DOI:
10.1039/C7RA03930A
(Paper)
RSC Adv., 2017,
7, 30886-30893
Selective C–H bond hydroxylation of cyclohexanes in water by supramolecular control†
Received
6th April 2017
, Accepted 30th May 2017
First published on 16th June 2017
Abstract
A new approach for selective hydroxylation of non-activated cyclohexanes using dioxirane generated in situ in water through supramolecular control has been developed. Using β-CD and γ-CD as the supramolecular hosts, selective hydroxylation of cyclohexane substrates, including trans/cis-1,4-, 1,3- and 1,2-dimethylcyclohexanes and trans/cis-decahydronaphthalene, was achieved in up to 54% yield in water. Furthermore, site-selective C–H bond hydroxylation of (+)-menthol was achieved by obstructing the approach of dioxirane to the C–H bond with higher steric hindrance through inclusion complexation with β-CD and γ-CD in water.
Introduction
Over the decades, significant advancements in the research area of C–H bond oxidation1–23 have been accomplished using stoichiometric oxidizing reagents,7,9–11 catalytic transition metal catalysts12–18 and biomimetic supramolecular complexes.2,19–23 Notably, selective oxidation of non-activated sp3 C–H bonds of alkanes is a very useful reaction because it offers a strategy with improved atom- and step-economy to transform inert C–H bonds into versatile functionalities for further synthetic elaborations.6 However, it is regarded as one of the most challenging tasks in organic chemistry as the functionalization of non-activated C–H bonds generally requires harsh reaction conditions, and the highly reactive oxidizing agents employed exhibit low functional group tolerance.3,19,24 Furthermore, the presence of multiple C–H bonds with similar reactivity in a single molecule renders site-selective oxidation difficult.24 In the literature, synthetic methods have been developed for selective oxidation of non-activated sp3 C–H bonds on the basis of electronic, steric, and substrate-based directing effects.1–8 However, these approaches are limited by the inherent structural properties of the substrates. For example, less activated aliphatic C–H bonds could be selectively targeted for oxidation using “directing” groups strategically installed in the substrates.17,25 In view of the significance of C–H bond functionalization, there remains great interest in developing new strategies for selective sp3 C–H bond oxidation to target a diverse variety of substrates.
Oxidized cyclohexanes are commonly found in the structural skeletons of natural products such as artemisinin,19 taxanes,26 terpenes,27,28 and steroids.29 Thus, selective oxidation of cycloalkanes would be an appealing reaction for efficient synthesis of these structurally diverse oxidized cyclohexane-containing organic molecules.
Supramolecular host–guest chemistry provides promising approaches for achieving selectivity enhancement in a wide range of organic reactions.30–33 Generally, supramolecular hosts are able to preferentially position the target sites of the substrates in proximity to the reaction centre, leading to selective organic transformations. On the other hand, access to the non-target sites of the substrates for reaction could be obstructed through inclusion complex formation.
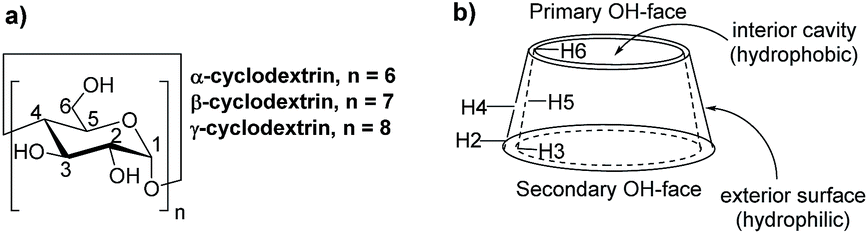
Cyclodextrins (CDs) are amphipathic molecules bearing a hydrophilic exterior surface and a hydrophobic interior cavity (Fig. 1). Due to the hydrophobic cavity of cyclodextrins and their derivatives, “host–guest” inclusion complex formation with a wide diversity of guest molecules through hydrophobic interactions has been extensively employed in supramolecular catalysis.34–40 Breslow reported a pioneering study on selective C–H bond oxidations of steroids using cyclodextrin-attached metalloporphyrins as catalysts.39,40 In 2003, we developed a cyclodextrin-ketoester as a supramolecular catalyst for stereoselective alkene epoxidation.37 Then, Bols and co-workers developed cyclodextrin ketones for organic oxidations.41–45 In 2012, we studied the site-selective oxidation of tertiary C–H bonds of 3,7-dimethyloctyl esters using cyclodextrins as the supramolecular host.38
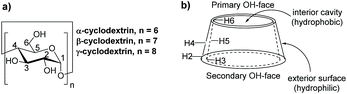 |
| | Fig. 1 General structure of cyclodextrins. | |
Because selective C–H bond hydroxylation of cyclohexanes using supramolecular control remains largely unexplored and cyclodextrin-promoted reactions are environmentally friendly using water as the solvent, we aim to realize site-selective oxidation of C–H bonds on cyclohexanes in water using cyclodextrins as supramolecular hosts, which would lead to interesting selectivity.46 Based on our previous studies on cyclodextrins,37,38,47 we developed a new approach for site-selective hydroxylation of cyclohexane derivatives through supramolecular control using cyclodextrins as the hosts. The inclusion models of trans-1,4-dimethylhexane (1) and (+)-menthol (9) with β-CD and γ-CD as well as the relationship between inclusion models and site-selective C–H bond hydroxylation of 9 have also been investigated. It is noteworthy that β-CD and γ-CD could act as reaction vessels such that the C–H bond hydroxylation could be performed in water without using organic solvents.
Results and discussion
The effect of different CDs on C–H bond hydroxylation of cyclohexanes
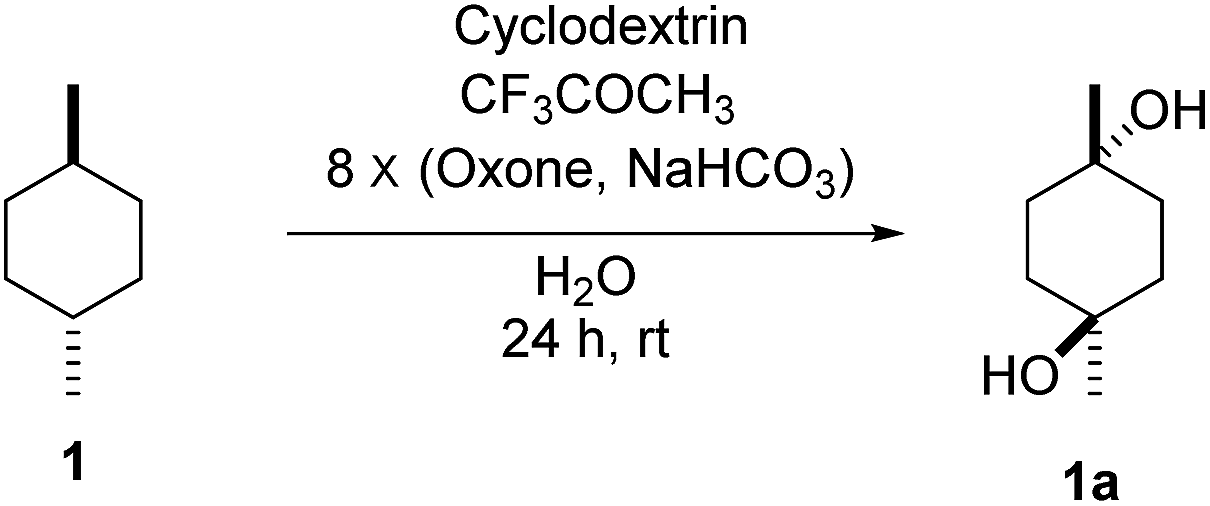
To study the efficiency of different CDs on C–H bond hydroxylation of cyclohexanes, trans-1,4-dimethylcyclohexane (1) was employed as the substrate (Table 1). Dioxirane generated in situ from 1,1,1-trifluoroacetone and Oxone was used as the oxidizing agent.48–59 Initially, the reaction was carried out at room temperature by stirring trans-1,4-dimethylcyclohexane (1) (1.0 mmol) with β-CD (1.1 mmol) and 1,1,1-trifluoroacetone (1.0 mmol) in water (50 mL) for 1 h, and then 8 portions of a mixture of Oxone (2.5 mmol × 8) and NaHCO3 (7.75 mmol × 8) were added within 7 h (one portion was added per hour). After stirring for an additional 16 h, the hydroxylation product 1a was isolated in 12% yield by flash column chromatography (Table 1, entry 1). Using γ-CD instead of β-CD as the supramolecular host, 1a was obtained in 32% yield (Table 1, entry 2). On using α-CD, only a trace amount of 1a was observed by TLC analysis (Table 1, entry 3). A control experiment was performed in water without cyclodextrin, and no hydroxylation product 1a was obtained, probably due to the low solubility of 1 in water (Table 1, entry 4). 1a was obtained in 16% yield when the reaction was conducted in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 CH3CN/H2O without cyclodextrins as the supramolecular host (Table 1, entry 5). These results suggest that β-CD and γ-CD could enhance the hydroxylation yield by acting as a reaction vessel and maximizing the dispersion of 1 through inclusion complex formation.60 Furthermore, β-CD and γ-CD could bind to 1 and dioxirane so as to place the two reactants in close proximity, stabilizing the transition state of the reaction and reducing the activation energy, and thus accelerating the reaction.61–65
:1 CH3CN/H2O without cyclodextrins as the supramolecular host (Table 1, entry 5). These results suggest that β-CD and γ-CD could enhance the hydroxylation yield by acting as a reaction vessel and maximizing the dispersion of 1 through inclusion complex formation.60 Furthermore, β-CD and γ-CD could bind to 1 and dioxirane so as to place the two reactants in close proximity, stabilizing the transition state of the reaction and reducing the activation energy, and thus accelerating the reaction.61–65
Table 1 Studies on the effect of different cyclodextrins towards C–H bond hydroxylation of trans-1,4-dimethylcyclohexane (1)a
|
| Entry |
Cyclodextrins |
Yield of 1ab (%) |
| Reactions were conducted with 1 (1.0 mmol), 1,1,1-trifluoroacetone (1.0 mmol) and cyclodextrin (1.1 mmol) in H2O (50 mL) at room temperature with Oxone (2.5 mmol × 8) and NaHCO3 (7.75 mmol × 8). Yield of isolated product. Observed by TLC analysis. CH3CN (25 mL) and H2O (25 mL). |
| 1 |
β-CD |
12 |
| 2 |
γ-CD |
32 |
| 3 |
α-CD |
Tracec |
| 4 |
Without CD (in H2O) |
0c |
| 5d |
Without CD (in CH3CN/H2O) |
16 |
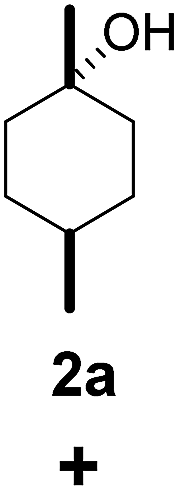
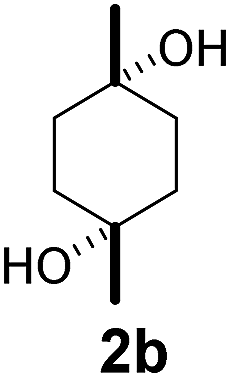
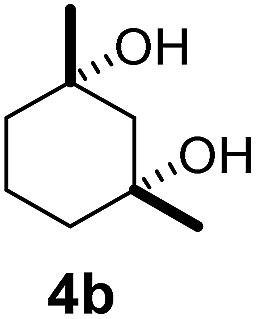
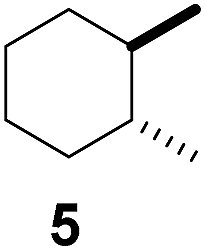
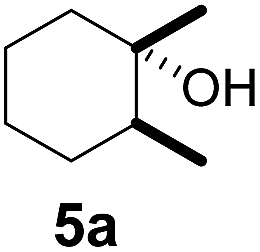
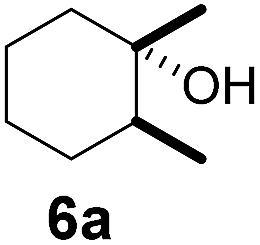
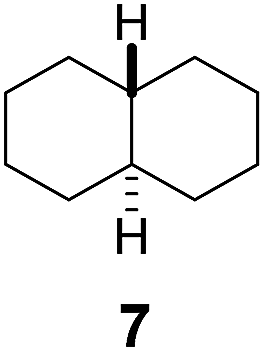
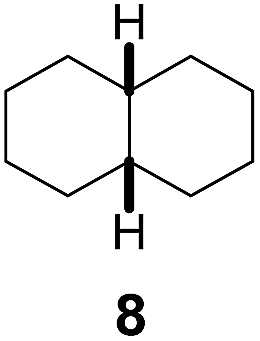
Subsequently, cis-1,4-dimethylcyclohexane (2), trans/cis-1,3- and 1,2-dimethylcyclohexane (3–6), and trans/cis-decahydronaphthalene (7 and 8) were employed as substrates. As shown in Table 2, dimethylcyclohexanes (1–6) can be oxidized by dioxirane generated in situ through complexation with β-CD or γ-CD to give the corresponding hydroxylation products. Notably, cis-1,4-dimethylcyclohexane (2) and trans/cis-1,3-dimethylcyclohexane (3 and 4) gave monohydroxylation and dihydroxylation products (Table 2, entries 2–4), while trans/cis-1,2-dimethylcyclohexane (5 and 6) and trans/cis-decahydronaphthalene (7 and 8) gave only monohydroxylation products (Table 2, entries 5–8). In the presence of β-CD, trans/cis-decahydronaphthalene (7 and 8) did not give monohydroxylation or dihydroxylation products. Good yields (54% and 40%) were achieved when γ-CD was used as a supramolecular host. These findings indicate that β-CD and γ-CD could act as reaction vessels through inclusion complexation of hydrophobic cyclohexanes in water. Moreover, these results showed that using γ-CD as the supramolecular host gave higher yields than using β-CD as the supramolecular host.
Table 2 Comparison of C–H bond hydroxylation of various disubstituted cyclohexanes with and without CDsa
1H NMR titration for binding of 1 to β-CD/γ-CD
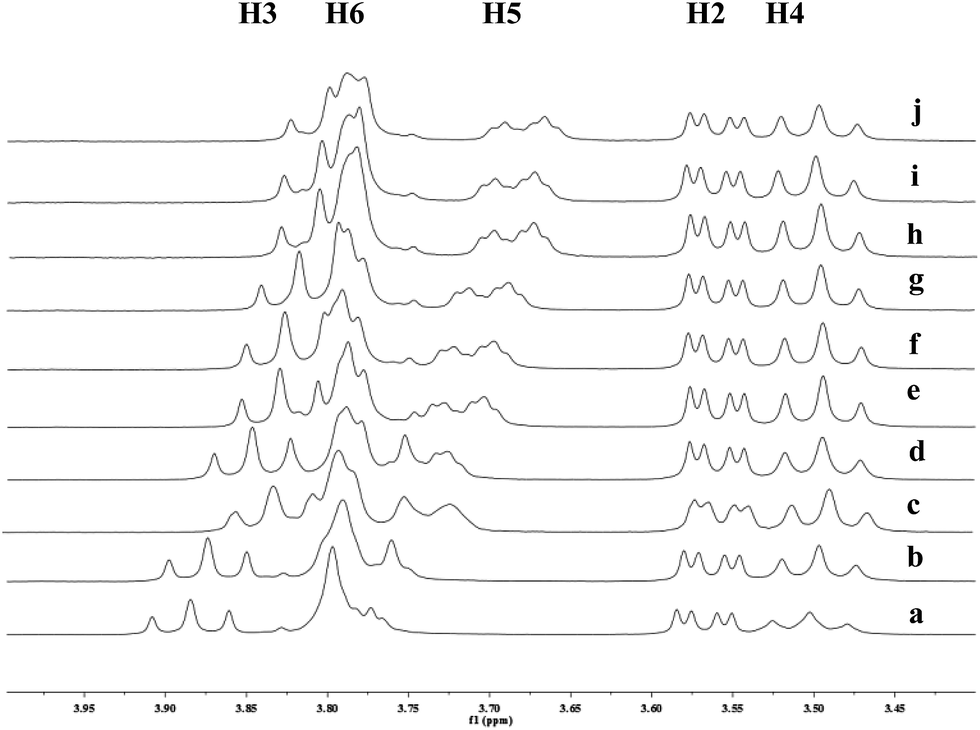
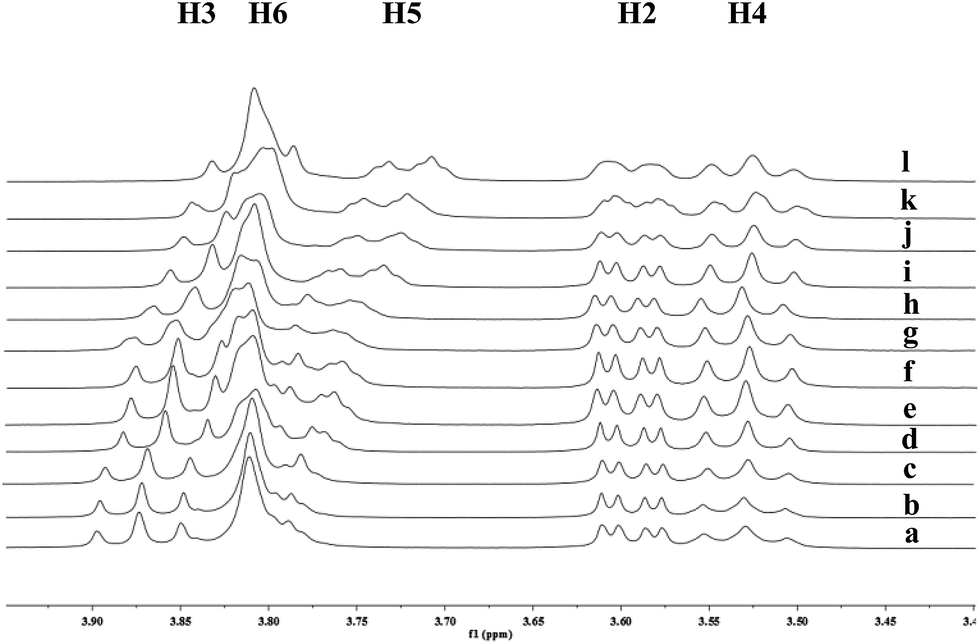
To investigate the effect of the cavity sizes of β-CD and γ-CD on C–H bond oxidation of cycloalkanes, we studied the inclusion complex formation between trans-1,4-dimethyl-cyclohexane (1) and β-CD or γ-CD by 1H NMR titration (Fig. 2 and 3).38,66–70 The signals of H3 and H5 of β-CD or γ-CD were shifted significantly upfield when the amount of 1 increased in the aqueous solution of β-CD or γ-CD, while the chemical shifts of H2 and H4 remained unchanged. These results indicate that 1 interacts with the hydrogens in the hydrophobic cavities of β-CD and γ-CD.
 |
| | Fig. 2 Partial 1H NMR spectra of a mixture of 1 and β-CD in D2O (signals of β-CD). Ratios of 1/β-CD: (a) 0:10, (b) 1:10, (c) 2:10, (d) 4:10, (e) 6:10, (f) 8:10, (g) 1:1, (h) 12:10, (i) 15:10, (j) 2:1. | |
 |
| | Fig. 3 Partial 1H NMR spectra of a mixture of 1 and γ-CD in D2O (signals of γ-CD). Ratios of 1/γ-CD: (a) 0:10, (b) 1:10, (c) 4:10, (d) 7:10, (e) 1:1, (f) 12:10, (g) 15:10, (h) 18:10, (i) 2:1, (j) 3:1, (k) 3.5:1, (l) 4:1. | |
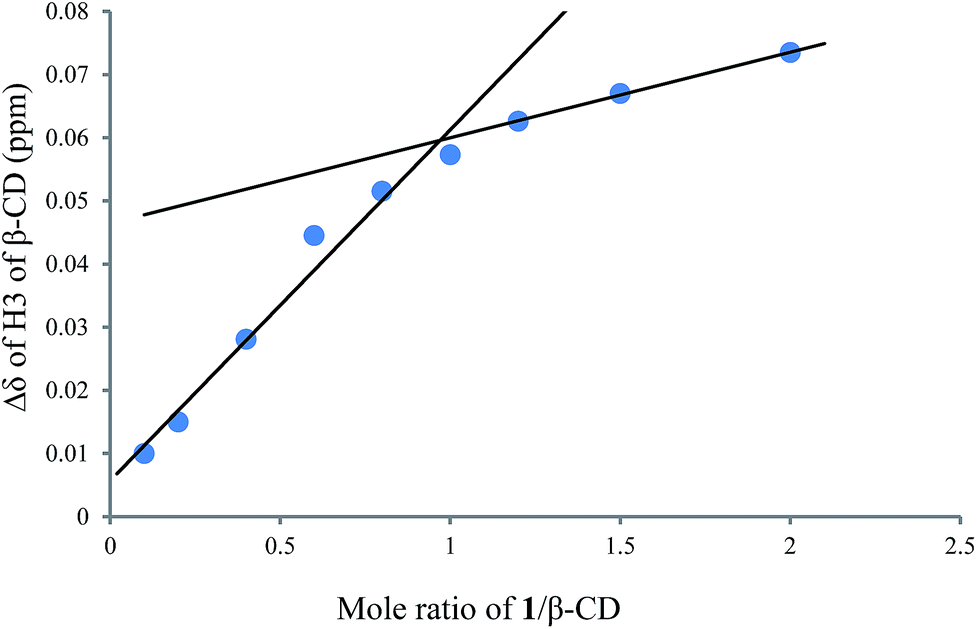
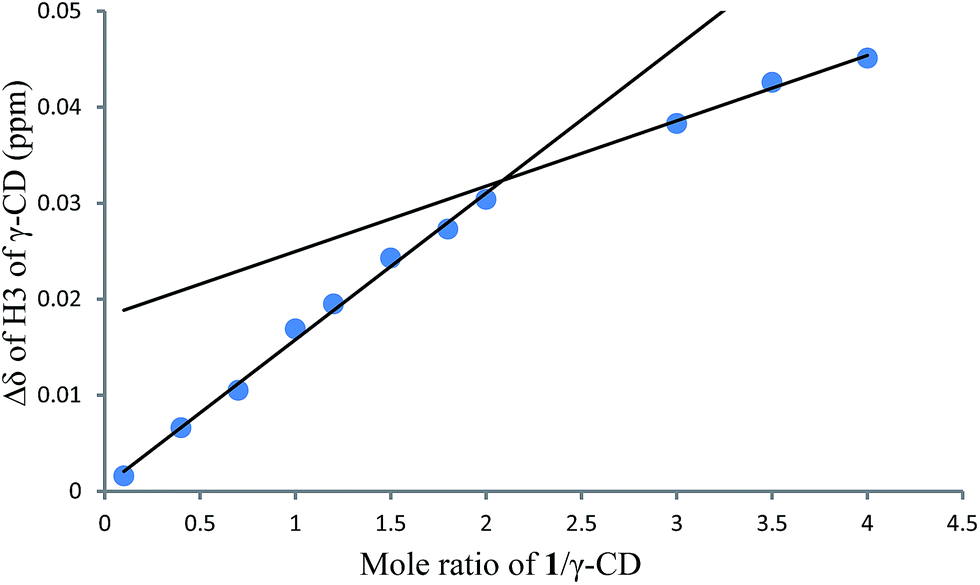
1H NMR titration curves were obtained by plotting the change in the chemical shift of H3 against the ratio of 1 to β-CD (Fig. 4) and the change in the chemical shift of H3 against the ratio of 1 to γ-CD (Fig. 5). The stoichiometry for the inclusion complex formation between β-CD and 1 was 1:1, while that of the inclusion complex formation between γ-CD and 1 was 1:2, as determined by extrapolating the curve.
 |
| | Fig. 4 1H NMR titration curve for 1 and β-CD. | |
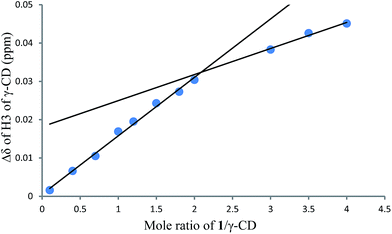 |
| | Fig. 5 1H NMR titration curve for 1 and γ-CD. | |
The effect of CDs on selective C–H bond hydroxylation of mixtures of trans/cis-cycloalkane isomers
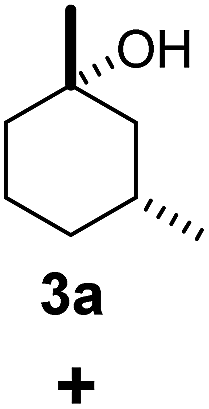
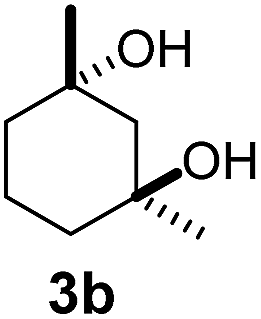
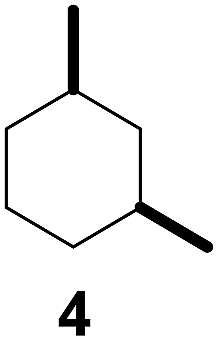
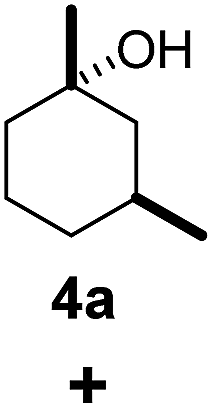
As both β-CD and γ-CD enhanced the selective C–H bond hydroxylation reactions of trans/cis-1,N-dimethylcyclohexanes in water, it is of interest to study the C–H bond hydroxylation selectivity in a mixture of trans and cis isomers. In this regard, a 1:1 mixture of trans- and cis-1,3-dimethylcyclohexane (3 and 4) was chosen to study the effect of CDs on the selectivity of C–H bond hydroxylation of the trans and cis isomers.
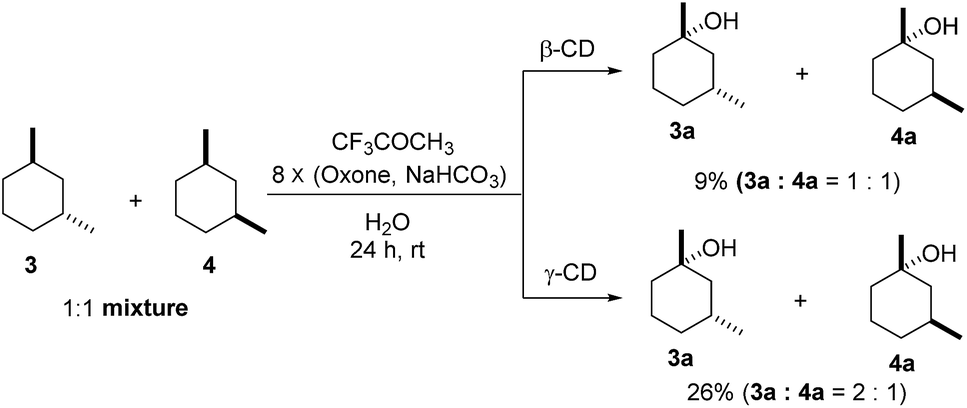
As shown in Scheme 1, when a 1:1 mixture of trans- and cis-1,3-dimethylcyclohexane (3 and 4) was subjected to the oxidation conditions with β-CD as the supramolecular host, the ratio of the corresponding products 3a and 4a was found to be 1:1. This result is consistent with the outcomes of the individual oxidations of trans- and cis-1,3-dimethylcyclohexane (3 and 4) under supramolecular control by β-CD. Using γ-CD as the supramolecular host, a 2:1 ratio of 3a and 4a was obtained. These findings indicate that γ-CD could provide enhanced isomer-selective C–H bond oxidation for 1,3-dimethylcyclohexane (3 and 4).
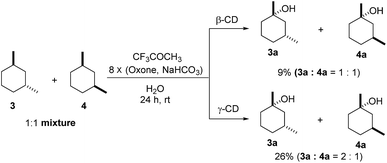 |
| | Scheme 1 Hydroxylation of a 1:1 mixture of 3 and 4 with β-CD and γ-CD as the supramolecular hosts. Reactions were conducted with a 1:1 mixture of 3 (0.5 mmol) and 4 (0.5 mmol), 1,1,1-trifluoroacetone (1.0 mmol), and β-CD or γ-CD (1.1 mmol) in water (50 mL) with Oxone (2.5 mmol × 8) and NaHCO3 (7.75 mmol × 8). | |
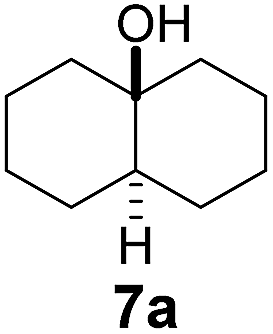
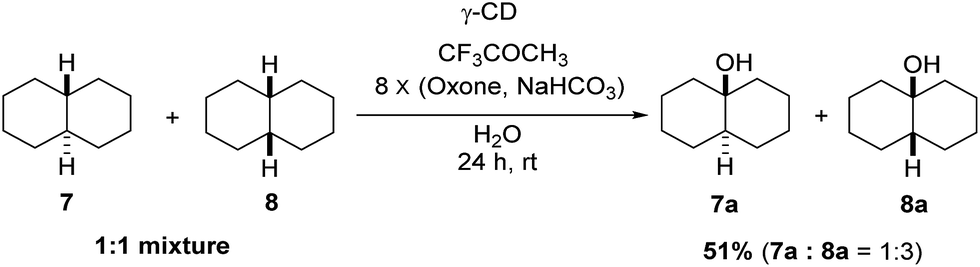
Using γ-CD as the supramolecular host, a 1:1 mixture of trans- and cis-decahydronaphthalene (7 and 8) was oxidized giving the corresponding products 7a and 8a in a 1:3 ratio (Scheme 2). In contrast, the ratio of 7a and 8a was 1:1.4, when 7 and 8 were individually oxidized in water with γ-CD as the supramolecular host.
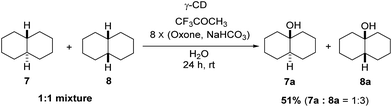 |
| | Scheme 2 Hydroxylation of a 1:1 mixture of 7 and 8 with γ-CD as the supramolecular host. Reactions were conducted with a 1:1 mixture of 7 (0.5 mmol) and 8 (0.5 mmol), 1,1,1-trifluoroacetone (1.0 mmol), and γ-CD (1.1 mmol) in water (50 mL) with Oxone (2.5 mmol × 8) and NaHCO3 (7.75 mmol × 8). | |
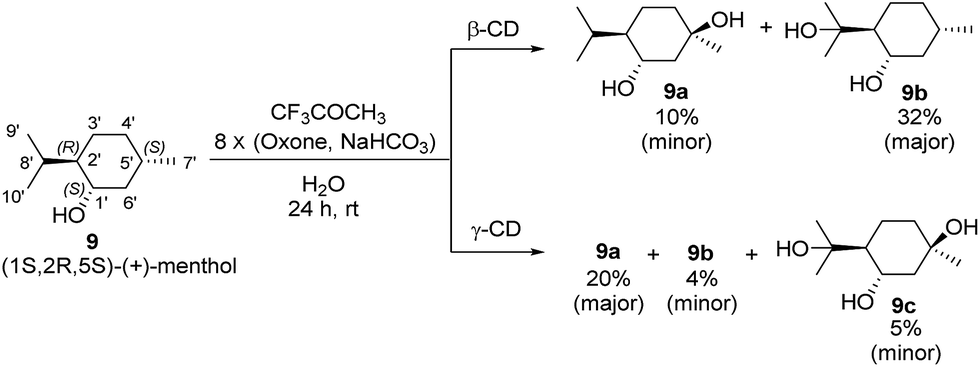
The effect of CDs on site-selective C–H bond hydroxylation of (1S,2R,5S)-(+)-menthol (9)
Menthol is an easily accessible chiral monoterpene possessing a cyclohexane skeleton with different types of C–H bonds that is an ideal substrate for studying C–H bond hydroxylation. To investigate the effects of β-CD and γ-CD on site-selective C–H bond hydroxylation, (1R,2R,5S)-(+)-menthol (9), which bears three tertiary C–H bonds at C2′, C5′ and C8′, was chosen as the substrate.
The reaction was carried out at room temperature by stirring 9 (1.0 mmol) with β-CD or γ-CD (1.1 mmol) and 1,1,1-trifluoroacetone (1.0 mmol) in water (50 mL) for 1 h, and then adding 8 portions of a mixture of Oxone (2.5 mmol × 8) and NaHCO3 (7.75 mmol × 8) within 7 h (one portion was added per hour). After stirring for an additional 16 h, products 9a and 9b were isolated by flash column chromatography (Scheme 3).
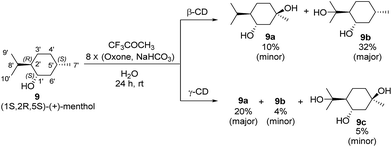 |
| | Scheme 3 Hydroxylation of (+)-menthol (9) with β-CD and γ-CD as supramolecular hosts. Reactions were conducted with 9 (1.0 mmol), 1,1,1-trifluoroacetone (1.0 mmol), and β-CD or γ-CD (1.1 mmol) in water (50 mL) with Oxone (2.5 mmol × 8) and NaHCO3 (7.75 mmol × 8). The yields were calculated based on the conversion of 9. | |
As shown in Scheme 3, using β-CD as the supramolecular host, the C5′-hydroxylated product 9a (10% yield; minor) and the C8′-hydroxylated product 9b (32% yield; major) were obtained with 20% conversion. Using γ-CD as the supramolecular host, significantly increased conversion (50%) was achieved, and the product distribution was reversed with respect to 9a (20% yield; major) and 9b (4% yield; minor). In addition, a dihydroxylation product 9c (5% yield) was obtained when γ-CD was used.
1H NMR titration experiments for binding of 9 with β-CD and γ-CD
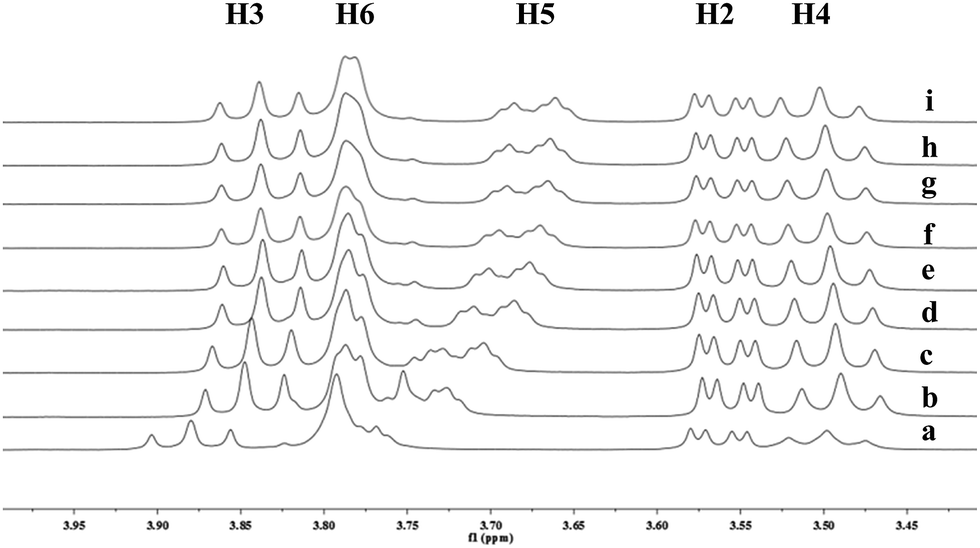
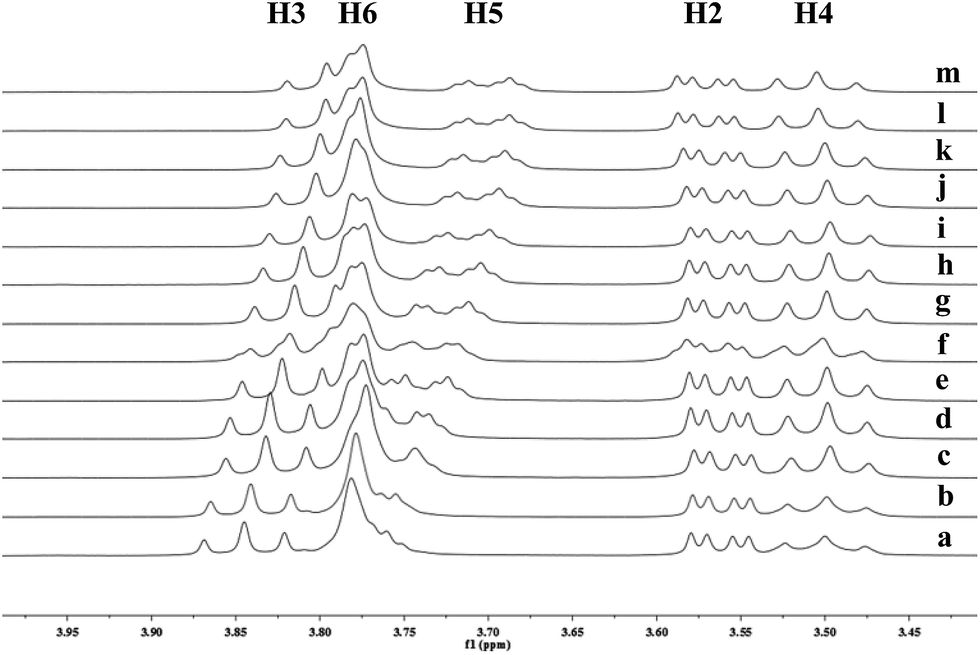
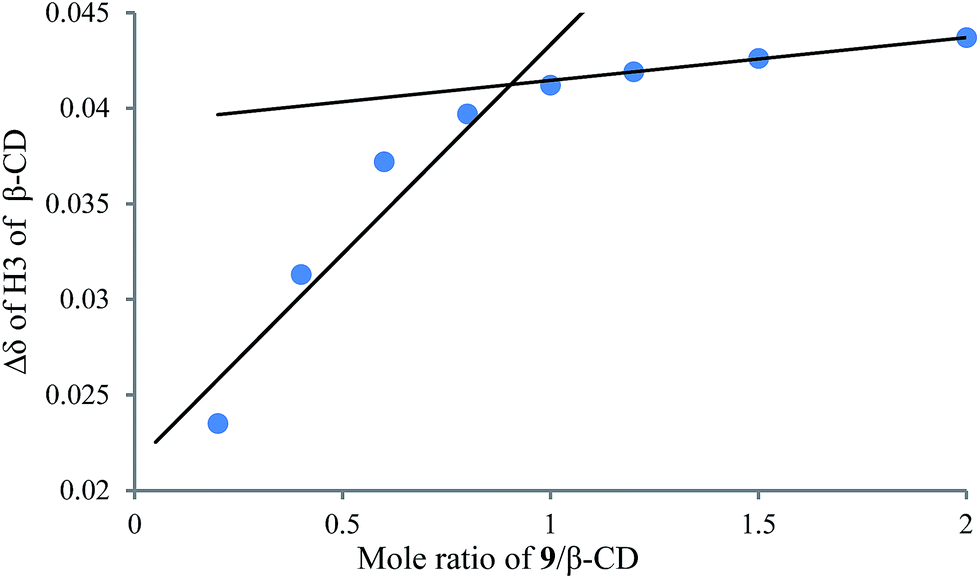
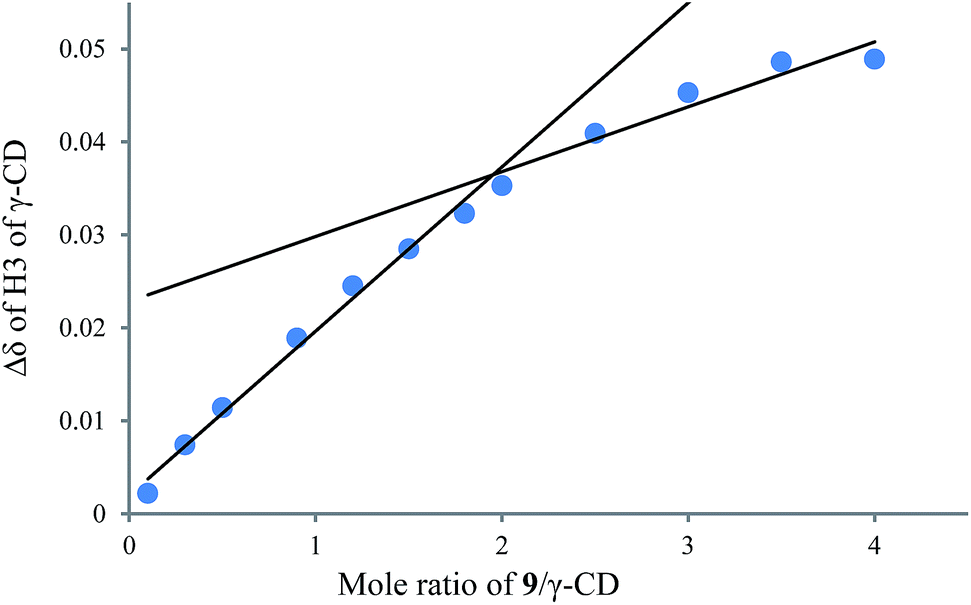
To understand the different product distributions in the hydroxylation of 9 using β-CD and γ-CD as the supramolecular hosts, we studied inclusion complexation between 9 and β-CD or γ-CD by 1H NMR titration. The peaks of H3 and H5 of the CDs showed significant upfield shifts when the amount of 9 increased in the aqueous solution of CDs (Fig. 6 and 7). Two 1H NMR titration curves were plotted based on the change in the chemical shift of H3 against the ratio of 9/β-CD (Fig. 8) and 9/γ-CD (Fig. 9). The stoichiometry of the inclusion complex between β-CD and 9 was found to be 1:1 and that between γ-CD and 9 was 1:2, as determined by extrapolating the curves.
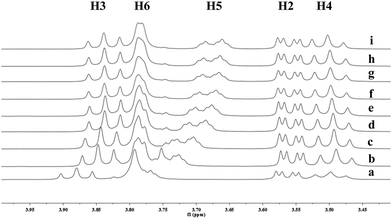 |
| | Fig. 6 Partial 1H NMR spectra of a mixture of 9 and β-CD in D2O (signals of β-CD). Ratios of 9/β-CD: (a) 0:10, (b) 2:10, (c) 4:10, (d) 6:10, (e) 8:10, (f) 1:1, (g) 12:10, (h) 15:10, (i) 2:1. | |
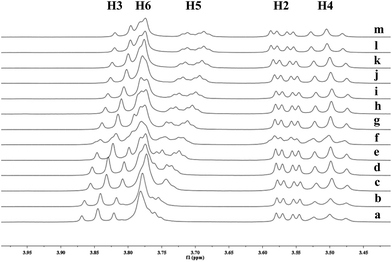 |
| | Fig. 7 Partial 1H NMR spectra of a mixture of 9 and γ-CD in D2O (signals of γ-CD). Ratios of 9/γ-CD: (a) 0:10, (b) 1:10, (c) 4:10, (d) 7:10, (e) 1:1, (f) 12:10, (g) 15:10, (h) 18:10, (i) 2:1, (j) 2.5:1, (k) 3:1, (l) 3.5:1, (m) 4:1. | |
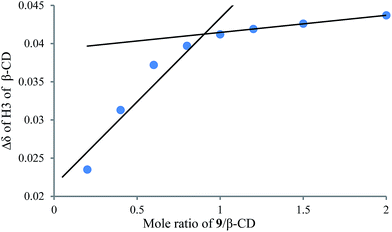 |
| | Fig. 8 1H NMR titration curve for 9 and β-CD. | |
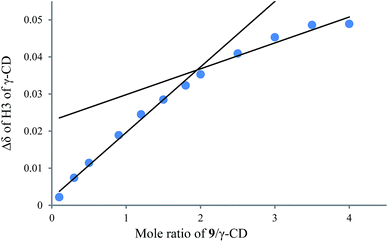 |
| | Fig. 9 1H NMR titration curve for 9 and γ-CD. | |
Studies on the binding geometry of (+)-menthol (9) with β-CD and γ-CD through 2D ROESY experiments
To further investigate the effect of β-CD and γ-CD on site-selective C–H bond oxidation of (+)-menthol (9), 2D ROESY experiments were conducted to determine the binding geometry of 9 with β-CD and γ-CD.
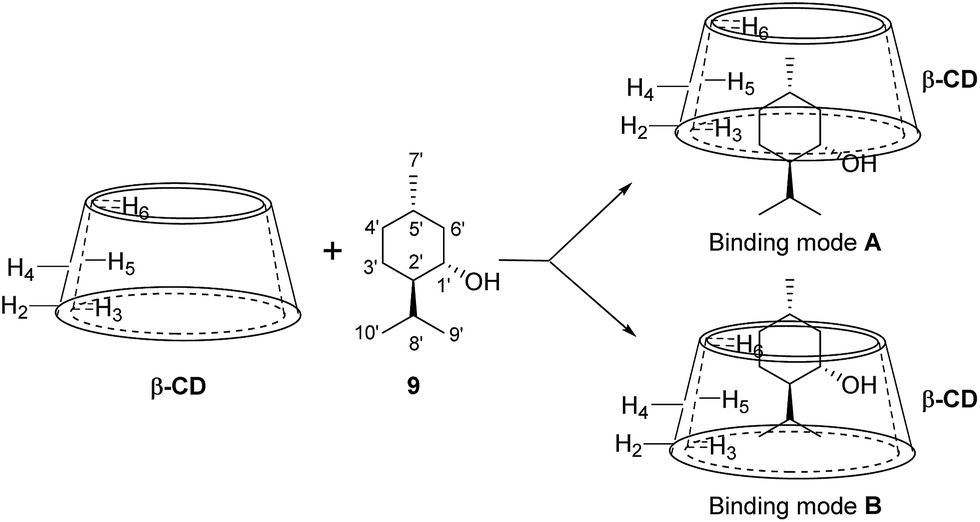
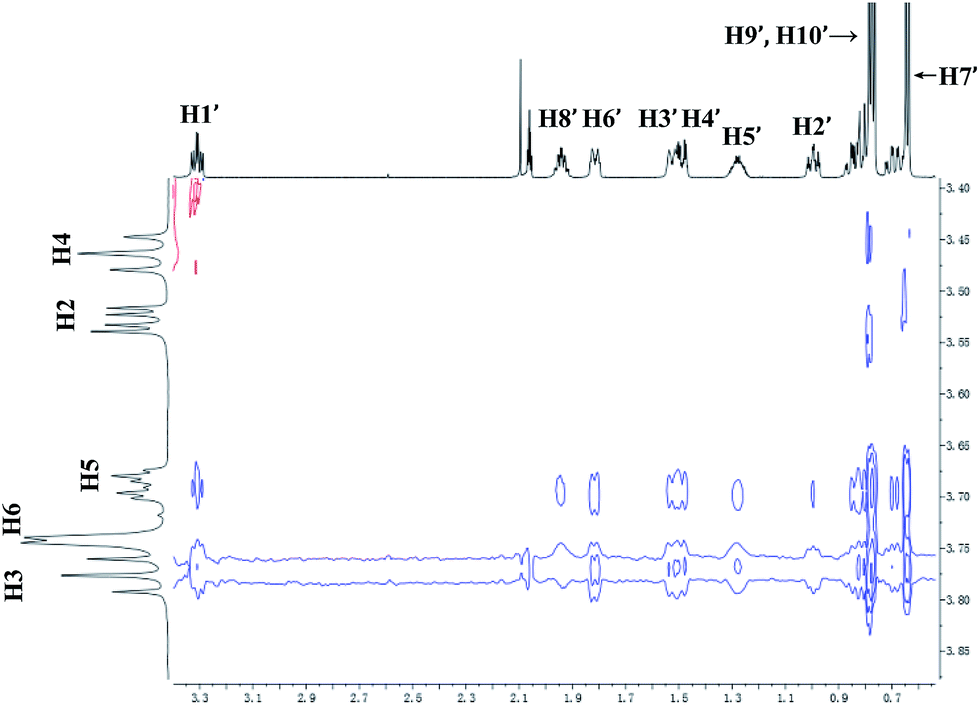
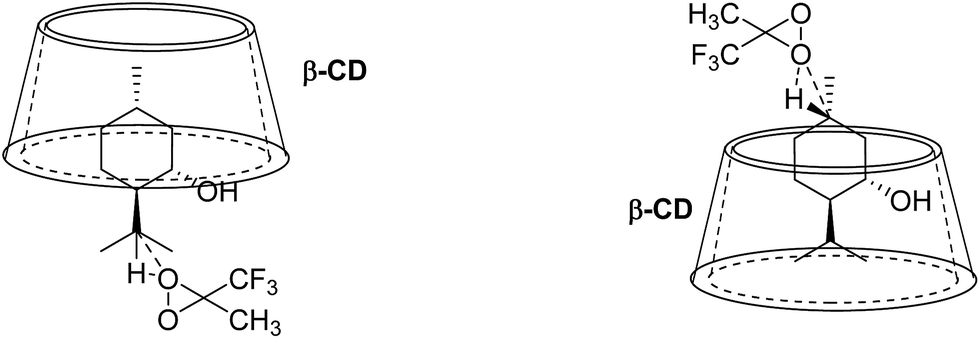
The 2D ROESY spectrum for the binding between 9 and β-CD showed that 9 has NOE correlation signals with hydrogens H3, H5 and H6 of β-CD (Fig. 10), which suggested that 9 was located inside the cavity of β-CD. As shown in Fig. 10, the 2D ROESY spectrum regarding the binding between 9 and β-CD showed that H9′ and H10′ of 9 have strong NOE correlation signals with H3 and H5 of β-CD and weak NOE correlation signals with H6 of β-CD. Moreover, strong NOE correlation signals of H7′ of 9 with H5 of β-CD and weak NOE correlation signals of H7′ of 9 with H6 of β-CD also appeared on the ROESY spectrum. These NOE correlation signals and the 1:1 stoichiometry of the complexation between 9 and β-CD suggested that β-CD most likely binds to 9 in two possible binding models: (1) the terminal methyl (H7′) of 9 located deep in the β-CD cavity with the terminal isopropyl (H9′, H10′) of 9 outside of the cavity and close to the secondary OH-face (Fig. 11, binding mode A); and (2) the terminal isopropyl (H9′, H10′) of 9 located deep in the β-CD cavity with the terminal methyl (H7′) of 9 outside of the cavity and close to the primary OH-face (Fig. 11, binding mode B). Further analysis of the strength of the NOE correlation signals indicated that β-CD prefers to bind to 9 in binding mode A rather than binding mode B.
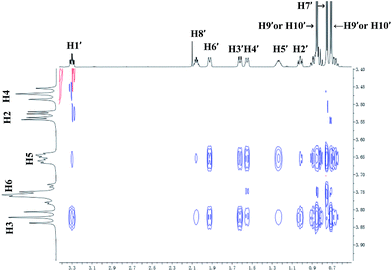 |
| | Fig. 10 Partial contour plot of the 600 MHz 2D ROESY spectrum for binding of (+)-menthol (9) to β-CD in D2O. | |
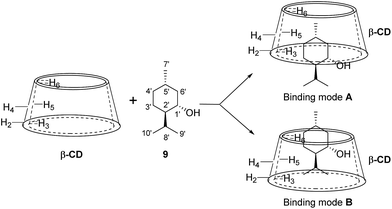 |
| | Fig. 11 Proposed binding geometry for the inclusion of (+)-menthol (9) in β-CD. | |
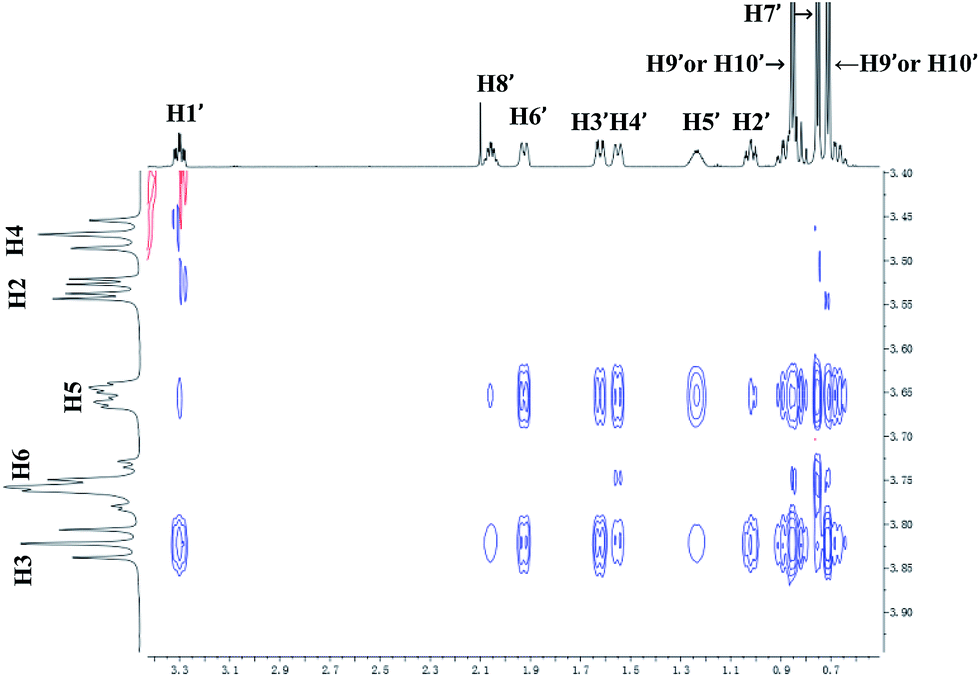
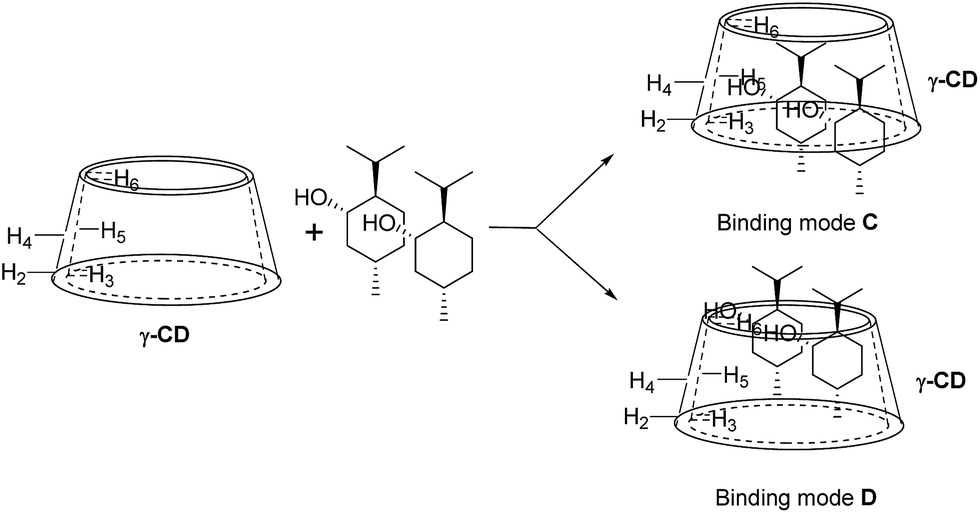
For 9 and γ-CD, the 2D ROESY spectrum (Fig. 12) showed that H7′ of 9 has NOE correlation signals with H1′, H8′ of 9 and H5′ of 9 have NOE correlation signals with H9′, H10′ of 9. The 2D ROESY spectrum also showed that H3, H5, and H6 of γ-CD have strong NOE correlation signals with H7′, H9′, and H10′ of 9; H6 of γ-CD has NOE correlation signals with H2′, H3′, H5′, H7′, H9′, and H10′ of 9; and H5 of γ-CD has NOE correlation signals with H4′, H5′, H6′, and H8′ of 9. Given the 2:1 stoichiometry of the complexation between 9 and γ-CD, the possible binding modes involve two molecules of 9 encased in the cavity of γ-CD. Therefore, two possible binding modes between γ-CD and 9 were proposed: (1) the terminal isopropyl (H9′, H10′) of 9 inserted deep into the γ-CD cavity with the terminal methyl (H7′) of 9 outside of the cavity and close to the secondary OH-face (Fig. 13, binding mode C); and (2) the terminal methyl (H7′) of 9 inserted deep into the γ-CD cavity with the terminal isopropyl (H9′, H10′) of 9 outside of the cavity and close to the primary OH-face (Fig. 13, binding mode D). The experimental results indicated that binding mode C is the favored binding geometry.
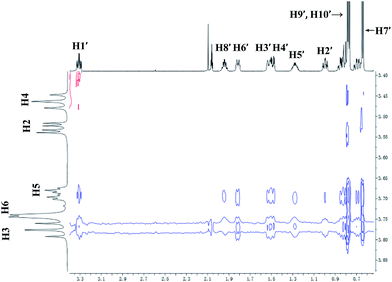 |
| | Fig. 12 Partial contour plot of the 600 MHz 2D ROESY spectrum for binding of (+)-menthol (9) to γ-CD in D2O. | |
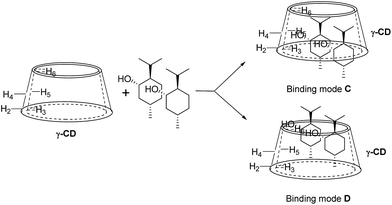 |
| | Fig. 13 Proposed binding geometry for the inclusion of (+)-menthol (9) in γ-CD. | |
Schematic diagrams for site-selective C–H bond oxidation of 9 with β-CD and γ-CD as supramolecular hosts
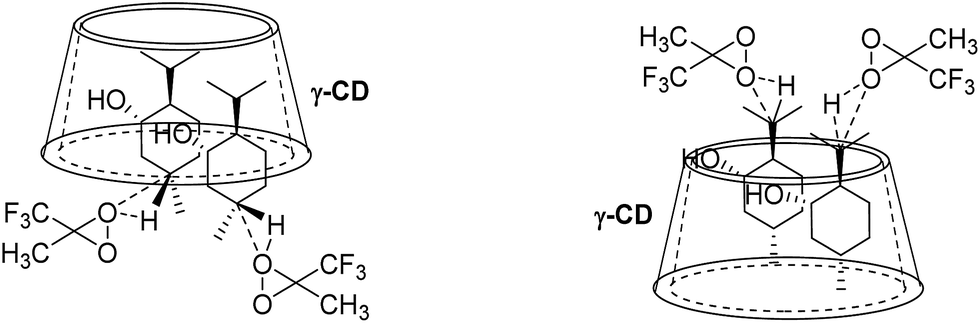
Based on the binding geometries of 9 to β-CD (Fig. 11), schematic diagrams for how β-CD affects the site-selectivity of C–H bond hydroxylation were proposed in Fig. 14. The preferred binding geometry of 9 to β-CD, in which the terminal methyl of 9 is located deep in the cavity of β-CD and the terminal isopropyl of 9 is exposed outside of the cavity, renders the C–H bond at position C8′ more easily subject to hydroxylation by dioxirane than that at position C5′. This leads to site-selectivity in the C–H bond hydroxylation. The proposed schematic diagrams are reasonably consistent with the experimental results (see Scheme 3, 9a/9b = 1:3) on the site-selectivity of C–H bond hydroxylation of 9 when using β-CD as the supramolecular host.
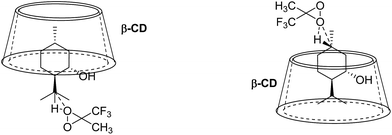 |
| | Fig. 14 Schematic diagrams for site-selective C–H bond oxidation of 9 in β-CD. | |
In contrast, for the binding of 9 to γ-CD, the preferred binding geometry is such that the C–H bond at position C5′ is more easily approached by dioxirane than that at position C8′, which gives 9a as the major product (Fig. 15). Thus, the binding geometries of 9 to γ-CD and steric hindrance lead to high site-selectivity in the C–H bond hydroxylation of 9 (9a:9b = 5:1).
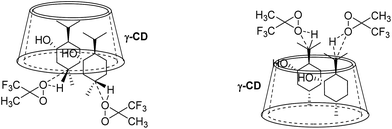 |
| | Fig. 15 Schematic diagrams for site-selective C–H bond oxidation of 9 in γ-CD. | |
Conclusions
In summary, we have investigated the hydroxylation of various cyclohexanes with dioxirane generated in situ through supramolecular control using β-CD and γ-CD as the hosts. The results indicated that CDs could act as reaction vessels for hydroxylation reactions performed in water without the use of organic solvents. The stoichiometries of inclusion complexation between β-CD or γ-CD and cyclohexanes were determined by 1H NMR titration. The effect of β-CD and γ-CD as supramolecular hosts on chemo-selective C–H bond hydroxylation of trans and cis isomers of disubstituted cyclohexanes was investigated. Site-selective C–H bond hydroxylation of (+)-menthol (9) was achieved through inclusion complex formation with β-CD and γ-CD in water.
Acknowledgements
The authors gratefully acknowledged the financial support by the National Natural Science Foundation of China (21272198), The Hong Kong Polytechnic University (4-BCA4) and the State Key Laboratory of Chirosciences.
Notes and references
- J. F. Hartwig, Acc. Chem. Res., 2017, 50, 549–555 CrossRef CAS.
- V. C. C. Wang, S. Maji, P. P. Y. Chen, H. K. Lee, S. S. F. Yu and S. I. Chan, Chem. Rev., 2017 DOI:10.1021/acs.chemrev.6b00624.
- T. Newhouse and P. S. Baran, Angew. Chem., Int. Ed., 2011, 50, 3362–3374 CrossRef CAS PubMed.
- S. R. Neufeldt and M. S. Sanford, Acc. Chem. Res., 2012, 45, 936–946 CrossRef CAS PubMed.
- T. Brückl, R. D. Baxter, Y. Ishihara and P. S. Baran, Acc. Chem. Res., 2012, 45, 826–839 CrossRef PubMed.
- W. R. Gutekunst and P. S. Baran, Chem. Soc. Rev., 2011, 40, 1976–1991 RSC.
- Y. Qin, L. Zhu and S. Luo, Chem. Rev., 2017 DOI:10.1021/acs.chemrev.6b00657.
- M. C. Feiters, A. E. Rowan and R. J. M. Nolte, Chem. Soc. Rev., 2000, 29, 375–384 RSC.
- B. H. Brodsky and J. Du Bois, J. Am. Chem. Soc., 2005, 127, 15391–15393 CrossRef CAS PubMed.
- M. Milan, M. Bietti and M. Costas, ACS Cent. Sci., 2017, 3, 196–204 CrossRef CAS PubMed.
- X. Li, X. Che, G.-H. Chen, J. Zhang, J.-L. Yan, Y.-F. Zhang, L.-S. Zhang, C.-P. Hsu, Y. Q. Gao and Z.-J. Shi, Org. Lett., 2016, 18, 1234–1237 CrossRef CAS PubMed.
- F. Kakiuchi and N. Chatani, Adv. Synth. Catal., 2003, 345, 1077–1101 CrossRef CAS.
- H. M. Davies, Angew. Chem., Int. Ed., 2006, 45, 6422–6425 CrossRef CAS PubMed.
- J. A. Labinger and J. E. Bercaw, Nature, 2002, 417, 507–514 CrossRef CAS PubMed.
- M. C. White, Science, 2012, 335, 807–809 CrossRef CAS PubMed.
- M. S. Chen and M. C. White, Science, 2010, 327, 566–571 CrossRef CAS PubMed.
- M. A. Bigi, S. A. Reed and M. C. White, Nat. Chem., 2011, 3, 216–222 CrossRef CAS PubMed.
- E. McNeill and J. Du Bois, J. Am. Chem. Soc., 2010, 132, 10202–10204 CrossRef CAS PubMed.
- K. Zhang, B. M. Shafer, M. D. Demars, H. A. Stern and R. Fasan, J. Am. Chem. Soc., 2012, 134, 18695–18704 CrossRef CAS PubMed.
- S. Das, C. D. Incarvito, R. H. Crabtree and G. W. Brudvig, Science, 2006, 312, 1941–1943 CrossRef CAS PubMed.
- J. Yang, B. Gabriele, S. Belvedere, Y. Huang and R. Breslow, J. Org. Chem., 2002, 67, 5057–5067 CrossRef CAS PubMed.
- B. R. Cook, T. J. Reinert and K. S. Suslick, J. Am. Chem. Soc., 1986, 108, 7281–7286 CrossRef CAS.
- L. Que Jr and W. B. Tolman, Nature, 2008, 455, 333–340 CrossRef PubMed.
- T. Gensch, M. N. Hopkinson, F. Glorius and J. Wencel-Delord, Chem. Soc. Rev., 2016, 45, 2900–2936 RSC.
- B. D. Dangel, J. A. Johnson and D. Sames, J. Am. Chem. Soc., 2001, 123, 8149–8150 CrossRef CAS PubMed.
- A. Mendoza, Y. Ishihara and P. S. Baran, Nat. Chem., 2011, 4, 21–25 CrossRef PubMed.
- T. J. Maimone and P. S. Baran, Nat. Chem. Biol., 2007, 3, 396–407 CrossRef CAS PubMed.
- K. Chen and P. S. Baran, Nature, 2009, 459, 824–828 CrossRef CAS PubMed.
- J. R. Hanson, Nat. Prod. Rep., 2010, 27, 887–899 RSC.
- J.-N. Rebilly, B. Colasson, O. Bistri, D. Over and O. Reinaud, Chem. Soc. Rev., 2015, 44, 467–489 RSC.
- Y. Murakami, J.-I. Kikuchi, Y. Hisaeda and O. Hayashida, Chem. Rev., 1996, 96, 721–758 CrossRef CAS PubMed.
- D. H. Leung, D. Fiedler, R. G. Bergman and K. N. Raymond, Angew. Chem., Int. Ed., 2004, 43, 963–966 CrossRef CAS PubMed.
- M. Yoshizawa, J. K. Klosterman and M. Fujita, Angew. Chem., Int. Ed., 2009, 48, 3418–3438 CrossRef CAS PubMed.
- R. Breslow and S. D. Dong, Chem. Rev., 1998, 98, 1997–2012 CrossRef CAS PubMed.
- K. Takahashi, Chem. Rev., 1998, 98, 2013–2034 CrossRef CAS PubMed.
- L. Marinescu and M. Bols, Curr. Org. Synth., 2010, 14, 1380–1398 CAS.
- W.-K. Chan, W.-Y. Yu, C.-M. Che and M.-K. Wong, J. Org. Chem., 2003, 68, 6576–6582 CrossRef CAS PubMed.
- Y. S. Fung, S. C. Yan and M. K. Wong, Org. Biomol. Chem., 2012, 10, 3122–3130 CAS.
- R. Breslow, X. Zhang, R. Xu, M. Maletic and R. Merger, J. Am. Chem. Soc., 1996, 118, 11678–11679 CrossRef CAS.
- R. Breslow, X. Zhang and Y. Huang, J. Am. Chem. Soc., 1997, 119, 4535–4536 CrossRef CAS.
- C. Rousseau, B. Christensen, T. E. Petersen and M. Bols, Org. Biomol. Chem., 2004, 2, 3476–3482 CAS.
- L. Marinescu, M. Mølbach, C. Rousseau and M. Bols, J. Am. Chem. Soc., 2005, 127, 17578–17579 CrossRef CAS PubMed.
- T. H. Fenger, L. G. Marinescu and M. Bols, Eur. J. Org. Chem., 2011, 2011, 2339–2345 CrossRef.
- T. H. Fenger, L. G. Marinescu and M. Bols, Org. Biomol. Chem., 2009, 7, 933–943 CAS.
- J. Bjerre, T. Hauch Fenger, L. G. Marinescu and M. Bols, Eur. J. Org. Chem., 2007, 2007, 704–710 CrossRef.
- C. I. Herrerías, X. Yao, Z. Li and C.-J. Li, Chem. Rev., 2007, 107, 2546–2562 CrossRef PubMed.
- T.-W. Hui, J.-F. Cui and M.-K. Wong, RSC Adv., 2017, 7, 14477–14480 RSC.
- W. Adam, R. Curci and J. O. Edwards, Acc. Chem. Res., 1989, 22, 205–211 CrossRef CAS.
- R. W. Murray, Chem. Rev., 1989, 89, 1187–1201 CrossRef CAS.
- S. E. Denmark and Z. Wu, Synlett, 1999, 1999, 847–859 CrossRef.
- Y. Shi, Acc. Chem. Res., 2004, 37, 488–496 CrossRef CAS PubMed.
- D. Yang, Acc. Chem. Res., 2004, 37, 497–505 CrossRef CAS PubMed.
- O. A. Wong and Y. Shi, Chem. Rev., 2008, 108, 3958–3987 CrossRef CAS PubMed.
- R. Curci, L. D'Accolti and C. Fusco, Acc. Chem. Res., 2006, 39, 1–9 CrossRef CAS PubMed.
- R. W. Murray, R. Jeyaraman and L. Mohan, J. Am. Chem. Soc., 1986, 108, 2470–2472 CrossRef CAS PubMed.
- G. Asensio, M. E. Gonzalez-Nunez, C. B. Bernardini, R. Mello and W. Adam, J. Am. Chem. Soc., 1993, 115, 7250–7253 CrossRef CAS.
- D. Yang, M.-K. Wong, X.-C. Wang and Y.-C. Tang, J. Am. Chem. Soc., 1998, 120, 6611–6612 CrossRef CAS.
- M.-K. Wong, N.-W. Chung, L. He and D. Yang, J. Am. Chem. Soc., 2003, 125, 158–162 CrossRef CAS PubMed.
- M.-K. Wong, N.-W. Chung, L. He, X.-C. Wang, Z. Yan, Y.-C. Tang and D. Yang, J. Org. Chem., 2003, 68, 6321–6328 CrossRef CAS PubMed.
- C. J. Abelt and J. M. Pleier, J. Org. Chem., 1988, 53, 2159–2162 CrossRef CAS.
- P. W. N. M. van Leeuwen, Supramolecular Catalysis, Wiley-VCH Verlag GmbH & Co. KGaA, 2008 Search PubMed.
- J. Kang and J. Rebek Jr, Nature, 1997, 385, 50–52 CrossRef CAS PubMed.
- D. M. Vriezema, M. Comellas Aragonés, J. A. A. W. Elemans, J. J. L. M. Cornelissen, A. E. Rowan and R. J. M. Nolte, Chem. Rev., 2005, 105, 1445–1489 CrossRef CAS PubMed.
- J. Lehn and C. Sirlin, J. Chem. Soc., Chem. Commun., 1978, 949–951 RSC.
- L. Xu, G. Fang and S. Li, RSC Adv., 2017, 7, 14046–14052 RSC.
- M. Zubiaur and C. Jaime, J. Org. Chem., 2000, 65, 8139–8145 CrossRef CAS PubMed.
- H.-J. Schneider, F. Hacket, V. Rüdiger and H. Ikeda, Chem. Rev., 1998, 98, 1755–1786 CrossRef CAS PubMed.
- D. Salvatierra, C. Jaime, A. Virgili and F. Sánchez-Ferrando, J. Org. Chem., 1996, 61, 9578–9581 CrossRef CAS.
- A. Botsi, K. Yannakopoulou, B. Perly and E. Hadjoudis, J. Org. Chem., 1995, 60, 4017–4023 CrossRef CAS.
- D. Salvatierra, X. Sánchez-Ruiz, R. Garduño, E. Cervelló, C. Jaime, A. Virgili and F. Sánchez-Ferrando, Tetrahedron, 2000, 56, 3035–3041 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra03930a |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab
*ab