
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Efficient total synthesis of neocryptolepine and synthetic access to 6-methylquinindoline from a common intermediate†
María V. Méndez ,
Daniel A. Heredia‡
,
Enrique L. Larghi,
Andrea B. J. Bracca and
Teodoro S. Kaufman*
,
Daniel A. Heredia‡
,
Enrique L. Larghi,
Andrea B. J. Bracca and
Teodoro S. Kaufman*
Instituto de Química Rosario (CONICET-UNR), Suipacha 531, S2002LRK, Rosario, SF, Argentina. E-mail: kaufman@iquir-conicet.gov.ar; Fax: +54-341-4370477 ext. 105; Tel: +54-341-4370477 ext. 105
First published on 30th May 2017
Abstract
A convenient approach toward the indoloquinolines neocryptolepine and 6-methylquinindoline from a common intermediate, is reported. Both sequences, designed for maximum use of accessible reagents and robust conditions, are straightforward and efficient. They involved the amidation of 2-aminobenzaldehyde (prepared by iron-mediated reduction of 2-nitrobenzaldehyde) with 2-nitrophenylacetic acid, followed by a K2CO3-assisted cyclization to form a 3-(2-nitrophenyl)quinolin-2-one as the common precursor. Me2CO3-mediated N-methylation of the lactam, reduction of the nitro moiety and final cyclization resulted in 55% overall yield of neocryptolepine, whereas cyclocondensation and N-methylation afforded 79% overall yield of 6-methyl quinindoline. Thus, the sequences toward the targets entailed two POCl3-promoted C–N bond forming reactions, two Fe-mediated nitro group reductions and two base-promoted transformations.
Introduction
Cryptolepis sanguinolenta (Lindl.) Schlachter (Periplocaceae) is a climbing vine, which grows in West and Central Africa, where it is locally known as nibima, kadze or gangamau, and has served Ghanian traditional healers as a folk medicine since ancestral times. The decoction of the roots of this plant have been used in the therapy against various fevers, including malaria, but also as treatment for hypertension, hepatitis, jaundice and inflammation.1The systematic phytochemical examination of C. sanguinolenta demonstrated that it is a rich source of indoloquinolines. The latter are a small and unique family of alkaloids, which feature two differently fused ring systems, an indole and a quinoline.2
Some of its monomeric members are neocryptolepine (1),3a cryptolepine (2),3b,c quindoline (3),3d quindolinone (4),3e cryptosanguinolentine (5),4a 11-isopropylcryptolepine (6),4b,c cryptolepinone (7),4d and quinindoline (8), as shown in Fig. 1.5a Several dimeric alkaloids are also known, including biscryptolepine (9),5b cryptoquindoline,4d cryptolepicarboline,6a cryptospirolepine,6b quindolinocryptotackieine,6c and cryptomisrine.6d
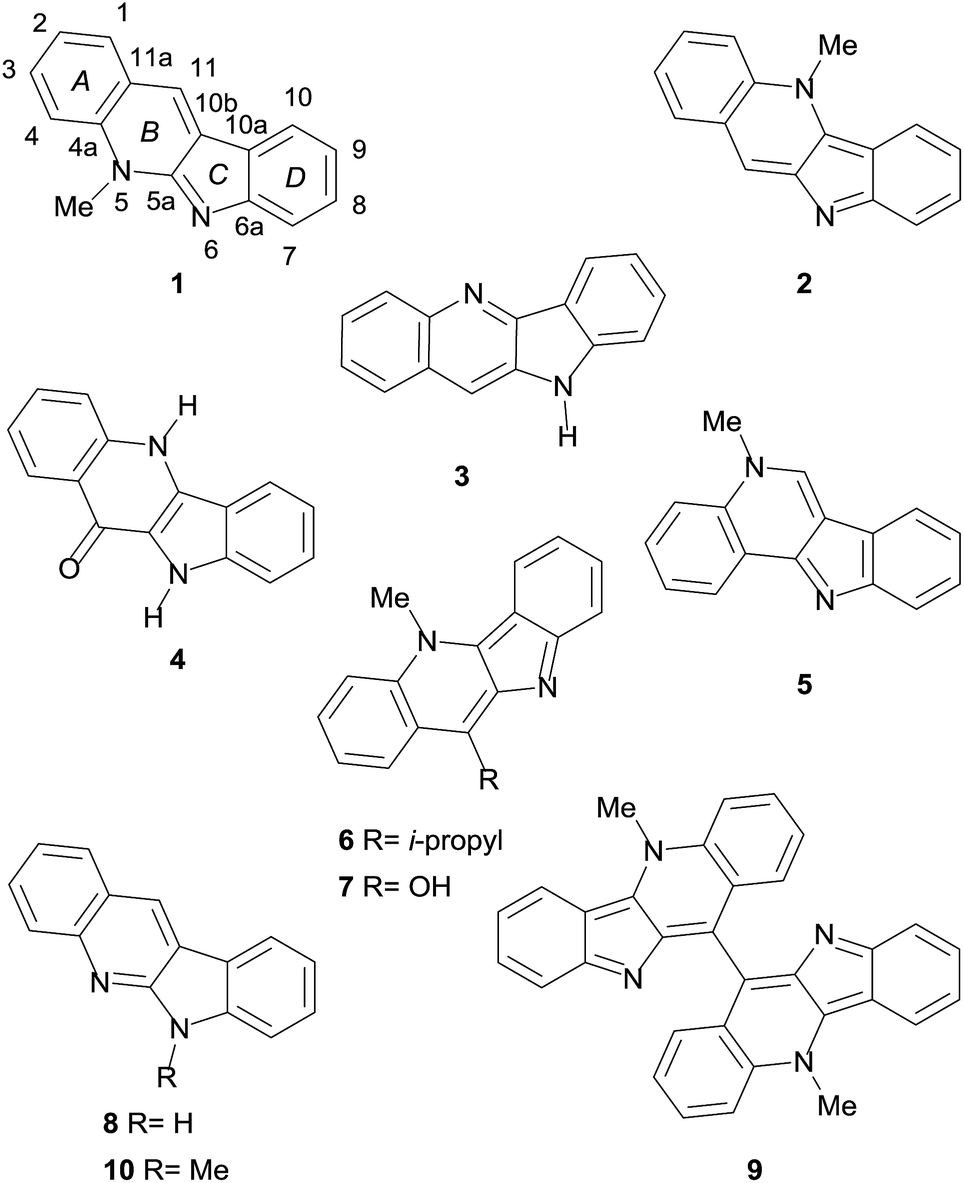 | ||
| Fig. 1 Chemical structures of representative examples of the indoloquinoline alkaloids. Neocryptolepine (cryptotackieine, 1), cryptolepine (2), quindoline (3), quindolinone (4), cryptosanguinolentine (5), 11-isopropylcryptolepine (6), cryptolepinone (7), quinindoline (8), biscryptolepine (9) and the unnatural 6-methylquinindoline (10). | ||
Although most of the indoloquinolines have been isolated from this shrub, there are some scattered reports disclosing additional sources of these heterocycles, including Cryptolepis triangularis N. E. Br., Sida acuta Burm. (Malvaceae), Microphilis guyanensis (A. DC) Pierre (Sapotaceae), Justicia betonica (Acanthaceae) and Genipa americana L. (Rubiaceae).7
The isolation of neocryptolepine (1) from the bark of the roots of C. sanguinolenta was reported independently and almost simultaneously in 1996, by the groups of Schiff, who termed the new alkaloid cryptotakieine,3a and Pieters.5b Curiously, this is not the only case of dual naming of the same alkaloid isolated from this plant.6c
In both instances, the structure of the natural product was elucidated through extensive NMR spectroscopic analyses, including 2D experiments,5b which conclusively proved that it is an isomer of cryptolepine and isocryptolepine,4a and unambiguously defined the site of N-methylation.
Interestingly enough, the structure of 1 conceited great interest and its synthesis was already described long before its isolation from natural sources.8a–d Furthermore, the natural product, whose skeleton bears key common features with those of the communesins,8e–g demonstrated to display cytotoxic and antimalarial activity, also acting as a DNA intercalator.
We have recently reviewed both, the synthesis and the biological activities of neocryptolepine,9 and classified the total syntheses of 1 in three groups, according to the compounds employed as key starting materials for their syntheses; these included those derived from indoles,10 quinolines11 and benzenoids.12
In this way, we found out that after its isolation the natural product has also attracted an unusually great amount of attention from the organic synthetic community, to the extent that approximately 15 total syntheses and 8 formal total syntheses13 of it have been reported to date; further, most of them have used quinindoline (8) as key intermediate.10a–c,11a,12a–f,13b–h However, in spite of the exceptionally important number of published total syntheses of 1, neither of them has been designed to take into account the care for the environment.
The development of efficient and versatile synthetic sequences toward complex molecules has become one of the most relevant goals of modern organic chemistry. Therefore, herein we wish to report a short total synthesis of neocryptolepine (1), which involves a new strategic approach for building the tetracycle and contains environmentally benign stages.
In addition, a synthesis of the isomeric 6-methylquinindoline (10) is also disclosed, employing a common synthetic intermediate and the same operational principles for both targets. Interestingly, there are only four previous records on the preparation of the non-natural compound 10.14
Results and discussion
Our approach toward 1 relied on the retrosynthetic analysis shown in Scheme 1. The initial rational disconnection was made on the N-5 substituent (Route a), assuming that it could be accessed by N-methylation under eco-conscious conditions, instead of using the more conventional but less safe Me2SO4 or MeI as alkylating agents, as in all previous syntheses. This revealed the synthetic need of 8a, the tautomer of quinindoline (8), which was conjectured as accessible through an intramolecular imination of the lactam 11.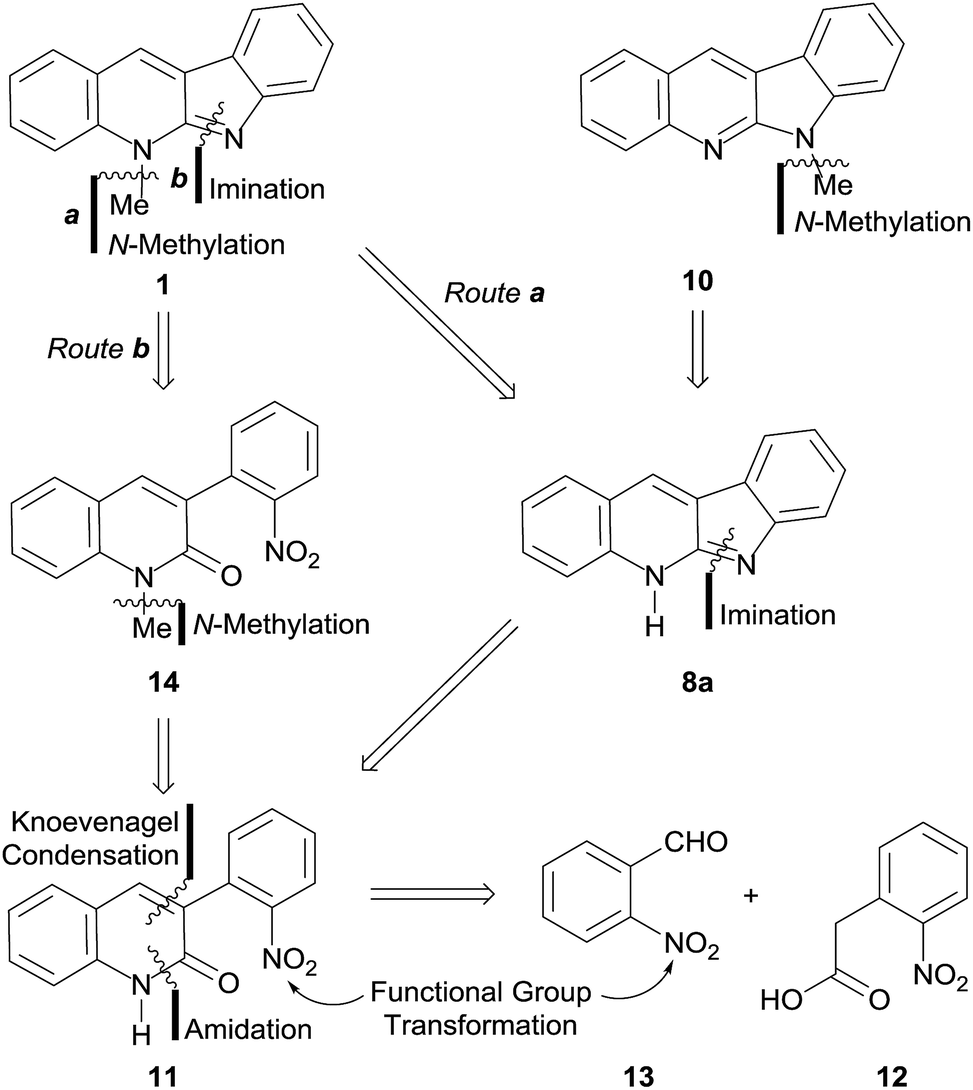 | ||
| Scheme 1 Retrosynthetic analyses of neocryptolepine (1) and 6-methyl quinindoline (10). | ||
In turn, the key intermediate 11 was structurally simplified by performing two successive strategic disconnections of its heterocyclic ring, which uncovered an amide precursor and further revealed 2-nitrophenylacetic acid (12) and 2-nitrobenzaldehyde (13), as the most logical starting materials.
On the other hand, a second set of disconnections was also deemed possible, by merely reversing the order of the initial disconnections. This strategy, which already proved fruitful in our hands,15 conveniently furnished an alternative path (Route b) toward the target.
In this case, the first disconnection was performed on the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond of the five-membered ring, unearthing 14, which was subjected to the retrosynthetic fission of the N-methyl bond, exhibiting the precursor lactam 11 as the common intermediate of both retrosynthetically proposed routes.
N bond of the five-membered ring, unearthing 14, which was subjected to the retrosynthetic fission of the N-methyl bond, exhibiting the precursor lactam 11 as the common intermediate of both retrosynthetically proposed routes.
In both cases, it was foreseen that functional group transformations would be required in order to accommodate the readily available nitro-derivatives proposed as starting materials to the needs of the synthetic sequences. However, this would be rewarded by completely avoiding the need of protective groups.
In this analysis, it was also hypothesized that 8a could also become a suitable precursor of 10, provided appropriately selective N-methylation conditions could be found.
The Route a, which pivoted on an A → AD → ABD → ABCD ring forming sequence, was initially explored. To that end, despite the required 2-aminobenzaldehyde 15 is commercially available, its elevated cost and reportedly high instability16 prompted for its synthesis in situ, which was undertaken as the first task.
Among the various protocols which were examined, the most successful one proved to be that which uses metallic iron in a refluxing hydro-alcoholic (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :90 v/v) medium, in the presence of a mild acid catalyst such as NH4Cl.17 Under these conditions, 96% of 15 was easily obtained, with no sensitive drop in yields when the reduction was performed on a gram scale (Scheme 2).
:90 v/v) medium, in the presence of a mild acid catalyst such as NH4Cl.17 Under these conditions, 96% of 15 was easily obtained, with no sensitive drop in yields when the reduction was performed on a gram scale (Scheme 2).
 | ||
| Scheme 2 Reagents and conditions: (a) Fe, NH4Cl, EtOH–H2O (9:1, v/v), reflux, 2 h (96%); (b) POCl3, pyridine, DMF (cat.), 12, PhCF3, r.t., 3 h (97%); (c) K2CO3, DMF, 100 °C, 1 h (99%); (d) Fe, AcOH, reflux, overnight (17, 99%) or Fe, NH4Cl, EtOH–H2O (9:1, v/v), reflux, 2 h (18, 99%); (e) Py·HCl, 240 °C, 10 min (70% from 17; 93% from 18) or POCl3, MeCN, 90 °C (94% from 18); (f) DMC, MgO, MW (170 °C), 30 min (89%). | ||
However, it was found that special care had to be placed in removing even the smallest traces of iron and its salts, as any of both promoted self-condensation of the product during concentration of the solutions containing the ortho-aminoaldehyde, through Schiff base formation.
After securing the supply of 15, its amidation with acid 12 was scrutinized, in a task that proved to be non-trivial. The use of carbodiimide coupling agents (DCC, EDCI) afforded only moderate yields of 16, partly due to self-condensation of 12,18a but also because of its rather poor solubility. On the other hand, the use of a mixed carboxylic anhydride strategy in THF (Table 1), with (Boc)2O as activating agent and Et3N (entry 1) or pyridine (entries 2–4) as bases,18b produced at best 26% of the expected amide 16.
|
||||
|---|---|---|---|---|
| Entry no. | Solvent | Coupling reagents (equivalents), temperature, time | Yielda (%) | |
| Step 1 | Step 2 | |||
| a Isolated yields of the product. | ||||
| 1 | THF | (Boc)2O, MgCl2, r.t., 1.2 h | Et3N, reflux, 15 h | 14 |
| 2 | THF | (Boc)2O, MgCl2, r.t., 1.2 h | Pyridine (2.0), reflux, 15 h | 26 |
| 3 | THF | (Boc)2O, MgCl2, r.t. 1.2 h | Pyridine (2.5) reflux, 15 h | 13 |
| 4 | THF | (Boc)2O, MgCl2, pyridine (2.5), reflux, 15 h | 20 | |
| 5 | PhMe | Ac2O, Et3N, 90 °C, 48 h | 28 | |
| 6 | PhMe | (COCl)2 (2.0), DMF (cat.), 0 °C to r.t. | Pyridine (3.0), 40 °C, 2.5 h | 34 |
| 7 | PhMe | SOCl2 (1.85), DMF (cat.), 50 °C, 5 h | Et3N (3.0), 50 °C, 28 h | 36 |
| 8 | PhMe | SOCl2 (1.85), DMF (cat.), 50 °C, 5 h | Pyridine (3.0), r.t., 1.5 h | 50 |
| 9 | PhMe | POCl3 (0.7), DMF (cat.), r.t., 1.5 h | Pyridine (3.0), r.t., 20 h | 27 |
| 10 | PhMe | POCl3 (0.7), DMF (cat.), pyridine (3.0), r.t., 48 h | 35 | |
| 11 | CH2Cl2 | POCl3 (0.7), DMF (cat.), pyridine (3.0), r.t., 15 h | 93 | |
| 12 | PhCF3 | POCl3 (0.7), DMF (cat.), pyridine (3.0), r.t., 3 h | 97 | |
The outcomes of these reactions were similar when carried out either in two stages (activation and condensation) or in a single stage (mixing all the reactants together at once). No substantially better performance was achieved with Ac2O in toluene (entry 5).23a
In the pharmaceutical industry, acid chlorides are by far the most common acylating agents for the preparation of drug candidate molecules,18c with oxalyl chloride and SOCl2 being preferred based on criteria of wide utility, scalability and greeneness.18d However, attempts at formation of the acid chloride with oxalyl chloride in toluene,18e and its further condensation with aniline 15 afforded at most 34% yield of 16, when pyridine was used as base (entry 6).
This result was substantially improved when SOCl2 (entries 7 and 8) was used, and yields as high as 50% could be obtained with pyridine as base. Surprisingly, however, no better yields were initially obtained when the less volatile POCl3 was employed as activating agent either in two-stage (entry 9) or single-stage (entry 10) setups.18f
An in-tube NMR monitoring experiment revealed that the first stage required catalytic amounts of DMF to speed up the formation of the acid chloride (ArCH2COCl as a singlet at δH = 4.54 ppm). The reaction resulted in H3PO4 as the only by-product (δ31P = 0.00 vs. δ31P = 3.19 ppm for POCl3) after complete conversion of the acid 12 was achieved; however, other phosphorus-containing reaction intermediates were also observed.19
Interestingly, the success of this experiment also confirmed that POCl3 afforded a transformation with better atom economy than SOCl2, minimizing reagent utilization and byproduct generation, and that the low solubility of 12 in toluene adversely affected the reaction performance.
It was also observed that addition of the base (Et3N or pyridine) all at once to the preformed acid chloride caused an almost immediate destruction of the latter, whereas dropwise or partial addition promoted self-condensation of the aminoaldehyde 15, compromising product yields.
The effects of overcoming the solubility problem on the yields was tested in a small-scale experiment run in CH2Cl2, where we were pleased to discover that this modification afforded 93% of the amide 16 (entry 11).
An eco-acceptable solution was implemented using benzotrifluoride as solvent replacement, which furnished 97% of the expected product (entry 12).20 The optimally accessed amide 16 was then submitted to an intramolecular Knoevenagel-type cyclization under K2CO3 promotion,21 to afford 99% of the desired 3-arylquinolone 11.
An attempt to reach the key intermediate 8 through a one pot reduction of the nitro moiety and subsequent in situ cyclization of the resulting product, was next made. Hence, 11 was exposed to iron powder in refluxing AcOH. However, this resulted in the generation of acetanilide derivative 17, which could not be cyclized with POCl3,11b,22 but afforded 70% of the expected product 8 upon exposure to molten pyridinium hydrochloride (Py·HCl).23
An effort to improve the performance of the synthesis was made, under the hypothesis that the free aniline should be a more suitable substrate for the cyclocondensation. Therefore, compound 11 was reduced with iron powder in EtOH–H2O, as for the synthesis of 15, providing to our delight the required intermediate aniline 18 in essentially quantitative yield.
Based on the previous experience, the use of the high temperature ionic liquid Py·HCl was deemed the most suitable approach toward cyclization of 18, which cleanly afforded the thermodynamic (more stable) amidine isomer 8 in 93% yield. The latter is a natural product, isolated from coal tar pitch and from Justicia betonica,5a,24 which has been repeatedly synthesized.25 However, quite a similar result (94% yield of 8) was obtained employing POCl3 as cyclization agent, under milder conditions.
Next, the final N-methylation stage was approached, with the use of a MgO-catalyzed methylation with dimethyl carbonate (DMC).26 The latter is a versatile and safe replacement of more hazardous alkylating agents, such as MeI, Me2SO4 and MeOTf, which have been widely used in the field of indoloquinoline chemistry; further, it does not produce harmful reaction waste.
However, and not without surprise, we were able to observe the smooth generation of a single less polar compound, in 89% yield, which turned out to be 6-methyl quinindoline (10), the unnatural isomer of 1 and one of our synthetic targets. Other alternatives were explored, including the use of DBU as catalyst; however, all of them met with the same result.27
The formation of 10 as the sole reaction product was unequivocally assessed by exhaustive examination of the 1H and 13C NMR spectra of the product, and their comparison with authentic spectra and literature data of both, 1 and 10.
The diagnostic 1H NMR resonance of the methyl moiety of 10 in CDCl3 was seen at δ 4.02 ppm, in agreement with the literature [δ 3.99,14b 3.97 (ref. 14b) and 3.82 (ref. 14a) ppm], whereas the 5-methyl group of the natural product was reported to resonate as a singlet in the narrow range of δ 4.34 ± 0.03 ppm. Interestingly, this analysis also revealed that at least once, these subtle spectral differences between 1 and 10 resulted in a case of incorrect assignment of product identity.14b
Although the intimate details of the mechanism of this unique transformation remain unknown, an operating mechanistic picture for the DBU-mediated transformation like that shown in Scheme 3 can be drawn, based on various literature precedents.
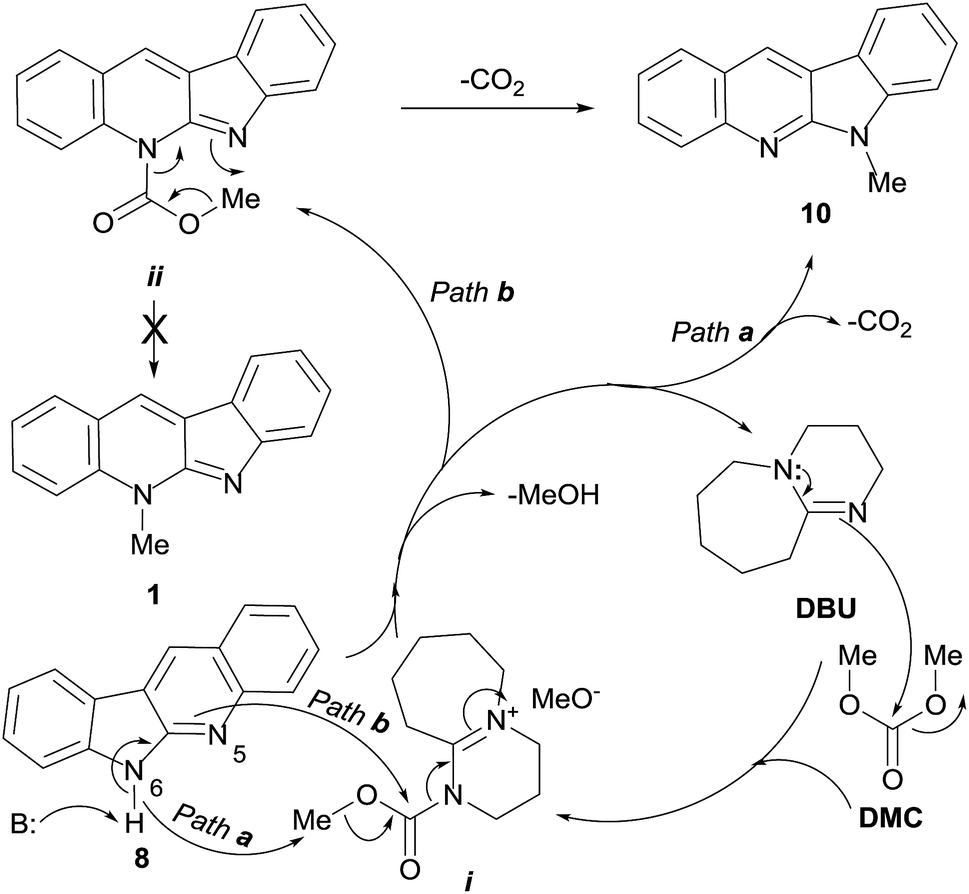 | ||
| Scheme 3 Proposed pathways for the DMC-mediated methylation of quinindoline (8), under DBU promotion, resulting exclusively in 6-methylquinindoline (10). | ||
Accordingly, it is reasonable to assume that the catalytic cycle is initiated by the nucleophilic attack of DBU to DMC, a recognized ambident electrophile.28 As previously proposed, at the reaction temperature (90 °C) this could generate the ion pair intermediate i.27 In turn, this highly activated species could act as methyl (Path a) or methoxycarbonyl (Path b) transferring agent.29
Quinindoline (8) is also an ambident nucleophile, which offers two possible sites of alkylation (N-5 and N-6). It can be postulated that in the presence of DBU as base, due to the relative basicities of both heteroatoms of the amidino moiety, N-6 may become the preferred position for the reaction (Path a), resulting in the methylated tetracycle 10, with concomitant loss of CO2 and MeOH, and regeneration of the catalyst.
Factors controlling analogous processes in other amidines have been discussed, and seem to be in agreement with the above findings.30 Further, there is ample precedent in the chemistry of indoloquinolines indicating that in unsubstituted derivatives, selective methylation of the pyridine ring takes place by reaction with MeI and Me2SO4 alone, whereas in the presence of bases, alkylation of the pyrrole ring becomes predominant or exclusive.31
Alternatively, it can be conjectured that the highly activated species i could act as a methoxycarbonyl transferring agent (Path b).29 Therefore, in the presence of quinindoline (8), this should result in the carbamate intermediate ii, formed under assistance of N-6 of the heterocycle 8, with concomitant loss of MeOH and regeneration of the catalyst.
It has been shown that methylcarbamates cannot be converted into the related N-methyl derivatives by mere heating. This would preclude the direct access of 1 from ii. However, intermediate ii could undergo an intramolecular thermally-assisted extrusion of CO2 that would result in the observed compound 10. Analogous mechanisms have been proposed for the organic base-mediated alkylation of other functional groups bearing acidic protons.32 On the other hand, the intermediacy of carbamates in these reactions has some precedents, and mixtures of carbamates and N-methyl derivatives have been obtained in many occasions.33
The exquisite and unusual selectivity of this N-6 methylation along Path b could also find an explanation based on the hard–soft acid–base theory. In this context, the formation of intermediate ii could be understood as the typical result of the attack of a hard nucleophile (N-5) to the hard carbonyl centre. This would bring about the production of a soft nucleophile and presumably lower the pKa of N-6, turning it capable of becoming involved in an intramolecular attack on the soft methyl group, to afford 10, with concomitant release of CO2.
Regardless the fine details of the mechanism, the reactivity of the nitrogen atoms of quinindoline is quite special and it has been shown that it can be modified by minor structural variations on the rings.34a,b Accordingly, the work of Mastalarz and coworkers is illustrative of the different outcomes of N-methylations of a substituted indoloquinoline with Me2SO4, MeI and DMC,34c and in agreement with our findings.
Ultimately, compound 10 was one of the projected synthetic targets and the reaction of 8 with DMC demonstrated to provide the suitably selective N-methylation conditions looked for in the planning stage. Therefore, focusing back on 1, the unexpected outcome of the DMC-mediated methylation of 8 prompted us to explore the Route b. Here, it became clear that efforts should be placed on transforming the lactam 11 into the natural product, with a view of simplifying the proposed synthesis.
Toward this end, the lactam was suspended in DMC to which a small amount of DMF was added in order to favor its solubility, and exposed to methylation under DBU catalysis. Delightfully, 95% of the more soluble N-methyl lactam 14 was obtained after heating overnight at 90 °C (Scheme 4). In turn, the second iron-mediated reduction of a nitro moiety was put in practice under the previously used conditions, affording 96% of the known aniline derivative 19.11b Finally, exposure of 19 to POCl3 in MeCN, to form a reactive 2-chloroquinolinium intermediate, followed by treatment of the latter with DIPEA, gave the expected tetracycle 1 in 63% yield. Its spectroscopic data were in full agreement with those of the literature for neocryptolepine.
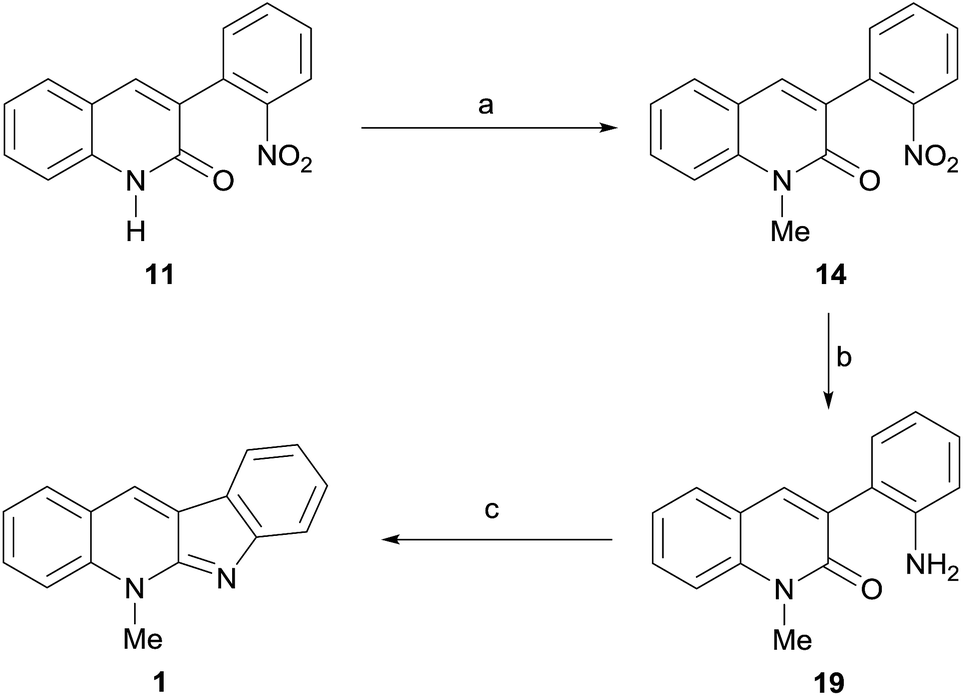 | ||
| Scheme 4 Reagents and conditions: (a) DMC, DMF, DBU, 90 °C, overnight (95%); (b) Fe, NH4Cl, EtOH–H2O (9:1, v/v), reflux, 3 h (96%); (c) 1. POCl3, MeCN, 90 °C, overnight; 2. DIPEA, 120 °C, overnight (63%). | ||
An original aspect is that the syntheses feature the environmentally benign DMC as methylating agent which, acting in two different scenarios, provides two different outcomes. Further, to the best of our knowledge, this is the first total synthesis of 1 which employs this alkylating agent; previous sequences relied on methylation steps with MeI or Me2SO4, which severely compromised their eco-friendliness.
Conclusions
We have developed very simple and efficient 5-step strategies for the total synthesis of the natural indoloisoquinoline alkaloid neocryptolepine (55% overall yield) and its non-natural isomer 6-methylquinindoline (79% overall yield) from 2-nitro phenylacetic acid and 2-aminobenzaldehyde, employing a 3-aryl quinolin-2-one common precursor.The syntheses were designed from the onset to be as straightforward and reagent-economic as possible, while complying with an original key retrosynthetic disconnection (C-11–C-10b). This is the first synthetic solution toward both targets featuring this strategy. Further, both targets were synthesized employing essentially the same reagents but inverting the order of some reaction steps, and unlike most previous approaches, the syntheses employed readily available starting materials and reagents.
The synthesis of neocryptolepine involved a couple of convenient iron-mediated reductions of aromatic nitro moieties to their corresponding anilines and two POCl3-assisted C–N bond forming reactions. A base-assisted cyclization and a base-promoted N-methylation with dimethyl carbonate (DMC) completed the set of reactions.
The strategy toward both heterocycles, which did not resort to the use of protecting groups, employed robust reactions and maximized the use of comparatively benign solvents. Being modular, differently substituted o-amino benzaldehydes and o-nitrophenylacetic acids could be used to prepare analogs. Hence, this contribution should pave the way for the elaboration of more complex analogs of the natural product and its congeners.
Experimental section
General information
All the reactions were carried out under dry nitrogen or argon atmospheres, employing oven-dried glassware. Anhydrous THF and Et2O were obtained from a M. Braun solvent purification and dispenser system; anhydrous DMF was obtained by heating the PA grade product over BaO for 4 h, followed by distillation under reduced pressure; anhydrous pyridine was prepared by sequential storage of the product for 7 days on NaOH pellets and atmospheric pressure distillation; anhydrous Et3N was prepared by distillation of the commercial product from CaH2; anhydrous solvents were stored in dry Young ampoules. EtOH refers to the 96° product. All other reagents were used as received.Flash column chromatographies were carried out with Merck's silica gel 60 H. The elutions were carried out with hexane/EtOAc or EtOAc/EtOH mixtures, under positive pressure and employing gradient of solvent polarity techniques. All new compounds gave single spots on TLC plates (silica gel 60 GF254) run in different hexane/EtOAc and EtOAc/EtOH solvent systems.
The chromatographic spots were detected by exposure to 254 nm UV light, followed by spraying with Dragendorff reagent (Munier and Macheboeuf modification),35 or with ethanolic p-anisaldehyde/sulfuric acid reagent and final careful heating of the plates for improving selectivity.
Apparatus
The melting points were measured on an Ernst Leitz Wetzlar model 350 hot-stage microscope and are reported uncorrected. The IR spectra were recorded with a Shimadzu Prestige 21 spectrophotometer, as solid dispersions in KBr disks or as thin films held between NaCl cells. The NMR spectra were acquired at 300.13 (1H), 121.5 MHz (31P) or 75.48 (13C) MHz on a Bruker Avance spectrometer in CDCl3, unless stated otherwise. The peak for CHCl3 in CDCl3 (δH = 7.26 ppm), H3PO4 (δ31P = 0.00 ppm) and the signal of CDCl3 (δC = 77.0 ppm) were used as the corresponding internal standards. Chemical shifts are reported in parts per million on the δ scale and J-values are given in Hertz. In special cases, 31P NMR, DEPT and 2D-NMR experiments (COSY, HMBC and HMQC) were also employed. Pairs of signals marked with asterisk (*) indicate that their assignments may be exchanged. The high-resolution mass spectra were obtained with a Bruker MicroTOF-Q II instrument (Bruker Daltonics, Billerica, MA). Detection of the ions was performed in electrospray ionization, positive ion mode. Microwave-assisted reactions were performed in a CEM Discover microwave oven.2-Aminobenzaldehyde (15)17c
2-Nitrobenzaldehyde (13, 500 mg, 3.3 mmol) was added to a refluxing mixture of clean iron powder (1020 mg, 18.3 mmol) and NH4Cl (120 mg, 2.2 mmol) in EtOH:H2O (9:1, v/v, 50 mL). The reaction mixture was stirred at reflux for 2 h under an argon atmosphere; then it was allowed to cool to room temperature, filtered and the residue was washed with EtOH. The filtrate was concentrated under reduced pressure and the resulting oily residue was redissolved in EtOAc (50 mL) and washed with saturated NaHCO3 solution (5 mL). The organic layer was dried (MgSO4) and concentrated to afford 15 (385 mg, 96%), as a stinky yellow oil, which slowly crystallized on standing to give a solid with a low melting point. IR (film, ν): 3459, 3443, 3418, 3333, 3321, 3282, 3100, 2916, 2848, 2700, 2357, 1643, 1591, 1557, 1479, 1454, 1396, 1317, 1205, 1155, 1026, 883, 748, 665 and 627 cm−1. 1H NMR: δ 6.11 (br s, w1/2 = 29.2, 2H, NH2), 6.64 (d, 1H, J = 8.4, H-3), 6.75 (ddd, J = 7.8, 7.2 and 1.0, 1H, H-5), 7.31 (ddd, J = 8.4, 7.2 and 1.6, 1H, H-4), 7.48 (dd, J = 7.8 and 1.6, 1H, H-6) and 9.87 (s, 1H, CHO). 13C NMR: δ 116.0 (C-3), 116.3 (C-5), 118.8 (C-1), 135.2 (C-4), 135.7 (C-6), 149.9 (C-2) and 194.0 (CHO).
N-(2-Formylphenyl)-2-(2-nitrophenyl)acetamide (16)
A solution of 2-nitrophenylacetic acid (12, 561 mg, 3.1 mmol), pyridine (0.67 mL, 8.36 mmol) and DMF (0.071 mL, 0.93 mmol) in PhCF3 (20 mL) was stirred 1 h at room temperature. Then, a solution of 2-aminobenzaldehyde (376 mg, 3.1 mmol) in PhCF3 (10 mL) was added to the mixture, which was successively treated dropwise at 0 °C with POCl3 (0.20 mL, 2.17 mmol). The mixture was stirred 3 h at room temperature, most of the solvent was recovered under reduced pressure, saturated Na2CO3 (10 mL) was added, and the mixture was stirred for additional 30 min. The organic products were extracted with EtOAc (4 × 15 mL), and the combined organic phases were washed with brine (10 mL), dried (MgSO4), and concentrated under reduced pressure. Chromatography of the residue furnished 16 (854 mg, 97%) as a yellow solid, mp 142–144 °C. IR (KBr, ν): 3285, 3100, 3050, 2930, 2850, 2700, 1813, 1740, 1607, 1490, 1450, 1369, 1240, 1215, 1059, 1043, 1030, 968, 885, 835, 797, 758 and 698 cm−1. 1H NMR: δ 4.18 (s, 2H, NHCOCH2), 7.22 (dt, J = 7.5 and 0.9, 1H, H-4), 7.49 (dd, J = 7.6 and 1.4, 1H, H-6′), 7.54 (dt, J = 7.8 and 1.7, 1H, H-4′), 7.57 (dt, J = 7.8 and 1.8, 1H, H-5), 7.65 (dd, J = 7.6 and 1.7, 1H, H-3), 7.67 (dt, J = 7.6 and 1.4, 1H, H-5′), 8.17 (dd, J = 8.1 and 1.2, 1H, H-3′), 8.69 (d, J = 8.4, 1H, H-6), 9.85 (s, 1H, CHO) and 11.21 (br s, w1/2 = 9.3, 1H, NHCOCH2). 13C NMR: δ 43.1 (NHCOCH2), 120.0 (C-6), 121.7 (C-2), 123.1 (C-4), 125.5 (C-3′), 128.9 (C-4′), 129.5 (C-1′), 133.8 (C-5′), 133.8 (C-6′), 135.9 (C-3), 136.2 (C-5), 140.7 (C-1), 149.0 (C-2′), 168.5 (NHCOCH2) and 195.5 (COH). HRMS m/z calcd for C15H13N2O4 285.0870 [M + H]+; found: 285.0861.3-(2-Nitrophenyl)quinolin-2(1H)-one (11)
A stirred solution of carboxamidobenzaldehyde 16 (197 mg, 0.69 mmol) in anhydrous DMF (7 mL) was treated with K2CO3 (447 mg, 3.45 mmol) and the reaction mixture was heated at 100 °C for 1 h under an argon atmosphere. Once warmed to room temperature, the reaction mixture was diluted with EtOAc (50 mL) and successively washed with a saturated solution of NH4Cl (10 mL) and water (10 mL). The organic phase was concentrated in vacuo to give 11 (184 mg, 99%), as an orange-brown solid, mp 320–321 °C (lit.: 317–318 °C).20a IR (KBr, ν): 3220, 2922, 2891, 2851, 1653, 1609, 1564, 1560, 1522, 1431, 1356, 1233, 1150, 955, 928, 785, 754, 710 and 631 cm−1. 1H NMR (DMSO-d6): δ 7.24 (t, J = 7.6, 1H, H-6), 7.35 (d, J = 8.2, 1H, H-8), 7.55 (t, J = 8.2, 1H, H-7), 7.64 (dd, J = 7.3 and 1.5, 1H, H-6′), 7.67 (dt, J = 8.0 and 1.5, 1H, H-4′), 7.76 (d, J = 7.6, 1H, H-5), 7.82 (t, J = 7.3, 1H, H-5′), 8.04 (d, J = 8.0, 1H, H-3′), 8.15 (s, 1H, H-4) and 11.97 (s, 1H, NH). 13C NMR (DMSO-d6): δ 115.5 (C-8), 119.9 (C-4a), 122.7 (C-6), 124.4 (C-3′), 128.8 (C-5), 129.9 (C-4′), 131.2 (C-7), 131.4 (C-3),* 131.5 (C-1′),* 132.7 (C-6′), 134.2 (C-5′), 138.1 (C-4), 139.0 (C-8a), 149.4 (C-2′) and 160.5 (C-2). HRMS m/z calcd for C15H11N2O3 267.0764 [M + H]+; found: 267.0768.Pyridine hydrochloride (Py·HCl)
Concentrated HCl (5.12 mL) was added dropwise to a vigorously stirred solution of pyridine (5 mL) in Et2O (30 mL) at room temperature. A white precipitate was immediately formed. The slurry thus obtained was filtered and the solid product was washed with Et2O, recrystallized from CHCl3/EtOAc and further washed with Et2O. The resulting purified material was dried under vacuum and stored into a desiccator.6H-Indolo[2,3-b]quinoline (quinindoline, 8)5a
:H2O (10:1 v/v, 3 mL) and the reaction mixture was stirred at reflux for 2 h under an argon atmosphere. Then, the solids were removed by filtration and the filtrate was concentrated under reduced pressure. The residue was redissolved in EtOAc (40 mL) and washed with saturated NaHCO3 solution (2 × 4 mL). The organic layer was dried (MgSO4) and concentrated in vacuo to afford 18 (44 mg, 99%) as an orange-brown solid. Mp 224–226 °C. IR (KBr, ν): 3100, 2953, 2918, 2849, 2359, 2150, 1715, 1607, 1574, 1495, 1471, 1429, 1396, 1321, 1258, 1119, 1050, 850, 787 and 743 cm−1. 1H NMR: δ 4.26 (br s, w1/2 = 31.9, 2H, NH2), 6.82 (dd, J = 8.1 and 1.3, 1H, H-3′), 6.88 (dt, J = 7.2 and 1.3, 1H, H-5′), 7.20–7.30 (m, 3H, H-4′, H-6′ and H-6), 7.32 (d, J = 8.3, 1H, H-8), 7.48 (dt, J = 7.5 and 1.2, 1H, H-7), 7.61 (dd, J = 7.8 and 1.2, 1H, H-5), 7.91 (s, 1H, H-4) and 11.68 (br s, w1/2 = 21.6, 1H, NH). 13C NMR δ 115.6 (C-8), 117.2 (C-3′),* 119.1 (C-5′),* 120.3 (C-5a), 123.0 (C-6), 123.8 (C-1′), 127.9 (C-5), 129.5 (C-4′), 130.5 (C-7), 131.4 (C-6′), 132.7 (C-3), 137.8 (C-8a), 141.4 (C-4), 145.9 (C-2′) and 162.6 (C-2). Without further purification, the cyclization procedure of Method A was applied on 18 (14 mg, 0.06 mmol), employing anhydrous Py·HCl (1.0 g). This afforded the tetracycle 8 (12.0 mg, 93%), as a yellow solid. Alternatively, POCl3 (0.046 mL, 0.49 mmol) was added dropwise to a stirred solution of 18 (38 mg, 0.16 mmol) in MeCN (2.2 mL) at 0 °C, the tube was sealed and the mixture was heated overnight at 90 °C. After evaporation of the solvent and the POCl3, the residue was treated with a solution of DIPEA (0.175 mL, 1.00 mmol) in MeCN (3 mL) and the reaction was heated overnight at 120 °C in a sealed tube. After evaporation of the solvent and the DIPEA, the residue was chromatographed to give 8 (33 mg, 94%), as a yellow solid. In both cases, the spectroscopic data of the product were in full agreement with those of the solid obtained employing Method A and with the literature.6-Methyl-6H-indolo[2,3-b]quinoline (6-methylquinindoline, 10)10b
A mixture of 8 (10 mg, 0.046 mmol), DMC (15 μL, 0.18 mmol), calcined MgO (7 mg, 0.18 mmol) and anhydrous DMF (1 mL) was poured in a sealed reaction vessel and heated in a microwave reactor at 170 °C for 30 minutes. The slurry was cooled to room temperature, the solids filtered off and washed with EtOAc (3 × 10 mL). The organic phase was washed with H2O (5 mL), dried (MgSO4) and concentrated under reduced pressure to afford 10 (9.5 mg, 89%), as an orange-brown solid, mp 78–81 °C (lit.: 83–87 °C).14d IR (KBr, ν): 3050, 2953, 2918, 2849, 2358, 2305, 2150, 1714, 1607, 1574, 1495, 1472, 1430, 1396, 1321, 1258, 1119, 1010, 786 and 743 cm−1. 1H NMR: δ 4.01 (s, 3H, N–CH3), 7.32 (ddd, J = 7.7, 7.4 and 0.9, 1H, H-9), 7.40–7.50 (m, 2H, H-2 and H-7), 7.60 (ddd, J = 7.5, 7.3 and 1.2, 1H, H-8), 7.72 (ddd, J = 8.5, 6.7 and 1.4, 1H, H-3), 8.02 (dd, J = 8.1 and 1.3, 1H, H-1), 8.14 (d, J = 8.5, 1H, H-10), 8.17 (d, J = 7.9, 1H, H-4) and 8.73 (s, 1H, H-11). 13C NMR: δ 27.8 (N–Me), 108.7 (C-7), 118.3 (C-10b), 120.0 (C-9), 120.4 (C-10a), 121.4 (C-4), 122.9 (C-2), 124.1 (C-11a), 127.4 (C-10 and C-11), 128.1 (C-8), 128.5 (C-1), 128.9 (C-3), 142.9 (C-6a), 146.8 (C-4a) and 152.8 (C-5a).1-Methyl-3-(2-nitrophenyl)quinolin-2(1H)-one (14)12h
A solution of 11 (130 mg, 0.48 mmol), DBU (7.2 μL, 0.048 mmol) and DMF (68 μL, 0.88 mmol) in DMC (0.68 mL, 8.1 mmol) was stirred overnight at 90 °C. After evaporation of the volatiles, the residue was chromatographed to give 14 (130 mg, 95%), as an amber solid, mp 141–143 °C. IR (KBr, ν): 3250, 3063, 2957, 2917, 2849, 2359, 2342, 2330, 1648, 1593, 1526, 1454, 1354, 1238, 1121, 1040, 945, 860, 789, 754 and 721 cm−1. 1H NMR: δ 3.75 (s, 3H, N–Me), 7.29 (dt, J = 7.5 and 1.0, 1H, H-6), 7.40 (d, J = 8.4, 1H, H-8), 7.46 (dd, J = 7.7 and 1.5, 1H, H-5), 7.54 (dt, J = 7.9 and 1.4, 1H, H-4′), 7.60–7.70 (m, 3H, H-5′, H-6′, H-7), 7.40 (d, J = 8.4, 1H, H-8), 7.81 (s, 1H, H-4) and 8.07 (dd, J = 8.1 and 1.3, 1H, H-3′). 13C NMR: δ 30.0 (N–Me), 114.3 (C-8), 120.5 (C-4a), 122.4 (C-6), 124.9 (C-3′), 129.0 (C-6′), 129.1 (C-4′), 130.9 (C-7), 131.5 (C-1′),* 131.9 (C-5), 132.0 (C-3), 133.1 (C-5′),* 136.4 (C-4), 140.0 (C-8a), 149.5 (C-2′) and 160.5 (CO).
3-(2-Aminophenyl)-1-methylquinolin-2(1H)-one (19)12g,h
A stirred solution of 14 (120 mg, 0.45 mmol) in EtOH:H2O (10:1 v/v, 8 mL) was treated with NH4Cl (16.8 mg, 0.315 mmol) and iron powder (138.6 mg, 2.48 mmol). The mixture was heated to reflux for 3 h, when it was allowed to cool to room temperature and filtered. The filtrate was diluted with brine (10 mL) and organic products were extracted with EtOAc (4 × 10 mL). The combined organic phases were washed with brine (10 mL), dried (MgSO4), and concentrated under reduced pressure. The residue was passed through a short plug of silica gel, affording 19 (102 mg, 96%), as a solid, mp 163–165 °C (lit.: 172–174 °C).11b IR (KBr, film): 3416, 3335, 3223, 2916, 2849, 2361, 2328, 1643, 1584, 1568, 1557, 1495, 1454, 1304, 1236, 1038, 945, 789, 750 and 656 cm−1. 1H NMR: δ 3.82 (s, 3H, N–Me), 4.18 (br s, 2H, NH2), 6.79 (d, J = 7.7, 1H, H-3′), 6.85 (dt, J = 7.7, 1H, H-4′), 7.15–7.21 (m, 1H, H-5′), 7.22 (dd, J = 7.4 and 1.2, 1H, H-6′), 7.28 (dt, J = 7.5 and 0.8, 1H, H-6), 7.41 (d, J = 8.5, 1H, H-8), 7.55–7.65 (m, 1H, H-7), 7.61 (d, J = 7.5, 1H, H-5) and 7.80 (s, 1H, H-4). 13C NMR: δ 30.1 (N–Me), 114.1 (C-8), 117.2 (C-3′), 119.0 (C-4′), 120.7 (C-4a), 122.5 (C-6), 124.5 (C-3), 129.0 (C-5), 129.4 (C-6′), 130.5 (C-7), 131.2 (C-5′), 132.6 (C-1′), 139.5 (C-4), 139.5 (C-8a), 145.8 (C-2′) and 161.2 (CO).
Neocryptolepine (1)
POCl3 (0.1 mL, 1.08 mmol) was added dropwise to a stirred solution of 19 (85 mg, 0.36 mmol) in MeCN (4 mL) at 0 °C, the tube was sealed and the mixture was heated overnight at 90 °C. After evaporation of the solvent and the POCl3, the residue was treated with a solution of DIPEA (0.35 mL, 6.48 mmol) in MeCN (4 mL) and the reaction was heated overnight at 120 °C in a sealed tube. After evaporation of the solvent and the DIPEA, the residue was chromatographed to give 1 (50 mg, 63%), as a deep orange solid, mp 104–106 °C (lit.: 104–106 °C).8b IR (KBr, ν): 3528, 3462, 3422, 3404, 3383, 3366, 2918, 2800, 2400, 1951, 1750, 1647, 1558, 1419, 1203, 1101, 1061 and 746 cm−1. 1H NMR: δ 4.37 (s, 3H, N–Me), 7.24 (ddd, J = 7.6, 7.4 and 1.1, 1H, H-9), 7.42–7.48 (m, 1H, H-2), 7.55 (ddd, J = 7.4, 7.2 and 1.2, 1H, H-8), 7.73–7.79 (m, 3H, H-3, H-4 and H-7), 8.00 (d, J = 7.9, 1H, H-1), 8.05 (d, J = 7.6, 1H, H-10) and 8.54 (s, 1H, H-11). 13C NMR: δ 33.3 (N–Me), 114.1 (C-4), 117.7 (C-7), 119.9.4 (C-9), 120.8 (C-11a), 121.0 (C-10), 121.9 (C-2), 124.0 (C-10a), 128.1 (C-11), 128.2 (C-10b), 129.3 (C-8), 130.0 (C-1), 130.4 (C-3), 137.0 (C-4a) and 155.4 (C-6a). The spectroscopic data of the synthetic compound were in full agreement with those of the literature.Acknowledgements
The authors gratefully acknowledge Secretaría de Ciencia y Tecnología de la UNR (SECyT-UNR), Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET), and Agencia Nacional de Promoción Científica y Tecnológica (ANPCyT), Projects BIO457, PIP No. 2012-0471 and PICT No. 2011-0455, respectively, for financial support. M. V. M. and D. A. H. also thank CONICET for their fellowships.Notes and references
- (a) A. A. Appiah, The Golden Roots of Cryptolepis sanguinolenta, in Chemistry and Quality - African Natural Plant Products: New Discoveries and Challenges, ACS, Washington, D.C., 2009, ch. 13, pp. 231–239 Search PubMed; (b) F. R. Irvine, in Woody Plants of Ghana, Oxford University Press, London, 1961 Search PubMed; (c) P. T. Parvatkar and S. G. Tilve, Bioactivities and synthesis of indoloquinoline alkaloids: cryptolepine, isocryptolepine and neocryptolepine, in Bioactive Heterocycles: Synthesis and Biological Evaluation, ed. K. L. Ameta, R. P. Pawar and A. J. Domb, Nova Science Publishers, 2012, ch. 10, pp. 218–233 Search PubMed.
- (a) P. T. Parvatkar, P. S. Parameswaran and S. G. Tilve, Curr. Org. Chem., 011, 15, 1036–1057 CrossRef; (b) J. Lavrado, R. Moreira and A. Paul, Curr. Med. Chem., 2010, 17, 2348–2370 CrossRef CAS PubMed; (c) L. R. Whittell, K. T. Batty, R. P. M. Wong, E. M. Bolitho, S. A. Fox, T. M. E. Davis and P. E. Murray, Bioorg. Med. Chem., 2011, 19, 7519–7525 CrossRef CAS PubMed; (d) J. Lavrado, G. G. Cabal, M. Prudêncio, M. M. Mota, J. Gut, P. J. Rosenthal, C. Díaz, R. C. Guedes, D. J. V. A. dos Santos, E. Bichenkova, K. T. Douglas, R. Moreira and A. Paulo, J. Med. Chem., 2011, 54, 734–750 CrossRef CAS PubMed.
- (a) M. H. M. Sharaf, P. L. Schiff Jr, A. N. Tackie, C. H. Phoebe Jr and G. E. Martin, J. Heterocycl. Chem., 1996, 33, 239–243 CrossRef CAS; (b) S. D. Ablordeppey, C. D. Hufford, R. F. Borne and D. Dwama-Badu, Planta Med., 1990, 56, 416–417 CrossRef CAS PubMed; (c) A. N. Tackie, M. H. M. Sharaf, P. L. Schiff Jr, G. L. Boye, R. C. Crouch and G. E. Martin, J. Heterocycl. Chem., 1991, 28, 1429–1435 CrossRef CAS; (d) D. Dwuma-Badu, J. S. K. Ayim, N. Y. Y. Fiagbe, J. E. Knapp, P. L. Schiff Jr and D. J. Slatkin, J. Pharm. Sci., 1978, 67, 433–434 CrossRef CAS PubMed; (e) R. C. Crouch, A. O. Davis, T. D. Spitzer, G. E. Martin, M. H. M. Sharaf, P. L. Schiff Jr, C. H. Phoebe and A. N. Tackie, J. Heterocycl. Chem., 1995, 32, 1077–1080 CrossRef CAS.
- (a) J.-L. Pousset, M.-T. Martin, A. Jossang and B. Bodo, Phytochemistry, 1995, 39, 735–736 CrossRef CAS; (b) C. E. Hadden, M. H. M. Sharaf, J. E. Guido, R. H. Robins, A. N. Tackie, C. H. Phoebe Jr, P. L. Schiff Jr and G. E. Martin, J. Nat. Prod., 1999, 62, 238–240 CrossRef CAS PubMed; (c) D. M. Fort, J. Litvak, J. L. Chen, Q. Lu, P. W. Phuan, R. Cooper and D. E. Bierer, J. Nat. Prod., 1998, 61, 1528–1530 CrossRef CAS PubMed; (d) A. Paulo, E. T. Gomes and P. J. Houghton, J. Nat. Prod., 1995, 58, 1485–1491 CrossRef CAS.
- (a) G. V. Subbaraju, J. Kavitha, D. Rajasekhar and J. I. Jimenez, J. Nat. Prod., 2004, 67, 461–462 CrossRef CAS PubMed; (b) K. Cimanga, T. De Bruyne, L. Pieters, M. Claeys and A. Vlietnick, Tetrahedron Lett., 1996, 37, 1703–1706 CrossRef CAS.
- (a) M. H. M. Sharaf, P. L. Schiff Jr, A. N. Tackie, C. H. Phoebe Jr, L. Howard, C. Myers, C. E. Hadden, S. K. Wrenn, A. O. Davis, C. W. Andrews, D. Minick, R. L. Johnson, J. P. Shockcor, R. C. Crouch and G. E. Martin, Magn. Reson. Chem., 1995, 33, 767–778 CrossRef CAS; (b) A. N. Tackie, G. L. Boye, M. H. M. Sharaf, P. L. Schiff Jr, R. C. Crouch, T. D. Spitzer, R. L. Johnson, J. Dunn, D. Minick and G. E. Martin, J. Nat. Prod., 1993, 56, 653–670 CrossRef CAS; (c) K. Blinov, M. Elyashberg, E. R. Martirosian, S. G. Molodtsov, A. J. Williams, A. N. Tackie, M. H. M. Sharaf, P. L. Schiff Jr, R. C. Crouch, G. E. Martin, C. E. Hadden, J. E. Guido and K. A. Mills, Magn. Reson. Chem., 2003, 41, 577–584 CrossRef CAS; (d) M. H. M. Sharaf, P. L. Schiff Jr, A. N. Tackie, C. H. Phoebe Jr, R. L. Johnson, D. Minick, C. W. Andrews, R. C. Crouch and G. E. Martin, J. Heterocycl. Chem., 1996, 33, 789–797 CrossRef CAS.
- (a) F. Ahmed, K. Toume, T. Ohtsuki, M. Rahman, S. K. Sadhu and M. Ishibashi, Phytother. Res., 2011, 25, 147–150 CrossRef CAS PubMed; (b) S. W. Yang, M. Abdel-Kader, S. Malone, M. C. M. Werkhoven, J. H. Wisse, I. Bursuker, K. Neddermann, C. Fairchild, C. Raventos-Suárez, A. T. Menendez, K. Lane and D. G. I. Kingston, J. Nat. Prod., 1999, 62, 976–983 CrossRef CAS PubMed; (c) E. Clinquart, Bull. Acad. R. Med. Belg., 1929, 9, 627–635 CAS.
- (a) S. J. Holt and V. Petrow, J. Chem. Soc., 1948, 922–924 RSC; (b) L. Kaczmarek, R. Balicki, P. Nantka-Namirski, W. Peczynska-Czoch and M. Modarsski, Arch. Pharm., 1988, 321, 463–467 CrossRef CAS; (c) W. Peczynska-Czoch, F. Pognan, L. Kaczmarek and J. Boratynski, J. Med. Chem., 1994, 37, 3503–3510 CrossRef CAS PubMed; (d) K. Kamienska-Trela, L. Kania and L. Kaczmarek, THEOCHEM, 1995, 347, 467–476 CAS; (e) S. P. Lathrop, M. Pompeo, W.-T. T. Chang and M. Movassaghi, J. Am. Chem. Soc., 2016, 138, 7763–7769 CrossRef CAS PubMed; (f) J. Belmar and R. L. Funk, J. Am. Chem. Soc., 2012, 134, 16941–16943 CrossRef CAS PubMed; (g) K. Popov, A. Hoang and P. Somfai, Angew. Chem., 2016, 128, 1833–1836 CrossRef.
- (a) A. B. J. Bracca, D. A. Heredia, E. L. Larghi and T. S. Kaufman, Eur. J. Org. Chem., 2014, 7979–8003 CrossRef CAS; (b) E. L. Larghi, A. B. J. Bracca, A. A. Arroyo Aguilar, D. A. Heredia, J. L. Pergomet, S. O. Simonetti and T. S. Kaufman, Curr. Top. Med. Chem., 2015, 17, 1683–1707 CrossRef.
- (a) R. Engqvist and J. Bergman, Org. Prep. Proced. Int., 2004, 36, 386–390 CrossRef CAS; (b) S. Sharma and B. Kundu, Tetrahedron Lett., 2008, 49, 7062–7065 CrossRef CAS; (c) P. T. Parvatkar and S. G. Tilve, Tetrahedron Lett., 2011, 52, 6594–6596 CrossRef CAS; (d) G. A. Kraus and H. Guo, Tetrahedron Lett., 2010, 51, 4137–4139 CrossRef CAS; (e) M. K. Vecchione, A. X. Sun and D. Seidel, Chem. Sci., 2011, 2, 2178–2181 RSC.
- (a) T. Dhanabal, R. Sangeetha and P. S. Mohan, Tetrahedron, 2006, 62, 6258–6263 CrossRef CAS; (b) G. Timari, T. Soos and G. Hajós, Synlett, 1997, 1067–1068 CrossRef CAS; (c) S. Hostyn, K. A. Tehrani, F. Lemière, V. Smout and B. U. W. Maes, Tetrahedron, 2011, 67, 655–659 CrossRef CAS.
- (a) C. Shi, Q. Zhang and K. K. Wang, J. Org. Chem., 1999, 64, 925–932 CrossRef CAS PubMed; (b) T.-L. Ho and D.-G. Jou, Helv. Chim. Acta, 2002, 85, 3823–3827 CrossRef CAS; (c) P. T. Parvatkar, P. S. Parameswarana and S. G. Tilve, Tetrahedron Lett., 2007, 48, 7870–7872 CrossRef CAS; (d) M. J. Haddadin, R. M. B. Zerdan, M. J. Kurth and J. C. Fettinger, Org. Lett., 2010, 12, 5502–8527 CrossRef CAS PubMed; (e) D. Basavaiah and D. Mallikarjuna Reddy, Org. Biomol. Chem., 2012, 10, 8774–8777 RSC; (f) P. Pitchai, M. Yathisateelan, A. Nepolraj and R. M. Gengan, Indian J. Biochem. Biophys., 2015, 54, 1290–1292 Search PubMed; (g) P. M. Fresneda, P. Molina and S. Delgado, Tetrahedron Lett., 1999, 40, 7275–7278 CrossRef CAS; (h) P. M. Fresneda, P. Molina and S. Delgado, Tetrahedron, 2001, 57, 6197–6202 CrossRef CAS; (i) T. H. M. Jonckers, S. van Miert, K. Cimanga, C. Bailly, P. Colson, M.-C. De Pauw-Gillet, H. van den Heuvel, M. Claeys, F. Lemière, E. L. Esmans, J. Rozenski, L. Quirijnen, L. Maes, R. Dommisse, G. L. F. Lemière, A. Vlietinck and L. Pieters, J. Med. Chem., 2002, 45, 3497–3508 CrossRef CAS PubMed; (j) M. Miller, J. C. Vogel, W. Tsang, A. Merrit and D. J. Procter, Org. Biomol. Chem., 2009, 7, 589–597 RSC.
- Formal total syntheses. From indoles: (a) F. Portela-Cubillo, J. S. Scott and J. C. Walton, J. Org. Chem., 2008, 73, 5558–5565 CrossRef CAS PubMed; (b) H. K. Kadam, P. T. Parvatkar and S. G. Tilve, Synthesis, 2012, 44, 1339–1342 CrossRef CAS; (c) G. S. M. Sundaram, C. Venkatesh, U. K. Syam Kumar, H. Ila and H. Junjappa, J. Org. Chem., 2004, 69, 5760–5762 CrossRef CAS PubMed; (d) P. T. Parvatkar, P. S. Parameswaran and S. G. Tilve, J. Org. Chem., 2009, 74, 8369–8372 CrossRef CAS PubMed; (e) R. Ghorbani-Vaghei and S. M. Malaekehpoor, Tetrahedron Lett., 2012, 53, 4751–4753 CrossRef CAS; (f) H. K. Kadam and S. G. Tilve, J. Heterocycl. Chem., 2016, 53, 2066–2069 CrossRef CAS; From quinolones: (g) B. Bogányi and J. Kámán, Tetrahedron, 2013, 69, 9512–9519 CrossRef. From benzenoids: (h) M. Alajarín, P. Molina and A. Vidal, J. Nat. Prod., 1997, 60, 747–748 CrossRef.
- (a) O. Amiri-Attou, T. Terme and P. Vanelle, Synlett, 2005, 3047–3050 CAS; (b) S. Ali, Y.-X. Li, S. Anwar, F. Yang, Z.-S. Chen and Y.-M. Liang, J. Org. Chem., 2012, 77, 424–431 CrossRef CAS PubMed; (c) Z. Yan, C. Wan, J. Wan and Z. Wang, Org. Biomol. Chem., 2016, 14, 4405–4408 RSC; (d) S. Badigenchala, V. Rajeshkumar and G. Sekar, Org. Biomol. Chem., 2016, 14, 2297–2305 RSC.
- S. O. Simonetti, E. L. Larghi, A. B. J. Bracca and T. S. Kaufman, Org. Biomol. Chem., 2012, 10, 4124–4134 CAS.
- (a) R. H. Dave and B. D. Hosangadi, Tetrahedron, 1999, 55, 11295–11308 CrossRef CAS; (b) A.-H. Li, E. Ahmed, X. Chen, M. Cox, A. P. Crew, H.-Q. Dong, M. Jin, L. Ma, B. Panicker, K. W. Siu, A. G. Steinig, K. M. Stolz, P. A. R. Tavares, B. Volk, Q. Weng, D. Werner and M. J. Mulvihill, Org. Biomol. Chem., 2007, 5, 61–64 RSC; (c) C. Zhang, C. Kanta De and D. Seidel, Org. Synth., 2012, 89, 274–282 CrossRef CAS.
- (a) S. Chandrappa, T. Vinaya, T. Ramakrishnappa and K. S. Rangappa, Synlett, 2010, 3019–3022 CAS; (b) B. Gabriele, G. Salerno, L. Veltri, M. Costa and C. Massera, Eur. J. Org. Chem., 2001, 4607–4613 CrossRef CAS; (c) X. Pang, Z. Lou, M. Li, L. Wen and C. Chen, Eur. J. Org. Chem., 2015, 3361–3369 CrossRef CAS.
- (a) S. Bhandari and S. Ray, Synth. Commun., 1998, 28, 765–771 CrossRef CAS; (b) G. Bartoli, M. Bosco, A. Carlone, R. Dalpozzo, E. Marcantoni, P. Melchiorre and L. Sambri, Synthesis, 2007, 3489–3496 CrossRef CAS; (c) J. S. Carey, D. Laffan, C. Thomson and M. T. Williams, Org. Biomol. Chem., 2006, 4, 2337–2347 RSC; (d) K. Alfonsi, J. Colberg, P. J. Dunn, T. Fevig, S. Jennings, T. A. Johnson, H. P. Kleine, C. Knight, M. A. Nagy, D. A. Perry and M. Stefaniak, Green Chem., 2008, 10, 31–36 RSC; (e) H. Ueki, T. K. Ellis, M. A. Khan and V. A. Soloshonok, Tetrahedron, 2003, 59, 7301–7306 CrossRef CAS; (f) R. J. Ternansky, C. L. Jordan, J. A. Eudaly and J. S. Kasher, J. Med. Chem., 1993, 36, 2332–2334 CrossRef CAS PubMed.
- M. L. Nielsen, J. V. Pustinger Jr and J. Strobel, J. Chem. Eng. Data, 1964, 9, 167–170 CrossRef CAS.
- (a) A. V. Aksenov, A. N. Smirnov, N. A. Aksenov, I. V. Aksenova, J. P. Matheny and M. Rubin, RSC Adv., 2015, 5, 8647–8656 RSC; (b) C. Capello, U. Fischer and K. Hungerbühler, Green Chem., 2007, 9, 927–934 RSC.
- (a) K. K. Park and J. Y. Jung, Heterocycles, 2005, 65, 2095–2105 CrossRef CAS; (b) M. Rowley, J. J. Kulagowski, A. P. Watt, D. Rathbone, G. I. Stevenson, R. W. Carling, R. Baker, G. R. Marshall, J. A. Kemp, A. C. Foster, S. Grimwood, R. Hargreaves, C. Hurley, K. L. Saywell, M. D. Tricklebank and P. D. Leeson, J. Med. Chem., 1997, 40, 4053–4068 CrossRef CAS PubMed.
- (a) D. V. Kravchenko, Y. A. Kuzovkova, V. M. Kysil, S. E. Tkachenko, S. Maliarchouk, I. M. Okun, K. V. Balakin and A. V. Ivachtchenko, J. Med. Chem., 2005, 48, 3680–3683 CrossRef CAS PubMed; (b) V. V. Mulwad and M. V. Lohar, J. Heterocycl. Chem., 2003, 42, 2567–2572 Search PubMed.
- (a) X. Bu, L. W. Deady and W. A. Denny, Aust. J. Chem., 2000, 53, 143–147 CrossRef CAS; (b) Ionic Liquids in Synthesis, ed. P. Wasserscheid and T. Welton, Wiley-VCH, Weinheim, Germany, 2nd edn, 2008, pp. 319–321 Search PubMed.
- R. Oberkobusch, Chem. Ber., 1953, 86, 975–978 CrossRef CAS.
- (a) S. Yu, Y. Li, X. Zhou, H. Wang, L. Kong and X. Li, Org. Lett., 2016, 18, 2812–2815 CrossRef CAS PubMed; (b) L. Shi and B. Wang, Org. Lett., 2016, 18, 2820–2823 CrossRef CAS PubMed; (c) A. Khorshidi and K. Tabatabaeian, J. Mol. Catal. A: Chem., 2011, 344, 128–131 CrossRef CAS; (d) C. Venkatesh, G. S. M. Sundaram, H. Ila and H. Junjappa, J. Org. Chem., 2006, 71, 1280–1283 CrossRef CAS PubMed.
- S. T. Gadge, A. Mishra, A. L. Gajengi, N. V. Shahi and B. M. Bhanage, RSC Adv., 2014, 4, 50271–50276 RSC.
- (a) R. Bernini, F. Crisante and M. C. Ginnasi, Molecules, 2011, 16, 1418–1425 CrossRef CAS PubMed; (b) W.-C. Shieh, S. Dell and O. Repic, Org. Lett., 2001, 3, 4279–4281 CrossRef CAS PubMed.
- (a) P. Tundo, L. Rossi and A. Loris, J. Org. Chem., 2005, 70, 2219–2224 CrossRef CAS PubMed; (b) P. Tundo, Pure Appl. Chem., 2001, 73, 1117–1124 CrossRef CAS.
- (a) E. Chrystiuk and A. Williams, J. Am. Chem. Soc., 1987, 109, 3040–3046 CrossRef CAS; (b) T. H. Black, S. M. Arrivo, J. S. Schumm and J. M. Knobeloch, J. Org. Chem., 1987, 52, 5425–5430 CrossRef CAS.
- (a) J. Fossey, A. Loupy and H. Strszelecka, Tetrahedron, 1981, 37, 1935–1941 CrossRef CAS; (b) M. F. Pereira, V. Thiéry and T. Besson, Tetrahedron Lett., 2007, 48, 7657–7659 CrossRef CAS; (c) M. J. Reyes, F. Delgado, M. L. Izquierdo and J. Alvarez-Builla, Tetrahedron, 2002, 58, 8573–8579 CrossRef; (d) H. Wu, F. Xue, X. Xiao and Y. Qin, J. Am. Chem. Soc., 2010, 132, 14052–14054 CrossRef CAS PubMed.
- (a) L. G. Mardenborough, X. Y. Zhu, P. Fan, M. R. Jacob, S. I. Khan, L. A. Walker and S. Y. Ablordeppey, Bioorg. Med. Chem., 2005, 13, 3955–3963 CrossRef CAS PubMed; (b) T.-L. Ho and D.-G. Jou, Helv. Chim. Acta, 2002, 85, 3823–3827 CrossRef CAS; (c) T. K. Lai, A. Chatterjee, J. Banerji, D. Sarkar and N. Chattopadhyay, Helv. Chim. Acta, 2008, 91, 1975–1983 CrossRef CAS.
- (a) W.-C. Shieh, S. Dell, A. Bach, O. Repic and T. J. Blacklock, J. Org. Chem., 2003, 68, 1954–1957 CrossRef CAS PubMed; (b) S. Ouk, S. Thiébaud, E. Borredon and B. Chabaud, Synth. Commun., 2005, 35, 3021–3026 CrossRef CAS.
- (a) T. N. Glasnov, J. D. Holbrey, C. O. Kappe, K. R. Seddon and T. Yan, Green Chem., 2012, 14, 3071–3076 RSC; (b) M. L. Laurila, N. A. Magnus and M. A. Staszak, Org. Process Res. Dev., 2009, 13, 1199–1201 CrossRef CAS; (c) H. Yan, L. Zeng, Y. Xie, Y. Cui, L. Ye and S. Tu, Res. Chem. Intermed., 2016, 42, 5951–5960 CrossRef CAS.
- (a) N. Vôute, D. Philp, A. M. Z. Slawin and N. J. Westwood, Org. Biomol. Chem., 2010, 8, 442–450 RSC; (b) Y.-L. Chen, H.-M. Hung, C.-M. Lu, K.-C. Li and C.-C. Tzeng, Bioorg. Med. Chem., 2004, 12, 6539–6546 CrossRef CAS PubMed; (c) H. Mastalarz, R. Jasztold-Howorko, F. Rulko, A. Croisy and D. Carrez, Arch. Pharm., 2004, 337, 434–439 CrossRef CAS PubMed.
- H. Wagner, S. Bladt and V. Rickl. Drug Analysis: A Thin Layer Chromatography Atlas, Springer, Heidelberg, Germany, 2nd edn. 2009, p. 360 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Selected spectra of intermediates and final product. See DOI: 10.1039/c7ra05349e |
| ‡ Present address: Departamento de Química, Universidad Nacional de Río Cuarto, Agencia Postal No. 3, X5804BYA, Río Cuarto, Argentina. |
| This journal is © The Royal Society of Chemistry 2017 |