
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOptical properties and structural phase transitions of W-doped VO2(R) under pressure†
Huafang Zhanga,
Zhou Guana,
Benyuan Chengb,
Quanjun Li*a,
Ran Liua,
Jing Zhanga,
Zhenxian Liuc,
Ke Yangd,
Tian Cui a and
Bingbing Liu*a
a and
Bingbing Liu*a
aState Key Laboratory of Superhard Materials, Jilin University, Changchun 130012, China. E-mail: liubb@jlu.edu.cn; liquanjun@jlu.edu.cn; Fax: +86-431-85168256; Fax: +86 18043176111; Tel: +86-431-85168256 Tel: +86 18043176111
bChina Academy of Engineering Physics, Mianyang, Sichuan 621900, China
cU2A Beam Line, Carnegie Institution of Washington, Upton, New York 11973, USA
dChinese Academy Sciences, Shanghai Institute Applied Physics, Shanghai 201204, China
First published on 21st June 2017
Abstract
We investigated the optical properties and structural phase transitions of W-doped VO2(R) nanoparticles under pressure based on in situ synchrotron X-ray diffraction (XRD) and infrared (IR) spectroscopy. The structural transition sequence follows VO2(R)–VO2(CaCl2-type)–VO2(Mx) and VO2(Mx)–VO2(Mx′) within metallic phases, in compression and decompression processes, respectively, demonstrating that the structural transition can be decoupled from the metal-insulator transition (MIT). VO2(R) and VO2(CaCl2-type) exhibit expected behavior of increased metallicity under pressure; surprisingly, VO2(Mx) shows gradually decreased metallicity with increasing pressure and VO2(Mx′) is still metallic under ambient conditions. We find that the reduced metallicity of VO2(Mx) is attributed to W-doping induced local structure distortion in the high-pressure region, while the metallic properties of VO2(Mx′) are associated with the enhancement of electron concentration due to the presence of W donors, which shifted the Fermi level toward the conduction band. The present results demonstrate that the structural transition is not the key factor in driving the metal-insulator transition, and provide an effective method for inducing MIT in VO2(Mx′).
Introduction
Vanadium dioxide (VO2) is a typical strongly correlated material with different polymorphs, including the tetragonal VO2(R), VO2(A) and VO2(C),1–3 and the monoclinic VO2(M1), VO2(B), and VO2(D).4–6 Changes in the structure and physical properties in the VO2 system can be induced by controlling temperature. For example, VO2 undergoes a reversible MIT at about 68 °C, accompanied with a structural transition from VO2(R) to VO2(M1), and huge changes in physical properties of electrical resistivity, infrared transmittance and magnetic susceptibility, which make this material potentially useful for intelligent devices.1,7,8 Moreover, VO2 also shows a reversible MIT between the semiconducting VO2(A) and a new metallic tetragonal structure (VO2(AH)) at about 162 °C, and the VO2(AH) transforms into the well-known metallic VO2(R) at about 450 °C.9,10 The semiconducting VO2(B) and VO2(D) transform directly into the metallic VO2(R) at about 500 °C and 320 °C, respectively.6,11 As the thermally driven MIT in the strongly correlated VO2 system is always accompanied by a structural transition, remarkable experimental and theoretical efforts have been devoted to addressing the role of structural transformation and electron–electron correlation in driving the MIT,12–16 but the mechanism is still under debate.Pressure, an important tuning parameter, which can effectively reduce atom distances and increase electron orbital overlaps, has great influence on the structure and physical properties on materials.17–19 In recent years, lots of high pressure investigations have been carried out on VO2 systems and found that VO2 in different polymorphs show unique structural phase transition and physical properties under pressure.20–32 The phase transition sequence of VO2(M1) follows M1–M1′–X/Mx, Mx–Mx′ in compression and decompression process, respectively,23,30–32 the M1′ phase is semiconductor within low pressure region but becomes metallic above 43.2 GPa. Different with VO2(M1), the VO2(A) and VO2(B) transform directly into an amorphous state under pressure,28,29 in particular, VO2(A) being metallic before amorphization due to the delocalization of V3d orbital electrons, whereas VO2(B) remains a semiconductor before and after amorphization. These studies on semiconducting VO2 polymorphs suggest that the MIT may occur without structural transition under pressure. In addition, the study on the structural transition of the metallic VO2(R) at 383 K indicates that VO2(R) transforms into an orthorhombic structure (VO2(CaCl2-type)) at about 13.3 GPa, and then starts to transform into a new monoclinic structure (VO2(X)) at about 38.6 GPa.30 According to previous study on metallic V2O3,33 another typical MIT system, for which the thermal-driven MIT always accompanied with a structure transformation from a corundum structure into a monoclinic phase, the corundum-to-monoclinic transformation occurs between two metallic phases under pressure, demonstrating that the structural transition can be decoupled from the MIT. These results suggest the possibility of decoupling MIT from structural transition in metallic VO2(R) phase under pressure, however, no efforts have been performed on the electrical study on the metallic VO2(R) under pressure.
As is known, the R-to-M1 transition temperature (Tc) can be reduced by doping with transition metal, such as W6+, Mo6+, and Nb5+.34–39 Among these, the most exciting prospective way would be the W-doping, which enabling precise and wide range control of Tc, with a nearly linear reduction rate of 18.4–26 °C/at%,34,37–39 providing experimental base for high pressure research on VO2(R). Infrared spectroscopy, as a powerful tool to probe the dynamics of charge carriers, has been widely used in the high-pressure study on strong correlated materials.40–43 In this study, we present the measurements of infrared properties and structure phase transitions of W–VO2(R) under pressure up to 63.9 GPa. We found a pressure induced rutile–orthorhombic–monoclinic transitions in three metallic phases, and obtained a metallic W–VO2(Mx′) (W–VO2(Mx′)) at ambient conditions.
Experimental
Synthesis
W–VO2(R) nanoparticles were synthesized through hydrothermal reaction combined with subsequent calcinations. In a typical procedure, 0.2275 g V2O5, 0.3938 g oxalic acid and 0.0824 g Na2WO4·2H2O were added to 100 ml deionized water, and followed by vigorous stirring until a pale-yellows solution was formed. The solution was transferred into a Teflon-lined autoclave with the filling ratio of 0.6, and held at 190 °C for 24 h. The precipitate was collected by centrifugal separation, washed with deionized water and alcohol and dried in vacuum freezing dryer. The resulted powers were calcined at 600 °C for 3 h in argon atmosphere.Characterization
Samples were characterized using transmission electron microscopy (TEM) (200 KV, HITACHI, H-81001V), and high-resolution transmission electron microscopy (HRTEM) (JEOL JEM-3010). High-pressure XRD and IR experiments were carried out by using diamond anvil cells (DACs). The T301 stainless-steel gaskets were preindented to 40–50 μm and drilled with a center hole of ∼100 μm as sample chamber. High-pressure XRD use a 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 methanol–ethanol mixture as pressure-transmitting medium. High-pressure IR measurements were carried out using pure sample. The ruby fluorescence technique was used to calibrate pressures for all experiments. The in situ high pressure synchrotron XRD measurements were performed at the BL15U beamline of Shanghai Synchrotron Radiation Facility (SSRF) (λ = 0.6199 Å). The high-pressure IR spectra were collected through a Bruker Vertex 80v FTIR spectrometer and a microscope/a Hyperion 2000 IR microscope equipped with a liquid nitrogen cooled MCT detector.
:1 methanol–ethanol mixture as pressure-transmitting medium. High-pressure IR measurements were carried out using pure sample. The ruby fluorescence technique was used to calibrate pressures for all experiments. The in situ high pressure synchrotron XRD measurements were performed at the BL15U beamline of Shanghai Synchrotron Radiation Facility (SSRF) (λ = 0.6199 Å). The high-pressure IR spectra were collected through a Bruker Vertex 80v FTIR spectrometer and a microscope/a Hyperion 2000 IR microscope equipped with a liquid nitrogen cooled MCT detector.
The optical reflectivity of sample–diamond interface Rsd(ω) was obtained by the following equation:44
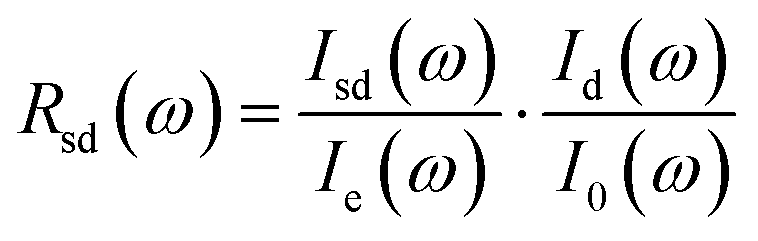 | (1) |
Results
Pressure-dependent IR spectra
The characterizations for the structure and morphology of synthesized W-doped samples are shown in Fig. S1,† all the diffraction peaks can be indexed to VO2(R), indicating that the W-doped samples are pure VO2(R) nanoparticles with average diameter of 100–200 nm. To investigate the electronic changes for W–VO2(R) nanoparticles under pressure, we performed high pressure mid-IR reflectivity Rsd(ω) spectra measurements (shown in Fig. 1). The data between 1700 cm−1 and 2700 cm−1 were cut out from Rsd(ω) spectra due to the absorption of diamond. At ∼1.9 GPa, Rsd(ω) is slightly lower than expected for sample–air interface due to the larger refractive index value in diamond.40,45 With increasing pressure, Rsd(ω) gradually increases up to 12.8 GPa, and then shows almost no change in the 12.8–30.2 GPa range. Surprisingly, Rsd(ω) starts to decrease above 30.2 GPa, and then decrease more subtle when the pressure is beyond 53.6 GPa. This unexpected behavior above 30.2 GPa has not been observed in VO2 system before. According to early study, the reflectivity in terms of dielectric constant (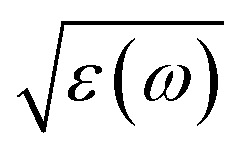 ) at normal incidence is given by45
) at normal incidence is given by45
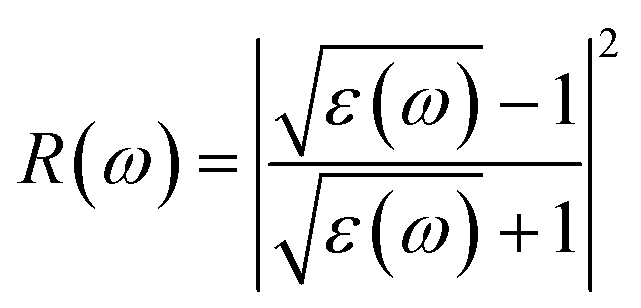 | (2) |
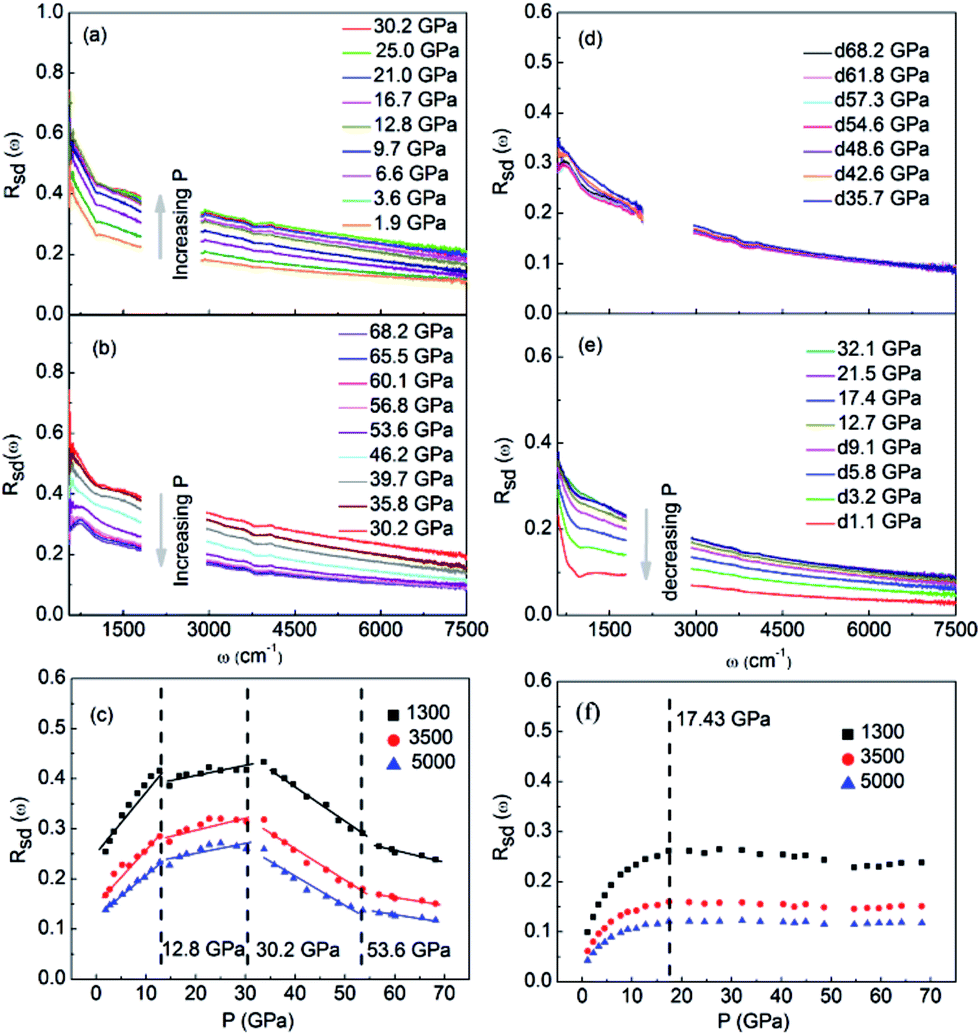 | ||
| Fig. 1 IR reflectivity spectra of W–VO2 nanoparticles upon compression in the pressure region of (a) 1.9–30.2 GPa and (b) 30.2–68.2 GPa, (c) pressure-reflectivity diagram of three different wave numbers (1300, 3500 and 3000 cm−1) upon compression. IR reflectivity spectra of W–VO2 nanoparticles upon decompression in the pressure region of (d) 68.2–35.7 GPa and (e) 32.1–1.1 GPa, (f) pressure-reflectivity diagram for three different wave numbers (1300, 3500 and 3000 cm−1) upon decompression. | ||
The 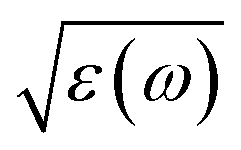 has the following form
has the following form
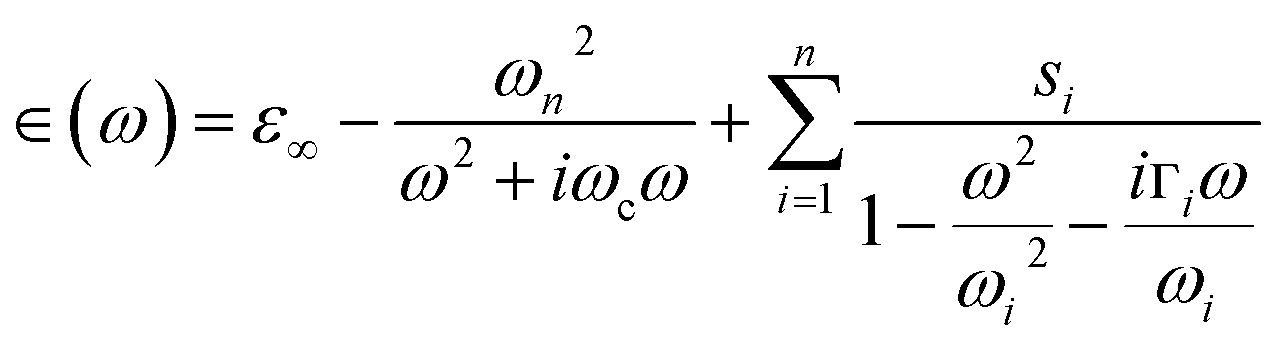 | (3) |
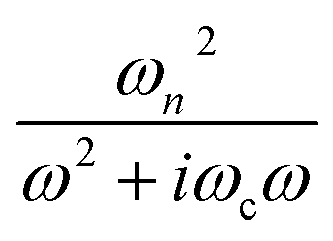 represents the contribution to ∈(ω) from free-electron, ωn and ωc are the carrier density parameter and collision frequency, respectively. The last term is the sum over Lorenz, classical oscillators that contribution to the nonconstant part of ∈(ω). In low-frequency, the reflectivity of the R phase is characterized by a continues rise due to the free-electronic contribution.46,47 The higher reflectivity in low-frequency region, indicates higher carrier density in measured samples. Therefore, the pressure dependence of the IR reflectivity at low frequencies gives the electronic evolution under pressure, qualitatively. As shown in Fig. 1c, the reflectivity at low frequencies increases gradually owing to a larger bandwidth up to 12.8 GPa, but show a noticeable change in slope at about 12.8 GPa, which could be related to the structural phase transition of VO2(R)-to-VO2(CaCl2-type) observed in early XRD study on pure VO2(R).30 When pressure is above 30.2 GPa, the reflectivity starts to decrease, but then decreases more sluggish with further compressing above 53.6 GPa, suggesting the metallicity decreases in W–VO2 under higher pressure. These results show pressure makes our sample more metallic only up to 30.2 GPa. According to early study, the IR spectroscopy decisively dependents on the configuration of atoms,48 therefore, the unexpected decrease of IR reflectivity should be mainly related to the accompanying change in crystal structure under pressure.
represents the contribution to ∈(ω) from free-electron, ωn and ωc are the carrier density parameter and collision frequency, respectively. The last term is the sum over Lorenz, classical oscillators that contribution to the nonconstant part of ∈(ω). In low-frequency, the reflectivity of the R phase is characterized by a continues rise due to the free-electronic contribution.46,47 The higher reflectivity in low-frequency region, indicates higher carrier density in measured samples. Therefore, the pressure dependence of the IR reflectivity at low frequencies gives the electronic evolution under pressure, qualitatively. As shown in Fig. 1c, the reflectivity at low frequencies increases gradually owing to a larger bandwidth up to 12.8 GPa, but show a noticeable change in slope at about 12.8 GPa, which could be related to the structural phase transition of VO2(R)-to-VO2(CaCl2-type) observed in early XRD study on pure VO2(R).30 When pressure is above 30.2 GPa, the reflectivity starts to decrease, but then decreases more sluggish with further compressing above 53.6 GPa, suggesting the metallicity decreases in W–VO2 under higher pressure. These results show pressure makes our sample more metallic only up to 30.2 GPa. According to early study, the IR spectroscopy decisively dependents on the configuration of atoms,48 therefore, the unexpected decrease of IR reflectivity should be mainly related to the accompanying change in crystal structure under pressure.
Upon decompression, Rsd(ω) exhibits subtle change up to 17.4 GPa (Fig. 1d and e). With further decompressing, Rsd(ω) starts to decrease significantly. When the applied pressure decreased to 1.1 GPa, the reflectivity is clearly lower than that of initial W–VO2(R) phase (Fig. 1a and e). Similar behaviors are observed in the pressure-reflectivity diagram for wave numbers at 1300, 3500 and 3000 cm−1. These results suggest that the metallicity is independent with pressure above 17.4 GPa, but decreases with pressure upon further decompressing.
For clarifying that whether W–VO2 nanoparticles in different pressure regions are metallic or not, we carried out high pressure transmittance spectra measurements on both W–VO2 and pure VO2 (shown in Fig. 2). The data between 1700 cm−1 and 2700 cm−1 were cut out from Rsd(ω) spectra due to the absorption of diamond. Compared with the transmittance spectra of the insulating VO2(M1) and the quenched VO2(Mx′) in pure VO2 (inset of Fig. 2), which present maxima in IR transmittance at around 1000 cm−1, there is no transmitted light in the whole spectrum in W–VO2 under various pressures. These results clearly identify that W–VO2 is metallic in whole pressure region.
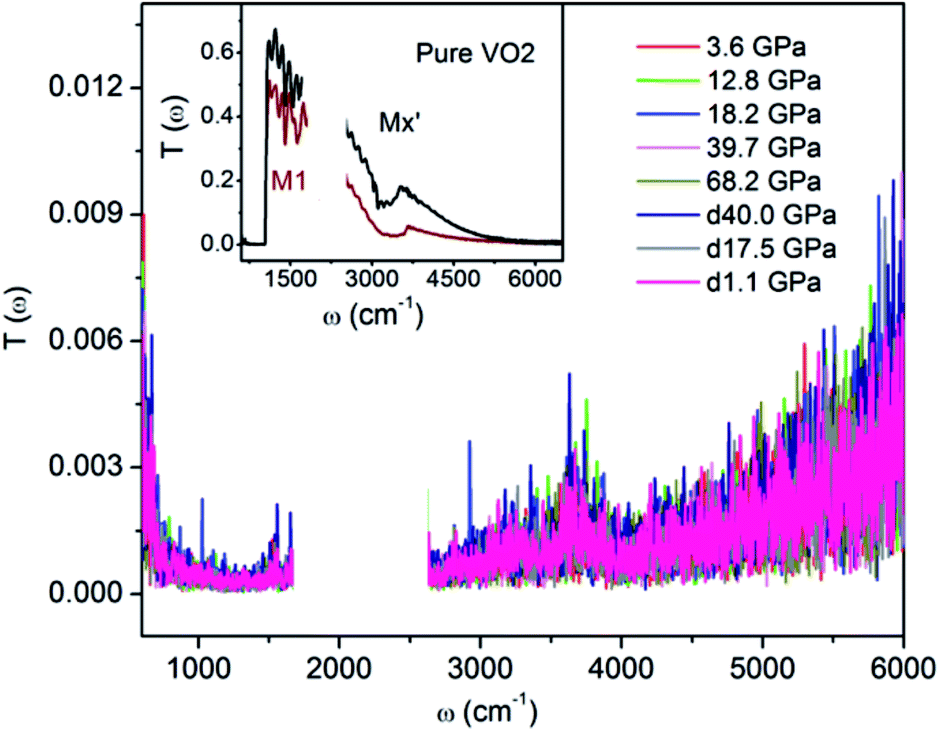 | ||
| Fig. 2 IR transmittance spectra of W–VO2 nanoparticles at selected pressures. Inset: IR transmittance spectrum of insulating M1 and Mx′ phases in pure VO2. | ||
Pressure-dependent XRD patterns
To have a better understanding of the changes in the metallicity of W–VO2(R) under pressure, we carried out high pressure XRD measurements in characterizing the crystal structural transition up to 63.9 GPa (shown in Fig. 3). In the low pressure range (0–13.5 GPa), all diffraction peaks shift toward smaller d-spacings due to lattice compression, without modifications of the overall diffraction pattern. Above 13.5 GPa, some diffraction peaks start to broaden or split (denoted by stars), indicating a structural transition from R into an orthorhombic CaCl2-type phase, which is consistent with previous study on pure VO2(R).30 With further increasing pressure above 31.3 GPa, several new peaks (marked with “#”) appear, suggesting the occurrence of the structural transition from CaCl2-type phase into a new high-pressure phase. At 63.9 GPa, the CaCl2-type phase completely transforms into the new high-pressure phase. The structure of the high-pressure phase is still unclear. The diffraction pattern of the high-pressure phase is clearly different from that of X phase in bulk VO2,30,31 but similar to that of the Mx phase in VO2 nanoparticles (Fig. 4a).32 Therefore, a monoclinic unit cell with space group P21/c was used to fit the measured XRD pattern at 63.9 GPa. As shown in Fig. 4b, the fitting gives relatively satisfying results, with Rwp = 0.48%, Rp = 0.36%. The lattice parameters are listed in Table 1. Note that the onset and finish pressures of the structural transition from the CaCl2-type to the high-pressure Mx phase agree well with the pressures of the slope change in IR reflectivity (Fig. 1c). This indicates the electric change of W–VO2 depends on its structural transition behaviors under high pressure.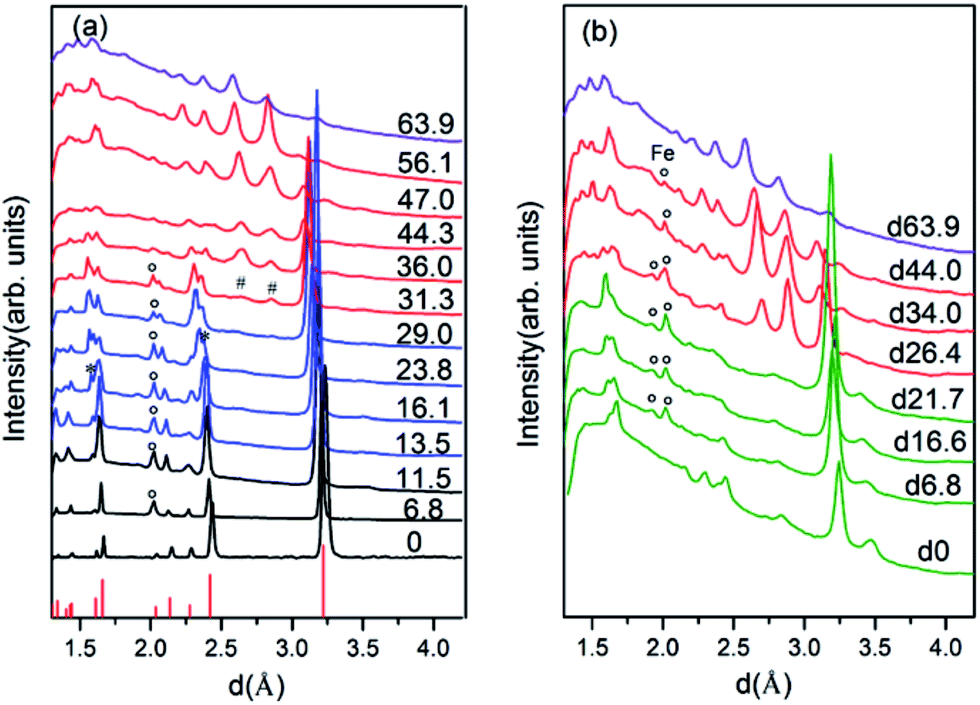 | ||
| Fig. 3 XRD patterns of W–VO2(R) nanoparticles at various pressures, (a) collected upon compression, (b) collected upon decompression. Diffraction peaks marked “*”, “#” and “°” originate from VO2(CaCl2-type), VO2(Mx) and Fe, respectively. The lines at the bottom of (a) correspond to the diffraction peaks of VO2(R) taken from PDF no. 79-1655. | ||
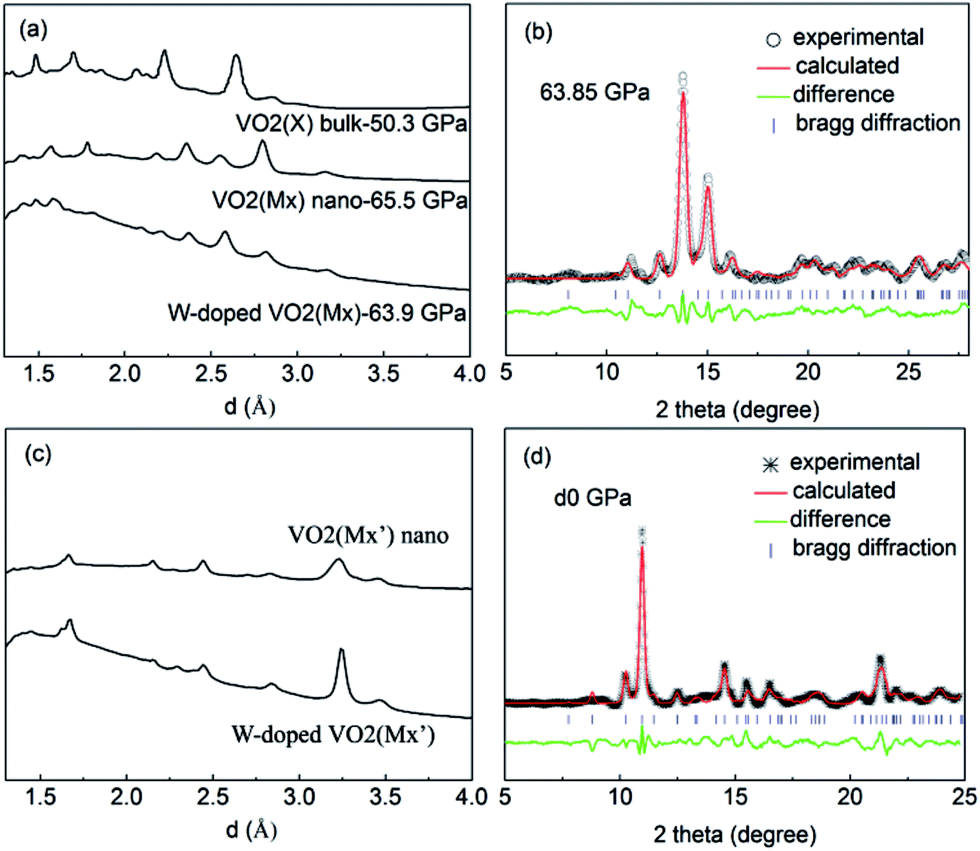 | ||
| Fig. 4 The XRD patterns of (a) W–VO2(Mx) nanoparticles, VO2(Mx) nanoparticles and bulk VO2(X), (c) W–VO2(Mx′) and VO2(Mx′) nanoparticles. Refinement results of (b) W–VO2(Mx) nanoparticles at 63.9 GPa, (d) W–VO2(Mx′) nanoparticles at ambient pressure. | ||
Upon decompression, a new peak appears at ∼3.09 Å when the pressure decreases to ∼44.0 GPa, indicating a structural transition from VO2(Mx) phase into a new structure (shown in Fig. 3b). With further decreasing pressure, all the diffraction peaks of VO2(Mx) phase gradually weaken and finally disappear at ∼21.7 GPa, showing that all the VO2(Mx) samples transform into the new phase. We note that this structural transition process is rather sluggish, with the two phases coexist spanning over 20 GPa, demonstrating that this structural transformation is kinetically hindered. The XRD pattern of the released sample is similar to that of Mx′ phase in pure VO2 nanoparticles32 (shown in Fig. 4c), therefore, a monoclinic structure with the same space group P21/c were used to refine it. The results of Rietveld refinements are shown in Fig. 4d, with Rwp = 0.50%, Rp = 0.35%, and the obtained cell parameters are listed in Table 1. Both of the cell parameters and cell volume V are close to those of pure counterparts,32 suggesting that W-doping induces only subtle modifications in crystal structure of the released sample. It is interesting that the quenched W–VO2 samples are metallic at ambient conditions. Thus, these results demonstrate the W-doping induced MIT in VO2(Mx′) without structural phase transition. The structural phase transitions of rutile–orthorhombic–monoclinic and monoclinic–monoclinic in metallic phases in compressing and decompressing processes, respectively, and the doping-induced MIT in VO2(Mx′) indicate that the structural transition can be decoupled from MIT.
Discussion
The pressure dependence of reflectivity in the R phase (Fig. 1c) indicates that pressure makes the R phase more metallic. Within the 0–13.5 GPa range, the isotropic compression in the R phase resulted in a continuous decrease in cell volume (Fig. 3). So the W–VO2(R) follows the expected behavior of increased metallicity due to larger bandwidth.33 In the pressure range of 12.8–30.2 GPa, the reflectivity is high and show subtle changes within the crystal structure of the CaCl2-type phase, indicating that CaCl2-type phase show better metallicity than the R phase. It is worth noting that the metallicity gradually decreases with further compressing above 30.2 GPa, accompanied with a structural transformation from the CaCl2-type phase into the high-pressure Mx phase, indicates that the high-pressure Mx phase in W–VO2 nanoparticles is less metallic. As the W–Mx phase has the same crystal structure with pure the Mx phase, we made a comparison on the metallicity of these samples (shown in Fig. 5). The reflectivity of the W–Mx is much lower than that of the pure Mx, suggesting that the W-doping makes Mx phase less metallic. Although the W–Mx and pure Mx phases show similar XRD patterns, we note that the diffraction peaks in the W–Mx phase is broader and weaker than that of the pure Mx phase (Fig. 4a), moreover, the lattice parameter c and the cell volume V of the W–Mx phase are larger than that of pure Mx phase (Table 1). These results suggest that W-doping induced disorder of local structure was probably formed in W–Mx. According to previous study, the W-doping induced disorder in VO2(R) results in an obvious increase in resistivity.49 So the poor metallicity in the W–Mx phase is likely related to W-doping induced disorder of local structure.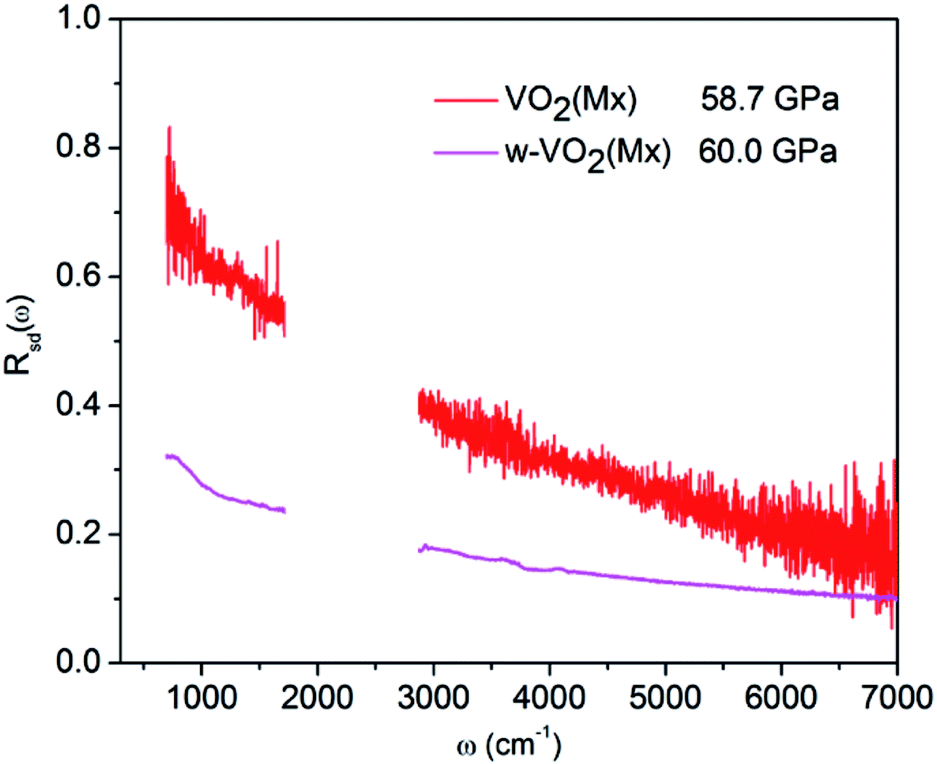 | ||
| Fig. 5 IR reflectivity spectra of the Mx phase for W-doped and pure VO2 nanoparticles. | ||
Upon decompression, the structural transition from the W–Mx to W–Mx′ phase starts from ∼44 GPa and completed at about 21.7 GPa, whereas the metallicity is independent of applied pressure above 17.4 GPa, suggests that the metallicity of these two phases are stable in the high pressure region. When released to ambient pressure, the W–Mx′ phase shows remarkable decreased metallicity but is still metallic, in contrast to the insulator Mx′ phase in pure VO2 nanoparticles (shown in the inset of Fig. 2). These results indicates that W-doping induced MIT occurs in the Mx′ phase. Compared with the W-doping induced MIT in M1 phase, which accompanied with a structural transformation from the M1 to R phase, the MIT in the Mx′ phase occurs without structural transition. Previous studies have indicated that the presence of W donors enhance the electron concentration in VO2,50–53 which could shifts the Fermi level toward conduction band and resulting in the MIT. These results not only pave an effective method for modifying electronic properties of VO2 new structure, but also provide further insight into the MIT of VO2.
Conclusion
We have investigated the structural phase transitions and optical properties on W-doped VO2(R) nanoparticles under pressure. Compared with the temperature-dependent study on VO2, where rutile–monoclinic structural transition always coincides with MIT, the structural transitions of rutile–orthorhombic–monoclinic occur within metallic phases under pressure. Moreover, we find a W-doping induced MIT in VO2(Mx′) without structural transitions. We suggest that the decreased metallicity in Mx phase under pressure can be attributed to the W-doping induced local structure distortion, and the MIT of Mx′ phase may results from the enhancement of electron concentration due to the presence of W donors, which shifted the Fermi level toward the conduction band. Present results demonstrate that structural transition is not the key factor in driving metal-insulator transition, and paves an effective method for modifying electronic properties of VO2 new structure.Acknowledgements
This work was financially supported by the NSFC (11374120 and 51320105007), National Basic Research Program of China (2011CB808200), Program for Changjiang Scholars and Innovative Research Team in University (No. IRT1132), and the Cheung Kong Scholars Programme of China.References
- F. J. Morin, Phys. Rev. Lett., 1959, 3, 34–36 CrossRef CAS.
- F. Théobald, J. Less-Common Met., 1977, 53, 55–71 CrossRef.
- D. Hagrman, J. Zubieta, C. J. Warren, L. M. Meyer, M. M. J. Treacy and R. C. Haushalter, J. Solid State Chem., 1998, 138, 178–182 CrossRef CAS.
- G. Andersson, Acta Chem. Scand., 1954, 8, 1599–1606 CrossRef CAS.
- F. Théobald, R. Cabala and J. Bernard, J. Solid State Chem., 1976, 17, 431–438 CrossRef.
- L. Liu, F. Cao, T. Yao, Y. Xu, M. Zhou, B. Qu, B. Pan, C. Wu, S. Wei and Y. Xie, New J. Chem., 2012, 36, 619–625 RSC.
- K. W. Lee, J. J. Kweon, C. E. Lee, A. Gedanken and R. Ganesan, Appl. Phys. Lett., 2010, 96, 243111 CrossRef.
- J. Zhou, Y. Gao, Z. Zhang, H. Luo, C. Cao, Z. Chen, L. Dai and X. Liu, Sci. Rep., 2013, 3, 3029 CrossRef PubMed.
- T. Yao, Y. Oka and N. Yamamoto, J. Solid State Chem., 1994, 112, 196–198 CrossRef CAS.
- S. Zhang, B. Shang, J. Yang, W. Yan, S. Wei and Y. Xie, Phys. Chem. Chem. Phys., 2011, 13, 15873–15881 RSC.
- X. Xiao, H. Cheng, G. Dong, Y. Yu, L. Chen, L. Miao and G. Xu, CrystEngComm, 2013, 15, 1095–1106 RSC.
- R. M. Wentzcovitch, W. W. Schulz and P. B. Allen, Phys. Rev. Lett., 1994, 72, 3389–3392 CrossRef CAS PubMed.
- M. M. Qazilbash, M. Brehm, B. G. Chae, P. C. Ho, G. O. Andreev, B. J. Kim, S. J. Yun, A. V. Balatsky, M. B. Maple, F. Keilmann, H. T. Kim and D. N. Basov, Science, 2007, 318, 1750–1753 CrossRef CAS PubMed.
- J. B. Goodenough, J. Solid State Chem., 1971, 3, 490 CrossRef CAS.
- M. W. Haverkort, Z. Hu, A. Tanaka, W. Reichelt, S. V. Streltsov, M. A. Korotin, V. I. Anisimov, H. H. Hsieh, H. J. Lin, C. T. Chen, D. I. Khomskii and L. H. Tjeng, Phys. Rev. Lett., 2005, 95, 196404 CrossRef CAS PubMed.
- T. C. Koethe, Z. Hu, M. W. Haverkort, C. Schussler-Langeheine, F. Venturini, N. B. Brookes, O. Tjernberg, W. Reichelt, H. H. Hsieh, H. J. Lin, C. T. Chen and L. H. Tjeng, Phys. Rev. Lett., 2006, 97, 116402 CrossRef CAS PubMed.
- C. C. Chen, A. Herhold, C. Johnson and A. Alivisatos, Science, 1997, 276, 398–401 CrossRef CAS PubMed.
- R. E. Cohen, I. I. Mazin. and D. G. Isaak, Science, 1997, 275, 654–657 CrossRef CAS PubMed.
- A. Togo, F. Oba and I. Tanaka, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 134106 CrossRef.
- C. H. Neuman, A. W. Lawson and R. F. Brown, J. Chem. Phys., 1964, 41, 1591–1595 CrossRef CAS.
- C. N. Berglund and A. Jayaraman, Phys. Rev., 1969, 185, 1034–1039 CrossRef CAS.
- E. Arcangeletti, L. Baldassarre, D. Di Castro, S. Lupi, L. Malavasi, C. Marini, A. Perucchi and P. Postorino, Phys. Rev. Lett., 2007, 98, 196406 CrossRef CAS PubMed.
- M. Mitrano, B. Maroni, C. Marini, M. Hanfland, B. Joseph, P. Postorino and L. Malavasi, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 184108 CrossRef.
- C. Marini, E. Arcangeletti, D. Di Castro, L. Baldassare, A. Perucchi, S. Lupi, L. Malavasi, L. Boeri, E. Pomjakushina, K. Conder and P. Postorino, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 235111 CrossRef.
- M. Baldini, P. Postorino, L. Malavasi, C. Marini, K. W. Chapman and H. k. Mao, Phys. Rev. B: Condens. Matter Mater. Phys., 2016, 93, 245137 CrossRef.
- X. Zhang, J. Zhang, F. Ke, G. Li, Y. Ma, X. Liu, C. Liu, Y. Han, Y. Ma and C. Gao, RSC Adv., 2015, 5, 54843–54847 RSC.
- W. P. Hsieh, M. Trigo, D. A. Reis, G. Andrea Artioli, L. Malavasi and W. L. Mao, Appl. Phys. Lett., 2014, 104, 021917 CrossRef.
- B. Cheng, Q. Li, H. Zhang, R. Liu, B. Liu, Z. Yao, T. Cui, J. Liu, Z. Liu, B. Sundqvist and B. Liu, Phys. Rev. B: Condens. Matter Mater. Phys., 2016, 93, 184109 CrossRef.
- Y. Wang, J. Zhu, W. Yang, T. Wen, M. Pravica, Z. Liu, M. Hou, Y. Fei, L. Kang, Z. Lin, C. Jin and Y. Zhao, Nat. Commun., 2016, 7, 12214 CrossRef CAS PubMed.
- L. Bai, Q. Li, S. A. Corr, Y. Meng, C. Park, S. V. Sinogeikin, C. Ko, J. Wu and G. Shen, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 91, 104110 CrossRef.
- H. Zhang, Q. Li, B. Cheng, Z. Guan, R. Liu, B. Liu, Z. Liu, X. Li, T. Cui and B. Liu, RSC Adv., 2016, 6, 104949–104954 RSC.
- Q. Li, H. Zhang, C. Lin, F. Tian, J. S. Smith, C. Park, B. Liu and G. Shen, J. Alloys Compd., 2017, 709, 260–266 CrossRef CAS.
- Y. Ding, C. C. Chen, Q. Zeng, H. S. Kim, M. J. Han, M. Balasubramanian, R. Gordon, F. Li, L. Bai, D. Popov, S. M. Heald, T. Gog, H. K. Mao and M. van Veenendaal, Phys. Rev. Lett., 2014, 112, 056401 CrossRef PubMed.
- Q. Gu, A. Falk, J. Wu, L. Ouyang and H. Park, Nano Lett., 2007, 7, 363–366 CrossRef CAS PubMed.
- Z. P. Wu, A. Miyashita, S. Yamamoto, H. Abe, I. Nashiyama, K. Narumi and H. Naramoto, J. Appl. Phys., 1999, 86, 5311–5313 CrossRef CAS.
- C. Piccirillo, R. Binions and I. P. Parkin, Eur. J. Inorg. Chem., 2007, 4050–4055 CrossRef CAS.
- P. Jin, S. Nakao and S. Tanemura, Thin Solid Films, 1998, 324, 151–158 CrossRef CAS.
- J. C. Rakotoniaina, R. Mokrani-Tamellin, J. R. Gavarri, G. Vacquier, A. Casalot and G. J. Calvarin, Solid State Chem., 1993, 103, 81–94 CrossRef CAS.
- T. Hörlin, T. Niklewski and M. Nygren, Mater. Res. Bull., 1972, 7, 1515–1524 CrossRef.
- L. Baldassarre, A. Perucchi, E. Arcangeletti, D. Nicoletti, D. Di Castro, P. Postorino, V. A. Sidorov and S. Lupi, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 245108 CrossRef.
- Y. Mita, D. Izaki, M. Kobayashi and S. Endo, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 100110 CrossRef.
- M. Matsunami, H. Okamura, A. Ochiai and T. Nanba, Phys. Rev. Lett., 2009, 103, 237202 CrossRef CAS PubMed.
- A. Perucchi, L. Baldassarre, P. Postorino and S. Lupi, J. Phys.: Condens. Matter, 2009, 21, 323202 CrossRef CAS PubMed.
- C. T. Seagle, D. L. Heinz, Z. Liu and R. J. Hemley, Appl. Opt., 2009, 48, 545–552 CrossRef CAS PubMed.
- H. W. Verleur, A. S. Barker and C. N. Berglund, Phys. Rev., 1968, 172, 788–798 CrossRef CAS.
- M. Kang, S. W. Kim, J. W. Ryu and T. Noh, AIP Adv., 2012, 2, 012168 CrossRef.
- M. M. Qazilbash, M. Brehm, G. O. Andreev, A. Frenzel, P. C. Ho, B. G. Chae, B. J. Kim, S. J. Yun, H. T. Kim, A. V. Balatsky, O. G. Shpyrko, M. B. Maple, F. Keilmann and D. N. Basov, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 075107 CrossRef.
- Y. Sun, S. Jiang, W. Bi, R. Long, X. Tan, C. Wu, S. Wei and Y. Xie, Nanoscale, 2011, 3, 4394–4401 RSC.
- K. Shibuya, M. Kawasaki and Y. Tokura, Appl. Phys. Lett., 2010, 96, 022102 CrossRef.
- X. Tan, T. Yao, R. Long, Z. Sun, Y. Feng, H. Cheng, X. Yuan, W. Zhang, Q. Liu, C. Wu, Y. Xie and S. Wei, Sci. Rep., 2012, 2, 466 Search PubMed.
- C. Wu, F. Feng, J. Feng, J. Dai, L. Peng, J. Zhao, J. Yang, C. Si, Z. Wu and Y. Xie, J. Am. Chem. Soc., 2011, 133, 13798–13801 CrossRef CAS PubMed.
- L. Q. Mai, B. Hu, T. Hu, W. Chen and E. D. J. Gu, Phys. Chem. B., 2006, 110, 19083–19086 CrossRef CAS PubMed.
- W. Choi, A. Termin and M. R. Hoffmann, J. Phys. Chem., 1994, 98, 13669–13679 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra04605g |
| This journal is © The Royal Society of Chemistry 2017 |