
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Carbon nanosphere–iron oxide nanocomposites as high-capacity adsorbents for arsenic removal†
Hui Sua,
Zhibin Ye *ab,
Nuri Hmidic and
Ramesh Subramaniana
*ab,
Nuri Hmidic and
Ramesh Subramaniana
aBharti School of Engineering, Laurentian University, Sudbury, Ontario P3E 2C6, Canada. E-mail: zye@laurentian.ca; zhibin.ye@concordia.ca
bDepartment of Chemical and Materials Engineering, Concordia University, Montreal, Quebec H3G 1M8, Canada
cGoldcorp Incorporated, Red Lake Gold Mines, 15 Mine Road, Box 2000, Balmertown, Ontario P0V 1C0, Canada
First published on 20th July 2017
Abstract
In the design of iron oxide-derived composite adsorbents for arsenic removal, the matrix selected for the encapsulation of iron oxide active material is critical to their arsenic adsorption performance. The ideal matrix should have a high surface area, high pore volume, and large pores that can accommodate the iron oxide nanoparticles while without causing the undesired pore filling or blockage. In this paper, we report the use of carbon nanospheres (size of ca. 28 nm) featuring high surface area, high pore volume, and hierarchical large mesopore/macropore structures resulting from nanosphere packing/aggregation as the matrix for the design of iron oxide composites. Iron oxide has been encapsulated into the carbon nanospheres with different contents (7–60 wt%). The composites have been systematically characterized for their structural, morphological, and textural properties, and investigated for their performance for arsenic adsorption. An optimum iron oxide content of 13 wt% has been established with high adsorption capacities of 416 and 201 mg g−1 achieved for As(III) and As(V), respectively, which are highest (for As(III)) or among the highest (for As(V)) reported thus far for iron oxide-based adsorbents. These are in contrast to the typically low adsorption capacities found with iron oxide composites involving other carbon-based matrices, such as activated carbon, carbon nanotubes, and mesoporous carbons. The results confirm the high potential of this class of composite adsorbents for arsenic removal. Meanwhile, the structure–performance relationship demonstrated herein is also of value to the further design of high-performance arsenic adsorbents.
Introduction
Arsenic is a persistent, bio-accumulative, toxic element, which is widely distributed in the Earth's crust with an average terrestrial content of about 5 g per ton.1 Arsenic contamination of natural water sources due to mineral leaching and/or anthropogenic activities has been recorded by World Health Organization as a first priority issue.1–3 Various techniques have been developed to remove arsenic from both natural and industrial water sources, including coagulation, adsorption, ion exchange, membrane filtration, biological remediation, etc. The advantages and disadvantages of each technique have been well presented in the existing literature.3–6 In particular, adsorption is most economical and efficient over other techniques, especially in the low arsenic concentration range. A broad spectrum of adsorbents has been extensively studied and developed for arsenic removal.3,4Iron oxide-derived adsorbents have received, in particular, enormous interest for arsenic removal due to their superior performance for arsenic adsorption while with low cost.7 Iron oxides in various forms have been studied, such as amorphous iron oxide,8,9 crystalline iron oxides,10–14 and various iron oxide nanostructures.15–22 Among the various forms of iron oxides, amorphous iron oxides show the highest adsorption capacity (as high as 260 and 200 mg g−1 for As(III) and As(V), respectively) due to its highest specific surface area,10 but with the difficulty (as fine powders) for separation and its tendency to form low-surface-area crystalline iron oxides during preparation.4 Crystalline iron oxides and iron oxide nanostructures generally show lowered adsorption capacity due to their relatively low surface area.12–22
To facilitate more convenient adsorbent separation, iron oxide-derived composite adsorbents with iron oxides loaded onto various matrices have also been extensively developed for arsenic adsorption.3,4 In this aspect, a careful selection of the appropriate matrices and the uniform dispersion of iron oxide are key to the performance of the resulting composite adsorbents. The desired matrices should have open, 3-dimensional, interconnected pore structures with accessible, high surface area and pore volume. Meanwhile, the pore size of the matrices should be significantly larger than that of the loaded iron oxide nanoparticles, particularly at high iron oxide content, so as to avoid or minimize pore blockages, which would otherwise affect the mass diffusion and consequently the adsorption performance. Typical matrices have included abundant ones, such as naturally occurring minerals,23 activated carbons,24 graphene oxide (GO),25–36 and cellulose,37 as well as some specially-synthesized ones, such as mesoporous carbons,38,39 carbon nanotubes,40 macroporous silica,41 etc. The majority of composite adsorbents reported to date show limited arsenic adsorption capacities, with significant room for improvements. One exception is the iron oxide–macroporous silica foam composite adsorbent reported by Yu et al.,41 which shows remarkably high adsorption capacity [320 and 248 mg g−1 for As(III) and As(V), respectively]. Therein, the encapsulation of iron oxide at high contents (up to 34.8 wt%) does not block the open pore structures of the macroporous silica foam substrate having large macropores.
In this article, carbon nanospheres (CNS) having unique hierarchical large meso-/macropore structures42 are chosen for the first time as the porous matrix to fabricate iron oxide nanocomposite adsorbents of high arsenic adsorption capacity. Iron oxide has been loaded onto the CNS matrix at different content (7–60 wt%). The resulting composites have been systematically characterized with various techniques for their compositional, structural, and textural properties. Their performance for arsenic adsorption has been evaluated. High adsorption capacities of 416 and 201 mg g−1 for As(III) and As(V), respectively, have been achieved, which are even better than or comparable to the best results demonstrated thus far in the literature for iron oxide-derived composite adsorbents.
Experimental
Materials
1,3-Diethynylbenzene (DEB, 97%, Aldrich), sodium dodecyl sulphate (SDS, ≥98.5%, Aldrich), palladium acetate (98%, Strem Chemicals), α,α′-bis(di-tert-butylphosphino)-o-xylene (97%, Strem Chemicals), methanol (ACS reagent, Fisher Scientific), methanesulfonic acid (99.5%, Aldrich), dichloromethane (HPLC grade, Aldrich), ammonium persulfate (98%, Aldrich), sulfuric acid (96.9 wt%, Fisher Scientific), iron(III) nitrate nonahydrate (≥98%, Aldrich), ethanol (ACS reagent, Fisher Scientific), were used as received without any additional purification. Deionized water was purified by a Barnstead/Synbron Nanopure II purification system.Sodium (meta)arsenite (NaAsO2, ≥90%, Aldrich) and sodium arsenate dibasic heptahydrate (Na2HAsO4·7H2O, ≥98%, Aldrich) were used as the source of As(III) and As(V), respectively. Fresh ammonium persulfate (APS) solution at 1.0 M was prepared in 2 M H2SO4. Fe(NO3)3 solution at 20 wt% was freshly prepared in ethanol. Standard stock solutions of As(III) and As(V) were prepared in deionized water at an arsenic concentration of 2000 mg L−1. The standard solutions with other different concentrations were prepared by diluting the standard stock solutions. The pH of the standard solutions was adjusted to desired values by adding an appropriate amount of HNO3 or NaOH solution (both at 1 M).
Synthesis of carbon nanospheres (CNS) and CO2-activated carbon nanospheres (A-CNS)
CNS was synthesized by a catalytic emulsion polymerization technique developed by our group.42 This technique involves the first synthesis of highly cross-linked poly(1,3-diethynylbenzene) nanospheres (average size of ca. 24 nm) by catalytic emulsion polymerization of DEB, followed by hydrothermal treatment and subsequent carbonization. For the emulsion polymerization, sodium dodecyl sulfate (SDS) (1.6 g) was dissolved in deionized water (31.2 g) in a round-bottom Schlenk flask, followed by the addition of DEB (3.72 g; 29.5 mmol). The mixture was sonicated for 15 min and was then stirred for 5 h at 60 °C. In the meantime, palladium acetate (6.62 mg; 0.03 mmol) and α,α′-bis(di-t-butylphosphino)-o-xylene (34.9 mg; 0.09 mmol) were dissolved in a mixture solvent containing 0.54 mL of dichloromethane and 0.06 mL of methanol to prepare a catalyst solution. Then the catalyst solution was injected into the flask, followed by the addition of five drops of methanesulfonic acid, to start the miniemulsion polymerization. The polymerization lasted overnight in N2 atmosphere under stirring at 450 rpm at 60 °C, rendering a dark brown emulsion dispersion of polymer nanospheres. A hydrothermal treatment of the resulting polymer nanospheres was subsequently undertaken. The emulsion was diluted and was hydrothermally treated in an autoclave at 220 °C overnight. The resulting hydrothermally treated polymer precipitates were collected by filtration, washed with a large amount of water, and dried under vacuum at 60 °C for 48 h, rendering the polymer nanospheres (2.77 g). Pyrolysis of the polymer nanospheres in a tube furnace at 800 °C for 1 h in a nitrogen atmosphere (preceded with heating from room temperature to 800 °C at a heating rate of 10 °C min−1) rendered CNS (1.65 g).A-CNS was prepared by CO2 activation of CNS at 900 °C for 3 h. In a tube furnace, CNS (1.65 g) was heated to 900 °C at a rate of 10 °C min−1 in a flowing N2 atmosphere. After reaching 900 °C, the atmosphere was changed from CO2 within 5 min and the temperature was maintained for 3 h. When the activation was finished, the atmosphere was switched back to N2 within 5 min, followed by natural cooling of the sample down to room temperature in the N2 atmosphere, rendering A-CNS (0.8 g).
Synthesis of iron oxide–CNS composites (FeOx–CNSs)
Iron oxide–CNS composites were synthesized by modifying a literature method reported for the preparation of iron oxide–mesoporous carbon nanocomposites.38 Surface oxidation of A-CNS with APS was first undertaken to render S-CNS with improved surface polarity for the loading of iron oxide. A-CNS (240 mg) was dispersed and stirred in 20 mL of 1.0 M acidic APS solution (in 2 M H2SO4) at 70 °C for 12 h. The solids were then filtered, washed with copious amounts of water and then methanol, and dried under vacuum at 60 °C overnight, rendering S-CNS (200 mg).The following is the procedure used for the synthesis of the representative iron oxide–CNS nanocomposite having an iron oxide content of 13 wt% (i.e., FeOx–CNS-13). S-CNS (200 mg) was dispersed in absolute ethanol (1.4 g), followed by the addition of a 20 wt% ethanolic solution of Fe(NO3)3 (152 mg, 374 mmol). After sufficient and subsequent solvent evaporation, the resulting powder was transferred into a small plastic ampoule. The ampoule was subsequently placed in a Teflon bottle containing an ammonia solution (∼10 mL at ∼14 wt%), but without direct contact of the powder with the ammonia solution. After sealing, the bottle was heated in an oven at 60 °C for 3 h to hydrolyze the metal precursor in situ. After cooling, the product was filtered and washed with small amounts of water and methanol. After drying at 60 °C under vacuum overnight, the solid product was pyrolyzed at 300 °C in a N2 atmosphere for 1 h to obtain FeOx–CNS-13 (0.2 g).
Other nanocomposites (FeOx–CNS-7, FeOx–CNS-28, and FeOx–CNS-60) having different contents of iron oxide (7, 28, and 60 wt%, respectively) were similarly prepared with the feed amounts of S-CNS and iron nitrate at different mass ratios (50 mg/19 mg, 100 mg/197 mg, and 50 mg/380 mg, respectively).
Characterization and measurements
Thermogravimetric analysis (TGA) of the various composites was carried out on a Q50 TGA from TA Instruments. Measurements were performed in an air atmosphere with a continuous airflow of 60 mL min−1 through the sample furnace and a continuous N2 flow of 40 mL min−1 through the balance compartment. In a typical measurement, the sample (10 mg) was heated to 100 °C at a rate of 10 °C min−1, held at 100 °C for 10 min, and then heated to 800 °C at a rate of 10 °C min−1. N2 sorption analysis was carried out with a Micromeritics ASAP 2020 physiosorption analyzer at 77 K to determine their Braunauer–Emmett–Teller (BET) specific surface area, pore volume, and pore size distribution. Before the sorption measurements, the samples were degassed under vacuum at 180 °C for at least 12 h. X-ray photoelectron spectroscopy (XPS) measurements of the samples were carried out on a Thermo Scientific Theta Probe XPS spectrometer. A monochromatic Al Kα X-ray source was used, with a spot area of 400 μm. The samples were run in a standard mode, i.e., all angles collected (60° angular acceptance) for the survey spectra, and for the region spectra. Wide-angle X-ray diffraction (XRD) patterns were recorded on an X'Pert Pro diffractometer with Cu radiation (wavelength 1.54 Å) at room temperature. Transmission electron microscopy (TEM) images were taken on a JEOL 2010F field emission electron microscope operated at 200 keV, equipped with energy-dispersive spectroscopy (EDS). The TEM samples were prepared by depositing a few drops of a dilute dispersion of the samples in methanol on holey grids, followed by drying. Dynamic light scattering (DLS) and zeta potential measurements of the dilute dispersions (0.1 mg mL−1) of the various samples were performed on a Brookhaven NanoBrook Omni Instrument at 25 °C. Fourier-transformed infrared (FTIR) spectra were obtained on a Thermo Scientific Nicolet 6700 Analytical FTIR spectrometer. The samples were prepared as pellets using spectroscopic-grade KBr. The arsenic concentration of the various aqueous solutions in the adsorption study was measured with an Analytik Jena 810 ICP-MS system with a detection limit of 50 μg L−1.Arsenic adsorption
Batch adsorption of arsenic with all adsorbents (A-CNS, S-CNS, or FeOx-CNS composites) was all undertaken with an adsorbent loading of 1.0 mg mL−1 at 23 °C. Typically, the adsorbent (1 mg) was dispersed in various As(III) or As(V) solutions (1 mL) at different known concentrations (50–2000 mg L−1), followed by magnetic stirring (300 rpm) for 24 h to achieve equilibrium. Afterwards, the dispersion was filtered with 0.2 μm syringe filter and the filtrate was measured with ICP-MS for equilibrium arsenic concentration and for the calculation of the amount of adsorbed arsenic to construct adsorption isotherms. The adsorption isotherms were fitted with Freundlich model (eqn (1)).| qe = k(Ce)1/n | (1) |
To obtain the adsorption kinetic curves, FeOx–CNS-13 (1 mg) was dispersed in a series of As(III) and As(V) solutions with the same volume (1 mL) and initial concentrations (950 mg L−1 for As(III) and 800 mg L−1 for As(V)). Each dispersion was stirred for a prescribed period of time (ranging from 15 min to 24 h) and was quickly filtered for measurement of the corresponding equilibrium concentration of As(III) and As(V), thus giving rise to the time-dependent adsorption uptake. The adsorption kinetics was fitted with the pseudo-second-order kinetic model (eqn (2) or (3)) based on which the initial adsorption rate was obtained (eqn (4)).
| dqt/dt = k2(qe − qt)2 | (2) |
| t/qt = 1/(k2qe2) + t/qe | (3) |
| V0 = k2qe2 | (4) |
Results and discussion
Synthesis and characterization of carbon nanosphere matrix
The carbon nanosphere matrix (S-CNS), employed herein for the construction of the composite adsorbents, was synthesized by CO2-activation of carbon nanospheres (CNS) obtained by a unique catalytic emulsion polymerization technique42 followed by surface oxidization. Scheme 1 summarizes the synthesis. The catalytic emulsion polymerization of a cross-linkable dialkyne monomer, DEB, rendered poly(DEB) polymer nanospheres of uniform sizes (average size of 27 nm with polydispersity index of 0.19 as per DLS characterization; see Fig. S1 in ESI†). Therein, each polymer nanosphere was constructed uniquely with highly cross-linked poly(DEB) networks. Carbonization of the polymer nanospheres following hydrothermal treatment rendered CNS.42 Having relatively low specific surface area (524 m2 g−1) and pore volume (0.84 cm3 g−1) as shown below, CNS, directly, was unsuitable as the matrix for iron oxide. CO2 activation was thus employed to yield A-CNS with dramatically enhanced surface area and pore volume. Therein, the enhanced porosity is generated by the gasification reaction of the carbon structure with CO2, rendering CO.43 Consisting predominantly of carbon, the surface of A-CNS was highly hydrophobic and incompatible with polar iron oxide. Wet chemical surface oxidation44 of A-CNS was further taken to render S-CNS with improved surface polarity in order for uniform encapsulation of iron oxide.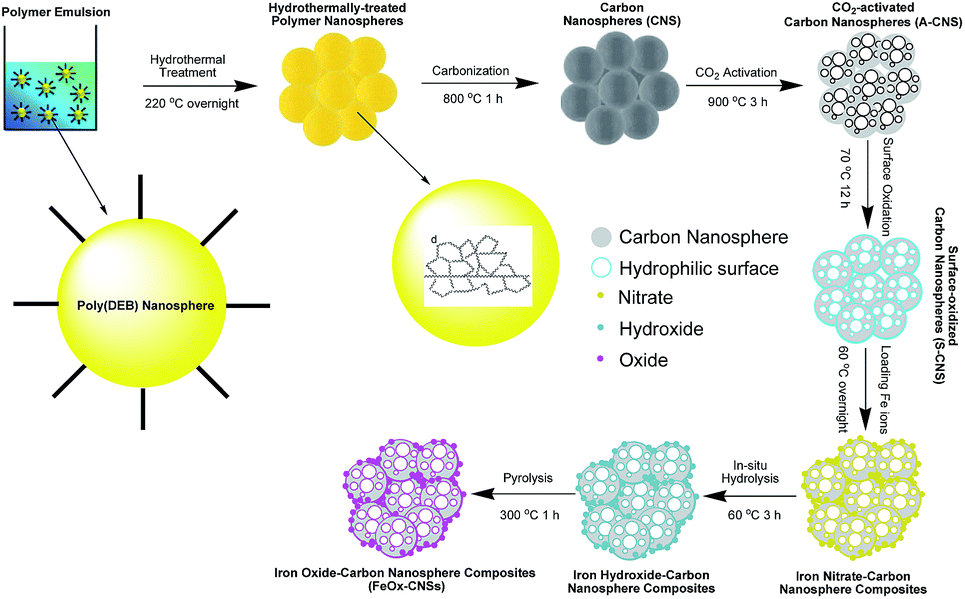 | ||
| Scheme 1 Schematic synthesis of carbon nanosphere matrix and the FeOx–CNS nancomposites. | ||
Fig. 1 shows TEM images of CNS, A-CNS, and S-CNS. CNS is comprised of aggregates of well-defined carbon nanospheres with average size of 24 nm (Fig. 1(a)). Following the CO2 activation, the nanosphere morphology is still well retained in A-CNS (Fig. 1(b)). The average nanosphere size in A-CNS is ca. 28 nm, which is slightly bigger than that in CNS, indicating the slight expansion of the carbon nanospheres due to mesopore/micropore generation within the nanospheres upon CO2 activation. Surface oxidation leads to slight structural collapse in S-CNS with fusion of some nanospheres observed; but the nanosphere morphology is still clearly retained (Fig. 1(c)).
 | ||
| Fig. 1 TEM of CNS (a), A-CNS (b), and S-CNS (c). | ||
N2 sorption analysis at 77 K was undertaken on the three carbon nanosphere samples to reveal their textural properties. Fig. 2(a) shows their N2 sorption curves, with the results summarized in Table 1. All three samples show typical type IV adsorption isotherm,45 with a slight uptake at low relative pressure (P/P0 < 0.05), and a sharp uptake at high relative pressure (P/P0 > 0.90 for CNS and A-CNS; P/P0 > 0.7 for S-CNS), indicating that all samples contain predominantly mesopores/macropores. All samples have distinct type H3 hysteresis loops,45 which is characteristic of slit-shaped pores. Relative to CNS, A-CNS has dramatically enhanced surface area (2271 vs. 524 m2 g−1) and pore volume (5.18 vs. 0.84 cm3 g−1) with the generation of a large quantity of new mesopores/macropores upon CO2 activation. However, upon further surface oxidation, the surface area and pore volume of S-CNS are reduced to 1073 m2 g−1 and 1.17 cm3 g−1, respectively, due to the slight unavoidable structural collapse. This is also accompanied with the reduction of the average meso-/macropore size from 41 nm for A-CNS to 20 nm for S-CNS. Despite this, S-CNS is suitable for the encapsulation of iron oxide given its high surface area/pore volume and large meso-/macropores.
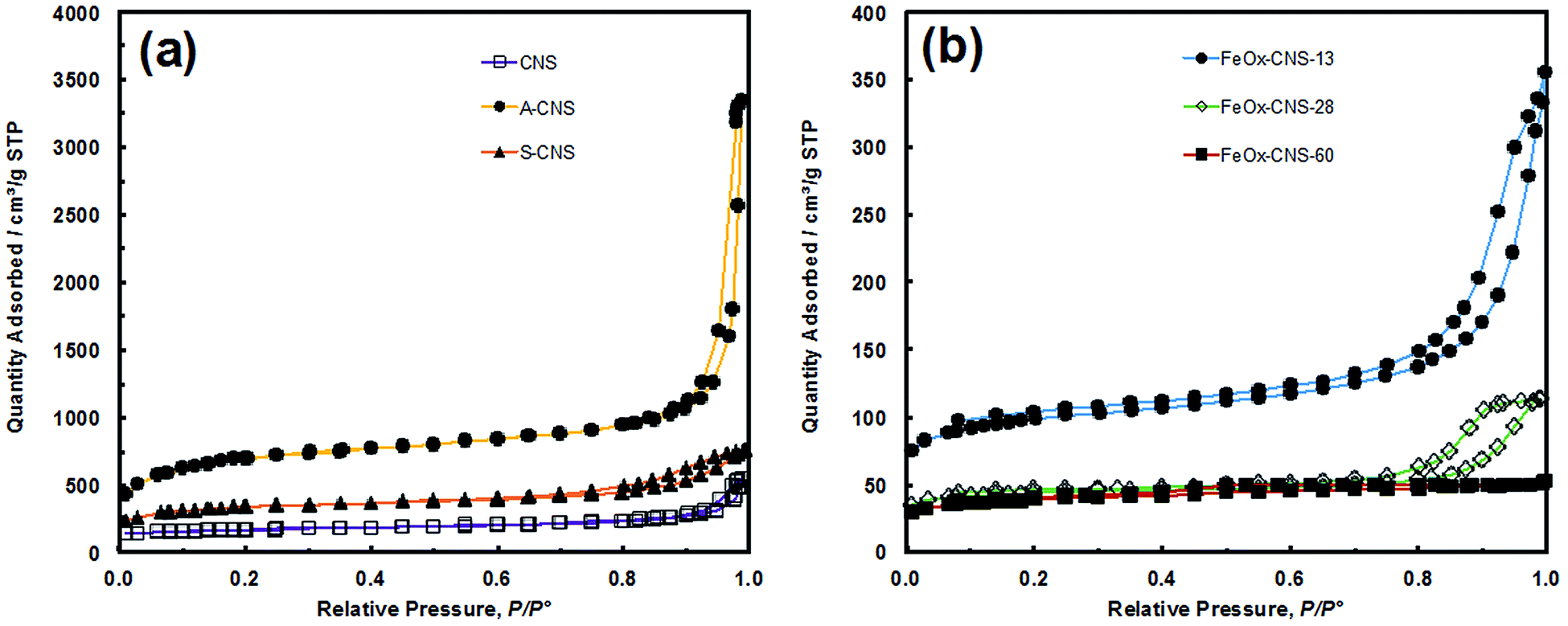 | ||
| Fig. 2 (a) N2 sorption isotherm of CNS, A-CNS and S-CNS, (b) N2 sorption isotherm of FeOx–CNS-13, FeOx–CNS-28 and FeOx–CNS-60. | ||
| Samples | Surface areaa (m2 g−1) | Pore volumeb (cm3 g−1) | Dmeso/macroc (nm) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| SBET | Smicro | % Smicro | Smeso/macro | Vtotal | Vmicro | % Vmicro | Vmeso/macro | ||
| a BET surface area (SBET), surface area of micropores (Smicro) and mesopores/macropores (Smeso/macro) determined with t-plot method; % Smicro denotes the percentage of surface area of micropores.b Total pore volume (Vtotal), micropore volume (Vmicro) and mesopore/macropore volume (Vmeso/macro) determined with t-plot method; % Vmicro denotes the percentage of micropore volume.c Average mesopore/macropore size (Dmeso/macro) determined from the N2 desorption data with NLDFT model. | |||||||||
| CNS | 524 | 292 | 56% | 232 | 0.84 | 0.15 | 18% | 0.69 | 32 |
| A-CNS | 2271 | 749 | 33% | 1522 | 5.18 | 0.41 | 8% | 4.77 | 41 |
| S-CNS | 1073 | 458 | 43% | 615 | 1.17 | 0.25 | 21% | 0.92 | 20 |
| FeOx–CNS-13 | 311 | 168 | 54% | 143 | 0.55 | 0.09 | 16% | 0.46 | 24 |
| FeOx–CNS-28 | 135 | 88 | 65% | 47 | 0.22 | 0.05 | 23% | 0.17 | 20 |
| FeOx–CNS-60 | 123 | 60 | 49% | 63 | 0.08 | 0.03 | 38% | 0.05 | |
All the three carbon nanosphere samples should have the characteristic hierarchical pore structures with the presence of both inter-sphere mesopores/macropores resulting from nanosphere aggregation/packing and intra-sphere micropores/mesopores. They are predominantly mesoporous/macroporous, with micropores contributing to only 8–21% of total pore volume (see Table 1). Fig. S2(a) in ESI† compares their mesopore/macropore size distribution curves. The 3-dimensional interconnected hierarchical mesopore/macropore structures of S-CNS, along with its high surface area and pore volume, is beneficial for rendering iron oxide composites with uniform encapsulation of iron oxide and in consequence high arsenic adsorption capacity.
XPS characterization (see Fig. S3 and S4 in ESI†) shows that S-CNS has a high content (22.8 atom%) of O, which is much higher than those (4.4 and 7.7 atom%, respectively) in CNS and A-CNS. Confirming the efficiency of surface oxidization, the high O content of S-CNS is beneficial for the subsequent even, uniform encapsulation of iron oxide. Fig. 3 compares their FTIR spectra. Both CNS and A-CNS show nearly featureless spectra, with only weak and broad bands in the frequency region of 1700–1000 cm−1 having two maxima at around 1570 cm−1 and 1149 cm−1. The broad bands can be assigned to the stretching vibrations of C–C and C–H bonds in aromatic carbon rings, respectively.46 New intense bands indicative of significant oxidation of the carbon surface are observed in the spectrum of S-CNS, which is in agreement with XPS results. The bands at 1733 and 1616 cm−1 can be assigned to asymmetric stretching vibrations of the newly formed –COOH carboxyl and –COO− carbonyl and/or –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O ketone units.44,47,48 The band at 1246 cm−1 can be attributed to asymmetric stretch of –C–C–C bridges in ketonic groups and/or to deformation vibrations of O–H in the carboxylic acid groups. In addition, the weak band at 667 cm−1 can be assigned to bending O–C–O vibrations of carboxyl units.41
O ketone units.44,47,48 The band at 1246 cm−1 can be attributed to asymmetric stretch of –C–C–C bridges in ketonic groups and/or to deformation vibrations of O–H in the carboxylic acid groups. In addition, the weak band at 667 cm−1 can be assigned to bending O–C–O vibrations of carboxyl units.41
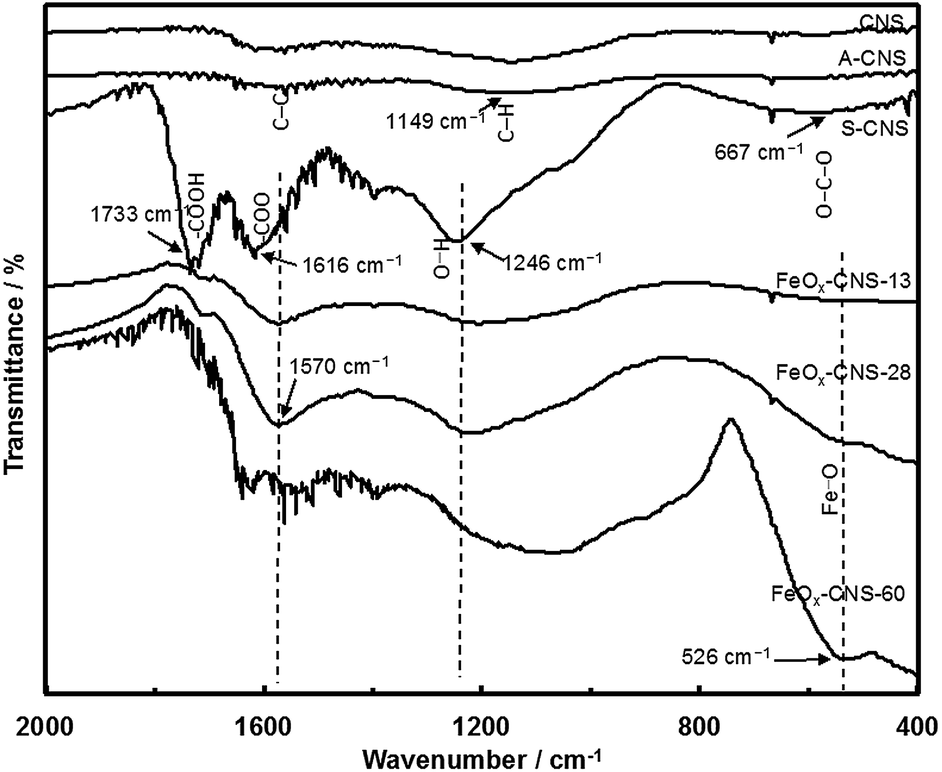 | ||
| Fig. 3 FTIR spectra of CNS, A-CNS, S-CNS, FeOx–CNS-13, FeOx–CNS-28, and FeOx–CNS-60. | ||
Synthesis and characterization of FeOx–CNS nanocomposites
FeOx–CNS nanocomposites were synthesized by loading iron nitrate precursors (Fe(NO3)3·9H2O) into the pore structures of S-CNS, followed by an in situ hydrolysis under ammonia atmosphere for the conversion of the iron nitrate precursors into hydroxides, and a pyrolysis step (at 300 °C) for the transformation of the hydroxides into oxides (Fe2O3 and/or Fe3O4) and the release of some porosity (see Scheme 1).38,49 In order to investigate the effects of the iron oxide content on the arsenic adsorption efficiency, four composites with varying iron oxide content were synthesized by tuning the feed ratio of S-CNS and iron nitrate (see Experimental section). Fig. 4 shows the TGA curves of the resulting composites in an air atmosphere, along with those of CNS, A-CNS, and S-CNS for comparison. Among the three carbon materials (CNS, A-CNS, and S-CNS), CNS shows the best thermal stability in air given its highest C content. It starts to show weight loss at around 450 °C and has the complete weight loss at around 600 °C with the peak weight loss at 587 °C. A-CNS starts to lose weight at ca. 300 °C and have complete weight loss at 500 °C with peak weigh loss temperature of 470 °C, due to its enhanced porosity and surface area for contact with air. S-CNS instead starts to lose weight at as early as 130 °C and the weight loss covers a broad temperature range of 130 to 600 °C, with the peak temperature of 560 °C. The weight loss at the low temperature region is attributed to the loss of the labile oxygen-containing functionalities upon heating.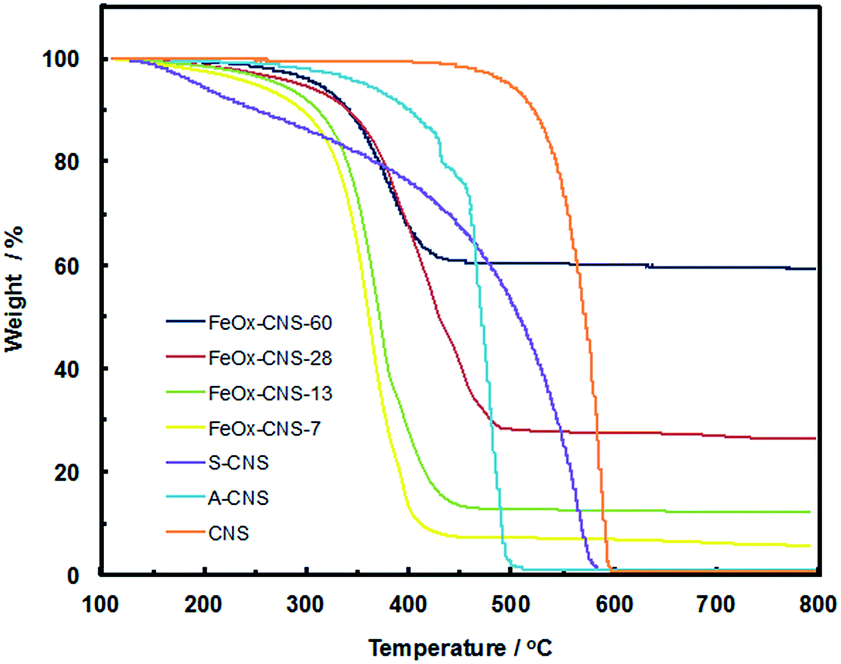 | ||
| Fig. 4 TGA curves of CNS, A-CNS, S-CNS, and FeOx–CNS composites in air. | ||
The FeOx–CNS nanocomposites all show weight loss within the temperature range of 220–480 °C, which should correspond primarily to the combustion of the carbon matrix. The peak maximum temperature is within the range of 360–400 °C, which is significantly reduced relative to that of S-CNS. This should be ascribed to the existence of iron oxide nanoparticles on the surfaces of the carbon matrix which act as catalysts for the carbon combustion since their exothermic oxidation takes place at lower temperatures.50 Different from the complete weight loss observed in the carbon samples, the composites show different residual char yield at 500 °C (7, 13, 28, and 60 wt%, respectively), corresponding to the content of iron oxide within the nanocomposites. Consequently, these composites are termed as FeOx–CNS-#, with the number (#) representing the mass percentage of iron oxide in the composites.
The composites were characterized with XPS and FTIR for their spectroscopic properties. The XPS spectra of FeOx–CNS-28 as the representative one are shown in Fig. S3 and S4 in ESI.† Relative to S-CNS, FeOx–CNS-28 has slightly reduced C and O content of 71.6 and 18.0 atom%, respectively, but with the significant presence of Fe at 4.0 atom%, confirming the incorporation of iron oxide. From its Fe2p spectrum and the deconvoluted peaks (Fig. S3(c) in ESI†), both Fe3+ and Fe2+ species are present in the composite. In particular, the presence of Fe3+ species is confirmed by the Fe2p3/2 peak at 712.58 eV and the associated satellite peak at 719.08 eV, Fe2p1/2 peak at 726.18 eV and the satellite peak at 732.68 eV; the Fe2+ species is confirmed by the Fe2p3/2 peak at 710.68 eV and the satellite peak at 715.78 eV, Fe2p1/2 peak at 724.28 eV and the satellite peak at 729.38 eV (see Fig. S3(c) in ESI†).51,52 The formation of Fe2+ can be attributed to the reducing ability of carbon matrix during the pyrolysis treatment in the synthesis.17 In particular, the Fe3+/Fe2+ ratio according to the Fe2p3/2 peaks is estimated to be 1.57![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, which is close to the ratio of 2:1 for Fe3O4.36 This indicates that the iron oxide present in the composite is primarily Fe3O4 and the presence of γ-Fe2O3 should be minor. The O1s spectrum (Fig. S4(a) in ESI†) can be well fitted to three peaks at 530.3, 531.6, and 533.6 eV, which are attributed to the binding energies of oxygen atoms in the O–Fe, carboxyl OC, and hydroxyl O–H bonds, respectively.53 The binding energy of C1s can be found at 284.8, 286.1, 287.7, and 288.8 eV (Fig. S4(b) in ESI†), in agreement with the binding energy values of C–C, C–O, CO and O–CO reported in the literature.54
:1, which is close to the ratio of 2:1 for Fe3O4.36 This indicates that the iron oxide present in the composite is primarily Fe3O4 and the presence of γ-Fe2O3 should be minor. The O1s spectrum (Fig. S4(a) in ESI†) can be well fitted to three peaks at 530.3, 531.6, and 533.6 eV, which are attributed to the binding energies of oxygen atoms in the O–Fe, carboxyl OC, and hydroxyl O–H bonds, respectively.53 The binding energy of C1s can be found at 284.8, 286.1, 287.7, and 288.8 eV (Fig. S4(b) in ESI†), in agreement with the binding energy values of C–C, C–O, CO and O–CO reported in the literature.54
FTIR spectra of FeOx–CNS-13, FeOx–CNS-28, and FeOx–CNS-60 are included in Fig. 3. The basic bands attributed to the vibrations of aromatic carbon rings, carbonyl and ketone units are still observed, indicating that the carbon matrix retains the basic carbon structure and the main active groups of the parent S-CNS. The incorporation of iron oxide in the nanocomposites can be confirmed with a new broad band in the low frequency region (750–400 cm−1) with the peak intensity at 526 cm−1 in FeOx–CNS-28 and FeOx–CNS-60 of significantly high iron oxide contents, which corresponds to the Fe–O vibration in Fe3O4 and γ-Fe2O3.39
Fig. 5 shows the XRD spectra of two representative composites, FeOx–CNS-13 and FeOx–CNS-60, along with those of A-CNS and S-CNS. In the spectra of all samples, two weak but broad peaks (indicated by arrows), attributed to the (002) and (100) peaks of graphitic structures, are found at around 24 and 43°, respectively.55 The weak and broad nature of the peaks suggests a low degree of graphitization with a low content of parallel single layers in these carbon materials, which is typical of amorphous carbon materials. Apart from the diffraction peaks of the carbon matrix, distinct diffractions arising from Fe3O4 and/or γ-Fe2O3 crystals (indicated by solid squares and open circles, respectively) can be clearly seen in the spectrum of FeOx–CNS-60 having the highest iron oxide content. These peaks can be indexed to the diffractions of Fe3O4 (JCPDS card no. 88-0315) and γ-Fe2O3 (JCPDS card no. 39-1346), which are indistinguishable.17 Herein, given the minor presence of γ-Fe2O3 in the composite according to the XPS results above, these diffraction peaks should thus result predominantly from Fe3O4. The average crystallite size (L) is evaluated from the full width at half maximum of the (311) peak at 2θ = 35.5° according to the Scherrer equation:
|
L = Kλ/βcosθ
| (5) |
 | ||
| Fig. 5 XRD patterns of A-CNS, S-CNS, FeOx–CNS-13, and FeOx–CNS-60. | ||
TEM characterization of two representative composites, FeOx–CNS-13 and FeOx–CNS-60, was conducted, with typical TEM images shown in Fig. 6 and S5 in ESI,† respectively. In FeOx–CNS-13, some nanospheres can still be observed (see Fig. 6(a)) though not as well defined as those in S-CNS. The presence of iron oxide nanoparticles in the composite can be confirmed from the high-resolution TEM (HRTEM) image shown in Fig. 6(b). In particular, the inset in Fig. 6(b) clearly demonstrates the crystalline lattices with a d-spacing of ∼2.5 Å, which matches well the d311 of Fe3O4/γ-Fe2O3.38 Fig. 6(c) shows a dark-field scanning transmission electron microscopy (DF STEM) image of FeOx–CNS-13. In DF-STEM images, heavy atoms such as Fe appear brighter compared to the light atoms (e.g., C or O). As shown in Fig. 6(c), uniformly distributed bright spots with sizes of around 3–4 nm, corresponding to the iron oxide nanoparticles, can be found in the dark domain of FeOx–CNS-13. The uniform dispersion of iron oxide nanoparticles in the carbon matrix is further confirmed by high-resolution elemental mapping for C, O, and Fe (Fig. 6(d)–(f), respectively), where the C, O, and Fe elements are quite uniformly distributed and faithfully correlated with the domain. Due to the significantly higher iron oxide content, the carbon nanosphere morphology is no longer observable in FeOx–CNS-60 (see Fig. S5(a) and (b) in ESI†) and its DF STEM image (Fig. S5(c) in ESI†) reveals much denser distribution of large iron oxide aggregates. Unlike the small, interspersed nanoparticles seen in FeOx–CNS-13, the iron oxide in FeOx–CNS-60 forms the inter-connected networks within the pores of the carbon matrix. Nevertheless, overall uniform distribution of the iron oxide within the composite is still maintained in FeOx–CNS-60 on the basis of the C, O, and Fe elemental maps (Fig. S5(d)–(f) in ESI†).
 | ||
| Fig. 6 TEM (a), HRTEM images (b), DF-STEM image (c) and the corresponding C (d) O (e) and Fe (f) elemental maps of FeOx–CNS-13. | ||
With the increase of iron oxide content, the composites show continuously reduced N2 sorption with much lowered surface area and pore volume relative to S-CNS (see Fig. 2(b) and Table 1). While both FeOx–CNS-13 and FeOx–CNS-28 still show the type IV isotherm with a hysteresis loop at the high relative pressure end (P/P0: 0.8–1), FeOx–CNS-60 of highest iron oxide content exhibits instead a type I isotherm with no distinct hysteresis loop (i.e., minor presence of mesopores/macropores). The surface area and pore volume decrease consistently from 311 m2 g−1 and 0.55 cm3 g−1 for FeOx–CNS-13 to 135 m2 g−1 and 0.22 cm3 g−1 for FeOx–CNS-28, and to 123 m2 g−1 and 0.03 cm3 g−1 for FeOx–CNS-60. These results indicate the significant pore filling and blockage upon the encapsulation of iron oxide within the carbon matrix, which become increasingly severe with the increase of the iron oxide content. In particular, such filling and blockage are reasoned to be more prevalent within mesopores/macropores since the percentages of mesopore/macropore surface and volume decrease while those for micropores increase (see Table 1) upon the encapsulation of iron oxide relative to S-CNS.
Arsenic adsorption with FeOx–CNS composites
A systematic investigation on the performance of the set of FeOx–CNS composite adsorbents in arsenic adsorption was undertaken. Batch equilibrium adsorption of both As(III) and As(V) was carried out at the initial arsenic concentrations of 50–2000 mg L−1 for As(III) (at pH = 8) and 50–1000 mg L−1 for As(V) (at pH = 3). The specific pH values are chosen herein since the composite adsorbents show optimum arsenic adsorption at the conditions according to the study on the effect of pH below. Due to the relatively low iron oxide contents in FeOx–CNS-7, FeOx–CNS-13, and FeOx–CNS-28, magnetic separation of the composites from the solution following arsenic adsorption was inefficient. As such, the separation was made by filtration. On the contrary, magnetic separation of FeOx–CNS-60 was spontaneous given its high iron oxide content.Fig. 7 shows the adsorption isotherms of the various composites at 23 °C, as well as those of A-CNS and S-CNS as control ones. The two control carbon nanosphere samples without containing iron oxide show similar isotherms for both As(III) and As(V), with S-CNS having slightly raised isotherms relative to A-CNS possibly due to its more hydrophilic surface. For the composites, the content of iron oxide shows a dramatic effect on their arsenic adsorption. Relative to the carbon samples, the incorporation of iron oxide at 7 and 13 wt% in FeOx–CNS-7 and FeOx–CNS-13 leads to significantly improved arsenic adsorption with increasingly raised isotherms for both As(III) and As(V). However, the further increase of iron oxide content to higher values of 28 and 60 wt% instead lowers the isotherms, which become close to those of the two control samples.
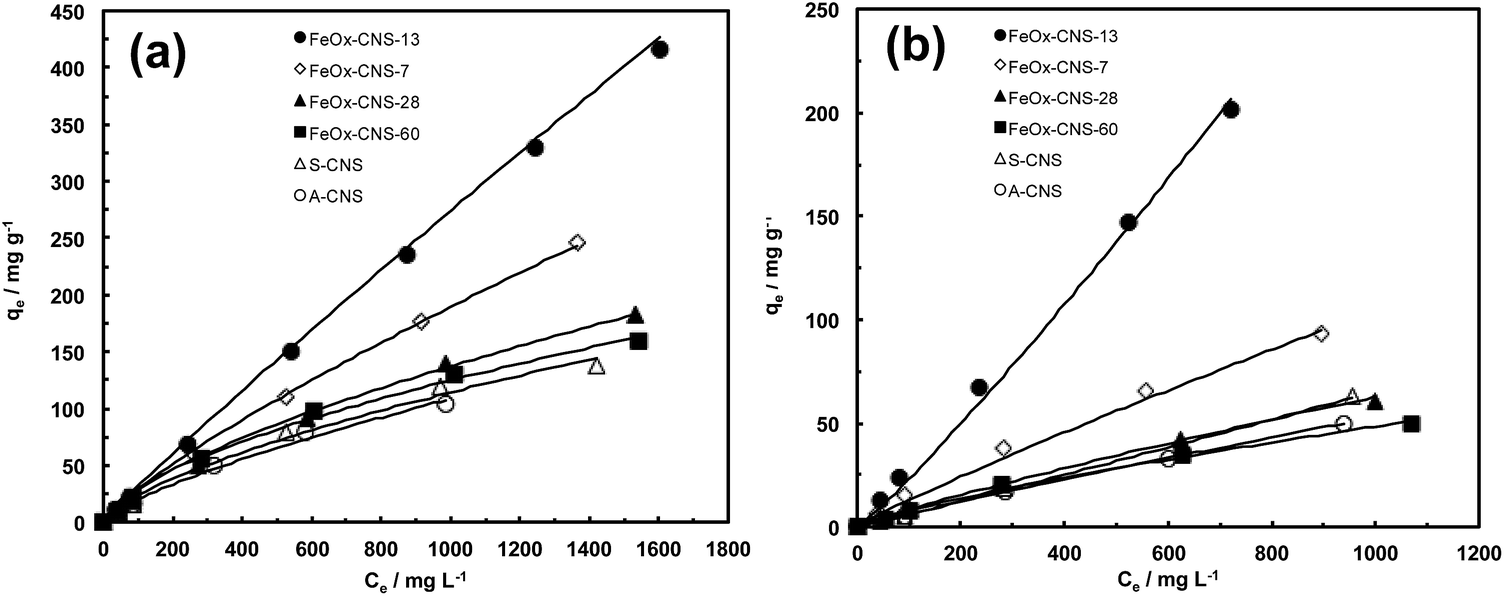 | ||
| Fig. 7 (a) As(III) adsorption isotherms (at pH = 8) and (b) As(V) adsorption isotherms (at pH = 3) of A-CNS, S-CNS, and FeOx–CNS composites. The adsorption isotherms are fitted the Freundlich adsorption model (solid line). | ||
All the isotherms have been found to follow Freundlich adsorption model (eqn (1)) with R2 values close to 1 (see Table S1 in ESI†). The Langmuir model instead does not give good fitting with low R2 values yielded. The adsorption capacity data (qmax), taken from the last point in each isotherm (i.e., highest adsorption uptake in each isotherm), are summarized in Table S2 in ESI.† With any given adsorbent herein, we find that the adsorption capacity for As(III) is always higher than that for As(V). This is beneficial and desirable because of the prevalence of more toxic As(III) in groundwater and wastewater.1 The two carbon nanosphere samples, A-CNS and S-CNS, show qmax of 104 and 138 mg g−1, respectively, for As(III) and 50 and 63 mg g−1, respectively, for As(V). These qmax data are significantly higher than the values reported for various activated carbons within the similar range of initial arsenic concentration,56 possibly due to the much higher accessible surface area of the carbon nanosphere samples as a result of their hierarchical pore structures.
The two composites having relatively low iron oxide contents, FeOx–CNS-7 and FeOx–CNS-13, show dramatically enhanced adsorption capacity, 246 and 416 mg g−1, respectively, for As(III), and 93 and 201 mg g−1, respectively, for As(V). In our best knowledge, the As(III) adsorption capacity of 416 mg g−1 for FeOx–CNS-13 is by far the highest among the various iron oxide-based adsorbents (see Table 2). It is even higher than the value of 320 mg g−1 for a high-performance γ-Fe2O3-macroporous silica composite adsorbent reported by Yu et al.41 Meanwhile, the As(V) adsorption capacity of 201 mg g−1 for FeOx–CNS-13 is also comparable to some best values (up to 248 mg g−1; see Table 2) reported.41 These capacity data are remarkable given the encapsulation of iron oxide at such low contents. Clearly, the small non-aggregated iron oxide nanoparticles interspersed uniformly on the porous carbon nanosphere matrix contribute to the dramatic enhancement in the adsorption capacity since their surface is more active than the carbon surface for arsenic adsorption. Increasing the iron oxide content within this range (<ca. 13 wt%) is thus beneficial for enhanced capacity.
| Iron oxide-based adsorbents | Adsorption capacity (mg g−1) | Reference | |
|---|---|---|---|
| As(III) | As(V) | ||
| Fe3O4 nanoparticles (12 nm) | 160 | 180 | 13 |
| Fe2O3 CAHNs | 137.5 | 16 | |
| Flowerlike α-Fe2O3 | 51 | 15 | |
| Hollow nestlike α-Fe2O3 | 58.6 | 75.3 | 21 |
| γ-Fe2O3 nanochains | 162 | 22 | |
| Bimetal iron(III)–titanium(IV) oxide | 85 | 14.3 | 8 |
| Fe–Cu binary oxide | 122.3 | 82.7 | 57 |
| Hollow Fe–Ce alkoxides | 266 | 206 | 58 |
| Fe3O4–RGO | 13.1 | 5.83 | 25 |
| Fe2O3–graphene nanoplatelet | 11.34 | 29 | |
| Fe2O3–cellulose | 23.16 | 32.11 | 37 |
| Fe2O3–ordered mesoporous carbon | 29.4 | 17.9 | 38 |
| Ce–Fe oxide@carbon nanotube | 28.74 | 30.96 | 40 |
| Fe2O3–carbon aerogel | 216 | 59 | |
| Fe2O3–diatomite | 60.65 | 81.16 | 60 |
| Fe2O3–macroporous silica | 320 | 248 | 41 |
| FeOx–CNS-13 | 416 | 201 | This study |
Relative to FeOx–CNS-13, the other two composites, FeOx–CNS-28 and FeOx–CNS-60, instead show much deteriorated adsorption capacity data (182 and 160 mg g−1, respectively, for As(III); 61 and 50 mg g−1, respectively, for As(V)) despite the higher iron oxide contents. Within this range of iron oxide content (ca. 28–60 wt%), the increase in iron oxide content is detrimental to arsenic adsorption. This should result from the formation of large iron oxide aggregates, which leads to filled/blocked pores and reduced active surface area for effective arsenic adsorption. Clearly, 13 wt% is approximately the optimum iron oxide content for this range of composite adsorbents. A similar trend of change in arsenic adsorption capacity has been noted in iron-impregnated granular activated carbon adsorbents, where the adsorption capacity increases with the iron content up to 6% and then decreases with further increase in the content.24
The adsorption kinetics of FeOx–CNS-13 was monitored at the initial arsenic concentration of 950 mg L−1 for As(III) (at pH = 8) and 800 mg L−1 for As(V) (at pH = 3). Fig. 8 shows the kinetic curves. For both curves, the adsorption can be divided into two stages, a rapid uptake within the first 30 min of contact and a slow uptake thereafter until equilibrium is reached. In particular, the majority of the arsenic uptake, 62 and 73% for As(III) and As(V), respectively, occurs within the first 30 min, indicating the very fast adsorption rate. The adsorption kinetics can be perfectly fitted with a pseudo-second-order model (eqn (2) or (3)) with the rate constant k2 of 0.000315 and 0.000504 g mg−1 min−1 for As(III) and As(V), respectively. The k2 values are high, in particular for As(III), when compared to other superior adsorbents reported in the literature. For example, the high-performance γ-Fe2O3–macroporous silica composite adsorbent reported by Yu et al. has the k2 values of 0.00015 and 0.0014 g mg−1 min−1 for As(III) and As(V), respectively, under similar conditions.41 These kinetic results demonstrate that, within the concentration ranges, the rapid removal of the arsenic species from wastewater can be achieved with FeOx–CNS-13.
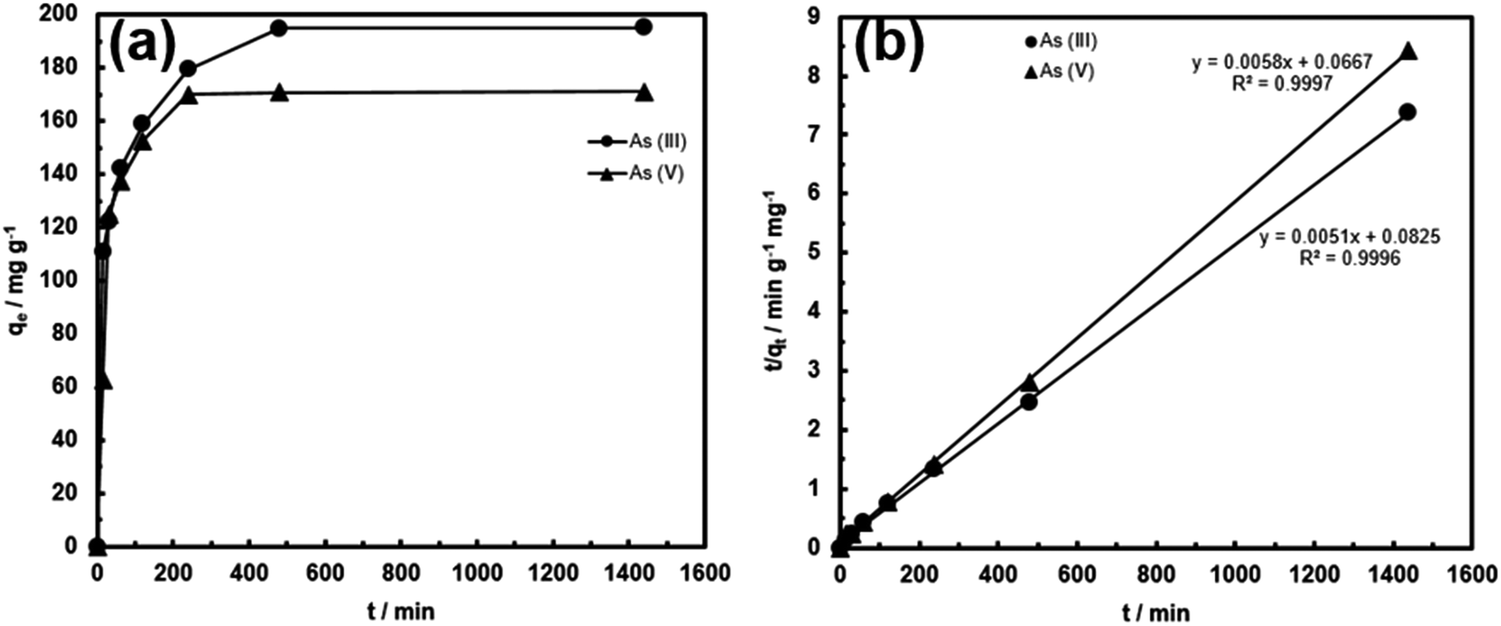 | ||
| Fig. 8 (a) As(III) and As(V) adsorption kinetic curves of FeOx–CNS-13; (b) fitting of the kinetic data with the pseudo-second-order model. | ||
It is expected that the pH of the aqueous medium should have a crucial influence on arsenic removal by the composite adsorbents, because it greatly affects the surface charge of the composites and arsenic species distribution.1–4 Thus, the adsorption of arsenic with FeOx–CNS-13 as a function of pH in a broad range has been investigated. Fig. 9 plots qe as a function of pH (within 2–10), which was undertaken at the initial As(III) and As(V) concentration of 1000 mg L−1 and the contact time of 24 h. Clearly, arsenic adsorption with FeOx–CNS-13 is strongly pH-dependent. With the increase of pH from 2 to 10, the As(V) uptake capacity remains nearly constant at 197–200 mg g−1 in the range of 2–4, followed by the continuous drop to 150 mg g−1 at pH = 10. The As(III) uptake shows an opposite trend. The maximum As(III) uptake (230 mg g−1) is found at pH = 8. Both increasing and decreasing the pH value lead to the reduction in adsorption capacity.
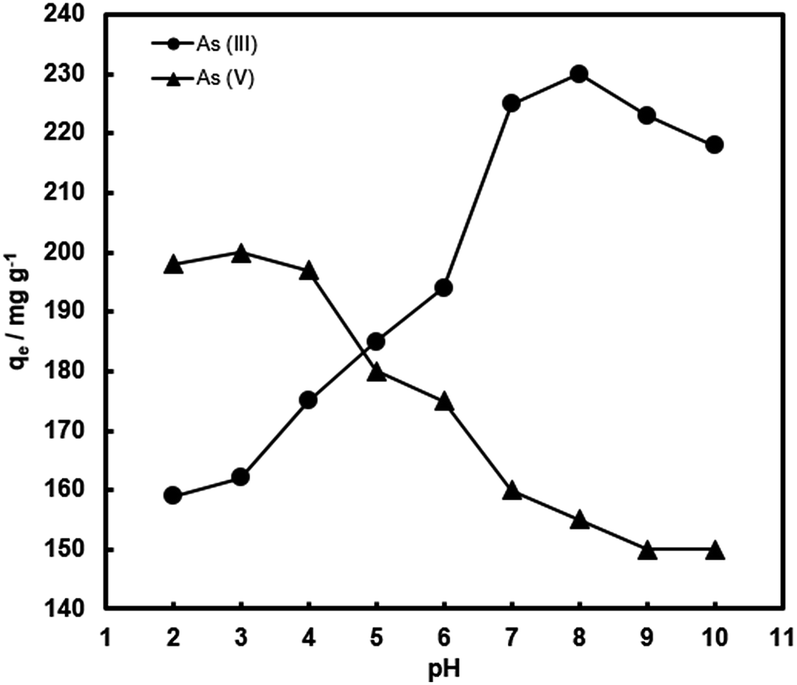 | ||
| Fig. 9 Arsenic uptake of FeOx–CNS-13 as a function of pH. The initial arsenic concentration is 1000 mg L−1. | ||
Though with minor variations in the optimum pH values for arsenic adsorption, similar trends of change for the pH effects have commonly been observed with various iron oxide-based adsorbents and can be explained by the changes in surface charge of the adsorbents and the arsenic speciation.9–11,14,23–26,28,34,37,40,41 The point of zero charge (pHPZC) of FeOx–CNS-13 is measured to be ≈2.8 (Fig. S6(a) in ESI†). The surface of FeOx–CNS-13 is positively charged at pH < pHPZC, and in the pH range of 2–2.8, the predominant species of As(V) is H2AsO4− with negative charge.2 The electrostatic interaction between positively charged FeOx–CNS-13 and negatively charged As(V) species results in the enhanced adsorption within this pH range. The deprotonation of surface hydroxyl groups occurs with the increase of pH, leading to a negatively charged surface for FeOx–CNS-13 after pH of 2.8, and the predominant species of As(V) is H2AsO4− from pH 3–6.5, and HAsO42− from after pH of 6.5, both with negative charge. It thus results in the reduced As(V) adsorption uptake. However, the adsorption of As(III) on FeOx–CNS-13 is different. The optimum pH for As(III) adsorption is 8. When the pH value is below 8, the arsenic removal efficiency significantly increases with the increase of pH, but decreases when pH is above 8. The solubilization and leaching of Fe species into the acidic solution at pH 2–6 (Fig. S6(b) in ESI†) is the primary reason for the reduced As(III) adsorption capacity with the decrease of pH in this range.25 At pH < 9.2, As(III) exists mainly as H3AsO3,2 indicating As(III) is adsorbed on FeOx–CNS-13 through a surface complexation mechanism.25 The removal efficiency drops at pH > 9 due to the ionization of H3AsO3 to H2AsO3− with negative charge, which results in the occurrence of more competition between arsenite and OH− anions. Moreover, the increasing coulombic repulsion between As(III) species and the negative surface of FeOx–CNS-13 may be another important reason for the decrease in As(III) uptake when pH > 9.
Conclusions
A set of FeOx–CNS nanocomposite adsorbents has been successfully synthesized by encapsulating iron oxide at different content (7–60 wt%) into a high-surface-area high-pore-volume carbon nanosphere matrix with hierarchical pore structure, S-CNS. Detailed characterizations of the nanocomposites have been undertaken. TEM results reveal the overall uniform distribution of the crystalline iron oxide within the nanocomposites. In FeOx–CNS-13, small iron oxide nanoparticles with sizes of 3–4 nm are interspersed uniformly on the carbon nanosphere matrix, with the retention of the hierarchical pore structures. On the contrary, aggregates of iron oxide are observed in FeOx–CNS-60 with severe pore filling and blockage. XPS and XRD results indicate that the iron oxide species is primarily crystalline Fe3O4. The adsorption of both As(III) and As(V) has been investigated with the nanocomposites. Best adsorption performance is achieved with FeOx–CNS-13 having the optimum iron oxide content, with superior adsorption capacity of 416 and 201 mg g−1 for As(III) and As(V), respectively, which are highest or among the highest reported thus far in the literature. Both reducing and increasing the iron oxide content lead to the reduced arsenic adsorption. Meanwhile, from the adsorption kinetics, FeOx–CNS-13 is also featured with high adsorption rate. The results confirm the high potential of FeOx–CNS-13 as superior adsorbents for arsenic removal from industrial wastewater streams.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The financial support from the Natural Science and Engineering Research Council (NSERC) of Canada (Grant #: EGP 491823-15 and EGP2 503876-16) and Goldcorp Inc. is greatly appreciated. HS also thanks Laurentian University for awarding incentive graduate scholarships.References
- W. R. Cullen and K. J. Reimer, Chem. Rev., 1989, 89, 713–764 CrossRef CAS.
- P. L. Smedley and D. G. Kinniburgh, Appl. Geochem., 2002, 17, 517–568 CrossRef CAS.
- D. Mohan and C. U. Pittman Jr, J. Hazard. Mater., 2007, 142, 1–53 CrossRef CAS PubMed.
- T. S. Y. Choong, T. G. Chuah, Y. Robiah, F. L. G. Koay and I. Azni, Desalination, 2007, 217, 139–166 CrossRef CAS.
- Z. Cheng, F. Fu, D. D. Dionysiou and B. Tang, Water Res., 2016, 96, 22–31 CrossRef CAS PubMed.
- M. Kobya, E. Demirbas and F. Ulu, J. Environ. Chem. Eng., 2016, 4, 1484–1494 CrossRef CAS.
- S. C. N. Tang and I. M. C. Lo, Water Res., 2013, 47, 2613–2632 CrossRef CAS PubMed.
- M. L. Pierce and C. B. Moore, Water Res., 1982, 16, 1247–1253 CrossRef CAS.
- K. P. Raven, A. Jain and R. H. Loeppert, Environ. Sci. Technol., 1998, 32, 344–349 CrossRef CAS.
- S. Dixit and J. G. Hering, Environ. Sci. Technol., 2003, 37, 4182–4189 CrossRef CAS PubMed.
- S. Luther, N. Borgfeld, J. Kim and J. G. Parsons, Microchem. J., 2012, 101, 30–36 CrossRef CAS.
- S. Yean, L. Cong, C. T. Yavuz, J. T. Mayo, W. W. Yu, A. T. Kan, V. L. Colvin and M. B. Tomson, J. Mater. Res., 2005, 20, 3255–3264 CrossRef CAS.
- C. T. Yavuz, J. T. Mayo, W. W. Yu, A. Prakash, J. C. Falkner, S. Yean, L. Cong, H. J. Shipley, A. Kan, M. Tomson, D. Natelson and V. L. Colvin, Science, 2006, 314, 964–967 CrossRef PubMed.
- T. Tuutijärvi, J. Lu, M. Sillanpää and G. Chen, J. Hazard. Mater., 2009, 166, 1415–1420 CrossRef PubMed.
- L.-S. Zhong, J.-S. Hu, H.-P. Liang, A.-M. Cao, W.-G. Song and L.-J. Wan, Adv. Mater., 2006, 18, 2426–2431 CrossRef CAS.
- F. Mou, J. Guan, Z. Xiao, Z. Sun, W. Shi and X.-A. Fan, J. Mater. Chem., 2011, 21, 5414–5421 RSC.
- F. Mou, J. Guan, H. Ma, L. Xu and W. Shi, ACS Appl. Mater. Interfaces, 2012, 4, 3987–3993 CAS.
- C.-Y. Cao, J. Qu, W.-S. Yan, J.-F. Zhu, Z.-Y. Wu and W.-G. Song, Langmuir, 2012, 28, 4573–4579 CrossRef CAS PubMed.
- B. Wang, H. Wu, L. Yu, R. Xu, T.-T. Lim and X. W. Lou, Adv. Mater., 2012, 24, 1111–1116 CrossRef CAS PubMed.
- T. Wang, L. Zhang, H. Wang, W. Yang, Y. Fu, W. Zhou, W. Yu, K. Xiang, Z. Su, S. Dai and L. Chai, ACS Appl. Mater. Interfaces, 2013, 5, 12449–12459 CAS.
- Z. Wei, R. Xing, X. Zhang, S. Liu, H. Yu and P. Li, ACS Appl. Mater. Interfaces, 2013, 5, 598–604 CAS.
- G. K. Das, C. S. Bonifacio, J. De Rojas, K. Liu, K. van Benthem and I. M. Kennedy, J. Mater. Chem. A, 2014, 2, 12974–12981 CAS.
- M. Jang, S.-H. Min, T.-H. Kim and J. K. Park, Environ. Sci. Technol., 2006, 40, 1636–1643 CrossRef CAS PubMed.
- Z. Gu, J. Fang and B. Deng, Environ. Sci. Technol., 2005, 39, 3833–3843 CrossRef CAS PubMed.
- V. Chandra, J. Park, Y. Chun, J. W. Lee, I.-C. Hwang and K. S. Kim, ACS Nano, 2010, 4, 3979–3986 CrossRef CAS PubMed.
- K. Zhang, V. Dwivedi, C. Chi and J. Wu, J. Hazard. Mater., 2010, 182, 162–169 CrossRef CAS PubMed.
- X.-L. Wu, L. Wang, C.-L. Chen, A.-W. Xu and X.-K. Wang, J. Mater. Chem., 2011, 21, 17353–17359 RSC.
- G. Sheng, Y. Li, X. Yang, X. Ren, S. Yang, J. Hu and X. Wang, RSC Adv., 2012, 2, 12400–12407 RSC.
- J. Zhu, R. Sadu, S. Wei, D. H. Chen, N. Haldolaarachchige, Z. Luo, J. A. Gomes, D. P. Young and Z. Guo, ECS J. Solid State Sci. Technol., 2012, 1, M1–M5 CrossRef CAS.
- J. Zhu, S. Wei, H. Gu, S. B. Rapole, Q. Wang, Z. Luo, N. Haldolaarachchige, D. P. Young and Z. Guo, Environ. Sci. Technol., 2012, 46, 977–985 CrossRef CAS PubMed.
- Y. Wang, Q. He, H. Qu, X. Zhang, J. Guo, J. Zhu, G. Zhao, H. A. Colorado, J. Yu, L. Sun, S. Bhana, M. A. Khan, X. Huang, D. P. Young, H. Wang, X. Wang, S. Wei and Z. Guo, J. Mater. Chem. C, 2014, 2, 9478–9488 RSC.
- G. Gollavelli, C.-C. Chang and Y.-C. Ling, ACS Sustainable Chem. Eng., 2013, 1, 462–472 CrossRef CAS.
- B. Paul, V. Parashar and A. Mishra, Environ. Sci.: Water Res. Technol., 2015, 1, 77–83 CAS.
- F. Yu, S. Sun, J. Ma and S. Han, Phys. Chem. Chem. Phys., 2015, 17, 4388–4397 RSC.
- J. Ding, B. Li, Y. Liu, X. Yan, S. Zeng, X. Zhang, L. Hou, Q. Cai and J. Zhang, J. Mater. Chem. A, 2015, 3, 832–839 CAS.
- H. Su, Z. Ye and N. Hmidi, Colloids Surf., A, 2017, 522, 161–172 CrossRef CAS.
- X. Yu, S. Tong, M. Ge, J. Zuo, C. Cao and W. Song, J. Mater. Chem. A, 2013, 1, 959–965 CAS.
- Z. Wu, W. Li, P. A. Webley and D. Zhao, Adv. Mater., 2012, 24, 485–491 CrossRef CAS PubMed.
- M. Baikousi, A. B. Bourlinos, A. Douvalis, T. Bakas, D. F. Anagnostopoulos, J. Tucek, K. Safarova, R. Zboril and M. A. Karakassides, Langmuir, 2012, 28, 3918–3930 CrossRef CAS PubMed.
- B. Chen, Z. Zhu, J. Ma, Y. Qiu and J. Chen, J. Mater. Chem. A, 2013, 1, 11355–11367 CAS.
- J. Yang, H. Zhang, M. Yu, I. Emmanuelawati, J. Zou, Z. Yuan and C. Yu, Adv. Funct. Mater., 2014, 24, 1354–1363 CrossRef CAS.
- V. K. Tiwari, Z. Chen, F. Gao, Z. Gu, X. Sun and Z. Ye, J. Mater. Chem. A, 2017, 5, 12131–12143 CAS.
- J. Pastor-Villegas and C. J. Durán-Valle, Carbon, 2002, 40, 397–402 CrossRef CAS.
- Z. Wu, P. Webley and D. Zhao, Langmuir, 2010, 26, 10277–10286 CrossRef CAS PubMed.
- K. S. W. Sing, D. H. Everett, R. A. W. Haul, L. Moscou, R. A. Pierotti, J. Rouquerol and T. Siemieniewska, Pure Appl. Chem., 1985, 57, 603–619 CrossRef CAS.
- A. Dandekar, R. Baker and M. Vannice, Carbon, 1998, 36, 1821–1831 CrossRef CAS.
- P. Bazula, A. Lu, J. Nitz and F. Schüth, Microporous Mesoporous Mater., 2008, 108, 266–275 CrossRef CAS.
- A. Kuznetsova, D. Mawhinney, V. Naumenko, J. Yates, J. Liu and R. Smalley, Chem. Phys. Lett., 2000, 321, 292–296 CrossRef CAS.
- Z. Wu, Q. Li, D. Peng, P. Webley and D. Zhao, J. Am. Chem. Soc., 2010, 132, 12042–12050 CrossRef CAS PubMed.
- G. Ennas, G. Marongiu and A. Musinu, J. Mater. Res., 1998, 14, 1570–1578 CrossRef.
- H. Kong, J. Song and J. Jang, Chem. Commun., 2010, 46, 6735–6737 RSC.
- G. Bhargava, I. Gouzman, C. Chun, T. Ramanarayanan and S. Bernasek, Appl. Surf. Sci., 2007, 253, 4322–4329 CrossRef CAS.
- R. Netterfield, P. Martin, C. Pacey, W. Sainty, D. Mckenzie and G. Auchterlonie, J. Appl. Phys., 1989, 66, 1805–1809 CrossRef CAS.
- S. Chao, Y. Takagi, G. Lucovsky, P. Pai, R. Custer, J. Tyler and J. Keem, Appl. Surf. Sci., 1986, 26, 575–583 CrossRef CAS.
- L. Xu, J.-W. McGraw, F. Gao, M. Grundy, Z. Ye, Z. Gu and J. Shepherd, J. Phys. Chem. C, 2013, 117, 10730–10742 CAS.
- J. Pattanayak, K. Mondal, S. Mathew and S. B. Lalvani, Carbon, 2000, 38, 589–596 CrossRef CAS.
- G. Zhang, Z. Ren, X. Zhang and J. Chen, Water Res., 2013, 47, 4022–4031 CrossRef CAS PubMed.
- B. Chen, Z. Zhu, S. Liu, J. Hong, J. Ma, Y. Qiu and J. Chen, ACS Appl. Mater. Interfaces, 2014, 6, 14016–14025 CAS.
- Y. Lin and J. Chen, J. Colloid Interface Sci., 2014, 420, 74–79 CrossRef CAS PubMed.
- Y. Du, H. Fan, L. Wang, J. Wang, J. Wu and H. Dai, J. Mater. Chem. A, 2013, 1, 7729–7737 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra06187k |
| This journal is © The Royal Society of Chemistry 2017 |