
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tetraphenylethene–diyne hybrid nanoparticles from Glaser-type dispersion polymerization†
Audrey Picard-Lafond,
Maxime Daigle and
Jean-François Morin *
*
Département de Chimie, Centre de Recherche sur les Matériaux Avancés (CERMA), Université Laval, 1045 Avenue de la Médecine, Pavillon Alexandre-Vachon, Québec, QC G1V 0A6, Canada. E-mail: jean-francois.morin@chm.ulaval.ca
First published on 20th July 2017
Abstract
Organic-based nanoparticles hold great potential for optoelectronics and biomedicine as they may provide optical properties in the visible range and notable advantages over inorganic counterparts. In this report, we exploit aggregation-induced emission (AIE) from the well-known tetraphenylethene (TPE) to yield photoluminescent polymer particles by a dispersion polymerization route. 1,1-Bis(4-ethynylphenyl)-2,2-diphenylethene is polymerized by a Glaser-type dispersion polymerization, providing nanoparticles of 170–600 nm with low polydispersity by simply varying monomer loadings. Optical characterization reveals emission bands at 500 nm and 600 nm, where UV irradiation of the particles causes the 500 nm band to increase relative to the 600 nm one. Structural characterization by 13C MAS-NMR allows confirming that this change upon UV irradiation is not induced by a chemical modification, thus suggesting a physical change in the particle's morphology. Altogether, the results of this work allow enlarging the library of TPE-comprising particles, but also enhance the variety of particles synthesized from Glaser-type dispersion polymerization. The study also allows gaining insight into the photochemical stability of TPE-containing particles, where the non-destructive UV irradiation lets us plan the development of new polyyne/TPE hybrid systems.
Introduction
Many organic-based nanoparticles with optical activity in the visible region are being studied to replace the traditional semiconductor quantum dots (QDs). Although QDs present advantageous properties, their toxicity hampers their implementation in biological applications. Carbon nanoparticles are organic counterparts that have attracted much attention since their discovery in 2004 (ref. 1) due to their high stability, low cytotoxicity and their tuneable photoluminescence (PL) properties.2–4 However, their preparation often requires harsh conditions.2,3 In addition to carbon nanoparticles, conjugated polymer nanoparticles (CPNs) are also interesting counterparts to QDs due to their low cytotoxicity and tuneable optical properties for electronic devices and biological applications.5–7 CPNs can be produced from several polymerization processes occurring in disperse media. However, methods such as emulsion and re-precipitation are known to yield high polydispersity.5,8,9 On the other hand, the dispersion polymerization method is attractive as it yields polymer particles with precise and reproducible dimensions for many metal-catalyzed polycondensation reactions such as Suzuki–Miyaura, Sonogashira and Heck.10–12 Recently, our group has used a Pd/Cu-catalyzed Glaser-type coupling of alkyne-rich units in dispersion medium as a hybrid approach to yield carbon-rich nanoparticles with tunable size.13 The main drawback of the method is the difficulty of preparing materials that absorb or emit light in the visible range, which is the main focus of studying organic-based nanoparticles. Although photoluminescence could be induced by a simple click functionalization with a fluorescent ligand, our group has been focusing on hosting the photoluminescence to the core of these particles instead of their surface. The interest of an intrinsically luminescent core is to provide optical properties while keeping the clickable surface available for other purposes such as biocompatibility or electroactivity.Aggregation-induced emission (AIE) is an interesting concept to keep in mind while synthesizing optically active materials in their aggregated state, since it contravenes the opposite phenomenon of aggregation-caused quenching (ACQ).14 Recently, Tang's group reported the use of the dispersion polymerization to yield AIE-active conjugated polymer nanoparticles by means of geminal Suzuki cross-coupling.15 This application of AIE-active units inspired us to adapt our Glaser-type dispersion polymerization with an AIE-active compound instead of a modest benzene ring as a linker between polyyne units (Fig. 1A).13 Tetraphenylethene (TPE) is the simplest and best known AIE compound and should suit well to induce the looked-for photoluminescence. With this objective, we synthesized poly(1,1-bis-(4-ethynylphenyl)-2,2-diphenylethene) nanoparticles (P1) from the Pd/Cu-catalysed homocoupling of 1,1-bis(4-ethynylphenyl)-2,2-diphenylethene (compound 1, Scheme 1). This achievement allows refining the library of AIE-active polymer nanoparticles and their optical behavior, but also serves as a benchmark to study the chemical stability of the TPE-containing particles upon UV exposure.
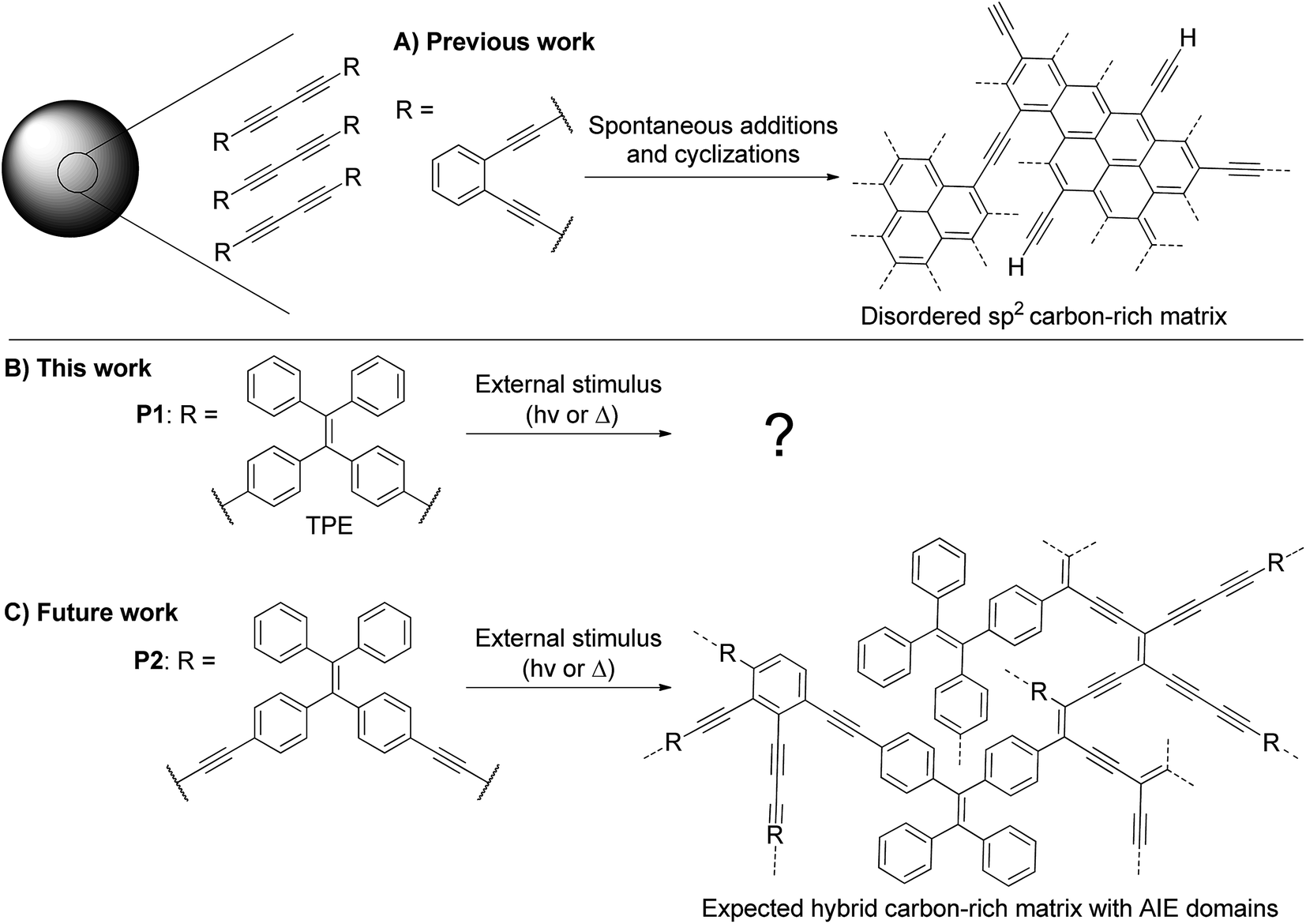 | ||
| Fig. 1 (A) Previously reported transformation of alkyne-rich particles with an ortho-substituted benzene ring as a linker to form carbon-rich particles.13 (B) Presented work on the transformation of diyne-containing TPE particles upon UV irradiation. (C) Upcoming work on the transformation of tetrayne-rich particles comprising TPE moieties in order to form a hybrid AIE/carbon-rich structure with distinct optical properties. | ||
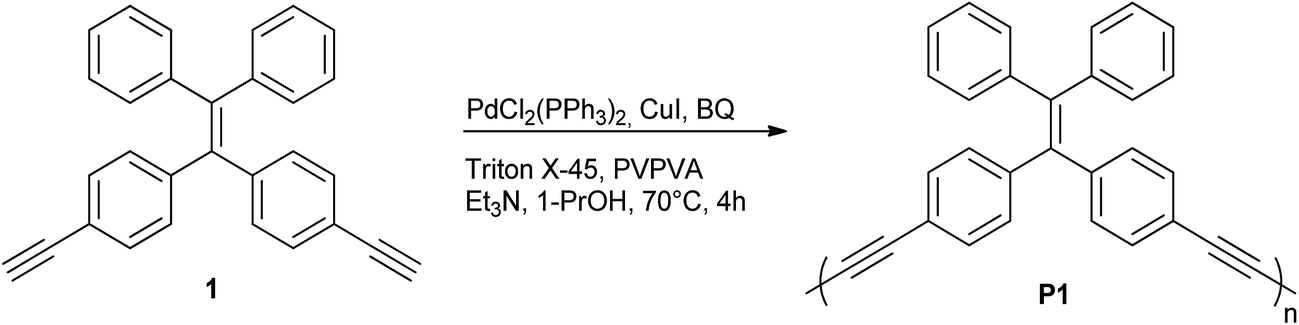 | ||
| Scheme 1 Synthesis of P1 particles from Glaser-type Pd/Cu-catalyzed homocoupling dispersion polymerization. | ||
Our group is highly interested in polyyne-containing materials as they are metastable and may polymerize when exposed to heat or UV light.16 However, the ethylenic double bond of TPE is also known to break under such UV exposure.17 Thus, the structure of P1 is ideal to study the photochemical stability of TPE-containing particles, since the internal diyne moieties are less likely to cause interference compared to longer polyynes. As an upcoming work, we plan on developing poly(1,1-bis(4-butadinylphenyl)-2,2-diphenylethene) nanoparticles (P2, Fig. 1C), which contain more reactive tetraynes in between the TPE units. With an appropriate stimulus, the conjugation of the system may be altered to modulate the photoluminescence properties of the nanoparticles (Fig. 1C).18,19 However, the behavior of TPE-containing particles under UV irradiation must first be investigated independently from the octatetrayne reactivity to ensure success of this long-term goal. It is in this perspective that P1 serves as a perfect primary candidate.
Herein, we report TPE-containing polymer particles (P1) as a new material whose size can be modulated from 170 to 600 nm with low polydispersity simply by varying the monomer loading. Furthermore, the study describes the structural and optical characterization of these particles as their fluorescence changes upon UV exposure without chemically disturbing their polymeric backbone.
Results and discussion
First, the monomer derivative (compound 1) required for the synthesis of P1 particles may be acquired in few synthetic steps as previously reported in the literature.20 To the best of our knowledge, it has never been applied to polymerization in dispersed media. As stated earlier, dispersion polymerization conditions suitable for a Glaser-type homocoupling of terminal alkynes have recently been reported by our group.13 These conditions include PdCl2(PPh3)2 and CuI as catalysts, poly(1-vinylpyrrolidone-co-vinyl acetate) (PVPVA) and Triton X-45 as stabilizers, 1-propanol as the reaction solvent, trimethylamine (Et3N) as a base and p-benzoquinone (BQ) as an oxidant. Although these conditions have only been applied for homopolymerization of 1,2-diethylbenzene, 1,2-bis(trimethylsilylbutadiynyl)benzene and 1,3-bis(trimethylsilylbutadiynyl)benzene, they were found to apply very well to 1 (Scheme 1). The TEM image shown in Fig. 2A testifies of the success of these conditions for the polymerization of 1 at a 6.4 mM concentration into P1 particles with an average diameter of 392 ± 21 nm. The size distribution histogram testifies of the uniformity of the particles' size with a polydispersity index (PDI) of 5% (Fig. S3†).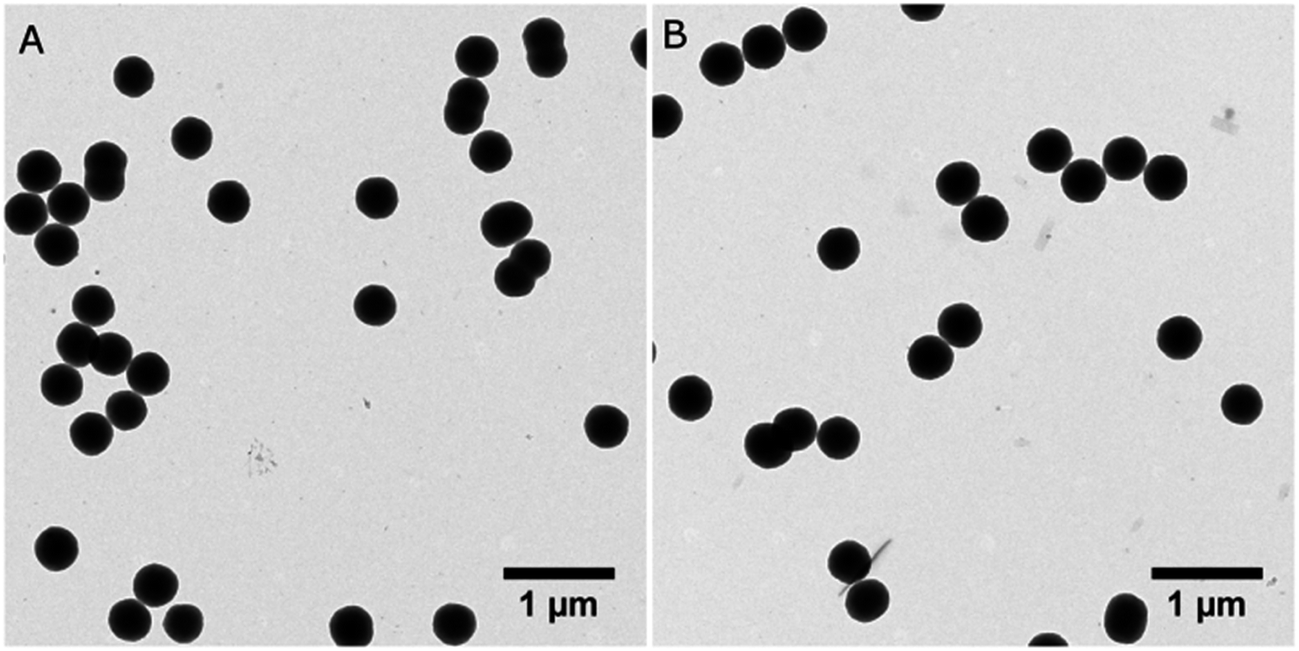 | ||
| Fig. 2 TEM images of P1 synthesized at a 6.4 mM concentration of 1 on (A) 102 mmol scale and (B) 1.02 mol scale. | ||
Dispersion polymerization is known to be a versatile method as it can yield sizes from 100 nm to multiple microns by simple adjustments of the synthetic parameters such as the amounts of monomer, base, catalysts, stabilizers, etc.21 Accordingly, we studied our system's capacity towards size control using monomer 1 concentrations below and over our initial 6.4 mM. The advantage of this Glaser-type coupling compared to heterocoupling polycondensations is the absence of stoichiometry imbalance between two co-monomers. This homocoupling particularity renders the weighting of milligram scale loadings less troublesome and thus allows testing lower concentrations in small scale synthesis. Fig. 3 shows our complete study of the effect of monomer concentration on the resulting particle size. One can observe the success of using low concentrations to yield P1 particles. Polymer particles synthesized from catalyzed polycondensation dispersion polymerizations are rarely reported at monomer concentrations below 2 mM.10–12,15 Our method was successful for a concentration as low as 1 mM to yield 172 ± 16 nm P1 particles. The PDI of the resulting sample is slightly higher (9%) but can still be considered reasonable. Our attempt to decrease the concentration to 0.5 mM did yield P1 particles but the size distribution of the sample is broad with a PDI of 26%. Besides, higher monomer concentrations up to 9.6 mM were also used for P1 synthesis. The overall results showed an increase in the average diameter with higher concentrations, as expected for most dispersion polymerization processes.22 Higher concentrations were not investigated further as it is known that bidispersity is often obtained from these syntheses.10,12,13,15 Nevertheless, the Glaser-type polymerization of 1 in dispersion conditions has proven successful for a broad range of concentrations to yield P1 with only slight polydispersity. TEM images and size distribution histograms of all tested concentrations are reported in Fig. S2 and S3.†
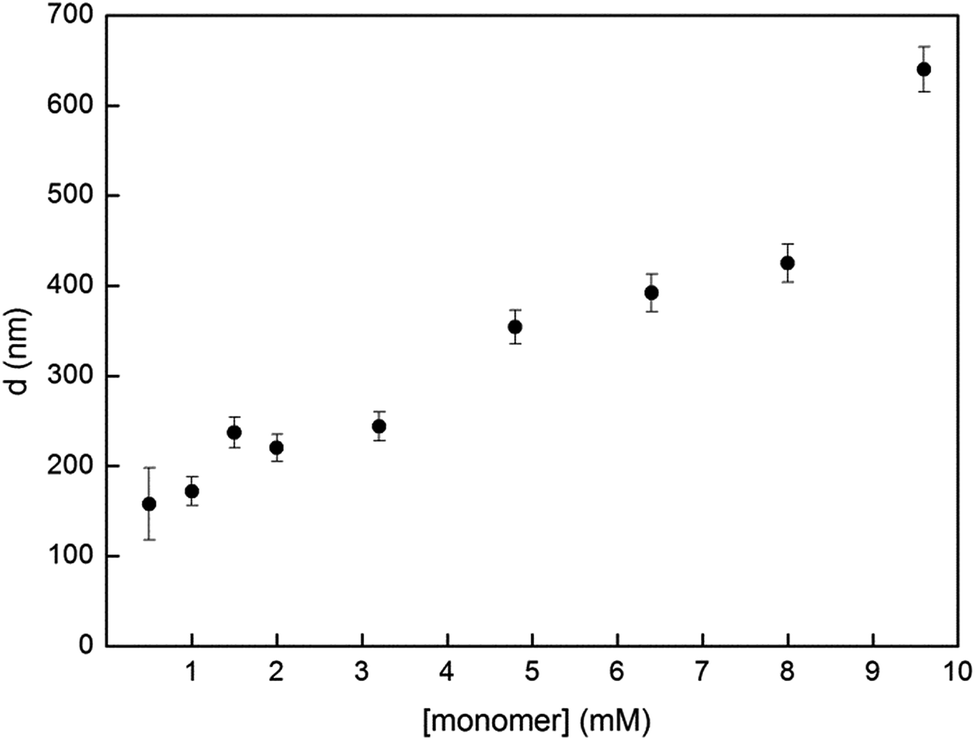 | ||
| Fig. 3 Particle diameter d of P1 as a function of monomer 1 concentration. Error bars represent the polydispersity as standard deviation from the mean diameter. More information regarding each data point is found in Fig. S2 and S3.† | ||
As stated previously, an objective of this work is to study the behavior of P1 under UV irradiation in order to assess the compatibility of TPE moieties with the tetrayne polymerization process considered for P2 in Fig. 1. Therefore, P1 is to be characterized by various techniques before and after a photochemical treatment to assess any change whatsoever. In order to do so, quite a large amount of material is required and the conditions originally intended for a 20–150 mmol scale must be adjusted to the mole level. Interestingly, a 10-fold scale-up of the polymerization procedure was successfully completed, yielding P1 particles of 406 ± 23 nm in diameter, representing a 6% PDI (Fig. 1B and S1†). XPS analysis of this sample confirmed that no residual catalyst is found in the resulting material, as it is composed of 90.2% of carbon, 8.2% of oxygen and 1.6% of nitrogen (Fig. S4†). Oxygen and nitrogen contents are most likely due to residual PVPVA incorporated to the particles.
Irradiation of P1 particles was completed by exposure of the dispersion to 300 nm light (Scheme 2). This treatment did not alter the shape nor the size of the particles (denoted P1′) as observed by TEM analysis (Fig. S5†). However, some changes were observed in the optical properties. The absorption spectra of 1, P1, and P1′ are presented in Fig. 4A. The monomer displays absorption bands at 208, 250 and 326 nm. As for P1, the sample has a strong absorption band centered at 415 nm, which decreases in intensity upon the UV treatment. Fluorescence spectra of 1, P1 and P1′ are provided in Fig. 4B. It must be mentioned that the spectrum of 1 was recorded in the solid-state, as its AIE characteristics implies low luminescence in solution. The aggregated state of monomer 1 presents a broad emission peak centered at 500 nm, whereas P1 shows an interesting feature of dual emission at 500 and 600 nm. This may suggest the presence of more than one species in the P1 backbone. Upon UV irradiation, the overall emission intensity of P1′ increases and the ratio of the two bands changes, providing a stronger contribution at 500 nm compared to the 600 nm band (Fig. S6†).
 | ||
| Scheme 2 Exposition of P1 to UV irradiation. | ||
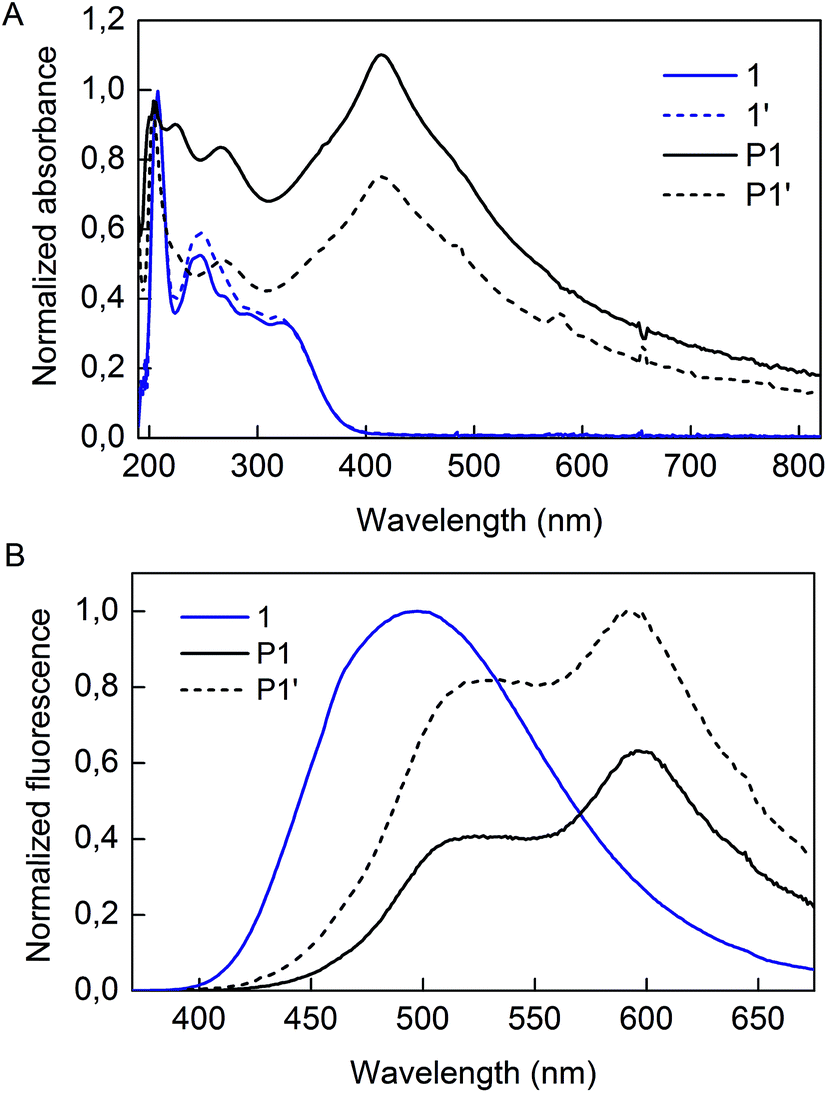 | ||
| Fig. 4 (A) Absorption spectra of 1, 1′, P1 and P1′ dispersed in 1-PrOH, (B) fluorescence spectra of 1, P1 and P1′. The spectrum of 1 is taken in the solid state with an excitation wavelength of 350 nm, whereas spectra of P1 and P1′ are taken as dispersion in 1-PrOH at an excitation wavelength of 380 nm. | ||
Our approach to investigate this optical alteration upon UV exposure is to assess whether it is caused by a chemical or structural change. First, to confirm whether alkyne polymerization or TPE side reactions occur in our UV treatment, monomer 1 was also subjected to UV irradiation and its spectrum was collected (denoted 1′ in Fig. 4A). Irradiation induces only a slightly minor intensity change for the 250 nm band. Also, elution of compound 1′ as a single spot on a TLC plate confirmed that no polymerization or decomposition of 1 occurred during the treatment. Thus, by the results of this minute experiment, we find it unlikely that the internal diynes or TPE moieties of P1 would react under UV exposure, considering the inertness of monomer 1 itself. Adding to this, the lack of new absorption bands upon irradiation of either 1 or P1 informs us that formation of new chemical species by the process is unlikely. Our hypothesis for the origin of the fluorescence alteration from P1 to P1′ is that a morphological change occurs inside the polymer backbone.
In order to confirm that the photoluminescence change from P1 to P1′ is associated to a morphological change rather than a chemical one, structural characterization techniques must be employed. Solid-state 13C MAS-NMR is an efficient technique for analysis of insoluble compounds and to prevent impacting the morphologies of samples upon solubilization. Fig. 5 shows the NMR spectra of P1 and P1′ particles (i.e. before and after UV irradiation). In the spectrum of P1, one can easily differentiate alkyne carbons (75.7 and 83.0 ppm) from the aromatics (115–140 ppm) and the alkene one (143.7 ppm) that are to be expected from the polymer backbone.20 Regarding the spectrum of P1′, no significant change in the ratio or broadness of the bands is observed as a result of the UV treatment. This can clearly confirm the optical changes in P1 arising from UV exposure are not induced by a chemical change. This, once again, leads us to anticipate a morphological change in the particles' backbone.
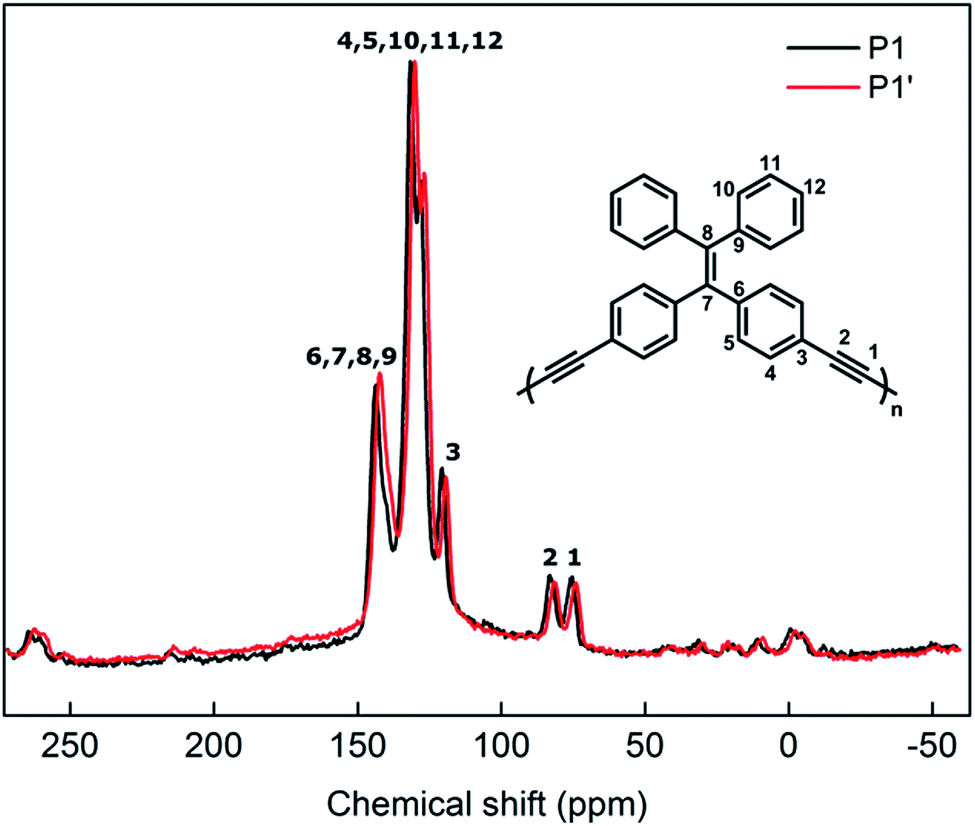 | ||
| Fig. 5 Solid-state MAS 13C NMR spectra of dried P1 and P1′. | ||
It has been reported previously that simple TPE derivatives show variable fluorescence by changing its morphology. Dong et al. reported that di(biphenyl-4-yl)diphenylethene can exhibit a bluer PL in its amorphous phase, whereas crystalline aggregates are redder emitters by 50 nm.23 Furthermore, its switching emission can be induced by solvent vapor, mechanical and thermal stimuli.24 These previously reported studies lead us to the hypothesis that an UV stimulus may also allow morphology changes in TPE aggregates and thus flexible PL properties.
While the process of dispersion polymerization yields quite spherical and amorphous nanoparticles thanks to the presence of stabilizers, crystalline spherulite particles have also been reported by this process.25 XRD analysis of P1 particles confirmed that the latter are indeed amorphous, which is corroborated by their fairly spherical shape (Fig. S7†). As for P1′ particles, they also show an amorphous XRD pattern, implying that no periodical arrangement arises from the UV treatment. Still, a certain degree of order may be induced by UV light at short distances. An important observation is that the ethylenic double bond of TPE can be broken by UV irradiation, leading to configurational isomerization.17 In our case, considering the symmetry of monomer 1, both isomers are equivalent and should not contribute to the formation of distinct species in the polymeric backbone. However, this breakable bond may allow enough intramolecular rotation to induce a slight structural change in the overall morphology, perceptible by enhancement of the overall fluorescence and increase of the 500 nm emission band to the detriment of the 600 nm one. Nonetheless, this hypothesis has yet to be confirmed by further characterizations as the mobility of the polymer chains in this aggregated state is not known.
Inspired by the effect of a thermal stimulus on simple TPE derivatives, we wondered if a thermal annealing of P1 would yield similar results as the ones obtained from our UV irradiation experiment. A dispersion of P1 in 1-PrOH was thus refluxed for 5 h and washed with pure 1-PrOH. TEM of the resulting samples confirmed that P1 particles have collapsed under this thermal treatment (Fig. S8†). Also, fluorescence measurements showed a decrease of the overall emission intensity and no variation in the PL bands ratio (Fig. S9†). Thus, the results obtained from UV irradiation seem unique as they alter the optical properties without breaking the spherical integrity of the particles.
In conclusion, the synthesis and characterization of P1 particles allowed us to achieve two distinct objectives. First, AIE-active nanoparticles can be obtained from a Glaser-type Pd/Cu-catalyzed homocoupling dispersion polymerization. TPE-containing particles from this process have never been reported before. Moreover, the size of the particles can be modulated from 170 to 600 nm with low polydispersity quite easily from monomer concentrations between 1.0 mM and 9.6 mM. Secondly, structural characterizations of P1 and P1′ confirm that no chemical changes occur under UV irradiation, evidencing that the polymeric backbone does not decompose or polymerize during the treatment. Regarding this study as a preliminary work for the synthesis of P2, it allows to settle that any polymerization obtained in this impending system will indeed be induced by octatetrayne moieties and not side reactions involving the TPE structure. This study also allowed investigating new optical properties arising from a morphological change following UV exposure. This observation may be analogous to the ones obtained for simple TPE-derived molecules exposed to thermal, mechanical and solvent vapor stimuli. Yet, more studies are to be undertaken to confirm this hypothesis, as more knowledge has to be collected regarding mobility of these polymer chains in aggregated states.
Experimental section
Chemicals and materials
Chemical reagents were purchased from Sigma-Aldrich Co. Canada and were used as received, excluding p-benzoquinone which was recrystallized in water before use. Solvents for general organic synthesis were obtained from Fisher Scientific and purified with a Solvent Purifier System (Vacuum Atmosphere Co., Hawthorne, USA). 1-Propanol (1-PrOH) and triethylamine (Et3N) used for Pd/Cu-catalyzed coupling reactions were degassed 20 min prior to use. All reactions were performed in dry glassware under positive nitrogen pressure. Analytical thin-layer chromatography was performed with silica gel 60 F254, 0.25 mm pre-coated TLC plates (Silicycle, Québec, Canada).Instrumentation
FT-IR was recorded in ATR mode using a Thermo-Nicolet Magna 850 infrared spectrometer equipped with Golden Gate. UV-visible absorption spectroscopy was performed with a Varian diode-array spectrophotometer (Cary 500 model). Fluorescence spectroscopy was performed using a Fluorolog 3, Jobin-Yvon Horiba. Transmission electron microscopy (TEM) images were taken using a JEOL apparatus, model JEM-1230. The samples were prepared from a particle dispersion dropped and dried onto a copper grid pre-coated with amorphous carbon. A PHI 5600-ci spectrometer (Physical Electronics U.S.A.) was used to perform XPS analysis. A standard aluminum X-ray source (1486.6 eV) was used for the survey spectrum (1400–0 eV). Charge neutralization was not applied. Detection was performed at 45° relative to the surface normal of a 0.5 mm2 area. The spectrometer work function was adjusted to give 285.0 eV for the main C 1s peak. Solid-state 13C MAS-NMR spectra were recorded at 25 °C using a 300 MHz (7.1 T) Bruker Advance spectrometer (Milton, Ontario, CA). The probe was tuned to 75.7 MHz for 13C detection. The powder samples were packed in 4 mm zirconia rotors and spun at 10 kHz. The spectra were collected with a pulse length of 4.1 μs for a 90° pulse with a recycle delay of 30 s. A total of 4475 and 500 scans were acquired for P1 and P1′ respectively. The chemical shifts were referenced relative to external adamantane (38.56 ppm). The powder X-ray diffraction (XRD) was recorded on Siemens X-rays Diffractometer (Model S3 D5000, Cu Kα, λ = 1.5406 Å) at 40 kV and 40 mA.Synthesis
Acknowledgements
This work was supported by NSERC through a Discovery Grant. We thank Richard Janvier (U. Laval) for help with TEM experiments, Rodica Plesu (U. Laval), Pierre Audet (U. Laval), François Paquet-Mercier (U. Laval), Denis Boudreau (U. Laval) and Pascale Chevalier (U. Laval) for their help in spectroscopic characterization. A. P.-L. thanks the NSERC and FQRNT for a M.Sc. scholarship.References
- X. Xu, R. Ray, Y. Gu, H. J. Ploehn, L. Gearheart, K. Raker and W. A. Scrivens, J. Am. Chem. Soc., 2004, 126, 12736–12737 CrossRef CAS PubMed.
- A. Kelarakis, MRS Energy & Sustainability, 2014, 1, 1–15 Search PubMed.
- H. Li, Z. Kang, Y. Liu and S.-T. Lee, J. Mater. Chem., 2012, 22, 24230–24253 RSC.
- O. Kozák, M. Sudolská, G. Pramanik, P. Cígler, M. Otyepka and R. Zbořil, Chem. Mater., 2016, 28, 4085–4128 CrossRef.
- J. Pecher and S. Mecking, Chem. Rev., 2010, 110, 6260–6279 CrossRef CAS PubMed.
- C. Wu and D. T. Chiu, Angew. Chem., Int. Ed., 2013, 52, 3086–3109 CrossRef CAS PubMed.
- A. Reisch and A. S. Klymchenko, Small, 2016, 12, 1968–1992 CrossRef CAS PubMed.
- M. C. Baier, J. Huber and S. Mecking, J. Am. Chem. Soc., 2009, 131, 14267–14273 CrossRef CAS PubMed.
- C. Wu, C. Szymanski, Z. Cain and J. McNeill, J. Am. Chem. Soc., 2007, 129, 12904–12905 CrossRef CAS PubMed.
- A. J. C. Kuehne, M. C. Gather and J. Sprakel, Nat. Commun., 2012, 3, 1–7 Search PubMed.
- N. Anwar, A. Rix, W. Lederle and A. J. C. Kuehne, Chem. Commun., 2015, 51, 9358–9361 RSC.
- S. Ciftci and A. J. C. Kuehne, Macromolecules, 2015, 48, 8389–8393 CrossRef CAS.
- A. Picard-Lafond and J.-F. Morin, Langmuir, 2017, 33, 5385–5392 CrossRef CAS PubMed.
- S. Chen, H. Wang, Y. Hong and B. Z. Tang, Mater. Horiz., 2016, 3, 283–293 RSC.
- T. Chen, H. Yin, Z.-Q. Chen, G.-F. Zhang, N.-H. Xie, C. Li, W.-L. Gong, B. Z. Tang and M.-Q. Zhu, Small, 2016, 12, 6547–6552 CrossRef CAS PubMed.
- E. T. Chernick and R. R. Tykwinski, J. Phys. Org. Chem., 2013, 26, 742–749 CrossRef CAS.
- Z. Yang, W. Qin, N. L. C. Leung, M. Arseneault, J. W. Y. Lam, G. Liang, H. H. Y. Sung, I. D. Williams and B. Z. Tang, J. Mater. Chem. C, 2016, 4, 99–107 RSC.
- Z. Zhao, H. Su, P. Zhang, Y. Cai, R. T. K. Kwok, Y. Chen, Z. He, X. Gu, X. He, H. H. Y. Sung, I. D. Williams, J. W. Y. Lam, Z. Zhang and B. Z. Tang, J. Mater. Chem. B, 2017, 1650–1657 RSC.
- M.-M. Xue, Y.-M. Xie, L.-S. Cui, X.-Y. Liu, X.-D. Yuan, Y.-X. Li, Z.-Q. Jiang and L.-S. Liao, Chem.–Eur. J., 2016, 22, 916–924 CrossRef CAS PubMed.
- Y. Liu, M. Gao, J. W. Y. Lam, R. Hu and B. Z. Tang, Macromolecules, 2014, 47, 4908–4919 CrossRef CAS.
- L. Parrenin, C. Brochon, G. Hadziioannou and E. Cloutet, Macromol. Rapid Commun., 2015, 36, 1816–1821 CrossRef CAS PubMed.
- C. M. Tseng, Y. Y. Lu, M. S. El-Aasser and J. W. Vanderhoff, J. Polym. Sci., Part A: Polym. Chem., 1986, 24, 2995–3007 CrossRef CAS.
- Y. Dong, J. W. Y. Lam, A. Qin, J. Liu, Z. Li, B. Z. Tang, J. Sun and H. S. Kwok, Appl. Phys. Lett., 2007, 91, 011111–011113 CrossRef.
- J. Shi, W. Zhao, C. Li, Z. Liu, Z. Bo, Y. Dong, Y. Dong and B. Z. Tang, Chin. Sci. Bull., 2013, 58, 2723–2727 CrossRef CAS.
- R. R. Rosencrantz, K. Rahimi and A. J. C. Kuehne, J. Phys. Chem. B, 2014, 118, 6324–6328 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra04513a |
| This journal is © The Royal Society of Chemistry 2017 |