
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAcylphloroglucinol and tocotrienol derivatives from the fruits of Garcinia multiflora†
Hui Liu‡
a,
Fei Gan‡a,
Shan Jinb,
Jun Lib,
Yu Chen*a and
Guangzhong Yang *bc
*bc
aCollege of Chemistry and Material Sciences, South-Central University for Nationalities, Wuhan 430074, P. R. China. E-mail: chenyuwh888@126.com; Fax: +86 27 6784 1196; Tel: +86 27 6784 1196
bSchool of Pharmaceutical Sciences, South-Central University for Nationalities, Wuhan 430074, P. R. China. E-mail: yanggz888@126.com
cNational Demonstration Center for Experimental Ethnopharmacology Education, South-Central University for Nationalities, Wuhan 430074, P. R. China
First published on 5th June 2017
Abstract
A new regioisomeric pair of garcimultine A (1) and B (2), one new tocotrienol derivative garcimultine C (8), together with six known compounds were isolated from the fruits of Garcinia multiflora. Their structures were determined by extensive spectroscopic techniques including NMR spectroscopy and mass spectrometry. The regioisomeric garcimultine A (1) and B (2) were not separated due to the rapid equilibration between the two isomeric forms. The compounds isolated were evaluated for their cytotoxicity against four cancer cell lines (HepG2, A549, SGC7901, MCF-7). All isolated compounds showed moderate cytotoxicity on four human tumor cell lines with IC50 values ranging from 4.06 to 35.61 μM.
1. Introduction
Polycyclic polyprenylated acylphloroglucinols (PPAPs) with a highly oxygenated bicyclo[3.3.1]nonane-2,4,9-trione or bicyclo[3.2.1]octane-2,4,8-trione core substituted with one or more prenyl or geranyl side chains have only been isolated from plants of the family Guttiferae so far. PPAPs have received considerable attention from natural products chemistry, synthetic chemistry, and pharmacology due to structural diversity and fascinating biological profiles.1–3 Garcinia multiflora Champ. is a traditional Zhuang medicine widely distributed in the south of China. The bark of this plant is used as a traditional medicine to stop bleeding, diminishing inflammation and relieving pain. The fruits are the only edible plant part. Fruits are picked during the period from July to December, which means that over a large part of the year fresh fruits are available, not only for their nutritional value (consisting of several vitamins, proteins, and minerals), but may also provide pharmacologically active compounds.4 Previous phytochemical investigations on the fruits led to isolation of seven new PPAPs with anti-inflammatory activity.5–7 Recently, Garmultin A is a polycyclic caged acylphloroglucinol isolated from the leaves and twigs of G. multiflora, which been highlighted in Hot off the Press articles in Natural Product Reports.8,9In the continuing search for antitumor natural products,10,11 a new regioisomeric pair of garcimultine A (1) and B (2), one new tocotrienol derivative garcimultine C (8), together with six known compounds were isolated from the fruits of G. multiflora. Herein, we report the isolation, structural elucidation, and biological activities of these isolated compounds.
2. Materials and methods
2.1 General experimental procedures
Optical rotations were determined in MeOH on a P-1020 digital polarimeter. (JASCO Co., Tokyo, Japan). UV spectra were obtained on a 2401PC spectrophotometer (Shimadzu Co., Tokyo, Japan). 1D and 2D NMR spectra were recorded on a Bruker AVANCEIII-500 MHz and a Bruker AV-400 MHz spectrometer (Bruker, Ettlingen, Germany) in DMSO-d6, C5D5N, acetone-d6 using tetramethylsilane (TMS) as an internal reference standard. Chemical shifts (δ) have been expressed in ppm and the coupling constants (J) have been given in Hz. High-resolution electrospray mass spectroscopy was performed on an Agilent G6230 TOF mass spectrometer (HR-ESI-MS) (Agilent Technologies Co. Ltd., California, America) and A waters Autospec Premier 776 mass spectrometer (HR-EI-MS) (Waters Technologies Co. Ltd., Massachusetts, America). High-performance liquid chromatography (HPLC) was conducted on an Ultimate 3000 HPLC system (Dionex Co., Sunnyvale, CA, USA) equipped with an Ultimate 3000 pump and Ultimate 3000 Variable Wavelength detector, as well as a semi-preparative YMC-Pack ODS-A column (250 × 10 mm, 5 μm) and a preparative YMC-Pack ODS-A column (250 × 20 mm, 5 μm) from YMC Co., Ltd (Kyoto, Japan), Column chromatography (CC) was conducted over silica gel (200–300 mesh and 300–400 mesh, Qingdao Haiyang Chemical Industry Co., Ltd., Qingdao, China). Chromatographic grade acetonitrile was purchased from Chang Tech Enterprise Co., Ltd (Taiwan, China). The cell lines were purchased from the cell bank of Chinese Academy of Sciences (Shanghai, China). 3-(4,5-Dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) was purchased from Biosharp Co. Ltd. (Hefei, Anhui, China). Doxorubicin hydrochloride was purchased from Aladdin Chemical Co. Ltd. (Shanghai, China). Dulbecco modified Eagle medium (DMEM), phosphate buffer saline (PBS) and penicillin–streptomycin solution were purchased from Hyclone Laboratories, GE healthcare life science (Logan, UT, USA). Fetal bovine serum (FBS) was purchased from Gibco, Life technologies (Grand Island, NY, USA). Reagent grade dimethyl sulfoxide (DMSO) was purchased from Vetec, Sigma Chemical Co. (St. Louis, MO, USA). The absorbance was read on a Multiskan GO microplate reader (Thermo Fisher Scientific Inc. Waltham, MA, USA).2.2 Plant material
The fruits of G. multiflora were purchased from Nanning, Guangxi Zhuang Autonomous Region, P. R. China and identified by Prof. Hongli Teng, Guangxi Zhuang medicine international hospital. The voucher specimen (2014091201) was deposited in the herbarium of School of Pharmaceutical Sciences, South Central University for Nationalities.2.3 Extraction and isolation
The dried fruits of G. multiflora Champ (5.2 kg) were powdered and extracted with 95% EtOH at room temperature for three times (each time for 24 h) to obtain EtOH extract 2.21 kg, and then successively partitioned with petroleum ether (P. E.), EtOAc and n-BuOH to get P. E. extract 125 g, EtOAc extract 166 g, n-BuOH extract 80 g. The P. E. extract (125 g) was chromatographed on a silica gel column (200–300 mesh) eluted successively with P. E./acetone gradient (50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 25:1, 10:1, 7:3, 1:1, 0:1) to obtain 6 fractions (Fr. 1–Fr. 6). Fr. 1 (7.96 g) was purified by recrystallization to afford the mixture of 1 and 2 (4.93 g). Fr. 5 (24.64 g) was purified by recrystallization to afford 4 (500 mg) and then Fr. 5 (24.14 g) was subjected to octadecylsilane CC eluting with H2O–MeOH (6:4, 1:1, 4:6, 3:7, 2:8, 0:1) to obtain 10 major fractions (Fr. 5.1–Fr. 5.10). Fr. 5.8 (14.7 g) was subjected to a silica gel column (300–400 mesh) eluted successively with P. E/CH2Cl2/MeOH gradient (20:5:0.1, 10:5:0.1, 5:5:0.1, 3:7:0, 0:0:1) to obtain 8 fractions (Fr. 5.8.1–Fr. 5.8.8). Fr. 5.8.6 (2.80 g) was purified by preparative HPLC (CH3CN–H2O, 98:2) to obtain 13 fractions (Fr. 5.8.6.1–Fr. 5.8.6.13). Fr. 5.8.6.1 (39.0 mg) was purified by semipreparative HPLC (CH3CN–H2O, 79:21) to afford 5 (3.6 mg) at tR 27.0 min. Fr. 5.8.6.3 (301 mg) was purified by recrystallization to afford 3 (101 mg) and then purified by semipreparative HPLC (CH3CN–H2O, 85:15) to afford 9 (16 mg) at tR 25.1 min. Fr. 5.8.6.8 (41.5 mg) was purified by semipreparative HPLC (CH3CN–H2O, 83:17) to afford 8 (3.1 mg) at tR 30.0 min. Fr. 5.8.6.9 (30.5 mg) was purified by semipreparative HPLC (CH3CN–H2O, 88:12) to afford 7 (10.1 mg) at tR 24.0 min. Fr. 5.8.6.11 (100 mg) was purified by semipreparative HPLC (CH3CN–H2O, 83:17) to afford 6 (7.70 mg) at tR 28.4 min.
:1, 25:1, 10:1, 7:3, 1:1, 0:1) to obtain 6 fractions (Fr. 1–Fr. 6). Fr. 1 (7.96 g) was purified by recrystallization to afford the mixture of 1 and 2 (4.93 g). Fr. 5 (24.64 g) was purified by recrystallization to afford 4 (500 mg) and then Fr. 5 (24.14 g) was subjected to octadecylsilane CC eluting with H2O–MeOH (6:4, 1:1, 4:6, 3:7, 2:8, 0:1) to obtain 10 major fractions (Fr. 5.1–Fr. 5.10). Fr. 5.8 (14.7 g) was subjected to a silica gel column (300–400 mesh) eluted successively with P. E/CH2Cl2/MeOH gradient (20:5:0.1, 10:5:0.1, 5:5:0.1, 3:7:0, 0:0:1) to obtain 8 fractions (Fr. 5.8.1–Fr. 5.8.8). Fr. 5.8.6 (2.80 g) was purified by preparative HPLC (CH3CN–H2O, 98:2) to obtain 13 fractions (Fr. 5.8.6.1–Fr. 5.8.6.13). Fr. 5.8.6.1 (39.0 mg) was purified by semipreparative HPLC (CH3CN–H2O, 79:21) to afford 5 (3.6 mg) at tR 27.0 min. Fr. 5.8.6.3 (301 mg) was purified by recrystallization to afford 3 (101 mg) and then purified by semipreparative HPLC (CH3CN–H2O, 85:15) to afford 9 (16 mg) at tR 25.1 min. Fr. 5.8.6.8 (41.5 mg) was purified by semipreparative HPLC (CH3CN–H2O, 83:17) to afford 8 (3.1 mg) at tR 30.0 min. Fr. 5.8.6.9 (30.5 mg) was purified by semipreparative HPLC (CH3CN–H2O, 88:12) to afford 7 (10.1 mg) at tR 24.0 min. Fr. 5.8.6.11 (100 mg) was purified by semipreparative HPLC (CH3CN–H2O, 83:17) to afford 6 (7.70 mg) at tR 28.4 min.
Garcimultine A (1) and B (2), white powder. [α]D = +79.4 (c = 0.336, MeOH); UV (MeOH) λmax nm (logε): 248 (4.30), 323 (3.79); 1H- and 13C-NMR: see Table 1. HR-EI-MS m/z: 568.3558 [M]+ (calcd for C38H48O4: 568.3553).
| No. | 1 | 2 | ||
|---|---|---|---|---|
| 1H-NMR | 13C-NMR | 1H-NMRa | 13C-NMR | |
| a The upfield 1H-NMR signals of 2 were overlapped with 1. | ||||
| 1 | 71.9 | 78.9 | ||
| 2 | 167.9 | 190.4 | ||
| 3 | 113.7 | 114.1 | ||
| 4 | 6.73 (1H, d, J = 10.0 Hz) | 115.0 | 6.61 (1H, d, J = 10.0 Hz) | 115.8 |
| 5 | 5.34 (1H, d, J = 10.0 Hz) | 125.3 | 5.48 (1H, d, J = 10.0 Hz) | 125.1 |
| 6 | 84.2 | 84.0 | ||
| 8 | 192.7 | 171.4 | ||
| 9 | 63.3 | 56.5 | ||
| 10 | 2.32 (2H, m) | 44.0 | 42.5 | |
| 11 | 1.62 (1H, m) | 49.1 | 48.9 | |
| 12 | 50.7 | 50.7 | ||
| 13 | 207.8 | 206.7 | ||
| 14 | 194.5 | 194.9 | ||
| 15 | 138.0 | 137.7 | ||
| 16 | 8.03 (1H, d, J = 8.0 Hz) | 129.5 | 7.94 (1H, d, J = 8.0 Hz) | 129.5 |
| 17 | 7.38 (1H, t, J = 8.0 Hz) | 128.9 | 7.32 (1H, t, J = 8.0 Hz) | 128.8 |
| 18 | 7.48 (1H, t, J = 8.0 Hz) | 133.1 | 7.38 (1H, t, J = 8.0 Hz) | 132.8 |
| 19 | 7.38 (1H, t, J = 8.0 Hz) | 128.9 | 7.32 (1H, t, J = 8.0 Hz) | 128.8 |
| 20 | 8.03 (1H, d, J = 8.0 Hz) | 129.5 | 7.94 (1H, d, J = 8.0 Hz) | 129.5 |
| 21 | 1.34 (3H, s) | 30.7 | 31.1 | |
| 22 | 0.58 (3H, s) | 28.7 | 29.6 | |
| 23 | 2.00 (1H, dd, J = 4.0, 14.0 Hz); 2.39 (1H, m) | 36.1 | 35.2 | |
| 24 | 2.99 (1H, m) | 44.9 | 45.3 | |
| 25 | 2.20 (1H, m); 2.27 (1H, m) | 34.0 | 34.8 | |
| 26 | 5.25 (1H, t, J = 7.0 Hz) | 126.0 | 5.30 (1H, t, J = 7.0 Hz) | 126.3 |
| 27 | 132.2 | 132.4 | ||
| 28 | 1.64 (3H, s) | 18.4 | 18.3 | |
| 29 | 1.69 (3H, s) | 26.2 | 26.2 | |
| 30 | 149.2 | 149.0 | ||
| 31 | 4.89 (1H, br s); 4.86 (1H, br s) | 113.3 | 4.93 (1H, br s); 4.72 (1H, br s) | 112.7 |
| 32 | 1.79 (3H, s) | 18.6 | 18.6 | |
| 33 | 2.45 (1H, m); 1.59 (1H, m) | 30.2 | 30.1 | |
| 34 | 5.00 (1H, t, J = 7.0 Hz) | 124.1 | 5.13 (1H, t, J = 7.0 Hz) | 124.2 |
| 35 | 133.0 | 132.4 | ||
| 36 | 1.64 (3H, s) | 18.5 | 18.5 | |
| 37 | 1.65 (3H, s) | 26.3 | 26.3 | |
| 38 | 1.70 (3H, s) | 24.1 | 23.1 | |
| 39 | 1.65 (3H, s) | 28.0 | 27.5 | |
Garcimultine C (8), white powder. [α]D = −26.1 (c = 0.216, MeOH); UV (MeOH) λmax nm (logε): 279 (3.86), 372 (3.46); 1H- and 13C-NMR: see Table 2. HR-ESI-MS (negative ion mode) m/z: 425.2713 [M − H]− (calcd for C27H37O4: 425.2692).
| No. | 1H-NMR | 13C-NMR | No. | 1H-NMR | 13C-NMR |
|---|---|---|---|---|---|
| 1 | 13 | 2.04–2.22 (2H, m) | 40.4 | ||
| 2 | 76.9 | 14 | 2.08–2.27 (2H, m) | 27.4 | |
| 3 | 1.80 (1H, m) | 31.1 | 15 | 5.28 (1H, m) | 125.1 |
| 1.87 (1H, m) | |||||
| 4 | 3.04 (2H, m) | 18.2 | 16 | 135.5 | |
| 4a | 117.2 | 17 | 2.04–2.22 (2H, m) | 40.4 | |
| 5 | 110.8 | 18 | 2.08–2.27 (2H, m) | 27.3 | |
| 6 | 7.86 (1H, s) | 111.3 | 19 | 5.22 (1H, t, J = 7.0 Hz) | 125.3 |
| 7 | 153.9 | 20 | 131.6 | ||
| 8 | 140.5 | 21 | 1.69 (3H, s) | 26.2 | |
| 8a | 153.8 | 22 | 1.60 (3H, s) | 18.1 | |
| 9 | 1.83 (1H, m) | 40.1 | 23 | 1.66 (3H, s) | 16.5 |
| 1.72 (1H, m) | |||||
| 10 | 2.25 (2H, m) | 23.0 | 24 | 1.66 (3H, s) | 16.4 |
| 11 | 5.28 (1H, m) | 125.1 | 25 | 1.35 (3H, s) | 24.0 |
| 12 | 135.6 | 26 | 10.85 (1H, s) | 188.1 |
2.4 Antiproliferative activity
According to the previously reported method,10 the antiproliferative activities against four human tumor cell lines (HepG2, A549, SGC7901, MCF-7) of isolated compounds were measured by the MTT assay. All the isolated compounds have been tested the antiproliferative activities against four human tumor cell lines (HepG2, A549, SGC7901, MCF-7) as summarized in Table 3.| Compounds | HepG2 | A549 | SGC7901 | MCF-7 |
|---|---|---|---|---|
| a Doxorubicin was used as positive control. | ||||
| Doxorubicina | 6.52 ± 0.13 | 14.03 ± 0.21 | 7.54 ± 1.11 | 4.40 ± 1.17 |
| 1 and 2 | 9.28 ± 2.36 | 8.77 ± 4.58 | 5.63 ± 1.19 | 6.92 ± 1.58 |
| 3 | 10.82 ± 0.81 | 5.70 ± 0.01 | 7.23 ± 2.81 | 21.03 ± 0.35 |
| 4 | 9.37 ± 0.28 | 4.06 ± 0.18 | 20.59 ± 0.98 | 14.44 ± 0.26 |
| 5 | 12.33 ± 4.31 | 16.78 ± 2.19 | 10.42 ± 0.57 | 18.99 ± 2.02 |
| 6 | 20.18 ± 3.58 | 16.23 ± 8.22 | 16.76 ± 0.26 | 35.61 ± 4.74 |
| 7 | 23.27 ± 4.33 | 30.58 ± 4.17 | 15.56 ± 1.39 | 32.55 ± 8.63 |
| 8 | >50 | >50 | >50 | >50 |
| 9 | 9.28 ± 1.34 | 12.47 ± 1.83 | 6.61 ± 0.12 | 14.68 ± 2.97 |
2.5 Energies of the compound conformation calculation
The 2D structures of molecule 1 and 2 were drawn in ChemDraw 13.0, and hydrogens were added using Openbabel 2.3.1. The energetics of different conformation of the molecule (1 and 2) were studied by Gaussian 09, a popular and widely used program for molecular mechanics calculations, semi-empirical quantum chemistry calculations and so on. The Gaussian calculation was performed using the following parameters: #HF/6-31G* SCF = tight Test Pop = MK opt.3. Result and discussion
3.1 Structural elucidation of isolated compounds
Garcimultine A (1) and B (2) were isolated as inseparable regioisomeric mixture and obtained as white amorphous powder. The ratio of garcimultine A (1) and B (2) is about 4:1 by HPLC analysis. Firstly, we discussed the structure elucidation of garcimultine A (1). Its molecular formula was determined as C38H48O4 by HR-EI-MS ([M]+ m/z 568.3558, calcd 568.3553), indicating 15 degrees of unsaturation. The 1H-NMR spectrum of 1 indictated the presence of an unsubstituted benzoyl group [δH 7.38 (2H, t, J = 8.0 Hz), 7.48 (1H, t, J = 8.0 Hz) and 8.03 (2H, d, J = 8.0 Hz)], a 2,2-dimethyl-2H-pyran moiety [δH 5.34 (1H, d, J = 10.0 Hz), 6.73 (1H, d, J = 10.0 Hz), 1.34 (3H, s), and 0.58 (3H, s)], two methyl signals [δH 1.65 (3H, s), 1.70 (3H, s)], a lavandulyl group and an isoprenyl group. The 13C NMR data, along with DEPT experiments, displayed 38 carbon signals corresponding for nine methyls, five methylenes (one olefinic carbon), eleven methines (nine olefinic carbons), and thirteen quaternary carbons (six olefinic carbons and three carbonyl carbons). The aforementioned functionalities accounted for 13 degrees of unsaturation, The remaining two degrees of unsaturation suggested compound 1 to be bicyclic. All the information mentioned above as well as the reported data of PPAPs-type compounds indicated 1 could be a PPAP derivative.12–15
A detailed comparison of the 1D NMR spectroscopic data with that of chamone II showed that the plane structure of 1 was the same as that of chamone II,15 which was further confirmed by HMBC correlations (see Fig. 2) from H-5 to C-3 (δC 113.7), H-4 to C-2 (δC 167.9) and C-8 (δC 192.7), and H2-23 to C-9 (δC 63.3) and C-13 (δC 207.8). Furthermore, an unusually high field methyl signal at δH 0.58 (3H, s) of 2,2-dimethyl-2H-pyran ring was observed in 1H-NMR spectrum of 1, which may be due to shielding effects from the unsubstituted phenyl group.13 Therefore, 2,2-dimethyl-2H-pyran ring was fused with the phloroglucinol moiety at C-2 and C-3 with the ether linkage at C-2. The major differences between the NMR data of 1 and chamone II were that the chemical shift of C-11 and Me-39 were both shifted upfield from δC 49.1 and 28.0 in 1 to δC 43.0 and 16.6 in chamone II, respectively, suggesting a β-orientation for the prenyl side chain located at position 11. The chemical shifts of Me-38 and Me-39 appeared at δC 24.1 and 28.0 respectively for 1 with an axial prenyl side chain on position C-11, whereas the gem-dimethyl group showed chemicals shifts at δC 24.7 and 16.6, respectively in chamone II. The upfield shift of the C-39ax signal resulted from γ-gauche interaction between Me-39 and the CH2-33 of the prenyl group.1 Therefore, the structure of 1 was determined as 11-epi-chamone II, named as garcimultine A.
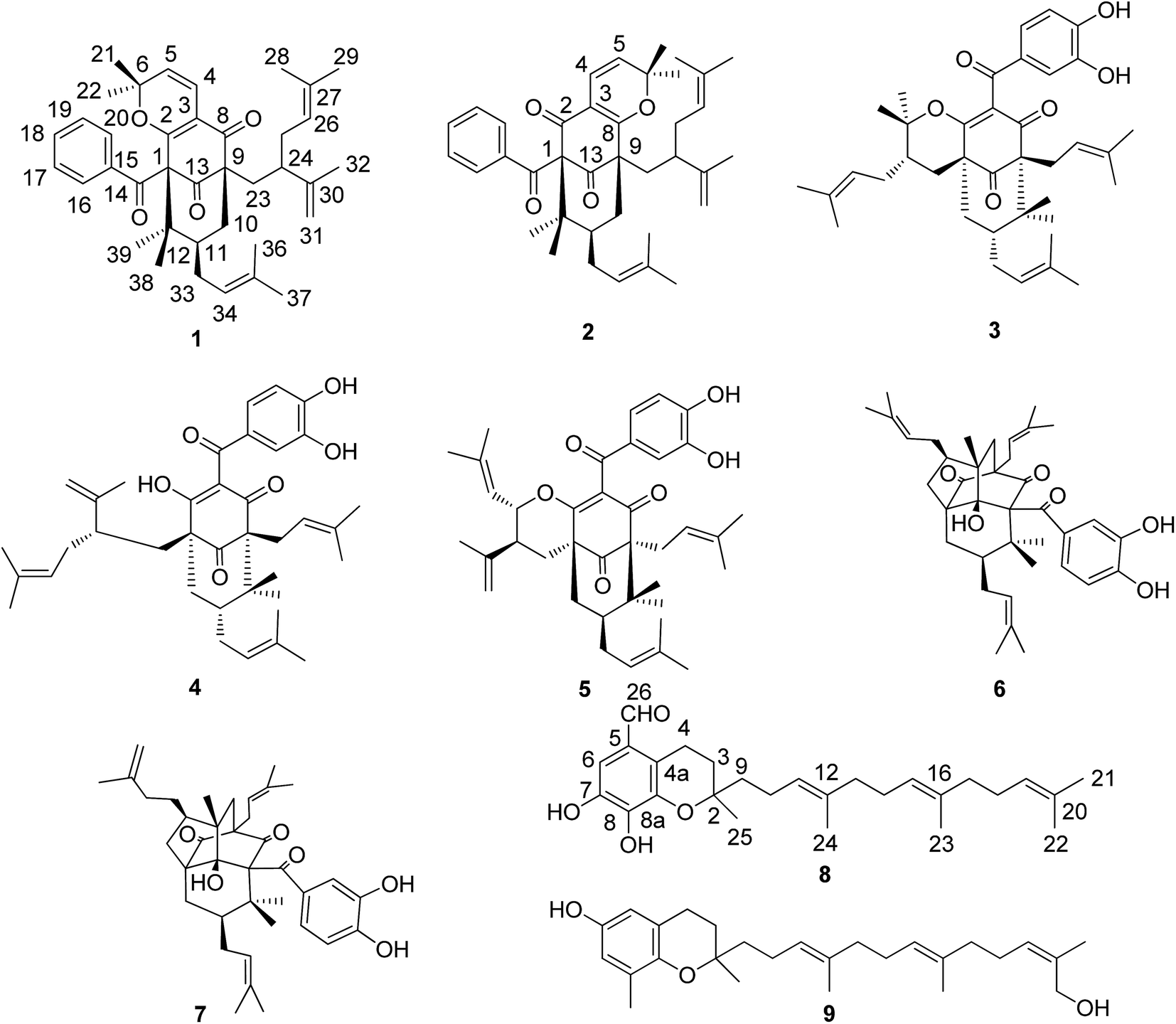 | ||
| Fig. 1 Structures of compounds 1–9. | ||
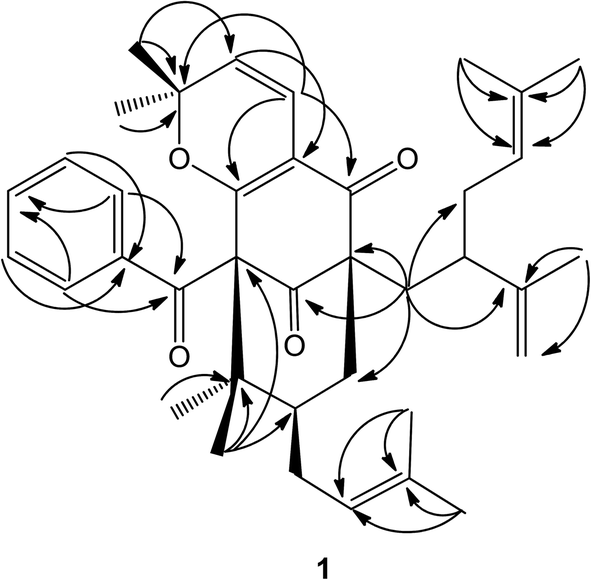 | ||
| Fig. 2 Selected HMBC for compound 1. | ||
Except for the 13C-NMR signals of 1 mentioned above, the remaining 38 carbon signals were attributed to 2. In comparison of 1H and 13C NMR data of 2 with those of 1, it was found that NMR data of 2 were almost identical with those of 1, suggesting that 2 is the regioisomer of 1. In the 13C NMR spectrum, the key differences between the 1 and 2 were that the chemical shift of C-1 was shifted downfield from δC 71.9 in 1 to δC 78.9 in 2, however, the chemical shift of C-9 was shifted upfield from δC 63.3 in 1 to δC 56.5 in 2. These differences could only be due to the modification of 2,2-dimethyl-2H-pyran ring fused with the phloroglucinol moiety at C-3 and C-8 in 2, which was the same as that of plukenetione G.13 Furthermore, the unusually high field methyl signal of 2,2-dimethyl-2H-pyran ring is absent in 2. Thus, the structure of 2 was determined as the regioisomer of 1, named as garcimultine B.
The regioisomeric mixture were well separated by HPLC (CH3CN:H2O = 98:2) to give garcimultine A at tR 16 min and B at tR 14 min. However, it was unsuccessful for the isolation of garcimultine A and B due to the rapid equilibration between the two isomeric forms. The major regioisomer garcimultine A was easily obtained by semipreparative HPLC. In order to investigate the stability of garcimultine A, the purity of garcimultine A was detected by HPLC every 2 hour (see Fig. 3). It was found that over a period of about 2 hours, garcimultine A was transformed to a mixture of 1 (96.74%) and 2 (3.26%). After 4 hours, the content of garcimultine B increased to 7.47%. With the extension of time, the content of garcimultine B increased gradually. After 12 hours, the equilibration between garcimultine A and B was achieved, in which the content of garcimultine B is not increased and kept about 18%. This equilibration appeared to be spontaneous and is likely to form the key quinone-methide intermediate (see Fig. 4). From those results, it suggested that 1 might be more stable than 2 due to the steric hindrance between Me-21, 22 and lavandulyl group in 2. Furthermore, we investigated the energetics of the regioisomeric 1 and 2. Quantum mechanical methods were employed to calculate their molecular energies at the HF/6-31G* level using the Gaussian 09 program. The result revealed that the conformational energy of 1 is lower than that of 2 by 2.01 kcal mol−1, suggesting that 1 is more stable in solution (see Fig. 5). The small energy difference demonstrated that the transition of 1 and 2 is relatively facile, but the 1 conformation is dominant.
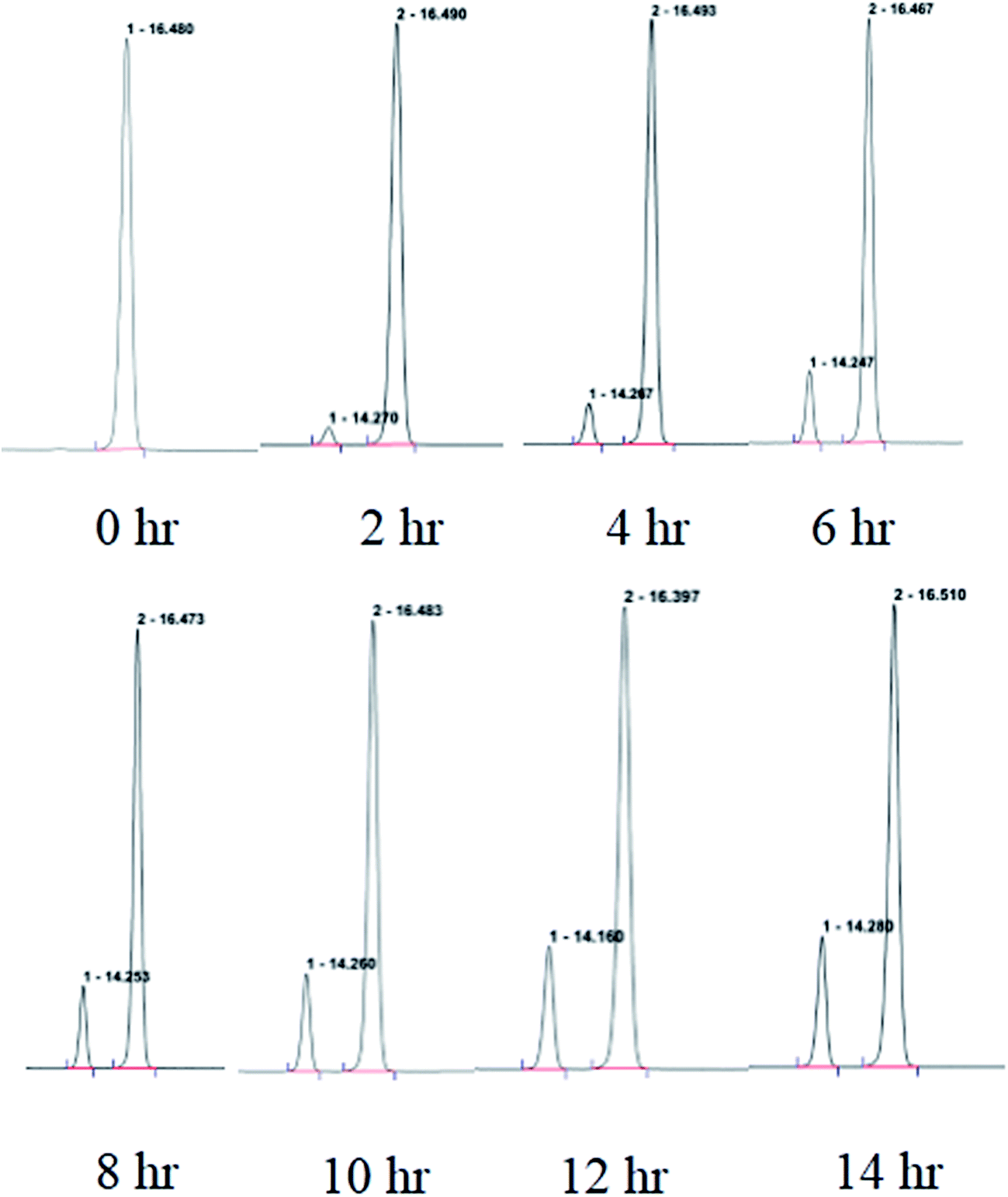 | ||
| Fig. 3 The purity of garcimultine A detected by HPLC every 2 hour. | ||
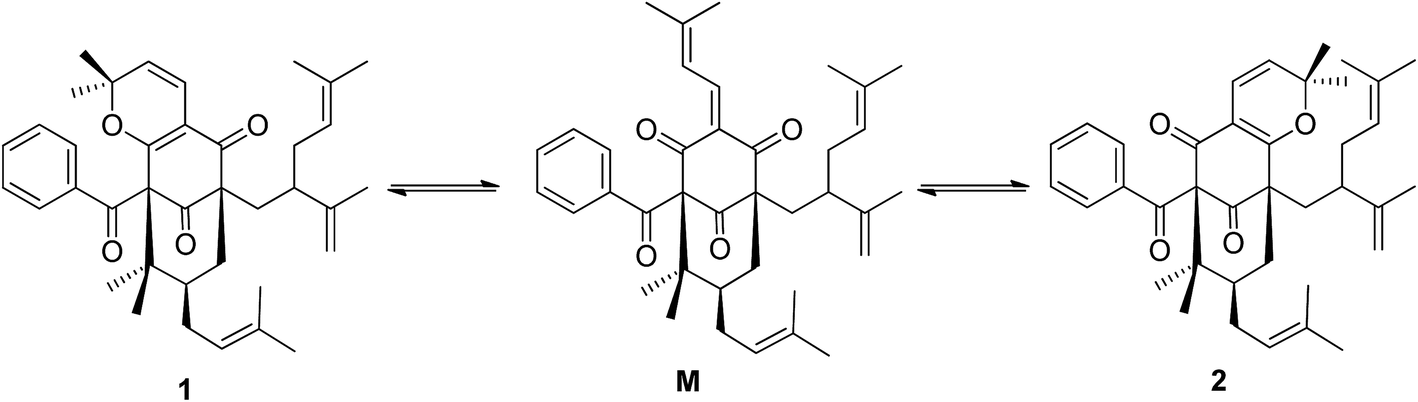 | ||
| Fig. 4 The equilibration between garcimultine A and B by the key quinone-methide intermediate M. | ||
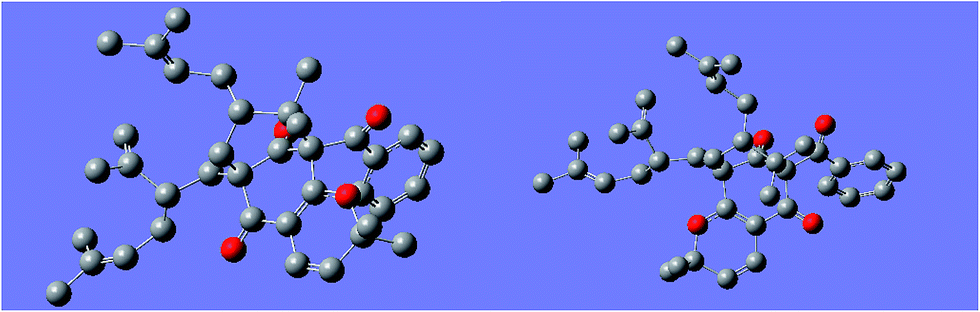 | ||
| Fig. 5 The minimum energy conformation of compound 1 (left) and 2 (right). | ||
Compound 8 was isolated as white amorphous powder and its molecular formula was established as C27H38O4 based on the [M − H]− quasimolecular ion peak observed in the HR-ESIMS spectrum. The 1H NMR spectrum of 8 displayed an isolated aromatic proton [δH 7.86 (1H, s)], four methylene protons of chroman ring [δH 1.80 (1H, m), 1.87 (1H, m) and 3.04 (2H, m)], three olefinic protons [δH 5.28 (2H, m) and 5.22 (1H, t, J = 7.0 Hz)], five methyl signals [δH 1.35 (3H, s), 1.60 (3H, s), 1.66 (6H, s), 1.69 (3H, s)], and an aldehyde group [δH 10.85 (1H, s)]. The 13C-NMR and DEPT spectrum indicated the presence of five methyls, eight methylenes, five methines (an aldehyde group), and nine quaternary carbons (eight olefinic carbons and one sp3 oxygenated carbon). From the above analysis of NMR data, it was suggested that compound 8 is a tocotrienol derivative similar to that of paucinervin D.16 Further comparison of the NMR spectroscopic data of 8 with those of paucinervin D revealed that an aldehyde group was located at C-5 in 8 instead of a methyl group in paucinervin D. This deduction was further confirmed by HMBC correlations (see Fig. 6) from H-6 (δH 7.86) to C-7 (δC 153.9), C-8 (δC 140.5) and C-26 (δC 188.1) and from H-26 (δH 10.85) to C-6 (δC 111.3) and C-4a (δC 117.2). As a result of the above information, the structure of 8 was deduced completely as showed in the Fig. 1.
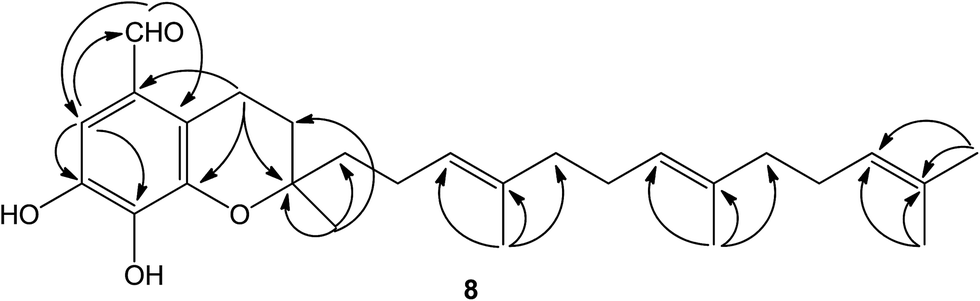 | ||
| Fig. 6 Selected HMBC for compound 8. | ||
The six known compounds including five PPAPs and one tocotrienol derivative were identified as isogarcinol (3),17 garcinol (4),18 garcicowin C (5),19 garcimulin A (6),20 garcimulin B (7),20 δ-tocotrienilic alcohol (9)21 by comparison of the spectroscopic data with those reported in the literature.
3.2 The antiproliferative activities of isolated compounds
The antiproliferative activities of all isolated compounds were evaluated on four human tumor cell lines (HepG2, A549, SGC7901, MCF-7). As shown in Table 3, the mixture of 1 and 2 showed a stronger inhibitory effect on all four cancer cells with IC50 values less than 10 μM. PPAP derivatives 3–7 and tocotrienol derivative 9 exhibited moderate inhibitory effect on all four cancer cells with IC50 values ranging from 4.06 to 35.61 μM. Compared with the positive control doxorubicin, compounds 3, 4 and 9 displayed more effectively against A549 cell line with IC50 values of 5.70, 4.06 and 12.47 μM, respectively. Compounds 3 and 9 also showed better cytotoxic activity than that of the positive control against SGC7901 cell line.4. Conclusions
In this study, chemical investigation on the fruits of G. multiflora yielded a new regioisomeric pair of garcimultine A (1) and B (2), one new tocotrienol derivative garcimultine C (8), together with six known compounds. Their structures were identified by extensive spectroscopic analyses. The regioisomeric garcimultine A (1) and B (2) were not separated due to the rapid equilibration between the two isomeric forms. To the best of our knowledge, it was the first time to report tocotrienol derivative from this plant. Compounds 3–9 were evaluated anti-proliferative activities against four human tumor cells (HepG2, A549, SGC7901, MCF-7). As a result, PPAPs derivatives (1–7) and tocotrienol derivative 9 displayed potential inhibitory activity against four human cancer cell lines. These findings support traditional applications of G. multiflora as healthy nutritional supplements or functional foods for cancer prevention.Acknowledgements
This work was financially supported by the National Science Foundation of China (No. 31370379) and Key projects of technological innovation of Hubei province (No. 2016ACA138). We wish to thank Shanghai Jiaotong University (Medicinal Bioinformatics Center, School of Medicine) for providing Maestro 9.0 (Schördinger) and Gaussian 09 software.References
- R. Ciochina and R. B. Grossman, Chem. Rev., 2006, 106, 3963–3986 CrossRef CAS PubMed.
- S. Kumar, S. Sharma and S. K. Chattopadhyay, Fitoterapia, 2013, 89, 86–125 CrossRef CAS PubMed.
- S. B. Wu, C. L. Long and E. J. Kennelly, Nat. Prod. Rep., 2014, 31, 1158–1174 RSC.
- B. Liu, X. B. Zhang, R. W. Bussmann, B. H. Hart, P. Li, Y. J. Bai and G. L. Long, Econ. Bot., 2017, 70, 417–430 Search PubMed.
- J. J. Chen, C. W. Ting, T. L. Hwang and I. S. Chen, J. Nat. Prod., 2009, 72, 253–258 CrossRef CAS PubMed.
- C. W. Ting, T. L. Hwang, I. S. Chen, M. H. Yen and J. J. Chen, Chem. Biodiversity, 2012, 9, 99–105 CAS.
- C. W. Ting, T. L. Hwang, I. S. Chen, M. J. Cheng, P. J. Sung, M. H. Yen and J. J. Chen, Chem. Biodiversity, 2014, 11, 819–824 CAS.
- D. S. Tian, P. Yi, L. Xia, X. Xia, Y. M. Fan, W. Gu, L. J. Huang, Y. B. David, Y. T. Di, C. M. Yuan and X. J. Hao, Org. Lett., 2016, 18, 5904–5907 CrossRef CAS PubMed.
- R. A. Hill and A. Sutherland, Nat. Prod. Rep., 2017, 34, 130–134 RSC.
- Y. Chen, F. Gan, S. Jin, H. Liu, S. J. Wu, W. T. Yang and G. Z. Yang, RSC Adv., 2017, 7, 17289–17296 RSC.
- Y. Chen, C. Tang, Y. Wu, S. S. Mo, S. Wang, G. Z. Yang and Z. N. Mei, Org. Biomol. Chem., 2015, 13, 6773–6781 CAS.
- B. Trusheva, M. Popova, H. Naydenski, I. Tsvetkova, J. G. Rodriguez and V. Bankova, Fitoterapia, 2004, 75, 683–689 CrossRef CAS PubMed.
- G. E. Henry, H. Jacobs, C. M. S. Carrington, S. McLean and W. F. Reynoldse, Tetrahedron, 1999, 55, 1581–1596 CrossRef CAS.
- A. L. M. Porto, S. M. F. Machado, C. M. A. Oliveira, V. Bittrich, M. C. E. Amaral and A. J. Marsaioli, Phytochemistry, 2000, 55, 755–768 CrossRef CAS PubMed.
- J. Lokvam, J. F. Braddock, P. B. Reichardt and T. P. Clausen, Phytochemistry, 2000, 55, 29–34 CrossRef CAS PubMed.
- X. M. Gao, T. Yu, F. S. F. Lai, Y. Zhou, X. Liu, C. F. Qiao, J. Z. Song, S. L. Chen, K. Q. Luo and H. X. Xu, Bioorg. Med. Chem., 2010, 18, 4957–4964 CrossRef CAS PubMed.
- G. Marti, V. Eparvier, C. Moretti, S. Susplugas, S. Prado, P. Grellier, P. Retailleau, F. Guéritte and M. Litaudon, Phytochemistry, 2009, 70, 75–85 CrossRef CAS PubMed.
- M. Iinuma, H. Tosa, T. Tanaka, S. Kanamaru, F. Asai, Y. Kobayshi, K. Miyauchi and R. Shimano, Biol. Pharm. Bull., 1996, 19, 311–314 CAS.
- G. Xu, W. L. T. Kan, Y. Zhou, J. Z. Song, Q. B. Han, C. F. Qiao, C. H. Cho, J. A. Rudd, G. Lin and H. X. Xu, J. Nat. Prod., 2010, 73, 104–108 CrossRef CAS PubMed.
- Y. M. Fan, P. Yi, Y. Li, C. Yan, T. Huang, W. Gu, Y. Ma, L. J. Huang, J. X. Zhang, C. L. Yang, Y. Li, C. M. Yuan and X. J. Hao, Org. Lett., 2015, 17, 2066–2069 CrossRef CAS PubMed.
- J. S. R. Teixeira, L. M. Moreira, M. L. S. Guedesb and F. G. Cruz, J. Braz. Chem. Soc., 2006, 17, 812–815 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra04208f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |