DOI:
10.1039/C7RA03884D
(Paper)
RSC Adv., 2017,
7, 30573-30581
Influence of pendant 2-[1,2,4]triazol-4-yl-ethylamine and symmetrical bis(pyrazol) ligands on dimensional extension of POM-based compounds†
Received
5th April 2017
, Accepted 2nd June 2017
First published on 14th June 2017
Abstract
Through utilizing pendant 2-[1,2,4]triazol-4-yl-ethylamine (tea) and two kinds of bis(pyrazol) ligand, five new polyoxometalate (POM)-based compounds [Cu3K2(tea)2(OH)4(H2Mo8O27)] (1), [Ag(tea)(β-H2Mo8O26)0.5] (2), [Ag(tea)(H2O)(γ-H2Mo8O26)0.5] (3), [Cu(H2bdpm)(β-Mo8O26)0.5] (4) and [Ag2(Hbhpe)(θ-Mo8O26)0.5] (5) (H2bdpm = 1,1′-bis(3,5-dimethyl-1H-pyrazol-4-yl)methane, H2bhpe = 1,2-bis(1H-pyrazolate)ethane) were synthesized under hydrothermal conditions and characterized by single-crystal X-ray diffraction. In compound 1, three CuII are fused by two tea and two OH− moieties to form a tri-nuclear cluster [Cu3(tea)2(OH)2]4+. The Mo8 anions link each other through sharing terminal O atoms to build an Mo-chain. The tri-nuclear clusters are further connected by an Mo-chain and K+ ions to form a 3D framework. In compound 2, two Ag+ ions are fused by two tea ligands to construct a bi-nuclear subunit, which are linked by β-Mo8 anions alternately to form a 1D chain. In compound 3, there exists a bi-nuclear Ag+ cluster [Ag2(tea)2]2+. The γ-Mo8 anions link bi-nuclear Ag+ clusters alternately and a 1D chain is constructed. Furthermore, each [Ag2(tea)2]2+ offers two additional N donors to connect two anions through Mo–N bonds. In compound 4, two 1D zigzag Cu–H2bdpm chains cover one 1D inorganic Mo-chain up and down to build a “hamburger-like” chain. The adjacent hamburger chains share the same Cu atoms to build a 2D structure. In compound 5, there exists a bi-nuclear [Ag2(Hbhpe)2]2+ cycle and a tetra-nuclear [Ag4(Hbhpe)4]4+ cycle. The bi-nuclear cycles and tetra-nuclear cycles alternately connect to form 1D metal–organic chains, which are further linked by θ-Mo8 anions to construct a 2D layer. The electrochemical and photocatalytic properties of compounds 1–5 are studied.
Introduction
Polyoxometalates (POMs) have attracted extensive attention as an emerging branch of inorganic chemistry because of their diverse structures1 and extensive applications in catalysis, medicine, magnetism, photochemistry and other fields.2 POMs can provide abundant O donors to connect transition metal complexes (TMCs) to construct metal–organic complex materials with various structures and properties.3 Among the abundant types of POM, some classical anions have received greater attention, such as the Keggin, Wells–Dawson, isopolymolybdate and Anderson anions. In addition, the octamolybdate-TMCs are an important branch. The octamolybdate anions have eight isomers: α, β, γ, δ, ε, ζ, η and θ,4 which can induce novel structures. Interestingly, two or more isomers can be captured in one octamolybdate-TMC compound.5 Furthermore, the octamolybdate anions may share the same O atoms to build new Mo-chains.6 Thus, the design and syntheses of octamolybdate-TMCs allow fascinating structures to be constructed to enrich the POM field. For example, Zubieta and co-workers have published numerous relevant papers concerning the use of octamolybdate ions as building blocks for the design of an extended network.7
In the synthetic strategy used for forming octamolybdate-TMCs, the choice of appropriate organic ligands is essential and important. Flexible organic molecules have become more popular in building high-dimensional frameworks, especially containing two symmetrical coordination groups.8 Organic ligands of this series contain flexibility and more coordination sites, conducive to constructing novel topologies. Up until now, a large number of flexible organic molecules containing two symmetrical coordination groups have been used, such as flexible bis(triazole),9 bis(imidazole)10 and bis(tetrazole) ligands.11 In our previous work, we have utilized a series of symmetric flexible bis(triazole) ligands to build POM-based compounds.12 On the other hand, pendant organic ligands containing only one coordination group are scarcely used in octamolybdate-based compounds. Compared with flexible ligands with two symmetrical coordination groups, the pendant ligands may induce structures with low dimensionalities.13 Thus, it is interesting and appealing to explore the influence of pendant ligand with one coordination group and flexible ligands with two symmetrical coordination groups on structural dimensionalities. In this work, we chose two kinds of ligand as the organic moiety with which to modify octamolybdate anions: pendant 2-[1,2,4]triazol-4-yl-ethylamine (tea) and the bis(pyrazol) ligands, 1,2-di(1H-pyrazol-4-yl)ethane (Hbhpe) and bis(3,5-dimethyl-1H-pyrazol-4-yl)methane (H2bdpm).
Herein, by introducing pendant tea and flexible Hbhpe and H2bdpm to the octamolybdate system, five new POM-based TMCs have been obtained: [Cu3K2(tea)2(OH)4(H2Mo8O27)] (1), [Ag(tea)(β-H2Mo8O26)0.5] (2), [Ag(tea)(H2O)(γ-H2Mo8O26)0.5] (3), [Cu(H2bdpm)(β-Mo8O26)0.5] (4) and [Ag2(Hbhpe)(θ-Mo8O26)0.5] (5). The electrochemical and photocatalytic properties of these five compounds were also studied.
Experimental
Materials and methods
All reagents and solvents for syntheses were purchased from commercial sources and used as received without further purification. Elemental analyses were achieved using a PerkinElmer 240C elemental analyzer, and Fourier-transform infrared (FT-IR) spectra were recorded on a Varian FT-IR 640 spectrometer (KBr pellets). Ultraviolet/visible (UV/Vis) absorption spectra were obtained using a SP-1601 UV/Vis spectrophotometer. Electrochemical measurements and data collection were performed using a CHI 440 electrochemical workstation connected to a Digital-586 personal computer. A conventional three-electrode system was used with a saturated calomel electrode (SCE) as the reference electrode and a Pt wire as the counter electrode. Modified carbon paste electrodes (CPEs) of the title compounds were used as the working electrodes.
X-ray crystallography
Crystallographic data for compounds 1–5 were collected on a Bruker SMART APEX II with Mo Kα (λ = 0.71073 Å) by ω and θ scan mode at 293 K. All structures were solved by direct methods and refined on F2 by full-matrix least squares using the SHELXL package.14 For compounds 1–5, the hydrogen atoms attached to water molecules were not located, but were included in the structure factor calculations, as well as disordered Ag1 and Ag2 atoms in compound 5. Detailed crystal data and structures refinement for 1–5 are given in Table 1. Selected bond lengths and angles are listed in Table S1. Crystallographic data for the structures reported in this paper have been deposited in the Cambridge Crystallographic Data Centre (CCDC), with CCDC numbers of 1531320 for 1, 1531323 for 2, 1531324 for 3, 1531321 for 4 and 1531322 for 5.†
Table 1 Crystal data and structure refinement for compounds 1–5
| R1 = ∑||Fo| − |Fc||/∑|Fo|. wR2 = {∑[w(Fo2 − Fc2)2]/∑[w(Fo2)2]}1/2. |
| Compounds |
1 |
2 |
3 |
4 |
5 |
| Formula |
C8H26N6Cu3K2Mo8O31 |
C4H10N4AgMo4O13 |
C4H12N4AgMo4O14 |
C11H16N4CuMo4O13 |
C8H10N4Ag2Mo4O13 |
| Fw |
1766.68 |
813.77 |
831.78 |
859.57 |
969.68 |
| Cryst. syst |
Triclinic |
Monoclinic |
Monoclinic |
Triclinic |
Triclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P21/n |
P21/n |
P |
P |
| a (Å) |
8.4612(7) |
10.1653(5) |
8.6207(3) |
8.224(6) |
9.8358(8) |
| b (Å) |
10.5659(11) |
12.4265(6) |
18.0125(7) |
10.388(7) |
10.7665(9) |
| c (Å) |
10.7372(9) |
12.5934(6) |
11.1084(4) |
12.836(9) |
11.2441(9) |
| α (°) |
100.957(2) |
|
|
84.460(14) |
116.8470(10) |
| β (°) |
102.211(2) |
99.9570(10) |
106.5790(10) |
80.618(15) |
91.7280(10) |
| γ (°) |
100.545(2) |
|
|
68.544(12) |
113.1800(10) |
| V (Å3) |
895.82(14) |
1566.83(13) |
1653.21(11) |
1006.2(12) |
944.71(13) |
| Z |
1 |
4 |
4 |
2 |
2 |
| Dc (g cm−3) |
3.256 |
3.441 |
3.334 |
2.831 |
3.402 |
| μ (mm−1) |
4.790 |
4.420 |
4.197 |
3.542 |
4.682 |
| F(000) |
829 |
1516 |
1556 |
818 |
900 |
| Total reflections |
6666 |
10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 966 966 |
11978 |
3942 |
6966 |
| Unique reflections |
4453 |
3904 |
4122 |
3344 |
4654 |
| Rin |
0.0160 |
0.0248 |
0.0223 |
0.0527 |
0.0168 |
| GOOF |
1.078 |
1.010 |
1.019 |
0.903 |
0.987 |
| Final R1a, wR2b [I > 2σ(I)] |
0.0396, 0.1203 |
0.0280, 0.0649 |
0.0270, 0.1044 |
0.0682, 0.1548 |
0.0388, 0.0856 |
| Final R1a, wR2b (all data) |
0.0470, 0.1248 |
0.0317, 0.0668 |
0.0302, 0.1082 |
0.1277, 0.1747 |
0.0530, 0.0923 |
Preparation of compounds 1–5
Synthesis of [Cu3K2(tea)2(OH)4(H2Mo8O27)] (1). A mixture of CuCl2 H2O (0.076 g, 0.446 mmol), KCl (0.02 g, 0.27 mmol), Na3(CrMo6O24H6)·8H2O (0.1 g, 0.081 mmol), tea (0.02 g, 0.098 mmol) and H2O (10 mL) was stirred for 40 min in air at room temperature. The pH value of the solution was adjusted to about 2.5 with 1.0 mol L−1 HCl and sodium hydroxide. The suspension was sealed in a Teflon-lined autoclave (25 mL) and kept at 160 °C for 5 days. Green clavate crystals of 1 (yield 48% based on Mo) were obtained. Elemental analysis (%) calc. for 1 C8H26N8Cu3K2Mo8O31 (1766.68): C 5.44, H 1.48, N 6.34. Found: C 5.49, H 1.43, N, 6.39. IR (KBr pellet, cm−1): 3086 (w), 2358 (w), 1626 (m), 1506 (m), 1436 (s), 1116 (w), 1018 (m), 931 (s), 879 (s), 745 (s), 627 (s).
Synthesis of [Ag(tea)(β-H2Mo8O26)0.5] (2). A mixture of AgNO3 (0.076 g, 0.086 mmol), [(n-C4H9)4N]2[Mo6O19] (0.1 g, 0.154 mmol), tea (0.1 g, 0.625 mmol) and H2O (10 mL) was stirred for 40 min in air at room temperature. The pH value of the solution was then adjusted to 3.2 with 1 M HNO3 and ammonium hydroxide. White block crystals of 2 were obtained. Elemental analysis (%) calc. for C4H10N4AgMo4O13 (813.77): C 5.90, H 1.24, N 6.88. Found: C 5.83, H 1.28, N 6.82. IR (KBr pellet, cm−1): 3111 (m), 2364 (m), 1637 (m), 1440 (m), 1328 (w), 1284 (w), 1205 (w), 1120 (w), 968 (s), 879 (s), 754 (s) and 565 (s).
Synthesis of [Ag(tea)(H2O)(γ-H2Mo8O26)0.5] (3). A mixture of AgNO3 (0.076 g, 0.086 mmol), [(n-C4H9)4N]2[Mo6O19] (0.1 g, 0.154 mmol), tea (0.02 g, 0.098 mmol) and H2O (10 mL) was stirred for 40 min in air at room temperature. The pH value of the solution was adjusted to about 2.6 with 1.0 mol L−1 HNO3 and ammonium hydroxide. The suspension was sealed in a Teflon-lined autoclave (25 mL) and kept at 160 °C for 5 days. Yellow-black crystals of 3 (yield 35% based on Mo) were obtained. Elemental analysis (%) calc. for 3 C4H12N4OAgMo4O13 (831.78): C 5.78, H 1.45, N 6.74. Found: C 5.71, H 1.48, N, 6.69. IR (KBr pellet, cm−1): 3390 (w), 2451 (w), 1626 (s), 1558 (s), 1500 (s), 1448 (s), 1340 (s), 1217 (s), 1112 (s), 1024 (s), 938 (s), 825 (s), 673 (s) and 554 (s).
Synthesis of [Cu(H2bdpm)(β-Mo8O26)0.5] (4). The compound 4 was prepared in the same way as compound 1 except that H2bdpm (0.1 g, 0.625 mmol) was used instead of tea. The pH value of the solution was then adjusted to 2.2 with 1.0 mol L−1 NaOH and HCl. Green clavate crystals of 4 (yield 30% based on Mo) were obtained. Elemental analysis (%) calc. for 4 C11H16N4CuMo4O13 (859.57): C 15.37, H 1.88, N 6.52. Found: C 15.31, H 1.92, N, 6.46. IR (KBr pellet, cm−1): 3340 (m), 2920 (w), 1634 (m), 1446 (s), 1271 (w), 1188 (m), 1039 (w), 970 (s), 880 (s), 761 (s) and 542 (s).
Synthesis of [Ag2(Hbhpe)(θ-Mo8O26)0.5] (5). Compound 5 was prepared in a similar way to 2, except that Hbhpe (0.02 g, 0.098 mmol) was used instead of tea. The pH value of the solution was adjusted to about 1.6 with 1.0 mol L−1 HNO3 and ammonium hydroxide. The suspension was sealed in a Teflon-lined autoclave (25 mL) and kept at 170 °C for 4 days. Pink rhombus crystals of 5 (yield 20% based on Mo) were obtained. Elemental analysis (%) calc. for 5 C8H10N4Ag2Mo4O13 (969.68): C 9.91, H 1.04, N 5.78. Found: C 9.85, H 1.08, N, 5.72. IR (KBr pellet, cm−1): 3327 (w), 2931 (w), 2355 (w), 1647 (m), 1560 (m), 1442 (m), 1286 (w), 1186 (w), 1031 (w), 964 (s), 871 (s), 766 (s) and 534 (s).
Preparation of 1- to 5-CPEs. Compound 1 modified carbon paste electrode (1-CPE) was synthesized as follows: graphite powder (100 mg) and compound 1 (90 mg) were mixed and ground together using an agate mortar and pestle for approximately 1 h to achieve the entire mixture. Then, 0.10 mL of the paraffin oil was added and stirred with a glass rod. The homogenized mixture was encapsulated to a 3 mm inner diameter hollow glass pipe with a length of 0.8 cm. The pipe surface was smoothed with weighing paper, and the electrical contact was assembled with the copper rod. In a similar process, 2-, 3- and 4-CPEs were synthesized with compounds 2, 3, 4 and 5.
Results and discussion
Structural description
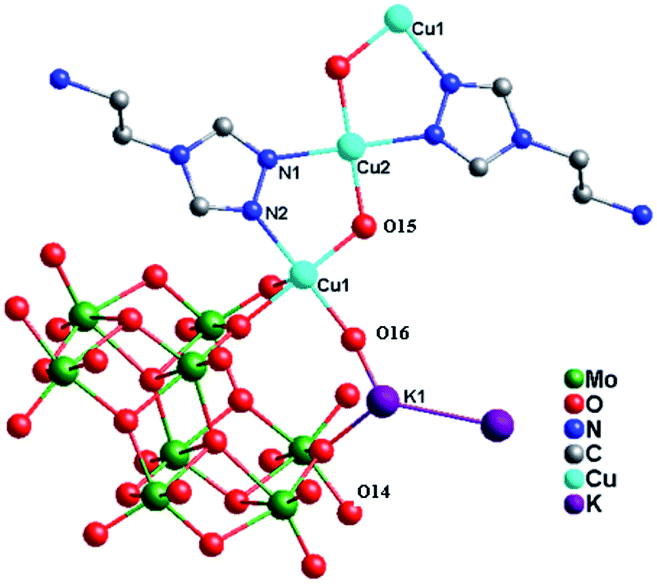
Crystal structure of compound 1. X-ray crystal structure analysis reveals that the asymmetric unit of compound 1 consists of one [Mo8O26]4− anion (abbreviated as Mo8), three Cu2+ ions, two tea ligands, two K+ cations and four hydroxy groups (Fig. 1). The valence sum calculations15 show that all Mo atoms are in the +VI oxidation state, while all Cu atoms are in the +II oxidation state.
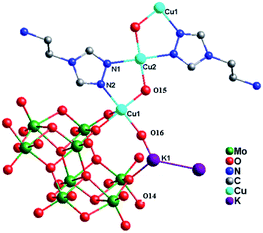 |
| | Fig. 1 Ball/stick view of the asymmetric unit of compound 1. The hydrogen atoms are omitted for clarity. | |
In compound 1, there are two crystallographically independent Cu2+ ions (Cu1 and Cu2), with two kinds of coordination geometries. Cu1 ion is six-coordinated by one N2 atom from one tea ligand, three O atoms (O8, O12 and O13) from two Mo8 anions and two hydroxy groups, adopting a distorted octahedral geometry. The Cu2 ion is also six-coordinated by two N1 atoms from two tea ligands, two O10 atoms from two Mo8 anions and two hydroxy groups. The Cu–N distances are 1.943(5) and 2.052(6) Å, while the Cu–O bond distances are in the range of 1.924(5)–2.394(5) Å. The N–Cu–N angle is 179.999(1) and the O–Cu–O angles are in the range 87.46(19)–180.00(1)° (Table S1†).
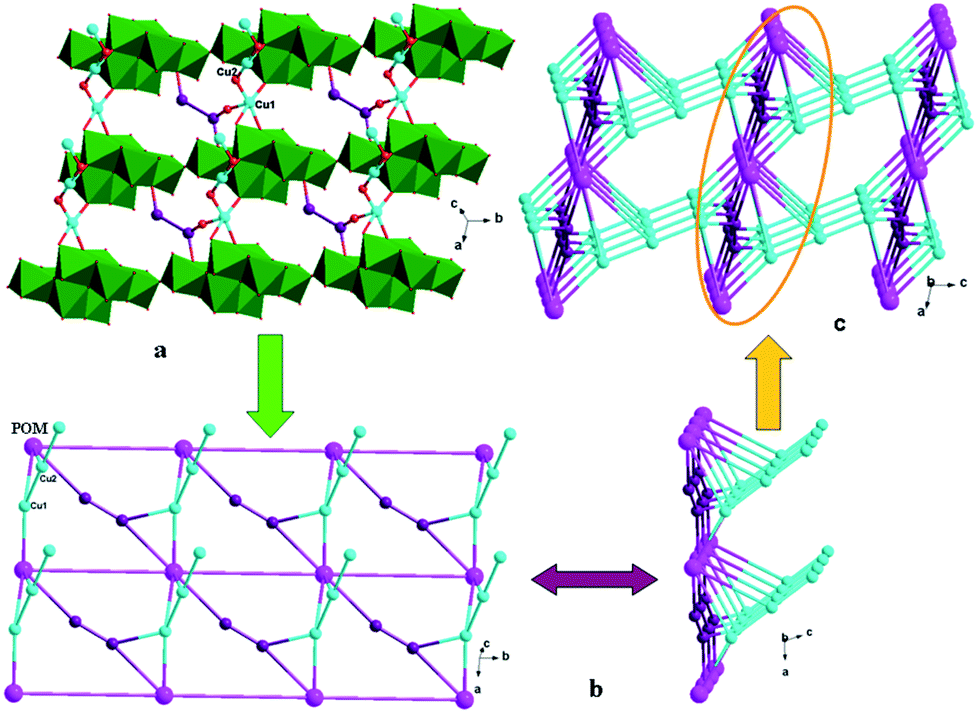
In compound 1, the tea ligand uses its two adjacent nitrogen donors to coordinate two Cu2+ ions. Two tea molecules are fused by three Cu atoms to form a linear tri-nuclear [Cu3(tea)2(OH)2]4+ subunit, in which two tea are located top and bottom (Fig. 1). This tri-nuclear cluster is a discrete unit. No dimensional extension of the metal–organic subunit is possible because the –NH2 group is difficult to link metal ions. Furthermore, the adjacent Mo8 anions share the same O14 atoms to form an infinite Mo-chain (Fig. 2). The tri-nuclear [Cu3(tea)2(OH)2]4+ clusters are linked by an Mo-chain and K+ atoms to form a 2D layer of 1 (Fig. 3a and b). Adjacent layers share the same Cu2+ ions to construct a 3D framework, as shown in Fig. 3c. In compound 1, the tri-nuclear metal–organic subunit is only a 0D cluster, with contributions from the Mo-chain and K+ ions to build a 3D structure.
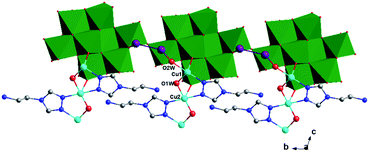 |
| | Fig. 2 1D structure of 1 containing Mo-chain (green polyhedron). | |
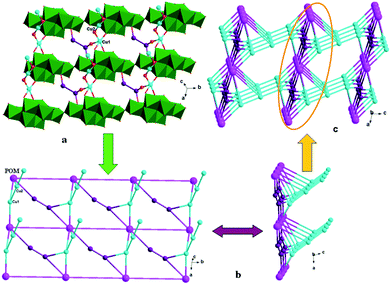 |
| | Fig. 3 (a) 2D grid-like layer of 1. (b) Schematic view of the 2D layer viewing along the different given directions. (c) 3D framework of 1. | |
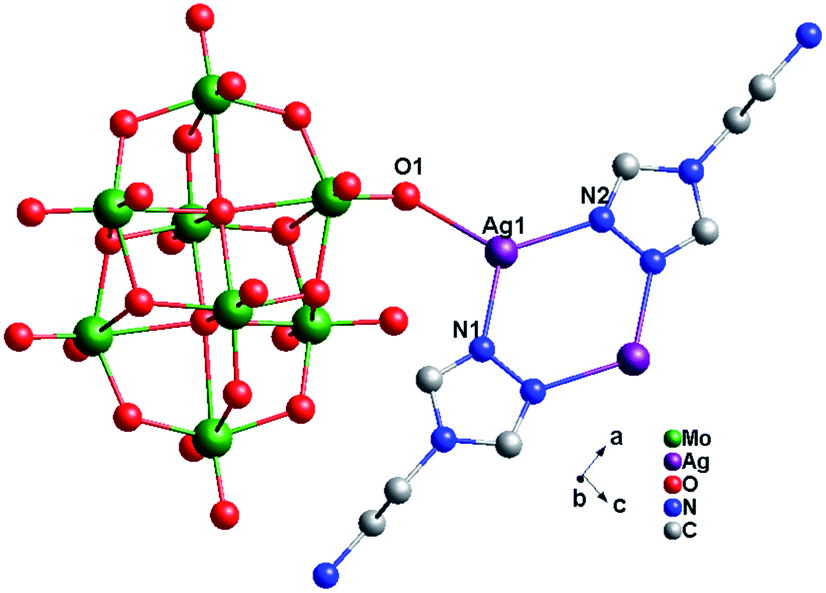
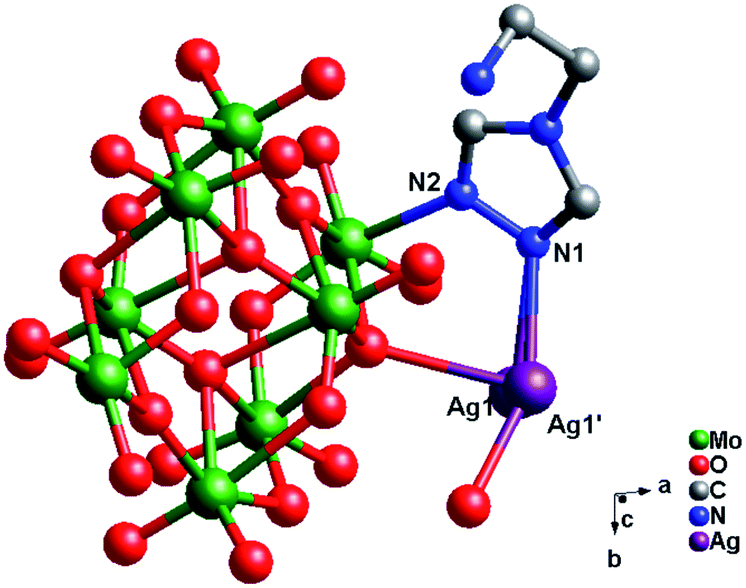
Crystal structure of compound 2. Single-crystal X-ray structure analysis reveals that compound 2 consists of half a β-Mo8 anion, one Ag+ ion and one tea ligand (Fig. 4). The β-Mo8 anion contains six {MoO6} octahedra. All Mo atoms are in the +VI oxidation state and the Ag atoms are in the +I oxidation state according to valence sum calculations.15 In order to balance the charge, two protons have been added in the formula of 2.
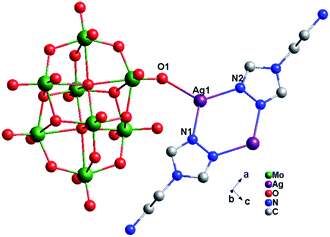 |
| | Fig. 4 Ball/stick view of the asymmetric unit of compound 2. The hydrogen atoms are omitted for clarity. | |
In compound 2, there exist only one crystallographically independent Ag+ ion. The Ag1 ion adopts a “Y”-type geometry, coordinated with two N atoms (N1 and N2) from one tea ligand and one O1 atom from one Mo8 anion. The Ag–N bond distances range from 2.221(4) to 2.253(4) Å, while the Ag–O bond distance is 2.320(3) Å and the N–Ag–N angle is 121.40(13)° (Table S1†).
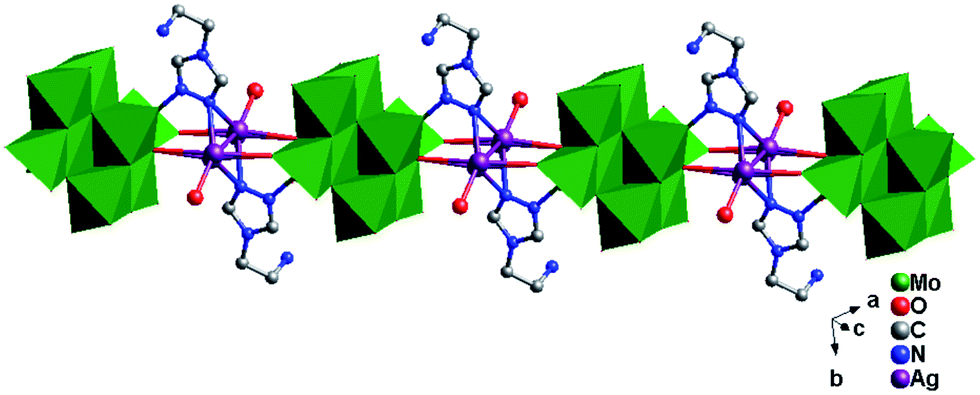
In compound 2, one tea ligand still provides two successive N atoms to link two Ag+ ions. Two tea molecule fuse two Ag+ ions and a 0D bi-nuclear cluster is constructed. The –NH2 still does not to coordinate with Ag+ ions and the metal–organic dimension is not extended. However, β-Mo8 plays the linking role in connecting adjacent bi-nuclear Ag+ clusters through Ag1–O1 bonds to build a 1D chain of 2, as shown in Fig. 5. Furthermore, the adjacent 1D chains are linked by hydrogen-bonding interactions (O5⋯Ag1 = 2.923 Å) to construct a 2D supramolecular layer (Fig. S1†).
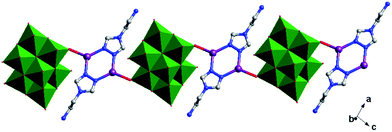 |
| | Fig. 5 1D chain of 2. | |
Crystal structure of compound 3. Single-crystal X-ray structure analysis reveals that compound 3 consists of half a γ-Mo8 anion, one Ag+ ion, one tea ligand and one coordinated water molecule (Fig. 6). The γ-Mo8 anion contains six {MoO6} octahedral and two {MoO5} subunits at both ends. All Mo atoms are also in the +VI oxidation state and the Ag atoms are in the +I oxidation state according to valence sum calculations.15 In order to balance the charge, two protons have been added in the formula of 3.
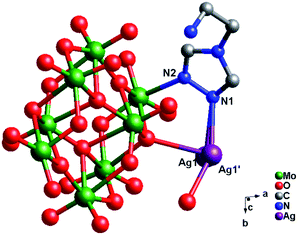 |
| | Fig. 6 Ball/stick view of the asymmetric unit of compound 3. The hydrogen atoms are omitted for clarity. | |
In compound 3, one Ag atom disorders into two (Ag1 and Ag1′), with each site half occupied. The Ag1 ion is four-coordinated by one N1 atom from one tea, two O atoms (O6 and O9) from two γ-Mo8 anions and one O atom from one coordinated water (O1W), adopting a distorted quadrangle geometry. Around the Ag+ atom, the Ag–O bond distances are in the range 2.266(4)–2.493(4) Å, while the Ag–N bond distances are in the range 2.347(5)–2.491(4) Å. The N(1)–Ag(1)–N angle is 114.14(12)°, while the N–N–Ag angles range from 110.2(3) to 123.8(3)° (Table S1†).
The tea in 3 also offers two N atoms (N1 and N2) to link two metal ions. However, unlike compound 2, only N1 links one Ag+ ion, while N2 links one Mo2 atom. Two tea offer two N1 atoms two fuse two Ag atoms, and a bi-nuclear Ag+ cluster [Ag2(tea)2]2+ is formed. The bi-nuclear Ag+ cluster through Ag–O9 and Ag–O6 bonds connect two adjacent γ-Mo8 to construct a 1D chain (Fig. 7). Furthermore, the left N2 donors in [Ag2(tea)2]2+ coordinate with two γ-Mo8 anions through two Mo2–N2 bonds. This linking style of Mo–N bonds is a structural feature of γ-Mo8 anion, which owns two {MoO5} subunits at both ends. The existence of Mo–N bonds strengthens the stability of 3. The adjacent 1D chains are still further linked by hydrogen-bonding interactions (O1W⋯O7 = 2.868 Å) to construct a 2D supramolecular layer (Fig. S2†).
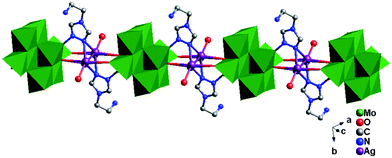 |
| | Fig. 7 1D organic–metal chain of 3. | |
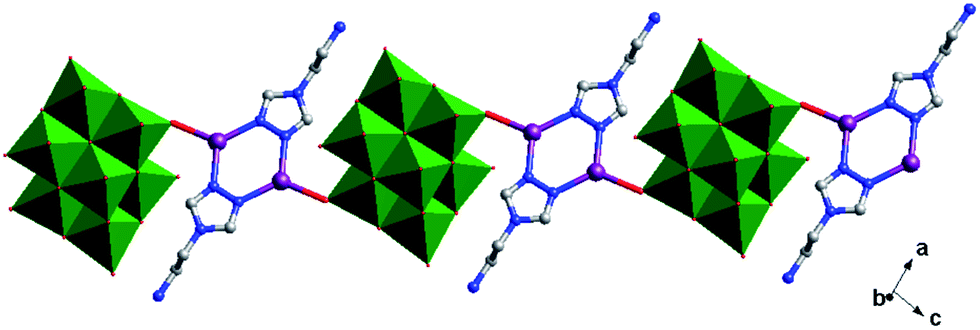
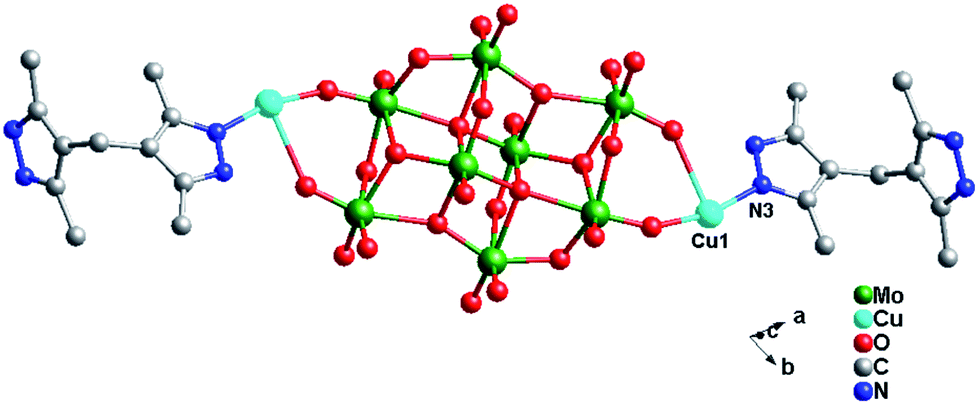
Crystal structure of compound 4. Single-crystal X-ray structure analysis reveals that compound 4 consists of half a β-Mo8 anion, one Cu2+ ion and one H2bdpm ligand (Fig. 8). Valence sum calculations15 show that all Mo atoms are in the +VI oxidation state and all Cu atoms are in the +II oxidation state. Compound 4 involves only one crystallographically independent Cu ion. Cu1 is six-coordinated with two N donors (N1 and N3) from two H2bdpm ligands and four terminal O atoms (O3, O7, O9 and O11) from three β-Mo8 anions. The Cu–O distances are in the range 1.948(10)–1.953(10) Å, while the Cu–N distances range from 1.955(14) to 1.956(14) Å. The O–Cu–O angle is 86.8(4)°, while the N–Cu–N angle is 93.1(6)° (Table S1†).
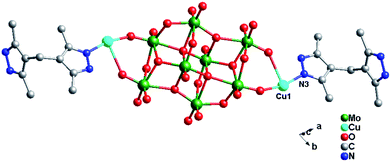 |
| | Fig. 8 Ball/stick view of the asymmetric unit of compound 4. The hydrogen atoms are omitted for clarity. | |
In compound 4, the adjacent β-Mo8 anions connect each other through sharing two bridging O12 atoms to build a inorganic Mo-chain (Fig. S3†). The H2bdpm ligand exhibits a single coordination mode only, offering two apical N donors to link two Cu2+ ions to construct a 1D zigzag Cu–H2bdpm chain (Fig. S4†). The H2bdpm with two symmetrical bis(pyrazole) groups induces the extensional metal–organic subunits. Two sets of Cu–H2bdpm chains cover the Mo-chain up and down, just like a “hamburger” chain, as shown in Fig. 9a. The adjacent hamburger chains are further linked by Cu1–O9 bonds to build a 2D layer, as shown in Fig. 5b and c.
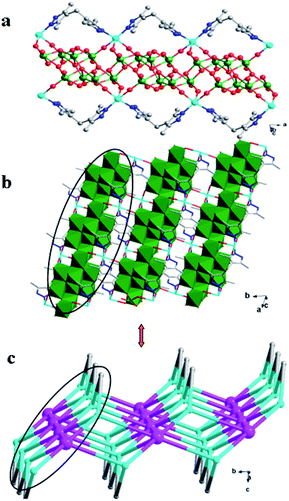 |
| | Fig. 9 (a) 1D “hamburger-like” chain of 4. (b) 2D layer of 4. (c) Schematic diagram of the 2D layer. | |
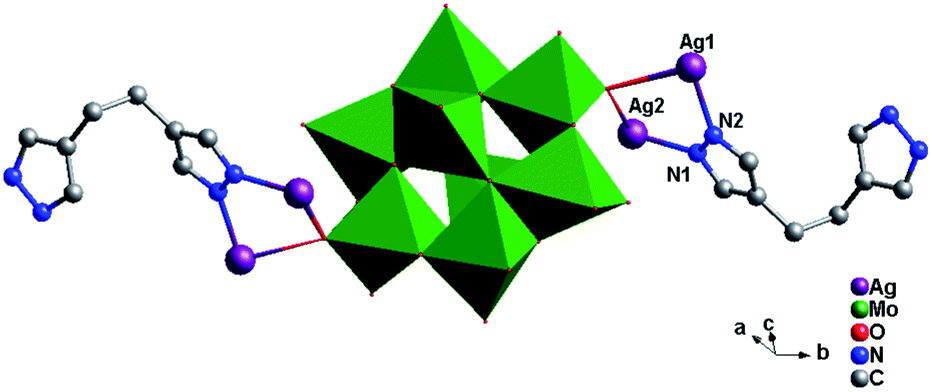
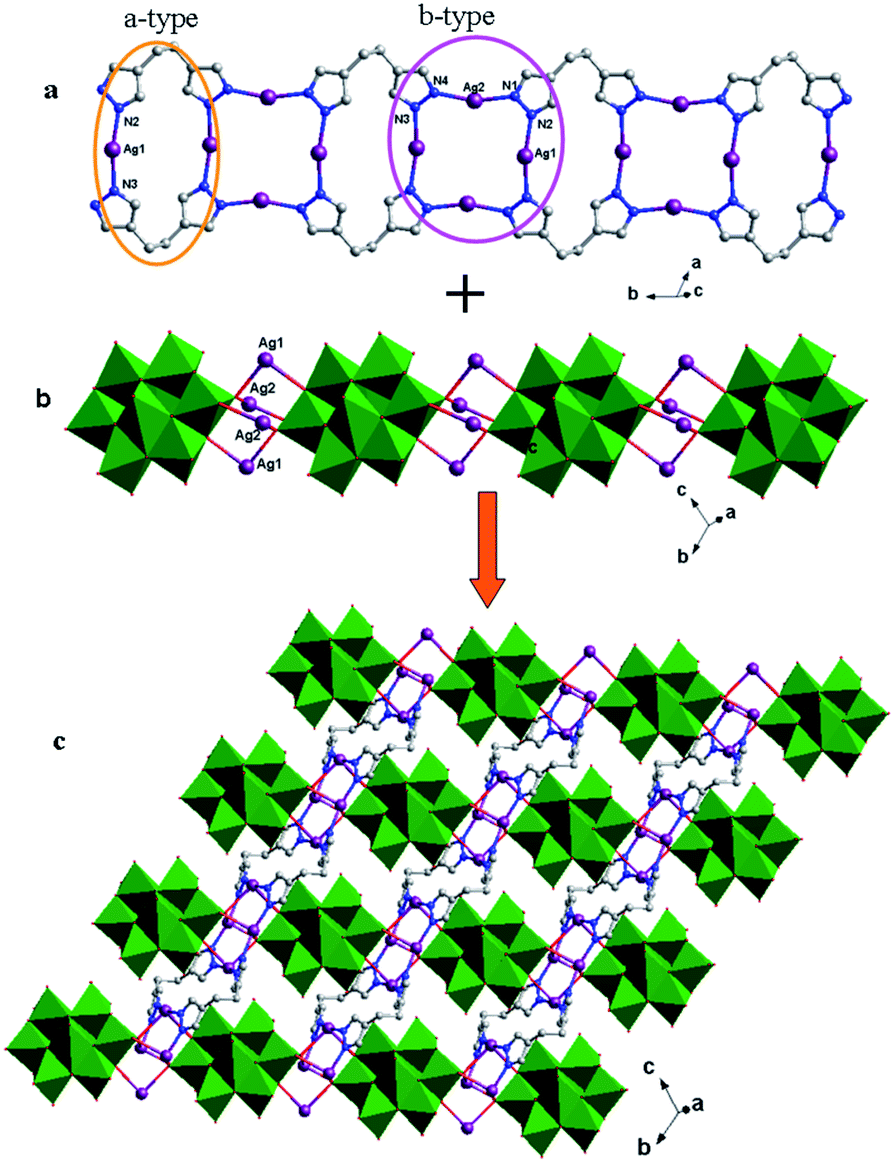
Crystal structure of compound 5. Single-crystal X-ray structure analysis reveals that compound 5 consists of half a θ-type Mo8 anion, one Hbhpe ligand and two Ag+ atoms (Fig. 10). The θ-Mo8 anion owns four {MoO6} octahedra, two {MoO5} and two {MoO4} subunits. All Mo atoms are in the +VI oxidation state and all Ag atoms are in the +I oxidation state according to valence sum calculations.15
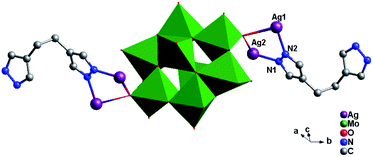 |
| | Fig. 10 Ball/stick view of the asymmetric unit of compound 5. The hydrogen atoms are omitted for clarity. | |
In compound 5, there are two crystallographically independent Ag ions (Ag1 and Ag2) with two different coordination geometries. Ag1 is six-coordinated with two N donors (N2 and N3) from two Hbhpe ligands and four terminal O atoms (O5, O6, O7 and O11) from three θ-Mo8 anions, with Ag–N bond distances of 2.208(5) and 2.212(5) Å, Ag–O bond distances of 2.670(5)–2.819(5) Å, and an N–Ag–N angle of 165.8(2)° (Table S1†). The Ag2 ion is four-coordinated with two N atoms (N1 and N4) from two Hbhpe ligands and two O atoms (O10 and O11) from two θ-Mo8 anions. The bond distances and angles around Ag2 are 2.155(5) and 2.160(6) Å for Ag–N, 2.511(5) and 2.617(5) Å for Ag–O and 160.8(2)° for N–Ag–N.
In compound 5, the Hbhpe ligand provides four N atoms to link four Ag+ ions, showing a “U” configuration. These two symmetrical pyrazole groups induce an extensional structure, a 1D cycle-connecting-cycle chain, as shown in Fig. 11a. In this chain, there are two types of cycle: a-type and b-type. The a-type cycle is a bi-nuclear one [Ag2(Hbhpe)2]2− with two Ag+ ions fused by two H2bhpe ligands. The b-type cycle exhibits a tetra-nuclear cycle, in which four Ag+ ions are aggregated by four ligands. The bi-nuclear [Ag2(Hbhpe)2]2− cycle and tetra-nuclear [Ag4(Hbhpe)4]4− cycle connect each other alternately through sharing the same H2bhpe ligands and a cycle-connecting-cycle chain is formed. The θ-Mo8 anions link the Ag+ atoms in tetra-nuclear cycles to build a 1D inorganic chain (Fig. 11b). The metal–organic cycle-connecting-cycle chain links the inorganic chain vertically through sharing the tetra-nuclear cycles. Furthermore, a 2D layer of 5 is constructed (Fig. 11c).
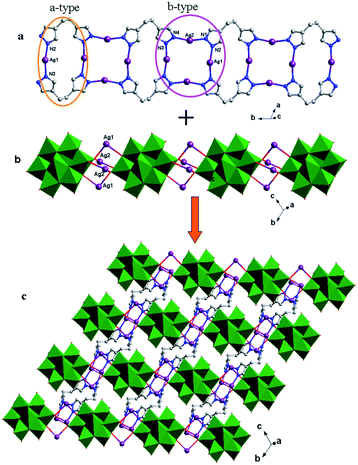 |
| | Fig. 11 (a) 1D metal–organic cycle-connecting-cycle chain of 5 with a-type (orange ellipse) and b-type cycles (purple ellipse) arranged alternately. (b) 1D inorganic chain of 5. (c) 2D layer with 1D metal–organic chains linking the inorganic chains vertically. | |
The influence of pendant tea and symmetrical bis(pyrazol) ligands on dimensional extension of metal–organic subunits. In recent years, pendant ligands involving only one coordination group and flexible ligands with two symmetrical coordination groups have attracted considerable attention for the construction of POM-TMCs. These two kinds of ligand show distinct coordination modes, which can induce different structures. In this work, we chose the pendant tea and symmetrical bis(pyrazole) ligands, H2bdpm and H2bhpe, to modify POM-Cu2+/Ag+ systems (Scheme 1). The tea owns one triazole group and one –NH2 group. However, the –NH2 unit is difficult to coordinate with metal ions, which prevent dimensional extension. Furthermore, the two N donors of tea are successive, forming low-dimensional multi-nuclear clusters. Compounds 1–3 have confirmed this point. In compound 1, a linear tri-nuclear [Cu3(tea)2(OH)2]4+ cluster has been built, showing a 0D metal–organic subunit. In compounds 2 and 3, bi-nuclear Ag clusters have been obtained, with no metal–organic extension. The H2bdpm and H2bhpe ligands have two symmetrical pyrazole groups, which can induce high-dimensional metal–organic structures, such as a 1D chain in 4 and a cycle-connecting-cycle chain in 5. Thus, using pendant and bis(pyrazole) ligands is a rational synthetic method to tune the dimensionality of metal–organic subunits in POM-based compounds.
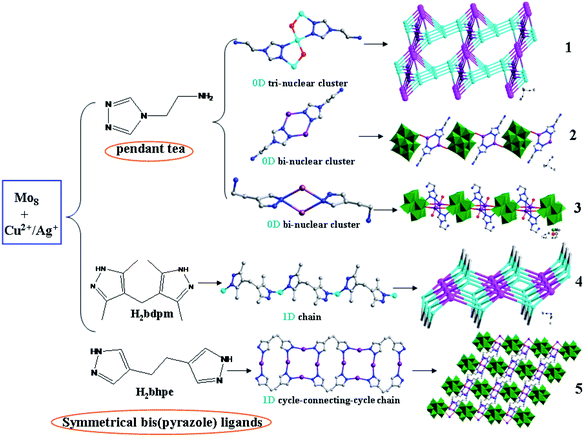 |
| | Scheme 1 Using pendant tea and symmetrical bis(pyrazole) ligands H2bdpm and H2bhpe to tune 0D and 1D metal–organic subunits in compounds 1–5. | |
FT-IR spectra
Fig. S5† shows the IR spectra of compounds 1–5. In the spectra, characteristic bands at 931, 879, 745, 627 and 1018 cm−1 for 1, 968, 879, 754 and 565 cm−1 for 2, 938, 825, 673 and 554 cm−1 for 3, 970, 880, 761 and 542 cm−1 for 4 and 964, 871, 766 and 534 cm−1 for 5, are attributed to ν(Mo–Ot), ν(Mo–Ob–Mo), ν(Mo–Oc–Mo),16 respectively. Bands in the regions of 1626–1116 cm−1 for 1, 1637–1120 cm−1 for 2 and 1626–1112 for 3, 1634–1188 cm−1 for 4 and 1647–1186 cm−1 for 5 are attributed to the tea, Hbhpe and H2bdpm ligands, respectively.
Voltammetric behavior of 1-CPE in aqueous electrolyte and its electrocatalytic activity
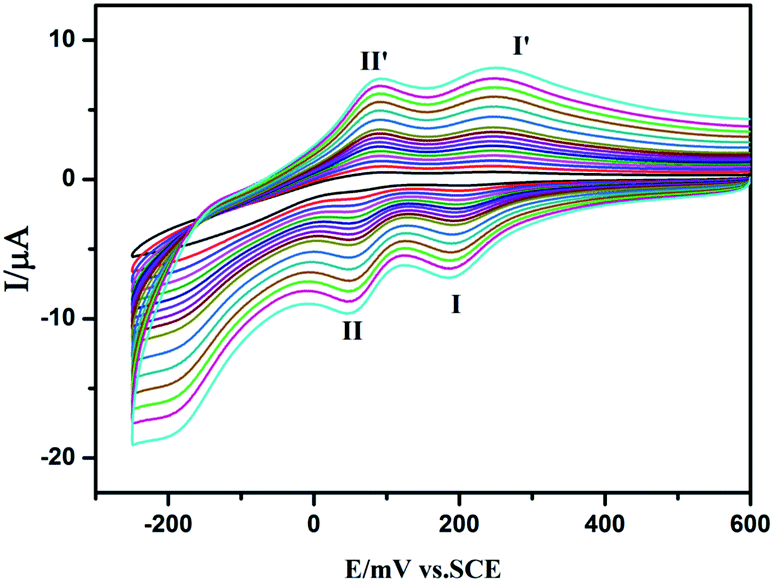
We studied the electrochemical properties of compounds 1–5. Owing to the similar electrochemical behaviors of compounds 1–5 modified carbon paste electrodes, 1-CPE has been used as a representative example to study the electrochemical properties. Cyclic voltammograms for 1-CPEs in 0.1 M H2SO4 + 0.5 M Na2SO4 aqueous solution at different scan rates are presented in Fig. 12. In the potential range from +600 to −250 mV, two reversible redox peaks I–I′ and II–II′ are seen, with half-wave potentials E1/2 = (Epa + Epc)/2 of +228 (I–I′), +70 (II–II′) mV (scan rate: 80 mV s−1). These two redox peaks should be ascribed to two consecutive two-electron process of Mo8.17 From 20 to 500 mV s−1 of scan rates, the peak potentials change gradually: the cathodic peak potentials shift towards the negative direction, while the corresponding anodic peak potentials shift to the positive direction.
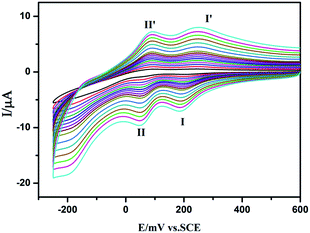 |
| | Fig. 12 Cyclic voltammograms of 1-CPE in 0.1 M H2SO4 + 0.5 M Na2SO4 aqueous solution at different scan rates (from inner to outer: 20, 40, 60, 80, 100, 120, 140, 160, 180, 200, 250, 300, 350, 400, 450 and 500 mV s−1, respectively). | |
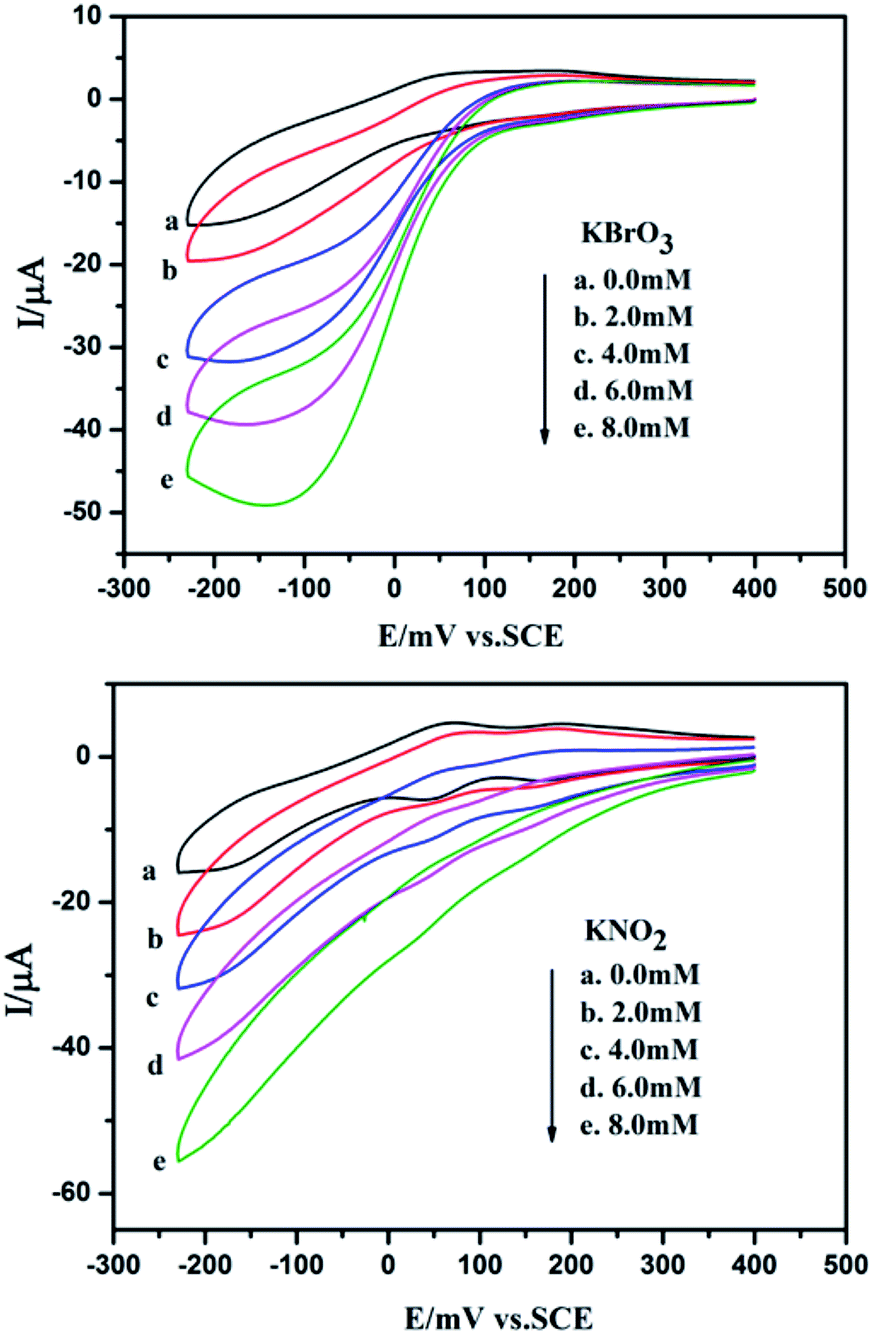
Fig. 13 shows cyclic voltammograms for the electrocatalytic reduction of nitrite and bromate at 1-CPE in 0.1 M H2SO4 + 0.5 M Na2SO4 aqueous solution. With the addition of bromate, the second reduction peak currents increase gradually, while the corresponding oxidation peak currents gradually decrease. However, the first redox peak remains almost unchanged. Furthermore, it can clearly be seen that with the addition of nitrite, both reduction peak currents gradually increased and the corresponding oxidation peak currents visibly decreased indicating that 1-CPE exhibits good electrocatalytic activity for the reduction of bromate and nitrite.
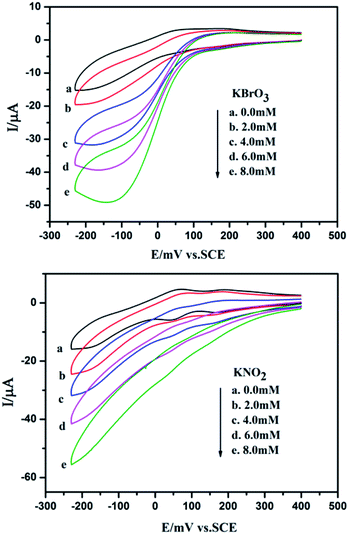 |
| | Fig. 13 Cyclic voltammograms of 1-CPE in 0.1 M H2SO4 + 0.5 M Na2SO4 containing 0 (a); 2.0 (b); 4.0 (c); 6.0 (d); 8.0 (e) mM nitrite/bromate. Scan rate: 200 mV s−1. | |
Photocatalytic activity
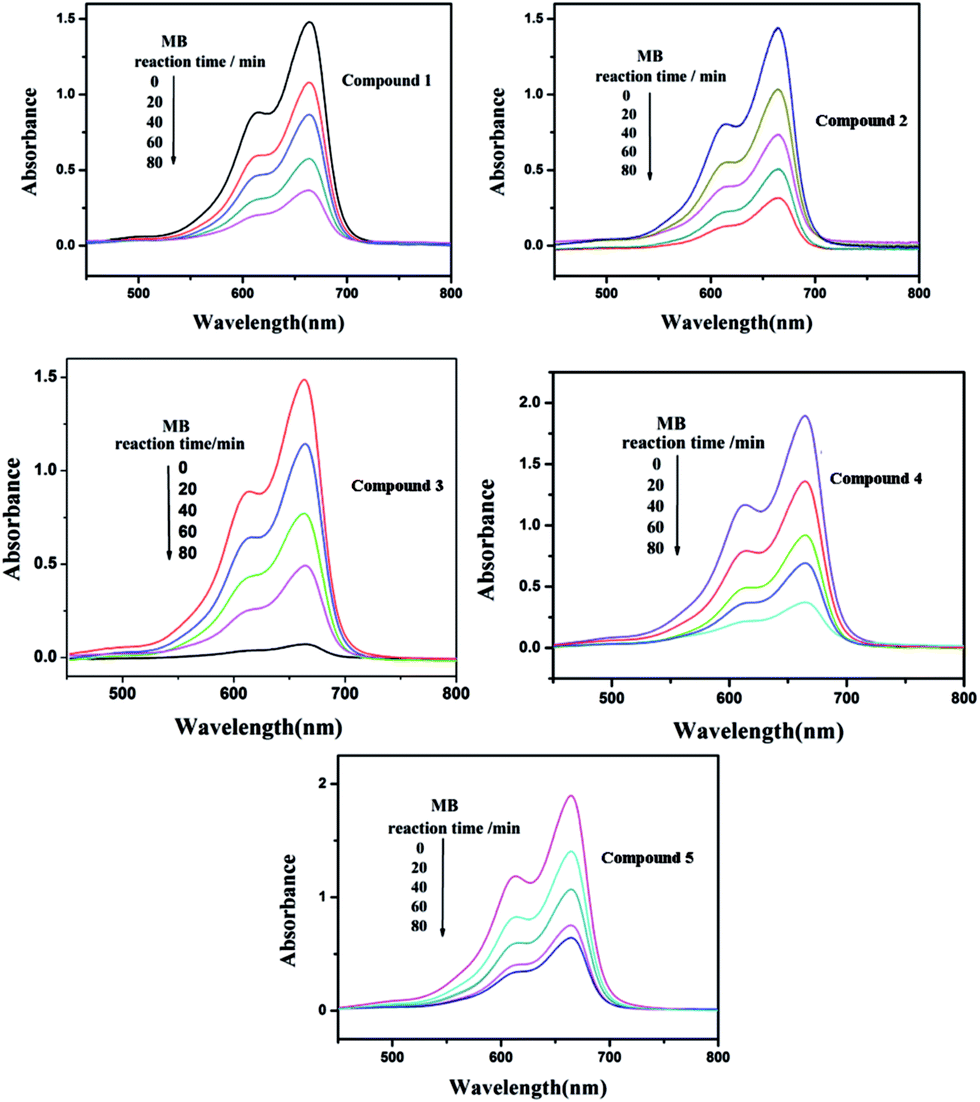
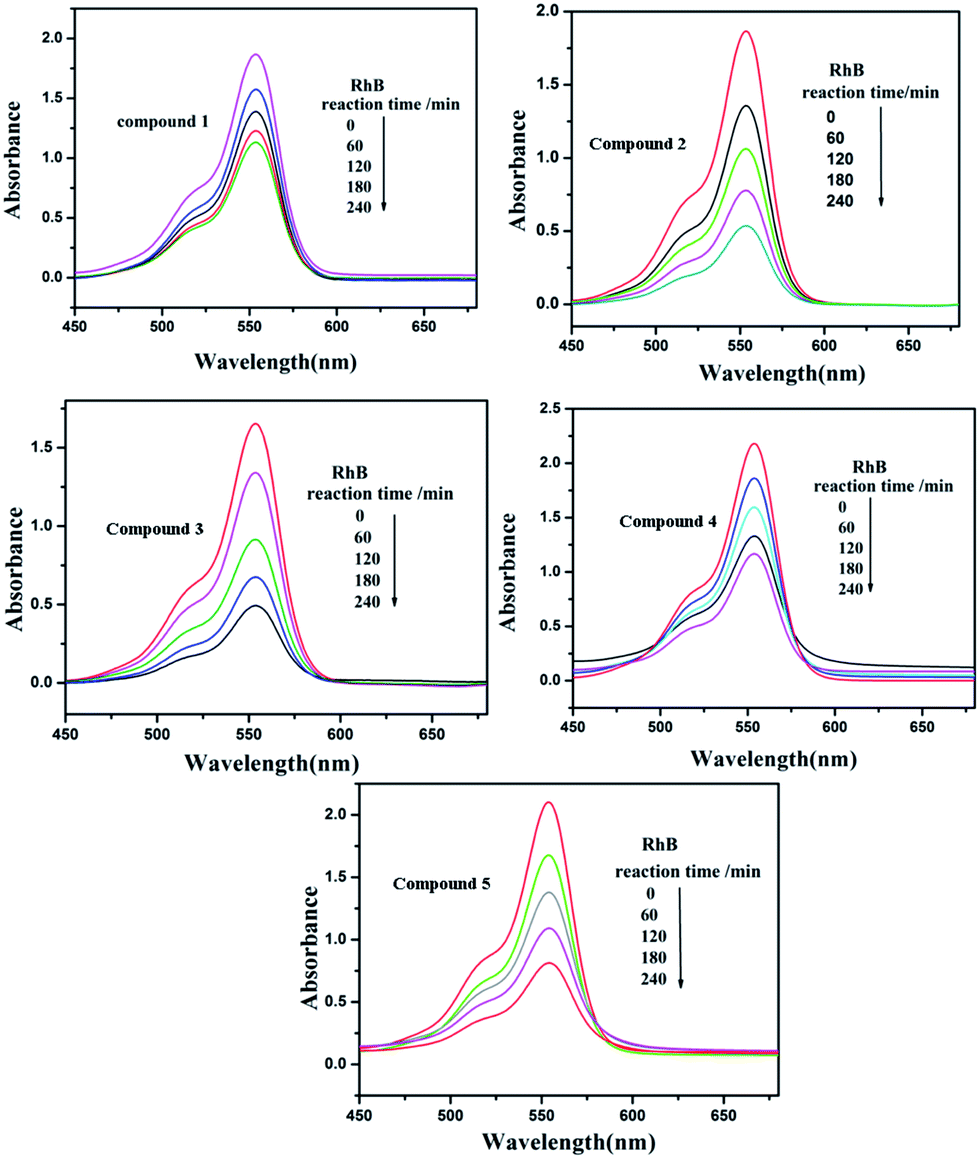
Under UV light irradiation, we selected two organic dyes, methylene blue (MB) and rhodamine B (RhB), as model pollutants in aqueous media to study the photocatalytic activities of compounds 1–5. In the process of photocatalysis, 150 mg of compounds 1–5 were decentralized in 0.02 mmol L−1 MB/RhB aqueous solution (90 mL) and magnetically stirred for about 10 min to ensure the equilibrium in the dark. Then the mixed solution was exposed to a UV Hg lamp with continuous stirring. At every interval (20 min for MB and 60 min for RhB), 5.0 mL samples were removed for analysis by UV-Vis spectrophotometer. We can clearly see that the percentage of MB degradation photocatalyzed by 1–5 increased clearly with increasing reaction time (Fig. 14). The conversions of MB are 75% for 1, 80.0% for 2, 95.2% for 3, 80.3% for 4 and 66.2% for 5, after 80 min (Fig. S6a†). Fig. 15 shows the photocatalytic degradation of RhB with the conversions of 39.5% for 1, 71.1% for 2, 70.3% for 3, 46.3% for 4 and 61.4% for 5, after 240 min (Fig. S6b†). This result shows that compounds 1–5 have good photocatalytic activities for the degradation of MB, while compounds 2, 3, 4 and 5 can act as good photocatalysts for the degradation of RhB.
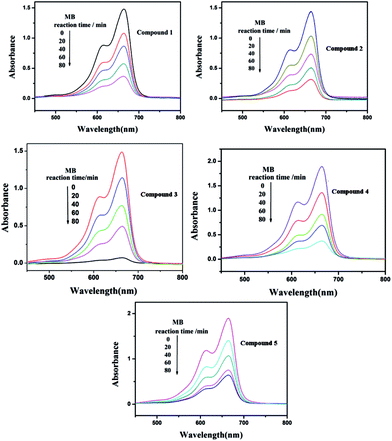 |
| | Fig. 14 Absorption spectra of the MB solution during the decomposition reaction under UV irradiation with the compounds 1–5 as the catalyst. | |
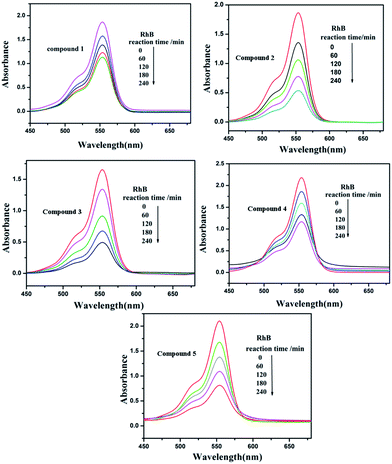 |
| | Fig. 15 Absorption spectra of the RhB solution during the decomposition reaction under UV irradiation with the compounds 1–5 as the catalyst. | |
Conclusions
In this paper, by using pendant tea and bis(pyrazole) ligands H2bdpm and H2bhpe, five new Mo8-based compounds have been synthesized under hydrothermal conditions. Single coordination group (tea) and two symmetrical bis(pyrazole) groups (H2bdpm and H2bhpe) induce 0D and 1D metal–organic subunits, respectively, in these five structures. The bis(pyrazole) ligands can induce extensional structures with ease. It is therefore rational to use pendant and bis(pyrazole) ligands to tune the metal–organic dimensionality of 1–5.
Note added after first publication
This article replaces the version published on 14th June 2017, which contained errors in the CCDC numbers.
Acknowledgements
Financial support of this research by the National Natural Science Foundation of China (No. 21571023, 21401010 and 21471021) and Talent-supporting Program Foundation of Education Office of Liaoning Province (LJQ2012097).
Notes and references
-
(a) R. Yu, X. F. Kuang, X. Y. Wu, C. Z. Lu and J. P. Donahue, Coord. Chem. Rev., 2009, 253, 2872 CrossRef CAS;
(b) J. W. Zhao, Y. Z. Li, L. J. Chen and G. Y. Yang, Chem. Commun., 2016, 52, 4418 RSC;
(c) T. Ueda, M. Ohnishi, M. Shiro, J. I. Nambu, T. Yonemura, J. F. Boas and A. M. Bond, J. Am. Chem. Soc., 2014, 136, 10941 CrossRef PubMed;
(d) A. Rubinstein, P. J. Lozanao, J. J. Carbó, J. M. Poblet and R. Neumann, J. Am. Chem. Soc., 2014, 136, 10941 CrossRef CAS PubMed;
(e) D. L. Long, E. Burkholder and L. Cronin, Chem. Soc. Rev., 2007, 36, 105 RSC.
-
(a) J. Q. Sha, J. W. Sun, M. T. Li, C. Wang, G. M. Li, P. F. Yan and L. J. Sun, Dalton Trans., 2013, 42, 1667 RSC;
(b) P. Kar, R. Haldar, C. J. Gómez-García and A. Ghosh, Inorg. Chem., 2012, 51, 4265 CrossRef CAS PubMed;
(c) Z. J. Liu, X. L. Wang, C. Qin, Z. M. Zhang, Y. G. Li, W. L. Chen and E. B. Wang, Coord. Chem. Rev., 2016, 313, 94 CrossRef CAS;
(d) J. J. Walsh, A. M. Bond, R. J. Forster and T. E. Keyes, Coord. Chem. Rev., 2016, 306, 217 CrossRef CAS.
-
(a) J. Zhou, X. Liu, R. Chen, H. P. Xiao, F. L. Hu, H. H. Zou, Y. Zhou, C. Liu and L. G. Zhu, CrystEngComm, 2013, 15, 5057 RSC;
(b) J. Zhou, J. W. Zhao, Q. Wei, J. Zhang and G. Y. Yang, J. Am. Chem. Soc., 2014, 136, 5065 CrossRef CAS PubMed.
-
(a) Z. G. Han, Y. Z. Gao and C. W. Hu, Cryst. Growth Des., 2008, 8, 1261 CrossRef CAS;
(b) S. L. Li, Y. Q. Lan, J. F. Ma, J. Yang, X. H. Wang and Z. M. Su, Inorg. Chem., 2007, 46, 8283 CrossRef CAS PubMed.
- Y. Q. Lan, S. L. Li, X. L. Wang, K. Z. Shao, D. Y. Du, H. Y. Zang and Z. M. Su, Inorg. Chem., 2008, 47, 8179 CrossRef CAS PubMed.
- Q. G. Zhai, X. Y. Wu, S. M. Chen, Z. G. Zhao and C. Z. Lu, Inorg. Chem., 2007, 46, 5046 CrossRef CAS PubMed.
-
(a) P. J. Zapf, C. J. Warren, R. C. Haushalter and J. Zubieta, Chem. Commun., 1997, 1543 RSC;
(b) P. J. Zapf, R. L. LaDuca, R. S. Rarig, K. M. Johnson and J. Zubieta, Inorg. Chem., 1998, 37, 3411 CrossRef CAS;
(c) J. R. D. DeBord, R. C. Haushalter, L. M. Meyer, D. J. Rose, P. J. Zapf and J. Zubieta, Inorg. Chim. Acta, 1997, 256, 165 CrossRef CAS;
(d) D. Hagrman, C. Zubieta, D. J. Rose, J. Zubieta and R. C. Haushalter, Angew. Chem., Int. Ed. Engl., 1997, 36, 873 CrossRef CAS;
(e) D. Hagrman, C. Sangregorio, C. J. O'Connor and J. Zubieta, J. Chem. Soc., Dalton Trans., 1998, 3707 RSC.
-
(a) J. Gu, X. E. Jiang, Z. H. Su, Z. F. Zhao and B. B Zhou, Inorg. Chim. Acta, 2013, 400, 210 CrossRef CAS;
(b) X. L. Wang, J. Li, A. X. Tian, G. C. Liu and H. Y. Lin, Sci. Sin.: Chim., 2011, 41, 806 CrossRef.
-
(a) B. X. Dong and Q. Xu, Inorg. Chem., 2009, 48, 5861 CrossRef CAS PubMed;
(b) X. L. Wang, X. J. Liu, A. X. Tian, J. Ying, H. Y. Lin, G. C. Liu and Q. Gao, Dalton Trans., 2012, 41, 9587 RSC.
-
(a) P. P. Zhang, J. Peng, A. X. Tian, H. J. Pang, Y. Chen, M. Zhu, D. D. Wang, M. G. Liu and Y. H. Wang, J. Mol. Struct., 2010, 984, 221 CrossRef CAS;
(b) B. X. Dong, J. Peng, C. J. Gómez-García, S. Benmansour, H. Q. Jia and N. H. Hu, Inorg. Chem., 2007, 46, 5933 CrossRef CAS PubMed.
-
(a) X. L. Wang, H. L. Hu, G. C. Liu, H. Y. Lin and A. X. Tian, Chem. Commun., 2010, 46, 6485 RSC;
(b) X. L. Wang, H. L. Hu, A. X. Tian, H. Y. Lin and J. Li, Inorg. Chem., 2010, 49, 10299 CrossRef CAS PubMed.
-
(a) A. X. Tian, J. Peng, J. Q. Sha, Z. G. Han, J. F. Ma, Z. M. Su, N. H. Hu and H. Q. Jia, Inorg. Chem., 2008, 47, 3274 CrossRef CAS PubMed;
(b) A. X. Tian, J. Ying, J. Peng, J. Q. Sha, Z. M. Su, H. J. Pang, P. P. Zhang, Y. Chen, M. Zhu and Y. Shen, Cryst. Growth Des., 2010, 10, 1104 CrossRef CAS.
- A. X. Tian, Y. L. Ning, J. Ying, X. Hou, Y. Tian, T. J. Li and X. L. Wang, Sci. Sin.: Chim., 2016, 46, 688 Search PubMed.
- G. M. Sheldrick, SHELXS-97, University of Göttingen, Germany, 1997 Search PubMed.
- I. D. Brown and D. Altermatt, Acta Crystallogr., Sect. B: Struct. Sci., 1985, 41, 244 CrossRef.
-
(a) W. G. Klemperer and W. Shum, J. Am. Chem. Soc., 1976, 98, 8291 CrossRef CAS;
(b) N. Xu, J. W. Zhang, X. L. Wang, G. C. Liu and T. J. Li, Dalton Trans., 2016, 45, 760 RSC;
(c) C. Rocchiccioli-Deltcheff, M. Fournier and R. Franck, Inorg. Chem., 1983, 22, 207 CrossRef CAS;
(d) R. Thouvenot, M. Fournier, R. Franck and C. Rocchiccioli-Deltcheff, Inorg. Chem., 1984, 23, 598 CrossRef CAS;
(e) C. Rocchiccioli-Deltcheff, R. Thouvenot and R. Franck, Spectrochim. Acta, 1976, 32, 587 CrossRef.
-
(a) D. Hagrman, C. Sangregorio, C. J. O'Connor and J. Zubieta, Dalton Trans., 1998, 22, 3707 RSC;
(b) F. Gruber and M. Jansen, Z. Anorg. Allg. Chem., 2010, 636, 2352 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: IR spectra, CV data and additional figures. CCDC 1531320 for 1, 1531323 for 2, 1531324 for 3, 1531321 for 4, 1531322 for 5. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra03884d |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *,
Huaiping Ni,
Xuebin Ji,
Yan Tian,
Guocheng Liu and
Jun Ying*
*,
Huaiping Ni,
Xuebin Ji,
Yan Tian,
Guocheng Liu and
Jun Ying*