- S. Ma, D. Yong, Y. Zhang, X. Wang and X. Han, Chem.–Eur. J., 2014, 20, 15580–15586 CrossRef CAS PubMed.
- S. Underwood and P. Mulvaney, Langmuir, 1994, 10, 3427–3430 CrossRef CAS.
- J. Liu, J. Alvarez, W. Ong, E. Román and A. E. Kaifer, J. Am. Chem. Soc., 2001, 123, 11148–11154 CrossRef CAS PubMed.
- N. Gao, J. Dong, H. Zhang, X. Zhou, G. Zhang and J. Eastoe, J. Colloid Interface Sci., 2006, 304, 388–393 CrossRef CAS PubMed.
- R. L. Whetten, J. T. Khoury, M. M. Alvarez, S. Murthy, I. Vezmar, Z. L. Wang, P. W. Stephens, C. L. Cleveland, W. D. Luedtke and U. Landman, Adv. Mater., 1996, 8, 428–433 CrossRef CAS.
- S. R. Johnson, S. D. Evans, S. W. Mahon and A. Ulman, Langmuir, 1997, 13, 51–57 CrossRef CAS.
- A. C. Templeton, M. J. Hostetler, E. K. Warmoth, S. W. Chen, C. M. Hartshorn, V. M. Krishnamurthy, M. D. E. Forbes and R. W. Murray, J. Am. Chem. Soc., 1998, 120, 4845–4849 CrossRef CAS.
- K. Vijaya Sarathy, G. U. Kulkarni and C. N. R. Rao, Chem. Commun., 1997, 537–538 RSC.
- L. O. Brown and J. E. Hutchison, J. Phys. Chem. B, 2001, 105, 8911–8916 CrossRef CAS.
- T. Zhang, J. Ge, Y. Hu and Y. Yin, Nano Lett., 2007, 7, 3203–3207 CrossRef CAS PubMed.
- S. O. Obare, N. R. Jana and C. J. Murphy, Nano Lett., 2001, 1, 601–603 CrossRef CAS.
- S. Gu, J. Onishi, E. Mine, Y. Kobayashi and M. Konno, J. Colloid Interface Sci., 2004, 279, 284–287 CrossRef CAS PubMed.
- Y. Rong, H. Z. Chen, G. Wu and M. Wang, Mater. Chem. Phys., 2005, 91, 370–374 CrossRef CAS.
- J. Ge, Y. Hu, T. Zhang and Y. Yin, J. Am. Chem. Soc., 2007, 129, 8974–8975 CrossRef CAS PubMed.
- H. J. Chen, L. Shao, Q. Li and J. F. Wang, Chem. Soc. Rev., 2013, 42, 2679–2724 RSC.
- A. Ohnuma, E. C. Cho, M. Jiang, B. Ohtani and Y. Xia, Langmuir, 2009, 25, 13880–13887 CrossRef CAS PubMed.
- J. Tian, J. Jin, F. Zheng and H. Zhao, Langmuir, 2010, 26, 8762–8768 CrossRef CAS PubMed.
- Y. Zhao, D. Yang, H. Hu, L. Chen, Y. Xu, L. Qu, P. Yang and Q. Zhang, Surf. Sci., 2016, 648, 313–318 CrossRef CAS.
- K. Takahashi, H. Kato, T. Saito, S. Matsuyama and S. Kinugasa, Part. Part. Syst. Charact., 2008, 25, 31–38 CrossRef CAS.
- N. Li, Q. Zhang, J. Liu, J. Joo, A. Lee, Y. Gan and Y. Yin, Chem. Commun., 2013, 49, 5135–5137 RSC.
- J. Liu, S. Qiao, H. Liu, J. Chen, A. Orpe, D. Zhao and G. Lu, Angew. Chem., Int. Ed., 2011, 50, 5947–5951 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional FTIR and DLS characterization. See DOI: 10.1039/c7ra03472e |
| ‡ These authors contribute equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |