
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mechanistic study on ligand-controlled copper-catalyzed regiodivergent silacarboxylation of allenes with carbon dioxide and silylborane†
Shu-Min Hana,
Xingdong Wangb,
Shenhua Miao*a,
Zhang-Yu Yuab and
Tao Liu *ab
*ab
aSchool of Chemistry and Chemical Engineering, Qufu Normal University, Qufu 273165, Shandong, China. E-mail: liutao_2005@126.com
bDepartment of Chemistry and Chemical Engineering, Jining University, Qufu 273155, Shandong, China
First published on 2nd June 2017
Abstract
Recently, Tsuji and co-workers reported a ligand controlled regiodivergent silacarboxylation of allenes with CO2 and a silylborane in the presence of a copper catalyst. Product P1 was generated in the rac-Me-DuPhos (LA) assisted system while product P2 was obtained in the PCy3 (LB) assisted system. To account for the observed regioselectivity, density functional theory (DFT) calculations have been carried out in the present study. We calculated the detailed mechanisms and analyzed electronic and steric factors in the selectivity-determining states TS3A, TS9A, TS2B, and TS5B to gain insight into the origin of the observed regioselectivity. For the LA-assisted system, the regioselectivity is predicted to be determined by the great steric effects of TS9A. The more stable geometry of TS3A causes P1 to be the major product. For the LB-assisted system, electronic effects can account for the regioselectivity. The stronger attractive interaction in TS5B compared with that in TS2B leads to the generation of P2.
1. Introduction
As useful building blocks in organic synthesis, organosilanes can undergo a good deal of silicon-assisted transformations.1 In particular, silylcupration of C–C multiple bonds (especially the allene derivatives) has become one of the most reliable and powerful procedures that forms both vinyl- and allyl-silanes.2 However, how to control the selectivity has become the serious problem since the addition reactions across unsaturated substrates often result in many regio- and stereo-isomers.2g,3 The first-row transition metals have often been used as catalysts to solve the problem,4 not only due to their considerably low price, but the differing electronic structures of 3d transition metals provide promise for developing significant molecular structures and uncovering new organometallic reactivities at a fundamental level.5 In addition, as a greenhouse gas providing serious harm to the environment, carbon dioxide (CO2) capture, utilization, and conversion have become hot topics in research.6Recently, Tsuji and co-workers reported a ligand controlled regiodivergent silacarboxylation of allenes with CO2 and a silylborane in the presence of a copper catalyst (Scheme 1).7 As shown in Scheme 1, the ratio of two different products P1 and P2 can be highly controlled by the proper choice of ligand. The major product is P1 in reaction condition A (when Cu(OAc) is used as the catalyst precursor, hexan as the solvent, and the rac-Me-DuPhos (LA) as the ligand). In contrast, the major product is changed to P2 when a mixture of CuCl/NaOAc was used as the catalyst precursor, THF as the solvent, and PCy3 (LB) as the ligand (reaction condition B, Scheme 1).
 | ||
| Scheme 1 Cu-catalyzed silacarboxylation of allenes with CO2 and silylborane reported by Tsuji group. | ||
To account for the observed regioselectivity of this reaction, a plausible mechanism has been proposed by Tsuji et al.7As shown in Scheme 2, a Cu catalyst precursor first reacts with one reactant R1 to form a silylcopper complex 1. Next, another reactant R2 coordinates to the Cu center of 1 via the terminal double bond and generates two isomers 2 and 5 according to the orientation of the unsymmetrical allene R2, which is followed by the subsequent alkene insertion to form the allylcopper complex 3 and vinylcopper complex 6, respectively. With CO2 inserting at the γ-position of 3, copper carboxylate complex 4 is afforded. Finally, through the σ-bond metathesis between 4 and R1, boron carboxylate P0–1 is produced, accompanied with the 1 regenerating. Through the conversion with H3O+ and Me3SiCHN2 further, P0–1 could afford the final product P1. In contrast, the insertion of CO2 into 6 generates intermediate 7, followed by the σ-bond metathesis with R1 to provide boron carboxylate P0–2 and regenerate 1. Finally, final product P2 could be obtained through the further conversion of P0–2 with H3O+ and Me3SiCHN2.
 | ||
| Scheme 2 Mechanisms of Cu-catalyzed silacarboxylation of allenes with CO2 and silylborane proposed by Tsuji group. | ||
Although a plausible mechanistic pathway has been proposed by Tsuji group, some key issues still need to be further discussed. (1) What is the detailed reaction mechanism? (2) How is the origin of this high ligand-controlled regioselectivity? Here we would like to report our detailed density functional theory (DFT) calculations on the reaction mechanisms in order to gain insight into the interesting experimental observations and distinct selectivity. We expect this work would help understand the detailed mechanisms and design new related reactions.
2. Computational details
In our calculations, the geometries of all intermediates and transition states were optimized at the B3LYP8 level of DFT in gas phase. A mixed basis set of 6-311+G(d) for Cu and 6-31G(d,p) for other atoms (BSI) was used, which has been proved to be appropriate for the copper-catalyzed reactions.9 Frequency analysis were carried out to ensure all stationary points as minima (zero imaginary frequencies) or transition states (one imaginary frequency) at the B3LYP/BSI level. When necessary, intrinsic reaction coordinates (IRC)10 calculations were applied for transition state to confirm it actually connecting the corresponding two minima. To consider solvent effects, we performed single-point energy calculations for all the gas-phase optimized structures with SMD11 solvent model at the M06 (ref. 12)/BSII (6-311+G(d) for Cu and 6-311++G(d,p) for other atoms) level. Natural bond orbital (NBO)13 charges were calculated for selected structures at the M06/BSII level. Free energies (kcal mol−1) obtained at the M06/BSII//B3LYP/BSI level were used in the following discussion. All the calculations were carried out with the GAUSSIAN 09 packages.143. Results and discussion
In this work, we first investigate the Cu/LA-catalyzed mechanisms leading to P0–1 and P0–2, respectively (reaction A). Then we study the reaction mechanisms catalyzed by Cu/LB (reaction B) to probe the regioselectivity observed experimentally.3.1 Cu/LA-catalyzed reaction mechanisms
The free energy diagrams for the reaction to lead to product P0–1 catalyzed by Cu/LA are calculated and shown in Fig. 1. Firstly, the precatalyst CuOAc (1A) coordinates with the ligand LA to form the active catalyst 2A by releasing the free energy of 57.0 kcal mol−1. Subsequently, the transmetalation of 2A with R1 occurs to generate a three-coordinate Cu(I) intermediate 3A and dissociate the complex B(pin)–OAc, through a concerted four-centered transition state TS1A related to σ-bond metathesis. This step is thermodynamically and kinetically feasible, with free energy decreasing of 6.8 kcal mol−1 as well as an attainable activation free energy (8.6 kcal mol−1). The terminal double bond of reactant R2 then coordinates to the Cu center of silylcopper intermediate 3A along with the dissociation of one arm of LA15–17 via the transition state TS2A and generates intermediate 4A with a barrier of 8.6 kcal mol−1. The subsequent alkene insertion step requires a low activation energy barrier of 7.5 kcal mol−1 via transition state TS3A to form the allylcopper intermediate 5A. The energy barrier is 14.4 kcal mol−1 for the migratory insertion step (3A → TS3A). In the next step, CO2 inserts at the γ-position of 5A to a provide copper carboxylate complex 7A via a six-membered-ring transition state TS4A with the activation energy barrier of 12.2 kcal mol−1. The stepwise pathway for the CO2 insertion step (CO2 first inserting into the C–Cu bond of 5A to give carboxylate intermediate 6A, which then undergoing a [1,3]-sigmatropic rearrangement to afford 7A) was also considered (see the red line in Fig. 1), but the calculated results indicates that such a proposal is inaccessible due to the forbidden high barrier (87.8 kcal mol−1). Finally, 7A combines with R1 to form intermediate 8A, which then produces boron carboxylate P0–1 and regenerates 3A via a σ-bond metathesis transition state TS7A with an attainable free energy barrier 13.3 kcal mol−1. Through the conversion with H3O+ and Me3SiCHN2 further, P0–1 could afford the final product P1. As shown in Fig. 1, the rate-determining step in reaction A to produce P0–1 is the alkene migratory insertion step (3A → TS3A) and the overall activation barrier is 14.4 kcal mol−1. | ||
| Fig. 1 Free energy diagrams to lead to P0–1 catalyzed by Cu/LA. The relative free energies and relative enthalpic energies (in parentheses) are given in kcal mol−1. | ||
As shown in Fig. 2, R2 can also coordinate with 3A via another conformation to produce intermediate 9A with a barrier of 12.2 kcal mol−1 (TS8A). An intramolecular migratory insertion then occurs to form the vinylcopper intermediate 10A through the transition state TS9A, which requires the activation energy barrier of 10.2 kcal mol−1. In the following step, CO2 inserts into the C–Cu bond of 10A to afford a copper carboxylate intermediate 11A through a four-membered-ring transition state TS11A with a free energy barrier of 11.6 kcal mol−1. Herein, another CO2 insertion transition state TS10A was excluded due to the higher free energy compared with TS11A (−6.1 vs. −12.9 kcal mol−1). Then, 11A binds up with R1 to form intermediate 12A, followed by the σ-bond metathesis (TS13A) to afford boron carboxylate P0–2 and regenerate 3A with an attainable free energy barrier 9.5 kcal mol−1. Finally, final product P2 could be obtained through the further conversion of P0–2 with H3O+ and Me3SiCHN2. It can be found from Fig. 2 that the alkene migratory insertion process (3A → TS9A) is also the rate-determining step with the overall activation barrier of 16.5 kcal mol−1 in reaction A leading to product P0–2.
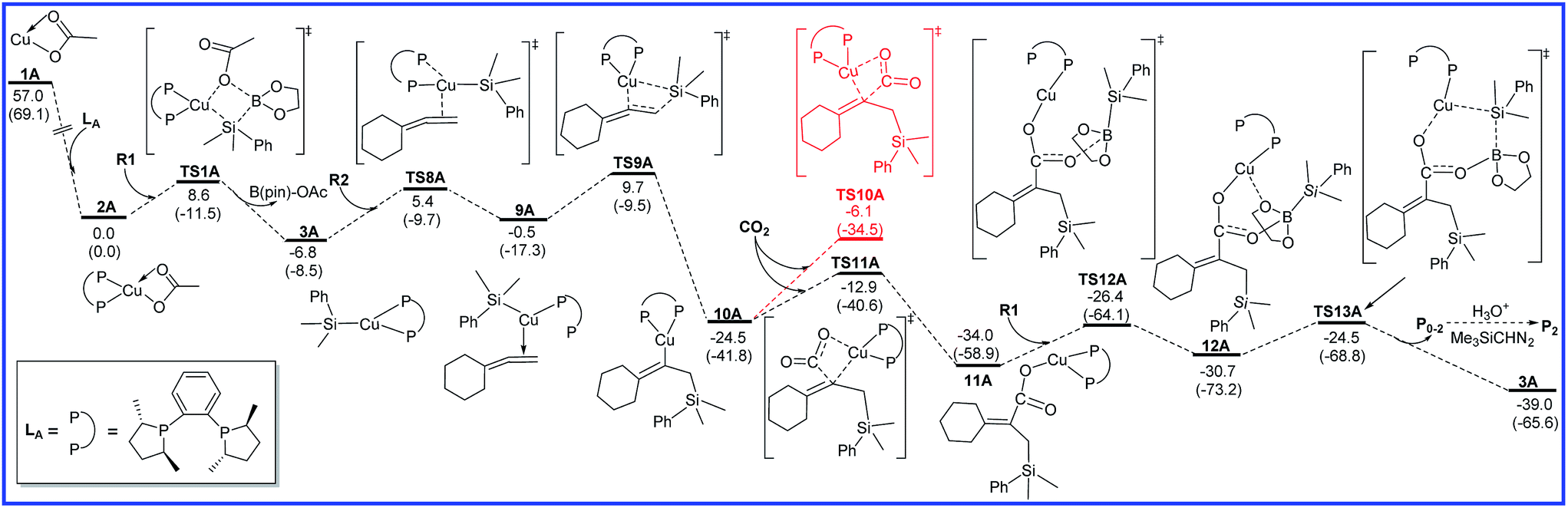 | ||
| Fig. 2 Free energy diagrams to lead to P0–2 catalyzed by Cu/LA. The relative free energies and relative enthalpic energies (in parentheses) are given in kcal mol−1. | ||
Taking the full process shown in Fig. 1 and 2 into account, the alkene migratory insertion step is the regioselectivity-determining step, and the competition between TS3A and TS9A determines the regioselectivity (P0–1 or P0–2). The calculated free energy difference (2.1 kcal mol−1) between TS3A and TS9A is reasonable agreement with the experimental major product P1.
3.2 Cu/LB-catalyzed reaction mechanism
The free energy diagrams for the reaction leading to P0–1 catalyzed by Cu/LB are shown in Fig. 3. At the entrance of the reaction, precatalyst PCy3CuCl (1B) transforms into the active catalyst PCy3CuOAc (2B) via the ligand substitution with the additive NaOAc. This process is exoergic by 3.1 kcal mol−1. 2B sequentially undergoes σ-bond metathesis with R1 to give the intermediate 3B with a barrier of 13.3 kcal mol−1. The transmetalation pathway of 1B with R1 was also considered, but the calculated high barrier rules out the possibility (see Fig. S1 in ESI†). Then, R2 inserts into the Cu–Si bond via transition state TS2B to form a allylcopper intermediate 5B. The energy barrier of insertion process is 13.5 kcal mol−1. The following CO2 insertion step requires the energy barrier of 17.9 kcal mol−1 to generate a copper carboxylate intermediate 6B via a six-membered-ring transition state TS3B. Finally, σ-bond metathesis of 6B with R1 occurs to afford boron carboxylate P0–1 and regenerate 3B with a free energy barrier 9.8 kcal mol−1. As shown in Fig. 3, the rate-determining step for the reaction leading to P0–1 catalyzed by Cu/LB is CO2 insertion (5B → TS3B), and the overall energy barrier is 17.9 kcal mol−1.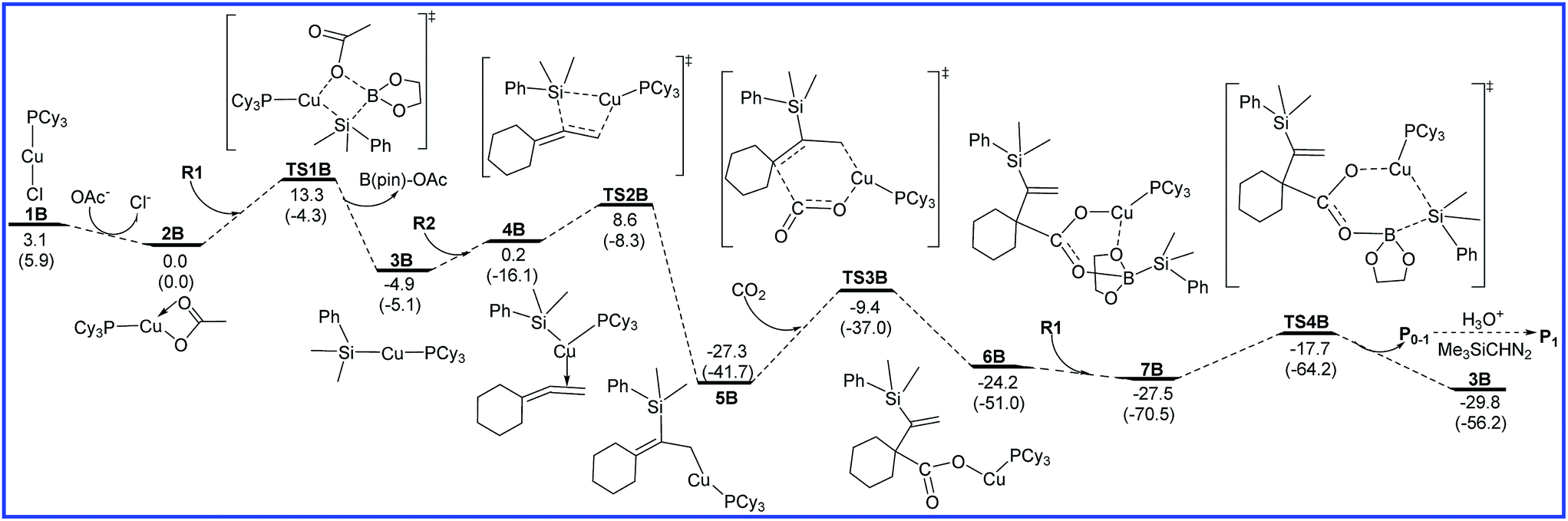 | ||
| Fig. 3 Free energy diagrams to lead to P0–1 catalyzed by Cu/LB. The relative free energies and relative enthalpic energies (in parentheses) are given in kcal mol−1. | ||
The free energy diagrams for the reaction leading to P0–2 catalyzed by Cu/LB are shown in Fig. 4. From intermediate 3B, the regioselectivity occurs. R2 inserts into the Cu–Si bond of 3B towards another orientation via the transition state TS5B to form the vinylcopper intermediate 9B with an energy barrier of 10.0 kcal mol−1. Then, CO2 inserts into the C–Cu bond of 9B through a four-membered-ring transition state TS6B to form a copper carboxylate intermediate 10B with a free energy barrier of 19.9 kcal mol−1. Another transition state TS7B for the CO2 insertion step was also calculated, but the higher relative free energy (6.1 kcal mol−1) indicates that this pathway is infeasible. Finally, 10B binds up with R1 to generate intermediate 11B, which is followed by the σ-bond metathesis (TS8B) to afford boron carboxylate P0–2 and regenerate 3B with an attainable free energy barrier 8.0 kcal mol−1. As shown in Fig. 4, for the reaction leading to P0–2 catalyzed by Cu/LB, the CO2 insertion process is found to be the rate-determining step with the overall activation barrier of 19.9 kcal mol−1 (9B → TS6B).
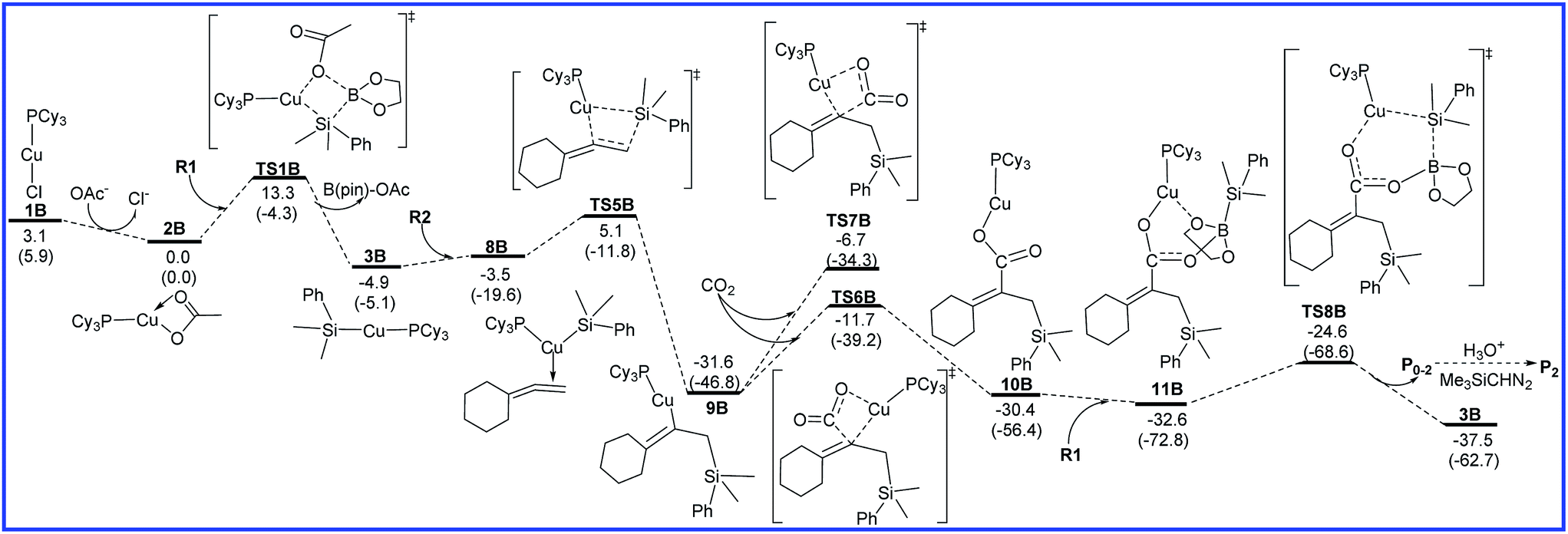 | ||
| Fig. 4 Free energy diagrams to lead to P0–2 catalyzed by Cu/LB. The relative free energies and relative enthalpic energies (in parentheses) are given in kcal mol−1. | ||
As shown in Fig. 3 and 4, the regioselectivity-determining steps to lead to P0–1 and P0–2 catalyzed by Cu/LB are 3B → TS2B (13.5 kcal mol−1) and 3B → TS5B (10.0 kcal mol−1), respectively. The relative free energy difference (3.5 kcal mol−1) between TS2B and TS5B is reasonable agreement with the experimental major product P2.
Other possible pathways were also considered. For example, in the transmetalation process of 1B and R1, the pathway that PCy3Cu moiety attacking toward B(pin) moiety, accompanied with the R1 moiety attacking toward Cl moiety was also considered, but the calculated results indicated that such a proposal is inaccessible (see Fig. S2 in ESI†). The inaccessible pathways that CO2 insertion prior to the alkene insertion step were also calculated (see Fig. S3 in ESI†). The impact of additive NaOAc on the regioselectivity was also calculated and ruled out (see Fig. S4 in ESI†).
In order to give a clear picture, the mechanistic scenarios for reactions A and B are summarized in Table 1. For reactions A and B, the regioselectivity-determining and rate-determining steps together with activation energy barriers are presented. It can be seen from the table that the regioselectivity-determining step is the reaction of 3A (3B) and R2 process, which is the divergence of P0–1 and P0–2. In addition, we can see all the reverse reactions of 3A (3B) and R2 are unachievable kinetically with too high activation energies from the preceding energy diagrams. Consequently, it is predictive that reactions of 3A (3B) and R2 are irreversible and thus this divergent step determines the major product (P1 or P2). In other words, the major product is determined by the relative stability of the two divergent transition states from reaction of 3A (3B) and R2.
| Reactions | Major product | Regioselectivity-determining stepa (ΔG‡1) | Rate-determining stepb (ΔG‡2) | ||||
|---|---|---|---|---|---|---|---|
| a ΔG‡1, Gibbs free activation energy of rate-determining step.b ΔG‡2, Gibbs free activation energy of rate-determining step. | |||||||
| Reaction A (Cu/LA) | P1 | Alkene insertion | 3A → TS3A | (14.4) | Alkene insertion | 3A → TS3A | (14.4) |
| 3A → TS9A | (16.5) | 3A → TS9A | (16.5) | ||||
| Reaction B (Cu/LB) | P2 | Alkene insertion | 3B → TS2B | (13.5) | CO2 insertion | 5B → TS3B | (17.9) |
| 3B → TS5B | (10.0) | 9B → TS6B | (19.9) | ||||
For Cu/LA-catalyzed reaction A, generation of P1 is calculated to be kinetically more favored than that of P2. The activation energy barrier difference between the two reactions is 2.1 kcal mol−1 (TS3A vs. TS9A). The rate-determining steps are same to the regioselectivity-determining steps which predicted to be the alkene insertion for the two reactions. For Cu/LB reaction B, P2 becomes the major product, and the activation energy difference between the two reactions of 3B + R2 is enlarged to 3.5 kcal mol−1. The rate-determining step is CO2 insertion process for the two reactions.
3.3 Origin of the regioselectivity
To gain insight into the origin of the observed regioselectivity, we checked both steric and electronic effects in the selectivity-determining transition states TS3A, TS9A, TS2B, and TS5B. The important bond distances and NBO charges for the four transitions states are shown in the Fig. 5.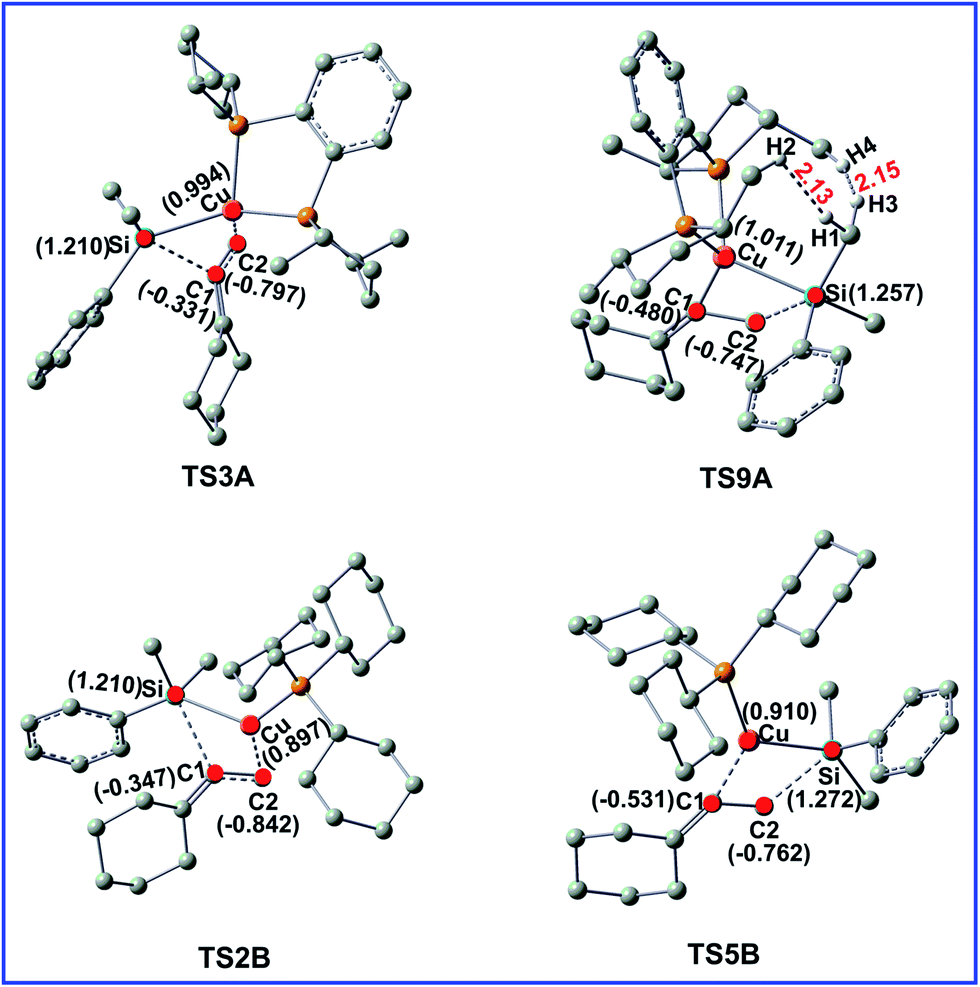 | ||
| Fig. 5 Optimized structures for the transition states TS3A, TS9A, TS2B, and TS5B. Some important bond distances are given in Å. The hydrogen atoms not participating in the reaction have been omitted for clarity. The values in parentheses are the related NBO charges (in e). | ||
For Cu/LA-catalyzed reaction A, the steric effects are the major factors to account for the regioselectivity. As shown in Fig. 5, the H1⋯H2 and H3⋯H4 distances in TS9A are 2.13 and 2.15 Å, respectively, which are less than the sums of the van der Waals radii of hydrogen (1.20 Å). Thus, the great steric effects destabilize TS9A and rule out the possibility to lead to P2 product in this reaction. In contrast, there are no obvious electronic effects difference in Cu/LA-catalyzed reaction (TS3A and TS9A).
For Cu/LB-catalyzed reaction B, there are no obvious steric bias both in TS2B and TS5B. Compared the corresponding NBO charges in Fig. 5, it can be found that the Cu atom become more negatively charged when the attached ligand LA is replaced by LB. As shown in Fig. 5, more negative C2 atom combines with the less positive Cu atom in TS2B, while combines with the more positive atom Si in TS5B. Obviously, the stronger attractive interaction caused by electronic factors in TS5B leads to the major product P2.
4. Conclusions
We have presented a detailed and plausible mechanism for the copper-catalyzed ligand controlled regiodivergent silacarboxylation of allenes with CO2 and silylborane by using DFT calculations. Two reactions are included in this study, reactions A (CuOAc as the catalyst and product P1 is favored with the bidentate LA ligand), and B (CuCl/NaOAc as the catalyst and product P2 is favored with the monodentate LB ligand). The rate-determining steps for the reactions to form products P1 and P2 catalyzed by Cu/LA both are the alkene insertion step, while rate-determining steps for the reactions catalyzed by Cu/LB both are the CO2 insertion step.The origin of the experimentally observed selectivity on different products has been analyzed based on the above mechanistic details. It is found that regioselectivity depends on the relative feasibility of alkene migratory insertion step. For Cu/LA-catalyzed system, the generation of product P1 is preferred mainly due to the great steric effects of TS9A that rule out the possibility to lead to P2. For Cu/LB-catalyzed system, electronic effects become the major factors to produce the major product P2. The in-depth understanding for the divergent reaction mechanisms and distinct regiochemistry could be beneficial in designing new related reactions.
Acknowledgements
This work was jointly supported by Natural Science Foundation of Shandong Province (No. ZR2014BM038), and Talent Team Culturing Plan for Leading Disciplines of University in Shandong Province.References
- (a) T. Liu and S. W. Bi, Organometallics, 2016, 35, 1114 CrossRef CAS; (b) R. Alam, T. Vollgraff, L. Eriksson and K. J. Szabó, J. Am. Chem. Soc., 2015, 137, 11262 CrossRef CAS PubMed; (c) M. Yus, J. C. González-Gómez and F. Foubelo, Chem. Rev., 2013, 113, 5595 CrossRef CAS PubMed; (d) I. Beletskaya and C. Moberg, Chem. Rev., 2006, 106, 2320 CrossRef CAS PubMed.
- (a) W. M. Yuan, S. Liu and S. M. Ma, Angew. Chem., Int. Ed., 2016, 55, 3140 CrossRef CAS PubMed; (b) J. Rae, Y. C. Hu and D. J. Procter, Chem.–Eur. J., 2014, 20, 13143 CrossRef CAS PubMed; (c) J. A. Calderone and W. L. Santos, Angew. Chem., Int. Ed., 2014, 53, 4154 CrossRef CAS PubMed; (d) Y. H. Xu, L. H. Wu, J. Wang and T. P. Loh, Chem. Commun., 2014, 50, 7195 RSC; (e) A. Barbero and F. J. Pulido, Acc. Chem. Res., 2004, 37, 817 CrossRef CAS PubMed; (f) M. Y. Wu, F. Y. Yang and C. H. Cheng, J. Org. Chem., 1999, 64, 2471 CrossRef CAS; (g) E. Langkopf and D. Schinzer, Chem. Rev., 1995, 95, 1375 CrossRef CAS.
- (a) M. Uehling, R. Rucker and G. Lalic, J. Am. Chem. Soc., 2014, 136, 8799 CrossRef CAS PubMed; (b) S. C. Yu and S. M. Ma, Angew. Chem., Int. Ed., 2012, 51, 3074 CrossRef CAS PubMed.
- (a) C. C. Xia, K. Wang, J. Xu, C. Shen, D. Sun, H. S. Li, G. D. Wang and P. F. Zhang, Org. Biomol. Chem., 2017, 15, 531 RSC; (b) T. Itoh, Y. Shimizu and M. Kanai, J. Am. Chem. Soc., 2016, 138, 7528 CrossRef CAS PubMed; (c) M. Pirnot, Y. Wang and S. Buchwald, Angew. Chem., Int. Ed., 2016, 55, 48 CrossRef CAS PubMed; (d) R. Bullock and M. Helm, Acc. Chem. Res., 2015, 48, 2017 CrossRef CAS PubMed; (e) K. Semba, T. Fujihara, J. Terao and Y. Tsuji, Angew. Chem., Int. Ed., 2013, 52, 12400 CrossRef CAS PubMed.
- (a) M. Usman, Z. H. Ren, Y. Y. Wang and Z. H. Guan, RSC Adv., 2016, 6, 107542 RSC; (b) Y. Y. Li, S. L. Yu, W. Y. Shen and J. X. Gao, Acc. Chem. Res., 2015, 48, 2587 CrossRef CAS PubMed; (c) B. Jung and A. H. Hoveyda, J. Am. Chem. Soc., 2012, 134, 1490 CrossRef CAS PubMed.
- (a) W. Y. Li, D. F. Huang and Y. J. Lyu, Org. Biomol. Chem., 2016, 14, 10875 RSC; (b) K. Nogi, T. Fujihara, J. Terao and Y. Tsuji, Chem. Commun., 2014, 50, 13052 RSC; (c) L. Zang and Z. Hou, Chem. Sci., 2013, 4, 3395 RSC; (d) I. Omae, Coord. Chem. Rev., 2012, 256, 1384 CrossRef CAS; (e) W. Wang, S. Wang, X. Ma and J. Gong, Chem. Soc. Rev., 2011, 40, 3703 RSC; (f) T. Fujihara, T. Xu, K. Semba, J. Terao and Y. Tsuji, Angew. Chem., Int. Ed., 2011, 50, 523 CrossRef CAS PubMed.
- Y. Tani, T. Fujihara, J. Terao and Y. Tsuji, J. Am. Chem. Soc., 2014, 136, 17706 CrossRef CAS PubMed.
- (a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS; (b) C. Lee, W. Yang and G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS; (c) P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623 CrossRef CAS.
- (a) R. Yuan and Z. Y. Lin, ACS Catal., 2014, 4, 4466 CrossRef CAS; (b) H. T. Zhao, Z. Y. Lin and T. B. Marder, J. Am. Chem. Soc., 2006, 128, 15637 CrossRef CAS PubMed; (c) H. T. Zhao, L. Dang, T. B. Marder and Z. Y. Lin, J. Am. Chem. Soc., 2008, 130, 5586 CrossRef CAS PubMed.
- (a) K. Fukui, J. Phys. Chem., 1970, 74, 4161 CrossRef CAS; (b) K. Fukui, Acc. Chem. Res., 1981, 14, 363 CrossRef CAS.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378 CrossRef CAS PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215 CrossRef CAS.
- (a) J. P. Foster and F. Weinhold, J. Am. Chem. Soc., 1980, 102, 7211 CrossRef CAS; (b) A. E. Reed and F. Weinhold, J. Chem. Phys., 1983, 78, 4066 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- (a) B. C. Hamann and J. F. Hartwig, J. Am. Chem. Soc., 1998, 120, 3694 CrossRef CAS; (b) M. Kawatsura and J. F. Hartwig, J. Am. Chem. Soc., 1999, 121, 1473 CrossRef CAS.
- M. N. Birkholz, Z. Freixa and P. W. van Leeuwen, Chem. Soc. Rev., 2009, 38, 1099 RSC.
- Q. H. Lu, B. Wang, H. Z. Yu and Y. Fu, ACS Catal., 2015, 5, 4881 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra03723f |
| This journal is © The Royal Society of Chemistry 2017 |