
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Straightforward synthesis of bis-tetraazacycloalkanes: towards new potential CXCR4 antagonists?†
Nicolas Sok *a,
Isabelle Baglinb,
Christelle Bassetc,
Fatima Fakkord,
Evelyne Kohlic,
Yoann Rousselinb,
Claire Bernhardb,
Frédéric Boschettie,
Christine Gozeb and
Franck Denat*b
*a,
Isabelle Baglinb,
Christelle Bassetc,
Fatima Fakkord,
Evelyne Kohlic,
Yoann Rousselinb,
Claire Bernhardb,
Frédéric Boschettie,
Christine Gozeb and
Franck Denat*b
aUniv. Bourgogne Franche-Comté, AgroSup Dijon, PAM UMR A 02.102, F-21000 Dijon, France. E-mail: nicolas.sok@agrosupdijon.fr
bInstitut de Chimie Moléculaire de l'Université de Bourgogne, UMR CNRS 6302, Univ. Bourgogne Franche-Comté, 9 Avenue Alain Savary, 21078 Dijon Cedex, France. E-mail: franck.denat@u-bourgogne.fr
cINSERM UMR 866, UFR des Sciences de Santé, Univ. Bourgogne Franche-Comté, Dijon, France
dCentre Hospitalier Universitaire, Dijon, France
eChematech S.A., 2 Rue Pauline Kergomard, 21000 Dijon, France
First published on 30th May 2017
Abstract
We report herein an efficient and general method for the synthesis of new bismacrocyclic compounds, structural analogues of biscyclam AMD3100, in which the two macrocycles are linked together through carbon atoms of the cycles. Several representatives of this new class of biscyclic derivatives were prepared by reacting C-aminomethyl-13aneN4 with aromatic dialdehydes. Preliminary in vitro studies were performed to evaluate the affinity of these compounds towards the co-receptor CXCR4.
1 Introduction
The CXCR4 chemokine receptor is a seven transmembrane helix protein, member of the G-protein-coupled receptor (GPCR) superfamily, whose own natural ligand is SDF-1α or CXCL12.1–4 The axis CXCR4/CXCL12 is implicated in the regulation of numerous biological processes in the human body that result in a variety of normal physiological responses, such as chemotaxis, cell survival and proliferation, intracellular calcium flux, and gene transcription. CXCR4 is also involved in many downstream pathways, such as inflammation,5 mobilisation of stem cells,6 and development of cancer cells metastasis.7,8 Furthermore, CXCR4 is a co-receptor for T-cell tropic strains of human immunodeficiency virus-1 (HIV-1) and allows fusion and entry of the virus into human white blood cells. Targeting specifically these receptors represents therefore a huge potential for many applications, such as characterization of a pathology, drug development, staging and restaging, following a therapy response, delineation and quantification of tumors in oncology. Nowadays, only one bis-cyclam derivative, AMD3100,9,10 is clinically used in oncology, in combination with GCSF (granulocyte colony stimulating factor) to mobilize haematopoietic stem cells and allow harvesting.11,12 Several AMD like compounds, differing by the size of the macrocycle and the nature of the linker, have been synthesized and studied,11,13–15 highlighting the tremendous importance of these parameters on the affinity of the bismacrocyclic compound for its target. Thus, it is of major interest to devise new synthetic routes for the construction of bismacrocyclic scaffolds analog to AMD3100, which could present better therapeutic properties.10,16,17In this study we report the synthesis of a series of new bis-tetraazacycloalkanes, in which the two macrocycles (13aneN4) are linked together through carbon atoms of the cycles. To this end, we decided to use C-aminomethyl macrocycles, already described by our group.18–20 Beside the fact that these precursors may lead to a new family of AMD analogues with enhanced properties, this approach is synthetically appealing since it represents a straightforward route towards bismacrocyclic compounds. Indeed, it is a very attractive alternative to the synthesis of AMD like derivatives in which the bridge between the two macrocycles is linked to nitrogen atoms of the cycles, which usually involves tedious purification steps and/or protection/deprotection sequences in order to differentiate one out of the four nitrogen atoms. We also report inhibition studies of anti-CXCR4 mAb binding and of SDF-1α chemotaxis activity by AMD3100 and its analogs.
2 Results and discussion
2.1 Synthesis
In previous work we have demonstrated that the primary amine function of C-aminomethyl-13aneN4 (1) readily reacts with one equivalent of aromatic aldehydes to form cyclic aminal derivatives involving an adjacent nitrogen atom of the cycle. Reduction of these aminal compounds with NaBH4 gave access to the corresponding macrocycles selectively alkylated on the pendant amino methyl group.20 It might be expected that such peculiar reactivity can be exploited to synthesize bismacrocycles by reacting the C-aminomethyl macrocycle (1) with dialdehydes. Indeed, by using 0.5 equivalent of various dialdehydes (isophthalaldehyde, terephthalaldehyde, pyridine-2,6-dicarbaldehyde, 2,2′-oxydibenzaldehyde), the corresponding bismacrocyclic bisaminal derivatives were obtained in good yields (up to 82%) (Scheme 1).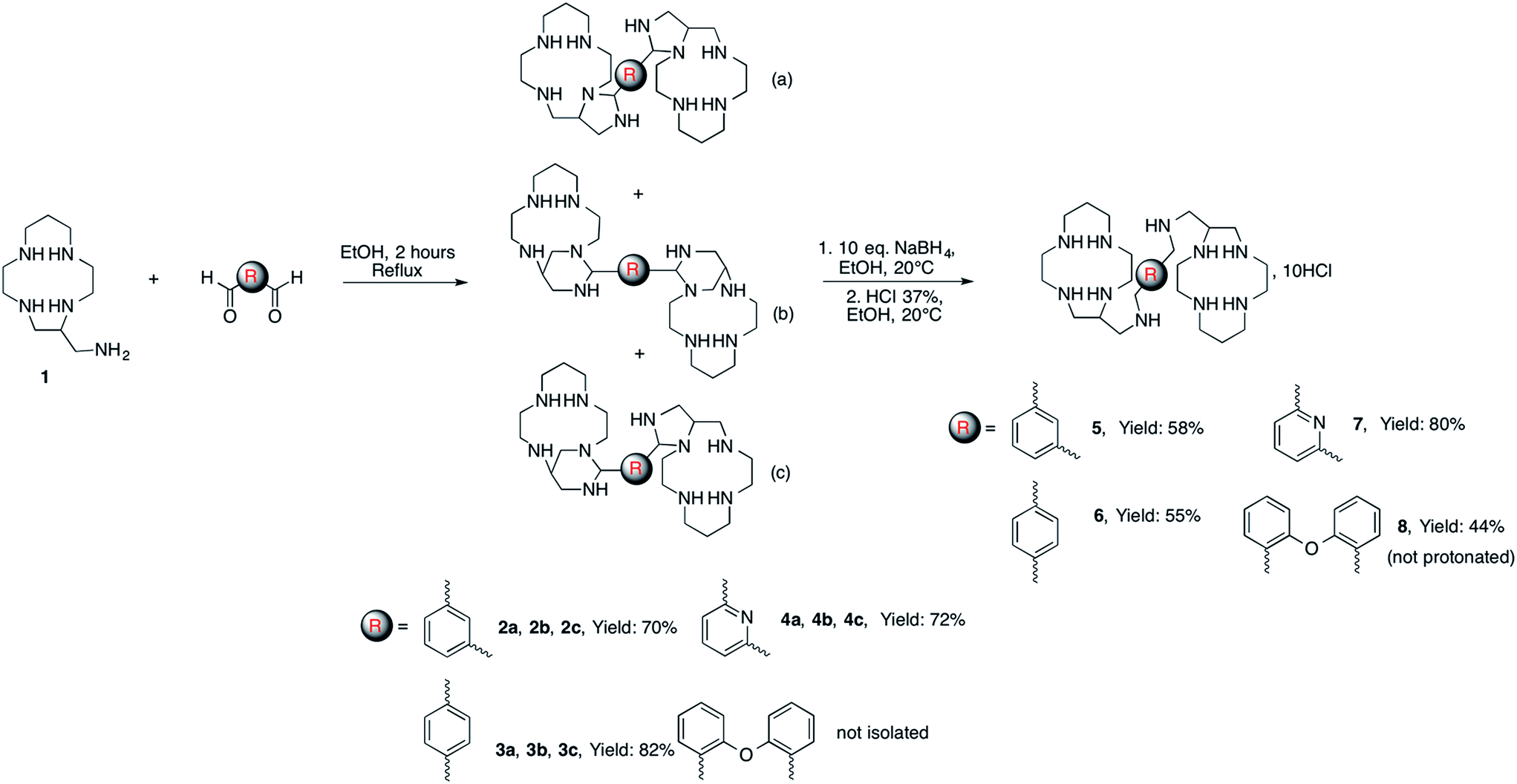 | ||
| Scheme 1 Synthesis of bistetraazacycloalkanes. | ||
The reduction of these compounds with NaBH4 followed by acidic treatment gave the hydrochloride salts of the aimed bis-tetraazacycloalkanes 5–8 as white powders.
The NMR spectra of the isolated bismacrocyclic bisaminal derivatives 2–4 are very complicated due to the formation of a mixture of different isomers. Monocrystals of pure 2a were prepared after slow crystallization of 2 in diethylether (Fig. 1, left). The structure of compound 4a was also determined by X-ray diffraction (Fig. 1, right). An exhaustive description of each compound is given in the ESI.†
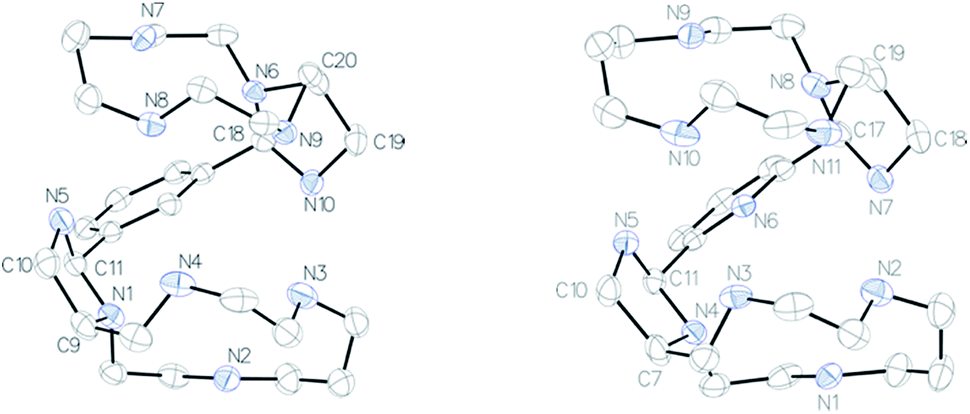 | ||
| Fig. 1 ORTEP views of compound 2a (left) and compound 4a (right). Thermal ellipsoids are drawn at 50% probability level. Water molecule is omitted for clarity. | ||
Both compounds crystallize identically, they present the same symmetry (monoclinic) and the same conformation with a water molecule in the asymmetric unit. A RMSDmax of 0.4 Å was found by superimposing both structures. Cremer–Pople parameters,21 which describe the conformation of a puckered ring in a quantitative mathematically well-defined manner, indicate that the five membered aminal rings have a slightly distorted chair geometry [compound 2a: N1–C9–C10–N5–C11, Q = 0.360(3) Å, ϕ = 312.4(4)°, N6–C18–N10–C19–C20, Q = 0.345(2) Å, ϕ = 226.6(4)°; compound 4a: N4–C7–C10–N5–C11, Q = 0.348(4) Å, ϕ = 314.1(7)°, N7, N7–C17–N8–C19–C18, Q = 0.363(4) Å, ϕ = 24.5(6)°]. These five membered rings impose the half conformation of a part of the polyazacycloalkane. The second shape of the molecule is governed by intra- and intermolecular hydrogen bonds (see ESI†).
The characterization of the three different isomers was not further investigated since they all give the same compound after opening the aminal bridges by NaBH4. The new bismacrocycles 5–8 were fully characterized using mass spectrometry, 1H and 13C NMR and elemental analysis. It is noteworthy that the number of signals in the 13C NMR spectra of these compounds is low. This feature can of course be explained by an expected symmetry of the molecule. However, the presence of two identical chiral carbon atoms in these bismacrocycles should give raise to the formation of three stereoisomers, i.e. a meso compound and a couple of enantiomers since the starting C-aminomethyl macrocycle 1 was used as a racemic mixture. The simplicity of the NMR spectra seems to prove that the reaction is stereospecific, leading to either the meso compound or a racemic mixture of the two enantiomers.
If the method described herein appears as a powerful route for the preparation of bis-13aneN4 derivatives, it must be noted that some unexpected results have been obtained with two other dialdehydes. When using ortho-phthalaldehyde, the formation of the bismacrocycle was not observed. NMR and mass spectrometry data suggest the formation of a [1 + 1] aminal adduct 9 containing a lactam moiety (Scheme 2).
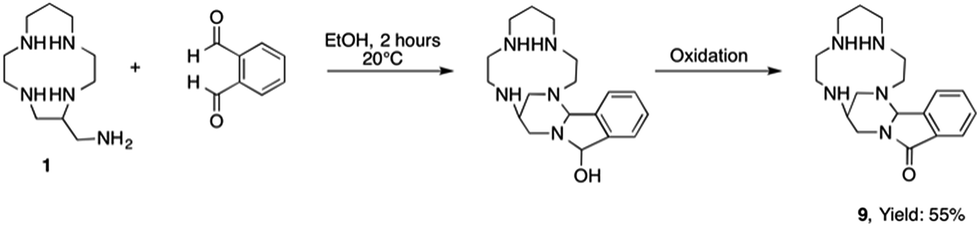 | ||
| Scheme 2 Synthesis of lactam derivative 9. | ||
The structure of compound 9 was confirmed by X-ray diffraction of the monocrystal grown from a hexane/dichloromethane (99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) mixture (Fig. 2). A 6-membered ring (N1–C9–C8–C10–N5–C11) with a slightly distorted chair conformation is formed [Cremer–Pople parameters: Q = 0.5717(19) Å, ϕ = 10.9(2)°, ϕ = 32.5(11)°]. An intramolecular hydrogen bond imposes the conformation of the nitrogen atoms N2 and N3.
:1) mixture (Fig. 2). A 6-membered ring (N1–C9–C8–C10–N5–C11) with a slightly distorted chair conformation is formed [Cremer–Pople parameters: Q = 0.5717(19) Å, ϕ = 10.9(2)°, ϕ = 32.5(11)°]. An intramolecular hydrogen bond imposes the conformation of the nitrogen atoms N2 and N3.
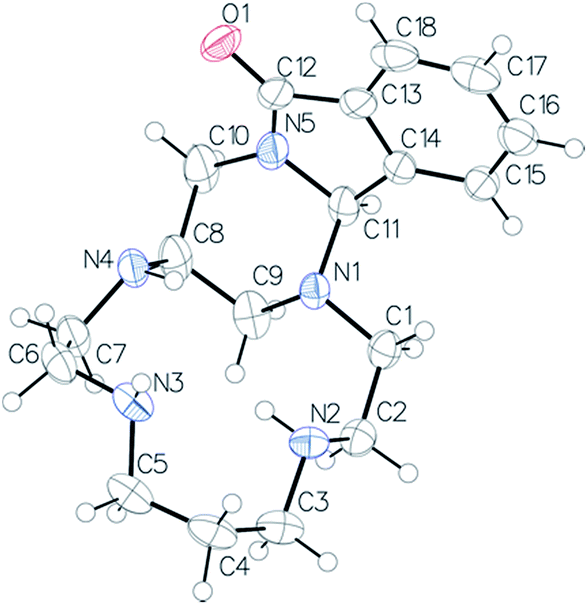 | ||
| Fig. 2 ORTEP view of compound 9. Thermal ellipsoids are drawn at 50% probability level. | ||
This reactivity can be explained by the close proximity of the two aldehyde functions in the rigid ortho-phthalaldehyde. Indeed, after the formation of the cyclic aminal resulting from the reaction of the primary amine of the starting macrocycle with one aldehyde, the second carbonyl group is attacked preferentially by the secondary nitrogen atom of the aminal cycle rather than by the primary amine of a neighbor macrocycle. This intramolecular reaction is thermodynamically favored by the formation of a five membered hemi-acetal ring which can not undergo a dehydration step but is oxidized into a lactam function, although no oxidizing reagent was used during the reaction, apart from the oxygen from air.
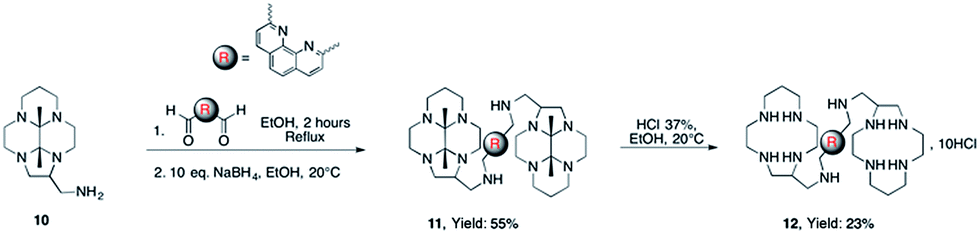
The expected bisaminal bismacrocycle was also not obtained when reacting C-aminomethyl-13aneN4 (1) with 2,9-dicarboxaldehyde-1,10-phenanthroline (2,9-DCP). Again, this could be attributed to intra- and/or intermolecular reaction of the aldehyde with different amine functions, giving side products. These results prompted us to devise another route for the preparation of the desired bismacrocycles. In order to avoid the intramolecular reaction of the second aldehyde with secondary amine functions, we decided to use as starting material the bisaminal protected macrocycle 10 which is actually the precursor of C-aminomethyl macrocycle 120 (Scheme 3).
 | ||
| Scheme 3 Synthesis of bismacrocycle 12. | ||
In this case the cyclic aminal cannot be formed and the reaction with 2,9-dicarboxaldehyde-1,10-phenanthroline followed by reduction with NaBH4 gives the protected bismacrocycle 11. This compound was finally treated by HCl 37% in ethanol to yield the aimed bis-macrocycle 12.
2.2 Biological studies
Cellular binding assays of new AMD3100 analogues to the CXCR4 receptor were investigated in a competition assay with an anti-CXCR4 monoclonal antibody (mAb) (12G5 conjugated to phycoerythrin). Jurkat cells, a T-lymphocyte cell line which expresses high level of CXCR-4 expression, were used and binding assays of new compounds 2(a–c), 3(a–c), 4(a–c), 5, 6, 7, and AMD3100 (as reference) to the cells were analysed by flow cytometry. On the whole, synthesized compounds were less efficient than AMD3100, which maximum inhibition concentration was 1 μg mL−1. Compounds 2(a–c), 3(a–c), 4(a–c) and 7, didn't show any inhibition potency. However, at 5 μg mL−1, the analogs 5 and 6 could inhibit the binding of the mAb (41.16% ± 8.1 and 54.83% ± 2.5, respectively) on Jurkat T cells (Fig. 3). This result was confirmed using DAUDI cells (data not shown). These preliminary results confirm the importance of both the nature of the macrocycle and of the bridge between the two macrocycles, and that small structural modifications can strongly reduce the affinity of the AMD analogues towards the CXCR4 receptor. Indeed, previous mutational analysis of the CXCR4 binding site for AMD3100 has identified amino acid Asp171 and Asp262 as key residues involved in the binding to the CXCR4 receptor22. The carboxylate groups of the receptor residues form three (one strong, one intermediate and one weak) hydrogen bonds with protonated cyclam of AMD3100.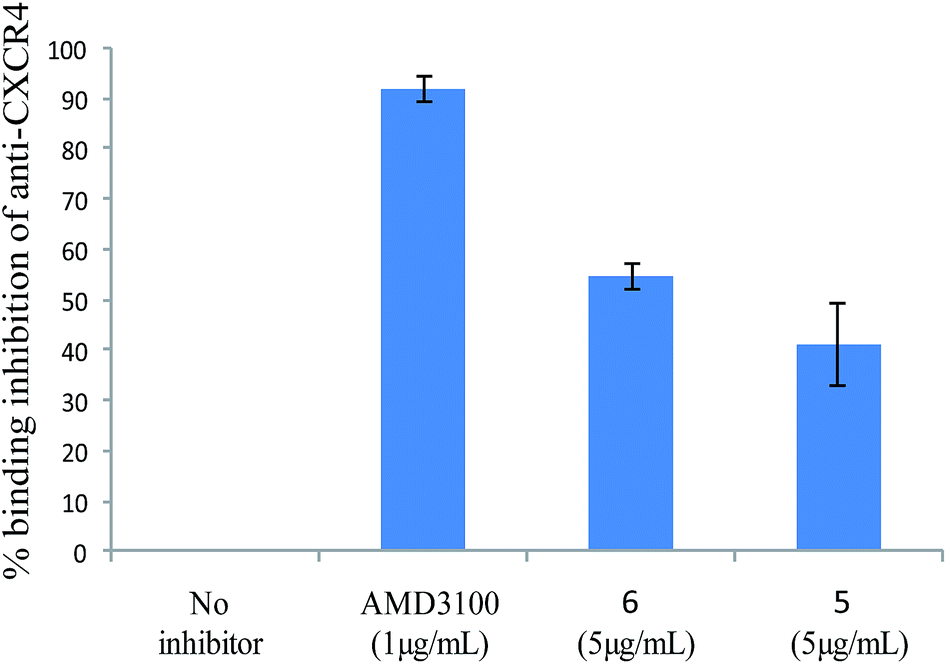 | ||
| Fig. 3 % inhibition of AMD3100, compared to 5 and 6. Results are the average of triplicate ± SE. | ||
Changing the geometry of the macrocycle and the spacer can really influence the binding of the compounds, yielding in a decrease of the resulting interactions for the receptor. Therefore, not surprisingly, 5 and 6, which are structurally more resembling to AMD3100, present the highest affinity for the CXCR4 receptor.
Since the interaction between SDF-1 and CXCR4 has been shown to direct tumor cells to organ sites with high levels of SDF-1 expression, the effect of AMD3100 and its analogs on inhibiting CXCL12-induced migration of Jurkat T cells was estimated by a classical chemotaxis assay. As expected, Jurkat T cells migrated in response to CXCL12, even if only 20% migrated because of a low % of Jurkat T cells bound the chemokine. After AMD3100 treatment (10 μg mL−1), chemotactic activity of Jurkat cells was totally inhibited. However, this property of AMD3100 to inhibit CXCL12-induced chemotaxis through CXCR4 was compromised by structural modifications as none of the analogs could strongly SDF-1-induced migration of Jurkat T cells (Fig. 4).
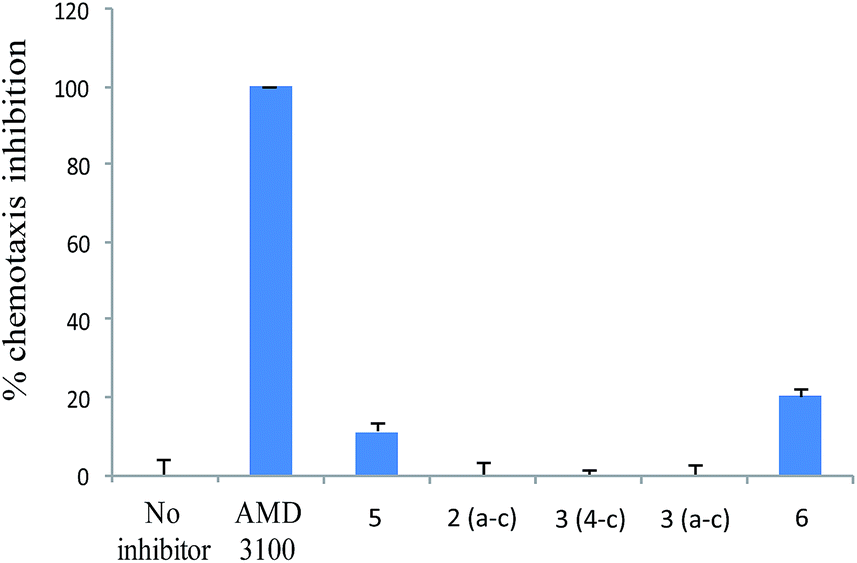 | ||
| Fig. 4 Inhibition of SDF-1 induced migration of Jurkat cells by AMD3100 and its analogs. Results shown are average of duplicate ± SE. This experiment is representative of two different experiments. | ||
3 Conclusion
The work described herein reports the synthesis of a new family of bis-tetraazacycloalkanes formed by two same 13aneN4 units connected by an aromatic spacer. The reaction of two equivalents of C-aminomethyl-13aneN4 with commercial dialdehydes in ethanol followed by reduction with NaBH4 resulted in the formation of bismacrocyclic compounds, structural analogs of AMD3100. In some cases, with ortho-phthalaldehyde or 2,9 DCP, unexpected lactam or side products were obtained. To prepare the aimed bismacrocycles, the protected macrocycle has been used instead of C-aminomethyl-13aneN4 in order to limit the formation of undesired compounds. Affinity assays with the CXCR4 receptor have been performed and showed an effective binding to the receptor for compounds 5 and 6. Further work is ongoing for the preparation of metal complexes of these bismacrocycles. Indeed, previous studies have shown that metal ions incorporation could increase the affinity of bismacrocycles such as AMD3100 or analogs for its receptor.23–254 Experimental section
4.1 Instruments
All analyses were performed at the “Plateforme d'Analyses Chimiques et de Synthèse Moléculaire de l'Université de Bourgogne”. High resolution and accurate mass measurements were carried out using a Bruker MALDI-TOF, microTOF-Q™ ESI-TOF (Electro Spray Ionization-Time of Flight) and a Thermo Scientific* LTQ Orbitrap mass spectrometer. 1H and 13C NMR spectra were recorded on a Bruker 300 and 500 NMR spectrometer. Chemical shifts were reported in parts per million using tetramethylsilane (TMS) as the internal standard. The following abbreviations are used; s: singlet, d: doublet, t: triplet, q: quartet, m: multiplet, b: broad.X-ray equipment and refinement are detailed in ESI.† Data CCDC 1476681, 1476682 and 1476683 contain the supplementary crystallographic data for compound 2a, 4a and 9 respectively.
4.2 Biologicals
4.3 Chemicals
Reagents, solvents and chemicals were purchased from Sigma-Aldrich and were used as supplied without further purification. Compounds 1 and 10 were purchased from Chematech SA.Acknowledgements
Support was provided by the CNRS, the University of Burgundy and the Conseil Régional de Bourgogne through the 3MIM Project. N. S. and C. B. thank the French Ministry of Research for PhD Grant.References
- D. Mukherjee and J. Zhao, Am. J. Cancer Res., 2013, 3, 46–57 CAS.
- M. Loetscher, T. Geiser, T. O'Reilly, R. Zwahlen, M. Baggiolini and B. Moser, J. Biol. Chem., 1994, 269, 232–237 CAS.
- I. D. Weiss and O. Jacobson, Theranostics, 2013, 3, 76–84 CrossRef CAS PubMed.
- J. Kuil, T. Buckle and F. W. B. van Leeuwen, Chem. Soc. Rev., 2012, 41, 5239–5261 RSC.
- M. Kucia, J. Ratajczak, R. Reca, A. Janowska-Wieczorek and M. Z. Ratajczak, Blood Cells, Mol., Dis., 2004, 32, 52–57 CrossRef CAS.
- Q. Ma, D. Jones, P. R. Borghesani, R. A. Segal, T. Nagasawa, T. Kishimoto, R. T. Bronson and T. A. Springer, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 9448–9453 CrossRef CAS.
- A. Muller, B. Homey, H. Soto, N. Ge, D. Catron, M. E. Buchanan, T. McClanahan, E. Murphy, W. Yuan, S. N. Wagner, J. L. Barrera, A. Mohar, E. Verastegui and A. Zlotnik, Nature, 2001, 410, 50–56 CrossRef CAS PubMed.
- W.-T. Choi, S. Duggineni, Y. Xu, Z. Huang and J. An, J. Med. Chem., 2012, 55, 977–994 CrossRef CAS PubMed.
- E. De Clercq, Nat. Rev. Drug Discovery, 2003, 2, 581–587 CrossRef CAS PubMed.
- B. Debnath, S. Xu, F. Grande, A. Garofalo and N. Neamati, Theranostics, 2013, 3, 47–75 CrossRef CAS PubMed.
- E. De Clercq, Biochem. Pharmacol., 2009, 77, 1655–1664 CrossRef CAS PubMed.
- S. Hatse, K. Princen, G. Bridger, E. De Clercq and D. Schols, FEBS Lett., 2002, 527, 255–262 CrossRef CAS PubMed.
- G. J. Bridger, R. T. Skerlj, D. Thornton, S. Padmanabhan, S. A. Martellucci, G. W. Henson, M. J. Abrams, N. Yamamoto, K. D. Vreese and E. De Clercq, J. Med. Chem., 1995, 38, 366–378 CrossRef CAS PubMed.
- H. C. Joao, K. De Vreese, R. Pauwels, E. De Clercq, G. W. Henson and G. J. Bridger, J. Med. Chem., 1995, 38, 3865–3873 CrossRef CAS PubMed.
- G. J. Bridger and R. T. Skerlj, in Advances in Antiviral Drug Design, ed. E. De Clercq, 1999, vol. 3, pp. 161–229 Search PubMed.
- T. Gaines, D. Camp, R. Bai, Z. Liang, Y. Yoon, H. Shim and S. R. Mooring, Bioorg. Med. Chem., 2016, 24, 5052–5060 CrossRef CAS PubMed.
- S. Poty, E. Gourni, P. Desogere, F. Boschetti, C. Goze, H. R. Maecke and F. Denat, Bioconjugate Chem., 2016, 27, 752–761 CrossRef CAS PubMed.
- F. Denat, Y. A. Diaz-Fernandez, P. Pallavicini, L. Pasotti, Y. Rousselin and N. Sok, Dalton Trans., 2009, 34, 6751–6758 RSC.
- P. Pallavicini, V. Amendola, G. Bergamaschi, G. Dacarro, F. Denat, F. Boschetti, M. Nikolantonaki, R. Gougeon, G. D'Alessio, A.-S. Viaux, L. Bertheau, S. Guyot and N. Sok, Eur. J. Inorg. Chem., 2016, 2016, 5106–5113 CrossRef CAS.
- Y. Rousselin, N. Sok, F. Boschetti, R. Guilard and F. Denat, Eur. J. Org. Chem., 2010, 9, 1688–1693 CrossRef.
- D. Cremer and J. A. Pople, J. Am. Chem. Soc., 1975, 97, 1354–1358 CrossRef CAS.
- C. W. Hendrix, A. C. Collier, M. M. Lederman, D. Schols, R. B. Pollard, S. Brown, J. B. Jackson, R. W. Coombs, M. J. Glesby, C. W. Flexner, G. J. Bridger, K. Badel, R. T. MacFarland, G. W. Henson and G. Calandra, J. Acquired Immune Defic. Syndr., 2004, 37, 1253–1262 CrossRef CAS PubMed.
- R. K. Singh and M. A. Khan, Int. J. Quantum Chem., 2011, 111, 4174–4185 CrossRef CAS.
- D. Oltmanns, S. Zitzmann-Kolbe, A. Mueller, U. Bauder-Wuest, M. Schaefer, M. Eder, U. Haberkorn and M. Eisenhut, Bioconjugate Chem., 2011, 22, 2611–2624 CrossRef CAS PubMed.
- A. Khan, G. Nicholson, J. Greenman, L. Madden, G. McRobbie, C. Pannecouque, E. De Clercq, R. Ullom, D. L. Maples, R. D. Maples, J. D. Silversides, T. J. Hubin and S. J. Archibald, J. Am. Chem. Soc., 2009, 131, 3416–3417 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1476681, 1476682 and 1476683. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra04218c |
| This journal is © The Royal Society of Chemistry 2017 |