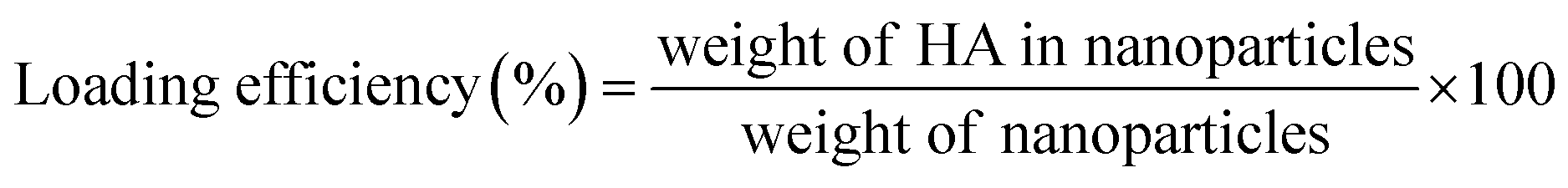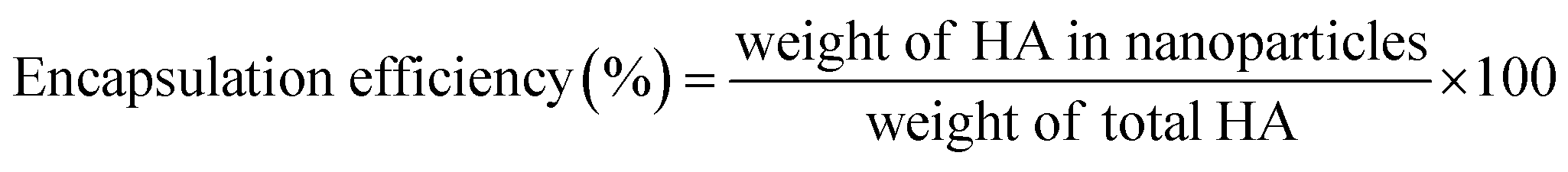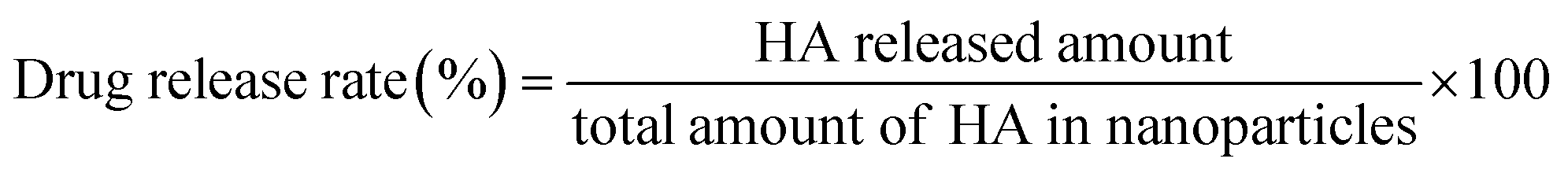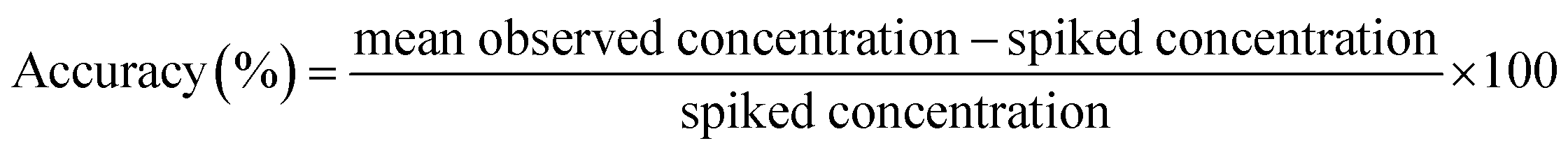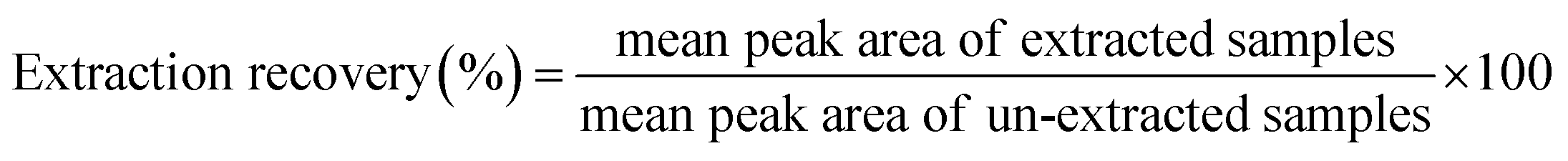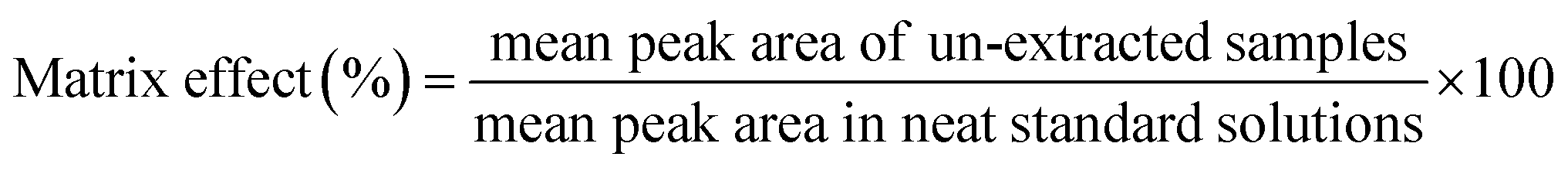DOI:
10.1039/C7RA04748G
(Paper)
RSC Adv., 2017,
7, 42073-42082
Poly(lactic-co-glycolic) acid nanoparticles improve oral bioavailability of hypocrellin A in rat
Received
27th April 2017
, Accepted 12th August 2017
First published on 30th August 2017
Abstract
Hypocrellin A (HA), a perihydroxylated polycyclic quinone isolated from the fungus of Shiraia bambusicola, exhibits a wide spectrum of biological activities in pre-clinical studies. However, poor water solubility is a major clinical constraint factor of HA, leading to unpredictable bioavailability. In this study, HA-loaded poly (lactic-co-glycolic acid) (PLGA) nanoparticles (PLGA/HA NPs) were prepared by the single-emulsion solvent-evaporation technique. Characterization of PLGA/HA NPs showed that HA was successfully encapsulated on the PLGA. The drug-loading content and encapsulation efficiency (EE) were 7.0% and 57.5%, respectively. The in vitro release profile demonstrated that HA was released more slowly from nanoparticles in pH 1.5 and pH 6.8 than in pH 7.4. Furthermore, the highest solubility of PLGA/HA NPs in aqueous solution was approximately 35.67-fold that of native HA (n-HA). PLGA/HA NPs had more superior stability compared with n-HA in physiological conditions. In this paper, a sample and sensitive LC-MS/MS method was validated for the first time to quantify HA in rat plasma. The pharmacokinetic parameters and bioavailability of HA between PLGA/HA NPs and n-HA were compared after oral administration in rat. The results implied that the relative bioavailability of PLGA/HA NPs was 2.67-fold that of n-HA, and PLGA/HA NPs had a longer half-life. Therefore, these results suggested that PLGA-blend nanoparticles improved the solubility, stability and bioavailability of HA, and could become a potential and promising carrier for the oral delivery of HA.
1. Introduction
Hypocrellin A (HA), an active compound isolated from Shiraia bambusicola that mainly grows in China and Japan, is a novel photosensitizer of high singlet oxygen quantum yield for photodynamic therapy (PDT).1–3 HA has many biological and pharmacological properties, such as antitumour,2,4,5 antiviral,6 antibacterial, and antileishmanial activities.7,8 The most effective activity of HA is anticancer, which can decrease A549 cell viability via PDT induced cell apoptosis.4 Moreover, it possesses excellent photodynamic antimicrobial activity.8 These findings suggested that HA may be used to treat various diseases. Thus far, HA has been almost wholly utilized as an external formulation for skin diseases such as white lesions of the vulva, vitiligo, tinea capitis, psoriasis and lichen amyloidosis.9 However, little information is currently available on the characterization of its pharmacokinetic properties; this information is necessary and helpful to develop new clinical HA preparations for oral and intravenous administration.
In addition, HA is a hydrophobic drug, and its solubility must be improved to maximize its potential bioavailability.10 One strategy to improve HA solubility is chemical modification. Typical chemical modification techniques included attempted salt formation, complexed with metal ions, and added cosolvents, hydrotropic agents and surfactants. These approaches increase solubility by altering a solvating environment, making it better suited for maintaining hydrophobic solutes in aqueous solution.11 Indeed, several research approaches have been reported to enhance HA solubility, including sulfonation of HA12 and chelation of HA with metal ions.13,14 However, most of the HA derivatives generated to date have exhibited less PDT efficacy than their parent; those water-soluble derivatives reduced cellular uptake and led to poor photodynamic activity in vitro, storage stability and biocompatibility.15
In recent years, nano drug delivery systems (NDDS) have provided remarkable advantages in enhancing water solubility and pharmacological activity of photosensitizers.16–20 As a biodegradable polymers, PLGA has been extensively used in NDDS for lower toxicity and excellent loaded-drug efficacy.17,19,20 PLGA improved loaded drugs the dispersibility, solubility, and absorption in vitro, and prolonged the half-life time and plasma circulation in vivo of loaded drugs.19,21 In addition, PLGA is degraded in the body into nontoxic lactic acid and glycolic acid, which are the normal components of Kreb's cycle, and subsequently eliminated as carbon dioxide and water without affecting the normal cellular functions.22
In previous work, we reported that HA-loaded PLGA nanoparticles (PLGA/HA NPs) improved the photodynamic anticancer effect.4 However, developed nanoparticle formulations need a thorough evaluation in terms of pharmacokinetic investigation. In fact, the physicochemical parameters of nanoparticles, such as particle size, hydrophobicity and surface modification, dramatically changed drug pharmacokinetics and impacted drug bioavailability and biodistribution.19 On the basis of these factors, in the present study, we further explored the in vitro release, solubility and stability of PLGA/HA NPs. Moreover, a sensitive liquid chromatography tandem mass spectrometry (LC-MS/MS) method with multiple reaction monitoring (MRM) was developed for the first time and validated for the determination of HA in rat plasma. Finally, we compared the pharmacokinetic parameters and bioavailability of HA between PLGA/HA NPs and n-HA after oral administration in rat.
2. Materials and methods
2.1 Chemicals
HA (98%) was isolated from the bodies of Shiraia bambusicola. Docetaxel (99.8%) was obtained from Aladdin Industrial Corporation (Shanghai, China). Poly (D,L-lactic-co-glycolic acid) (PLGA, L/G = 50/50, MW = 30 kDa) was obtained from Ji Nan Dai Gang Biomaterial Corporation (Shandong, China). Polyvinyl alcohol (PVA, MW = 31 kDa) and dimethyl sulfoxide (DMSO) were obtained from Sigma-Aldrich (MO, USA). Pepsin (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3000) and trypsin (1:250) were obtained from Solarbio Corporation (Beijing, China). Spectramax M2 Multiscan Spectrum was obtained from Molecular Devices Corporation (USA). Methanol and acetonitrile (HPLC grade) were purchased from Tedia Company (USA). HPLC-grade water produced by MilliQ System (Millipore, Paris, France) was utilized throughout the experiments. All other chemicals were of analytical grade and commercially available.
:3000) and trypsin (1:250) were obtained from Solarbio Corporation (Beijing, China). Spectramax M2 Multiscan Spectrum was obtained from Molecular Devices Corporation (USA). Methanol and acetonitrile (HPLC grade) were purchased from Tedia Company (USA). HPLC-grade water produced by MilliQ System (Millipore, Paris, France) was utilized throughout the experiments. All other chemicals were of analytical grade and commercially available.
2.2 Preparation of PLGA/HA NPs
PLGA/HA NPs were obtained using the single-emulsion solvent-evaporation technique previously reported by our group.4 Briefly, HA (10 mg) and PLGA (100 mg) were dissolved in acetone (2.5 mL) for 6 h at room temperature. The organic phase was rapidly injected into 50 mL PVA aqueous solution (0.5%, w/v) and homogenized using a high-speed homogenizer (IKA, Germany), resulting in an oil-in-water (O/W) solution. Next, the residual organic solvent was eliminated by mechanical stirring at 1500 rpm overnight. The particles were then recovered by centrifugation (Eppendorf Centrifuge 5418R, Germany) at 13500 rpm for 30 min at 4 °C, and washed twice with distilled water to remove the residual drugs and emulsifiers. Lastly, PLGA/HA NPs were added 2% sucrose as a freeze-dried adjuvant agent to avoid aggregation; PLGA/HA NPs were freeze-dried at −80 °C for 48 h and stored at −20 °C in the dark before use.
2.3 Drug loading efficiency (LE) and encapsulation efficiency (EE)
The account of HA loading and encapsulation in PLGA/HA NPs was investigated by HPLC. Chromatographic separation was performed on an Agilent Zorbax Extend-C18 column (4.6 × 150 mm, 5 μm; Agilent Technologies, Waldbronn, Germany). The mobile phase consisted of methanol (A) and water containing 0.1% formic acid (B) (90:10 v/v), pumped isocratic elution at a flow rate of 0.8 mL min−1. The sample injection volume was 10 μL, and the UV detector was used at a wavelength of 464 nm. One milligram of lyophilized PLGA/HA NPs was dissolved in 10 mL of methanol aimed to extract HA. The samples in methanol were ultrasonically mixed for 15 min to leach out HA entirely. Then, samples were centrifuged at 13500 rpm for 30 min, and supernatant was collected. An aliquot of 10 μL was used for HPLC analysis. A calibration curve was established by HA standard solutions. The calibration curve had good linearity (r2 = 0.999), ranging from 0.75–100 μg mL−1. The amount of HA loaded and encapsulated in PLGA/HA NPs was expressed as LE (1) or EE calculated as follows (2):| |
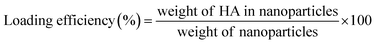 | (1) |
| |
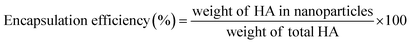 | (2) |
2.4 In vitro release profile
The release of HA from PLGA/HA NPs was carried by dissolving PLGA/HA NPs in artificial gastric (pH 1.5), intestinal (pH 6.8) and blood (pH 7.4) media. Those suspensions were kept at 37 °C and 100 rpm. The release of HA from PLGA/HA NPs was conducted by the dialysis membrane method. PLGA/HA NPs (10 mg) were divided in 2 mL of distilled water and sealed in a dialysis membrane bag (cut off MW = 12 kDa, Millipore, France). The dialysis bag containing nanoparticles was immersed in 100 mL artificial gastric (pH 1.5), intestinal (pH 6.8) and blood (pH 7.4) media with 0.5% (w/v) Tween-80 in a conical flask, followed by constant shaking at a speed of 100 rpm at 37 °C. At a determined time, 1 mL of release medium was withdrawn to analyse. The release medium was completely replaced with the same volume of fresh buffer solution. The released HA was measured by HPLC as mentioned above. All assays were carried out in triplicate.
The drug release rate (RR) was calculated according to the following formula (3):
| |
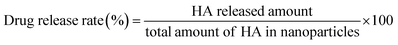 | (3) |
2.5 Solubility study and stability
To verify the solubility of n-HA and PLGA/HA NPs, excessive samples (n-HA and PLGA/HA NPs) were dissolved and dispersed into 10 mL double distilled water at 25 ± 2 °C, then acutely blended 30 s every 5 min. After 30 min, samples were taken out and filtered through a 0.22 μm Millipore membrane. The filtrate was diluted an appropriate number of times, and the absorbance was detected by UV-vis spectrophotometer at 464 nm. The n-HA and PLGA/HA NPs solubility was calculated by standard curve and dilution times. The chemical stability of HA was determined by exposing both n-HA and PLGA/HA NPs to physiological conditions, and the remaining HA present in the media was measured. Specifically, the n-HA and PLGA/HA NPs suspensions in artificial gastric (pH 1.5), intestinal (pH 6.8) and blood (pH 7.4) media were kept at 37 °C and 100 rpm. At designated time points (0, 1, 2, 4, 7, 10, 12 h), samples were removed and detected using Multiscan Spectrum at 464 nm. All of the analyses were expressed as the mean ± standard deviation (S.D.) of three replicates.
2.6 Animal
Sprague-Dawley rats were kept in an environmentally controlled breeding room and fed with standard laboratory food and water for two weeks before starting the experiments. The experimental protocol were conformed to the principles of the National Institutes of Health guide for the care and use of Laboratory Animals, (NIH publications no. 8023, revised 1978) and approved by the Animal Care and Institutional Animal Ethics Committee of Nanjing Normal University.
2.7 Chromatograph system and conditions for HA quantification
The LC-MS/MS analysis was conducted in positive ion ESI mode on a Waters Quattro Micro System equipped with a triple-quadrupole mass spectrometer connected to a liquid chromatograph (Waters Alliance). Chromatographic separation was performed on an Agilent Zorbax Extend-C18 column (4.6 × 150 mm, 5 μm; Agilent Technologies, Waldbronn, Germany). The column temperature was maintained at 28 °C.23 The mobile phase consisted of methanol (A), and water containing 0.1% formic acid (B) (90:10 v/v) was used for isocratic elution at 1 mL min−1, with a total run time per sample of approximately 5 min. The injection volume was 10 μL.
Mass spectrometric detection was performed on a Waters Quattro Micro triple-quadrupole mass spectrometer equipped with an electrospray ionization (ESI) interface. Both the analyte HA and internal standard (IS) docetaxcel were monitored under positive ion-multiple reaction monitoring (+MRM) mode with transitions of m/z 547.1 → 529 for HA, and m/z 830.7 → 549.4 for IS, respectively. Other parameters of the mass spectrometer were as follows: spray voltage, 3250 V; vaporizer temperature, 300 °C; and capillary temperature, 350 °C. Argon was used as the collision gas for collision-induced dissociation (CID), 3.2 × 10−3 mbar; solvent off gas flow rate, 400 L h−1; and gas curtain gas velocity, 50 L h−1. The auto sampler cooler was maintained at 4 °C.
The data were acquired and processed using Analyst version MassLynx software (Version 4.0). For quantification, the peak area ratios of the target ions of the HA were compared to those of the IS with weighted 1/X2 (where X = drug concentration) least squares calibration curves in which the peak area ratios of the calibration standards were plotted versus their concentrations.
2.8 Preparation of stock, standard and quality control samples
Stock solutions of HA (100 μg mL−1) and IS docetaxel (500 μg mL−1) were dissolved in methanol. The IS working solution was diluted with methanol to 2.5 μg mL−1. Standard solutions of HA in rat plasma were prepared by spiking with an appropriate volume of the serially diluted stock solutions, giving final concentrations of 15, 50, 100, 200, 500, 1 000, 1 500, and 2000 ng mL−1 for HA.
Quality control (QC) samples were prepared from blank plasma at high, medium and low concentrations of 1 500, 500, and 50 ng mL−1, respectively. All solutions were stored at −20 °C prior to analysis. In addition, all the samples containing the calibration samples, QC samples and plasma samples were processed and prepared according to the procedure described as follows.
2.9 Plasma sample preparation
The 100 μL of heparinized plasma sample (or containing 10 μL HA blank plasma) was aliquoted into a 1.5 mL polypropylene tube and mixed with 10 μL IS solution (2.5 μg mL−1) and 50 μL acetic acid. Then, 500 μL of acetonitrile was added, and the solution was vortexed for 5 min. Next, the solution was ultrasonically mixed for 10 min and then centrifuged at 16000 rpm for 10 min. After centrifugation, 400 μL of the supernatant was transferred to a clean vial and dried with concentrator plus (Eppendorf 5305, Germany). The residue was re-dissolved with 100 μL methanol: H2O (50:50, v/v), and a 10 μL aliquot of the sample was injected into LC-MS/MS for analysis.
2.10 Method validation
Methods were validated for specificity, linearity and sensitivity, accuracy and precision, extraction recovery and matrix effect according to the US Food and Drug Administration (FDA) guidance.
2.10.1 Specificity. The specificity of the method was evaluated by comparing the chromatograms of blank plasma from six different rats with the corresponding plasma samples spiked with HA and IS, as well as real samples collected from treated rats after intravenous (i.v.) administration for 5 min.
2.10.2 Linearity and sensitivity. Calibration curves were constructed by plotting the peak-area ratio of HA to the IS versus the concentrations of the analytes. It was achieved from eight-point calibration curves covering a concentration range from 15 to 2000 ng mL−1 HA in plasma samples, using a weighted least-squares linear regression (the weighting factor, 1/X2). The lower limit of quantification (LLOQ) was determined based on at least 10 times the signal-to-noise ratio at which the precision (expressed by relative standard deviation, RSD) and accuracy (calculated by relative error, RE) were lower than 20%.
2.10.3 Accuracy and precision. The intra-day accuracy and precision were investigated by determining six replicates of high, medium and low QC samples at concentrations of 1 500, 500 and 50 ng mL−1 on the same day. The inter-day accuracy and precision were studied by determining three levels of QC samples on three consecutive days. The accuracy calculated as follows (4):| |
 | (4) |
2.10.4 Extraction recovery and matrix effect. Extraction recovery of HA was determined by comparing the peak area of the HA spiked and extracted from blank plasma with those of extracted blank plasma spiked with the HA at the same concentration levels. Recoveries were determined at three QC concentrations for plasma (n = 6). Matrix effects were calculated by the mean peak area ratios of blank plasma samples spiked with HA after extraction divided by the injected working solution with HA at the same QC concentrations. The extraction recovery (5), matrix effects calculated as follows (6):| |
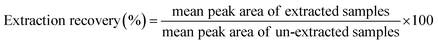 | (5) |
| |
 | (6) |
2.10.5 Stability. The stability of HA was tested by analysing six replicates of HA plasma samples at three QC concentrations under different conditions: 6 h exposure at room temperature, three freeze–thaw cycles at −20 °C, and one week storage at −20 °C. The stock solution stability of the HA was evaluated using three QC levels samples against freshly spiked solutions. If assay values were within acceptable limits of accuracy (±15% RE) and precision (≤15% RSD), samples were considered stable.
2.11 Pharmacokinetic study of HA and PLGA/HA NPs in rat
Eighteen Sprague-Dawley rats (male and female) with body weight of 200–220 g were divided into three groups (n = 6). The first group was i.v. administered n-HA (4 mg kg−1) in 5% glucose aqueous solution, the second group was orally administered n-HA (40 mg kg−1) prepared in 0.5% carboxymethyl cellulose sodium (CMC-Na) to form a suspension, and the third group was orally administered PLGA/HA NPs (containing HA 12.5 mg kg−1) in aqueous solution. All rats were fasted overnight before drug administration. Blood samples (300 μL) were drawn from the orbital vein into heparinized centrifuge tubes under anaesthesia at 0.0833 (only for first group), 0.25, 0.5, 1, 2, 3, 4, 6, 8, 10, 12, 24, and 48 h after dosing. The blood samples were centrifuged at 4000 rpm for 10 min. The supernatant liquor was collected and stored at −20 °C until used for analysis.
2.12 Pharmacokinetics data analysis
All in vitro analyses were expressed as the mean ± standard deviation (S.D.) and repeated three times. Furthermore, the in vivo pharmacokinetics results were described as the mean ± S.D. of six replicates. The pharmacokinetics parameters including the area under curve (AUC), t1/2 elimination, volume of distribution (Vd) and apparent clearance (CL) calculated using WinNonlin software (Pharsight 6.2, NC, USA) with compartmental analysis. Statistical analysis was performed using Graph Pad Prism (Graph Pad Inc, USA). Statistical comparison of mean values was evaluated by ANOVA test, and the results were considered statistically significant if P-value < 0.05.
3. Results and discussion
3.1 Preparation of PLGA/HA NPs
In our study, PLGA/HA NPs were prepared by emulsion solvent evaporation technique. As the linker in PLGA/HA NPs, PVA is a widely used stabilizer for PLGA polymers. The particle size of these nanoparticles was within the range of 20–200 nm reported by Qi et al.4 The drug loading content of the nanoparticles was 7.0%, and encapsulation efficiency of the nanoparticles was 57.5%. These characteristics did not differ significantly from previous experimental results. Particle size, loading and encapsulation efficiency influenced drug solubility, stability and dispersibility. Moreover, properties of PLGA, including concentration, molecular weight, and volume ratio, were related to drug delivery system.24
3.2 In vitro release profile
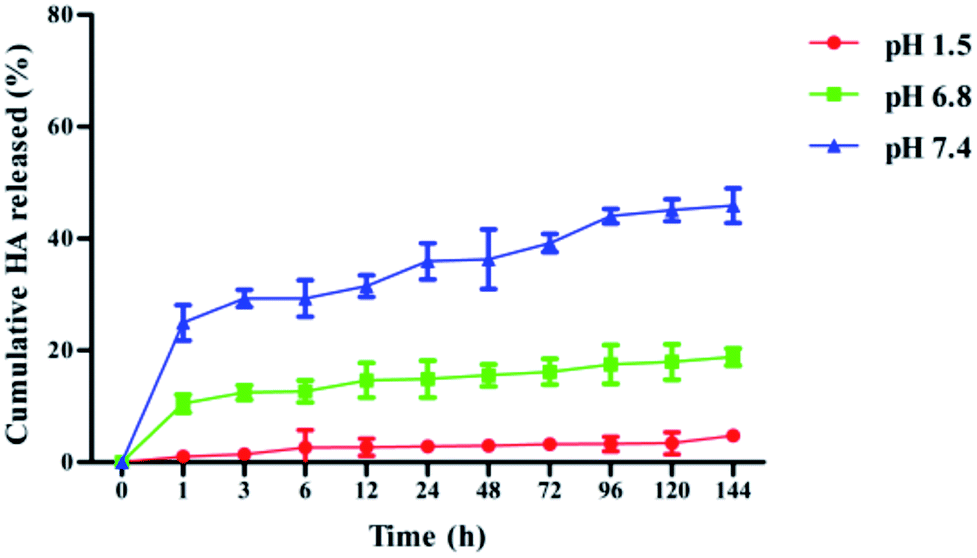
Several mechanisms were reported that were responsible for the drug release from the nanoparticles, including surface and bulk erosion, desorption and diffusion.25 Most of the drug-loaded nanoparticles presented a biphasic release pattern: there was an initial burst release followed by a sustained release of the drug. However, low molecular weight PLGA NPs have also been reported to exhibit a zero-order release profile, particularly with hydrophobic drug molecules.26 In our study, the dialysis bag diffusion technique was used to study the in vitro drug release of HA from PLGA/HA NPs in artificial gastric (pH 1.5), intestinal (pH 6.8) and blood (pH 7.4) biological conditions over a period of 144 h at 37 °C. The cumulative percentage release of HA from PLGA/HA NPs is depicted in Fig. 1. The in vitro drug release kinetics showed a biphasic release only in pH 7.4, with a fast rate of HA release to 29.3% within the initial 3 h. However, we observed that HA release in pH 1.5 and pH 6.8 exhibited zero-order release profiles. HA release was sustained in pH 1.5, pH 6.8 and pH 7.4 to a total of approximately 4.7%, 18.8% and 45%, respectively. At the early stage, the rapid release rate of HA from the nanoparticles may be due to partial absorption or precipitation on the surface of the nanoparticles. Later, HA showed a sustained slow release rate that mainly because diffuse from being encapsulated in nanoparticles.
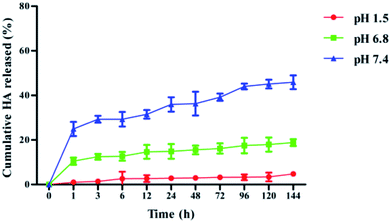 |
| | Fig. 1 Release rates of HA from PLGA nanoparticles in vitro in artificial gastric (pH 1.5), intestinal (pH 6.8) and blood (pH 7.4) biological conditions over a period of 144 h (mean ± S.D., n = 3). | |
3.3 Solubility and stability study
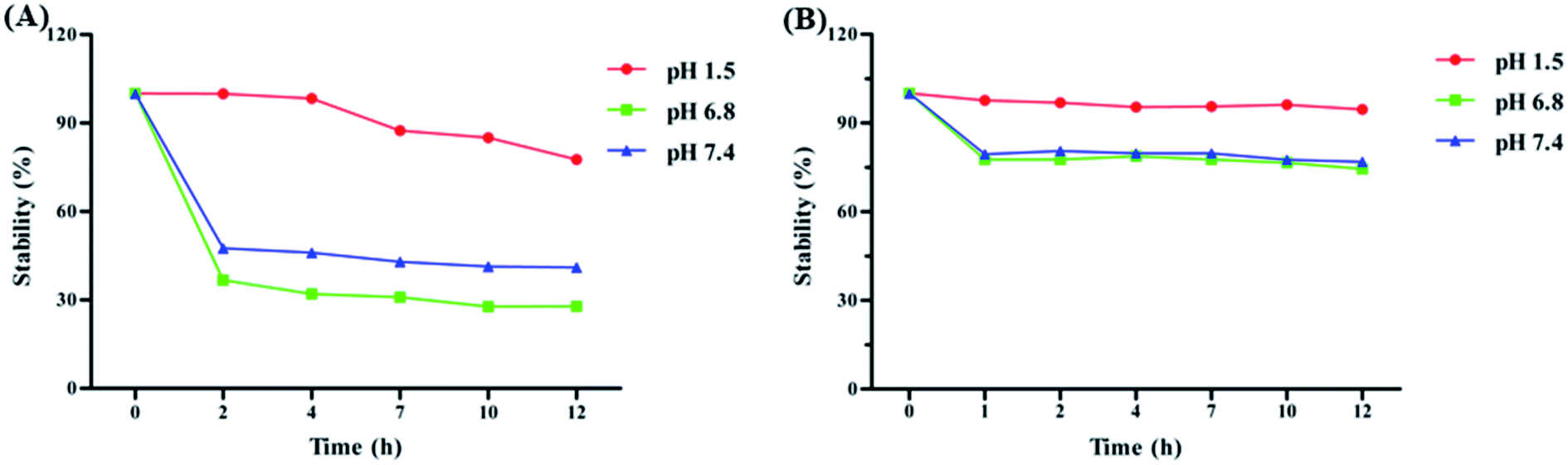
To investigate the solubility of HA and PLGA/HA NPs, samples were dissolved in double distilled water, forming a supersaturated solution. The results in Fig. 2 showed that the dissolved PLGA/HA NPs formed a red aqueous solution, while the n-HA was hydrophobic and hardly dissolved in aqueous solution, with a slightly visible soluble compound. The PLGA/HA NPs aqueous solution dissolved much more than did n-HA. The highest solubility of PLGA/HA NPs and n-HA was 643 μg mL−1 and 18.03 μg mL−1, respectively. The solubility of PLGA/HA NPs in aqueous solution was approximately 35.67-fold that of n-HA. In general, poorly water-soluble drugs often show low bioavailability when administered orally, because the absorption of the drugs in the gastrointestinal tract can usually be a rate-limiting step.27 Therefore, it is important to enhance drug solubility for improved bioavailability. PLGA could increase the drug solubility and adhesion by forming the stable and amorphous nanoparticles to eliminating the oversaturation of the difference between the particle size of nanoparticles.11,27 Moreover, PLGA polymer was commonly used to develop smaller particles with narrow poly dispersity.28 We used PLGA as the nano-carrier because it could distinctly encapsulate drugs into the pore of PLGA to increase hydrophobic drug water solubility. Instability and degradability of drug molecules in physiological pH conditions hindered drug delivery to target tissue.29 To study the stability properties of HA, n-HA and PLGA/HA NPs were incubated in artificial gastric (pH 1.5), intestinal (pH 6.8) and blood (pH 7.4) media. As shown in Fig. 3, HA was stable in pH 1.5 with the change of time, but rapid degradation of n-HA occurred in physiological pH 6.8 and pH 7.4. Nevertheless, PLGA/HA NPs were more stable under the same conditions, especially under pH 1.5. Consequently, as a drug carrier, PLGA improved the stability of HA in physiological pH conditions. PLGA protected the encapsulated HA against enzymatic and hydrolytic biodegradation, thereby preventing HA from breaking down from the gastrointestinal acid.30,31
 |
| | Fig. 2 Solubility of n-HA and PLGA/HA NPs in aqueous solution. | |
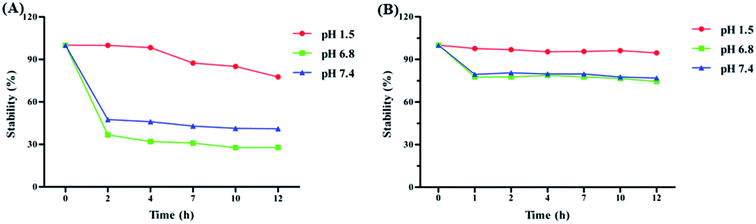 |
| | Fig. 3 The stability of n-HA (A) and PLGA/HA NPs (B) in artificial gastric (pH 1.5), intestinal (pH 6.8) and blood (pH 7.4) biological condition at 37 °C (mean ± S.D., n = 3). | |
3.4 Method development
3.4.1 Selection of IS. In LC-MS/MS analysis, it is important to select a suitable compound for the analyte and serve as an internal standard. Four types of compounds were investigated: 1,8-dihydroxy anthraquinone, lafutidine, p-hydroxy benzoic acid butyl ester and docetaxel. However, 1,8-dihydroxy anthraquinone, lafutidine, p-hydroxy benzoic acid butyl ester presented low recovery in plasma compared to HA with the same plasma preparation methods. In our study, docetaxel is a stable and commercialized compound with similar mass spectrometric, chromatographic condition and extraction recovery as HA. Thus, docetaxel is the best selection of IS for HA, and it is helpful when a significant matrix effect appeared.
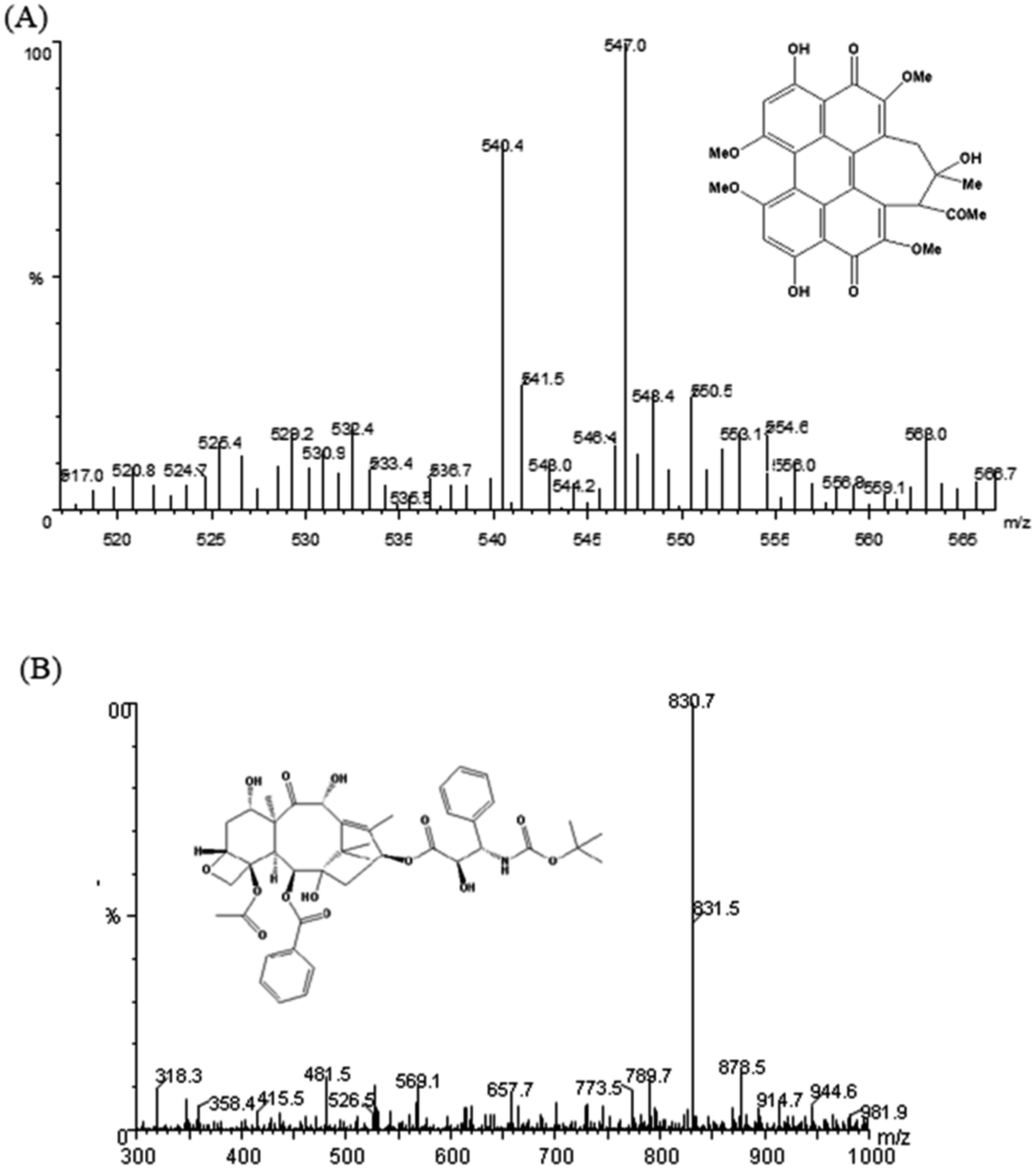
3.4.2 Optimization of the mass condition and chromatographic condition. Given that rat plasma is a complex matrix, a sensitive and stable LC-MS/MS condition was first optimized to quantify HA in plasma samples. First, the standard solution of HA and the IS were injected into the mass spectrometer. The ionization of HA and IS in positive ion and negative ion modes was performed on an ESI ion source and compared in terms of the intensity of produced ions. Finally, HA and IS were analysed in positive ion mode, for the intensity was stronger. In addition, the MRM mode was selected for its better sensitivity and specificity relative to the single ion monitoring (SIM) mode. Thus, the parameters of MS/MS were optimized for the two MRM transitions of m/z 547.1 → 529 for HA Fig. (4A), and m/z 830.7 → 549.4 for IS Fig. (4B), respectively. The satisfactory resolution of HA and IS was achieved with methanol and water containing 0.1% formic acid (90:10 v/v) with gradient elution at flow rate of 1.0 mL min−1 on an Agilent Zorbax Extend-C18 column (4.6 × 150 mm, 5 μm). Under the present chromatographic condition, the run time of each sample was 5 min, and the retention time was 3.38 min, 1.78 min for HA and IS, respectively.
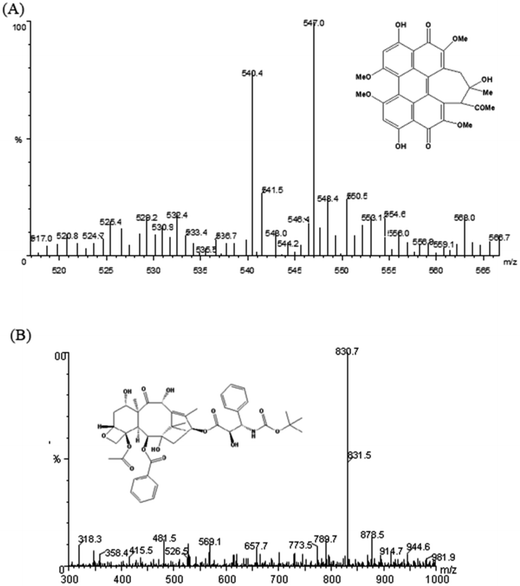 |
| | Fig. 4 Product ion mass spectra of HA (A) and IS docetaxel (B) in positive mode. | |
3.4.3 Sample preparation. To attain clean samples with elimination of endogenous interferences, different extraction procedures were conducted, such as solid phase extraction (SPE), liquid–liquid extraction (LLE) and protein precipitation (PP). The SPE method and LLE method were evaluated for extraction of HA and IS, and undeniably produced the cleanest extracts with high sensitivity, but resulted in lower extraction recovery of HA. However, the PP method was the fastest and simplest method resulting in a clean extract. The PP method also resulted in the high extraction recovery of HA and IS. Acetonitrile was adopted for sample preparation because it offered lower background noise and favourable separation. Indeed, acetic acid was also tested to achieve the maximum recovery of HA. In our study, 10, 20, 30, 40, and 50 μL of acetic acid were tested. Finally, an acetic acid volume of 50 μL was the best selection for HA and IS recovery.
3.5 Method validation
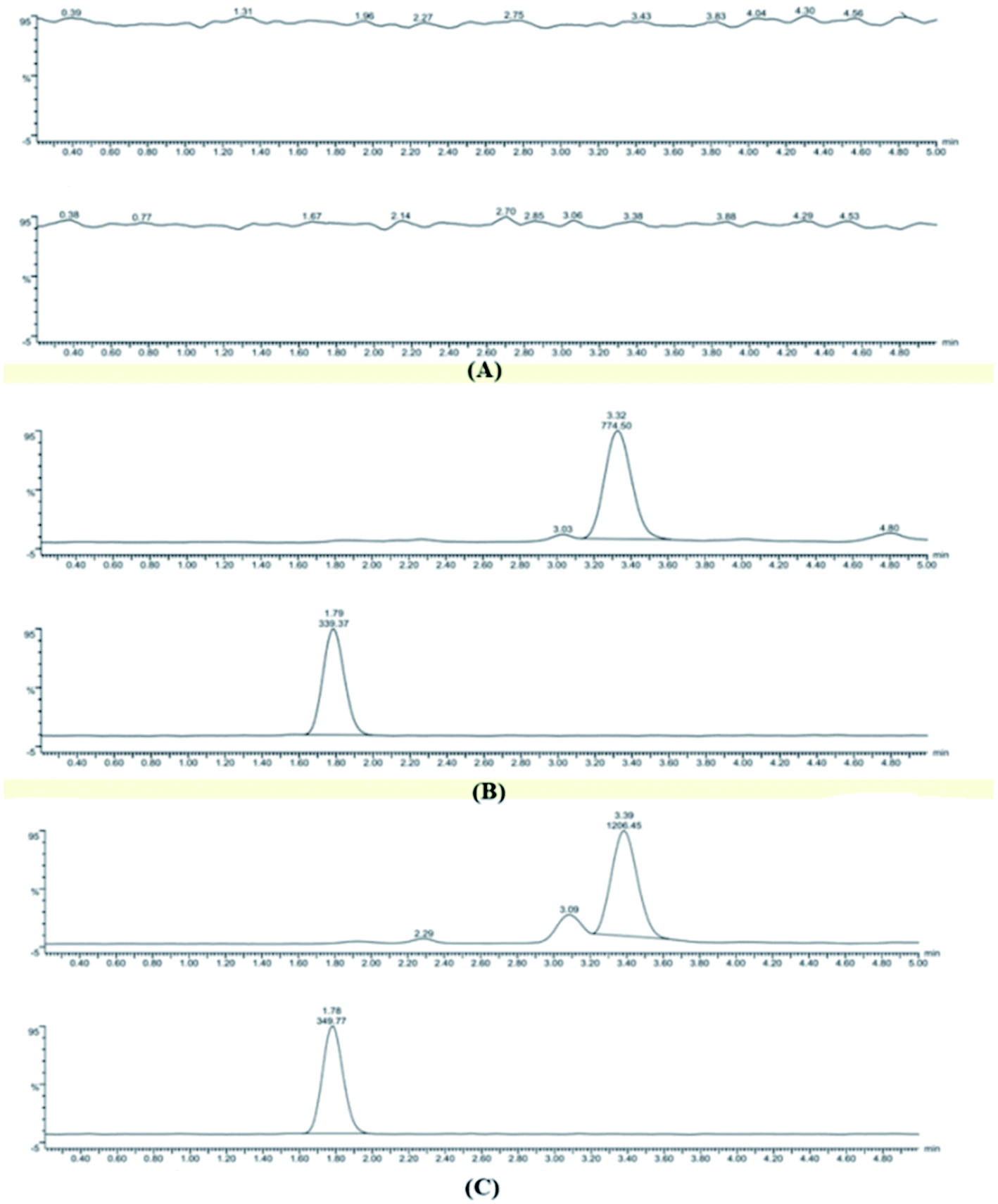
3.5.1 Specificity. The specificity study aimed to investigate whether endogenous substances in plasma would interfere with the analysis of HA and IS. As shown in Fig. 5, this test included a blank plasma sample (Fig. 5A), plasma containing HA and IS (Fig. 5B), and a plasma sample obtained after i.v. administration for 5 min (Fig. 5C). The results suggested that no endogenous constituents were eluted at the retention time of HA (3.48 min) or IS (1.78 min), and the total run time was 5 min.
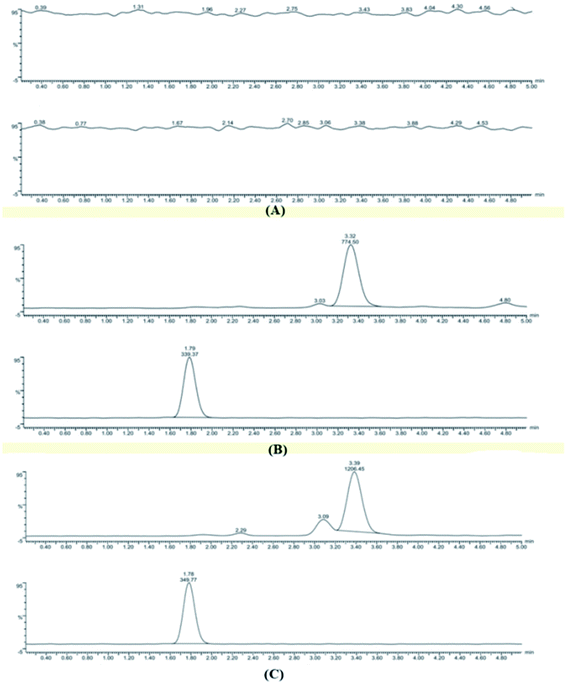 |
| | Fig. 5 Representative MRM chromatograms of HA and IS in plasma: a blank plasma sample (A); a blank plasma sample spiked with HA and IS (B); and a plasma sample collected after i.v. administration 5 min(C). | |
3.5.2 Linearity and sensitivity. In the current analytical method, the calibration curves contained eight points, covering a concentration range from 15 to 2000 ng mL−1, in rat plasma with correlation coefficient R2 > 0.9986 for HA. The LLOQ (signal-to-noise ratio > 10) of HA in this method was 10 ng mL−1.
3.5.3 Accuracy and precision. The intra-day and the inter-day accuracy and precision date of HA in biological matrices are presented in Table 1. The intra-day and the inter-day precision (RSD, %) values were ≤10.5% and ≤15%, respectively. The intra-day and the inter-day accuracy (%) values were in the range of 93.27–98.87% and 94.6–98.87%, respectively. All these results indicated that the method was reproducible and reliable for the determination of HA in rat plasma.
Table 1 Precision, accuracy, extraction recovery and matrix effect for HA in rat plasma (n = 3)
| Concentration (ng mL−1) |
Intra-daya |
Inter-dayb |
Recoveryc (%) |
Matrix effectc (%) |
| Precision (RSD, %) |
Accuracy (RE, %) |
Precision (RSD, %) |
Accuracy (RE, %) |
| Plasma samples were analyzed three times on the same day. Plasma samples were run on each of three validation. Values reported as mean ± S.D.; P < 0.05. |
| 50 |
10.5 |
−1.13 |
15 |
5.4 |
102 ± 0.28 |
99.5 ± 0.22 |
| 500 |
1.85 |
−1.78 |
8.9 |
2.2 |
105.7 ± 0.08 |
99 ± 0.34 |
| 1500 |
3.12 |
−6.73 |
9.3 |
1.0 |
110 ± 0.15 |
95 ± 0.05 |
3.5.4 Extraction recovery and matrix effect. The extraction recovery and matrix effect results of HA are summarized in Table 1. The extraction recovery of HA from low, medium, and high QC samples was in the range from 102 ± 0.28 to 110 ± 0.15%. The matrix effect of HA was from 95 ± 0.05 to 99.5 ± 0.22%, which was considered negligible or insignificant matrix effects.
3.5.5 Stability. The stability of HA in plasma was evaluated under different conditions (short-term, freeze/thaw, long term stability). The results are shown in Table 2. HA was stable in rat plasma and processed samples under different storage conditions.
Table 2 The results of bench top stability, freeze–thaw stability, long term stability of HA in rat plasma (n = 3)
| Stability test |
Nominal concentration in QC samples (ng mL−1) |
Measured mean concentration in stability QC samplesa (ng mL−1) |
| Values reported as mean ± S.D.; P < 0.05. |
| Bench top stability (room temperature, 6 h) |
50 |
55.0 ± 0.13 |
| 500 |
491.1 ± 0.02 |
| 1500 |
1399.3 ± 0.05 |
| Freeze–thaw stability (after 3 cycles, −20 °C) |
50 |
48.5 ± 0.05 |
| 500 |
511.3 ± 0.02 |
| 1500 |
1548.1 ± 0.11 |
| Long term stability (7 days, −20 °C) |
50 |
40.3 ± 0.03 |
| 500 |
407.0 ± 0.02 |
| 1500 |
1192.2 ± 0.06 |
3.6 Pharmacokinetic study
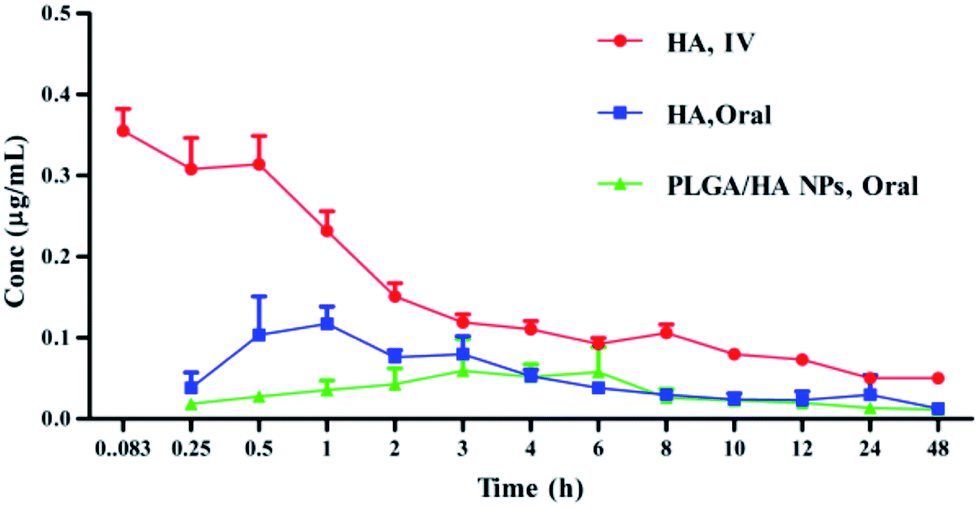
The validated LC-MS/MS method was first applied to the pharmacokinetic study of HA in plasma after oral administration (40 mg kg−1) of n-HA (first group), i.v. administration (4 mg kg−1) of n-HA (second group) and oral administration (12.5 mg kg−1) of PLGA/HA NPs (third group). The mean HA plasma concentration–time profiles were expressed by a two-compartment model with a weight of 1/C2 in Fig. 6. The relevant pharmacokinetic parameters are shown in Table 3.
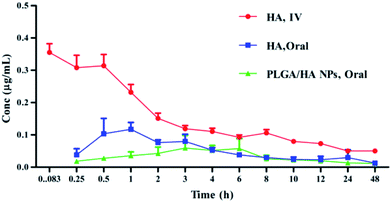 |
| | Fig. 6 Plasma mean concentration–time curves of HA after i.v. administration of 4 mg kg−1, oral administration of 40 mg kg−1, and PLGA/HA NPs after oral administration of 12.5 mg kg−1 (n = 6). | |
Table 3 Pharmacokinetic parameters of n-HA, PLGA/HA NPs oral administration and n-HA after i.v. administration in rat (n = 6)a
| Parameters |
n-HA oral |
n-HA i.v. |
PLGA/HA NPs oral |
| Values reported as mean ± S.D. (n = 6). Cmax: peak concentration; Tmax: time to reach peak concentration; t1/2α: mean distribution half-life; t1/2β: mean elimination half-life; AUC0−t: area under the plasma concentration–time curve; Vd: apparent volume of distribution; CL: clearance; F: oral bioavailability; P < 0.05. |
| Dose (mg kg−1) |
40 |
4 |
12.5 |
| Cmax (ng mL−1) |
110.3 ± 0.274 |
337.12 ± 66.28 |
56 ± 0.0233 |
| Tmax (h) |
1.03 ± 0.274 |
— |
2.822 ± 1.0835 |
| t1/2a (h) |
0.1245 ± 7.0158 |
— |
0.8797 ± 0.8148 |
| t1/2α (h) |
0.9732 ± 0.5887 |
0.2814 ± 3.545 |
2.553 ± 0.1228 |
| t1/2β (h) |
6.649 ± 0.0058 |
2.564 ± 0.0943 |
18 ± 0.303 |
| AUC0−t (ng h mL−1) |
1158.2 ± 171.01 |
991.67 ± 219.4 |
969.69 ± 230.24 |
| Vd (L) |
141.87 ± 37.92 |
17.01 ± 4.413 |
182.31 ± 13.475 |
| CL (L h−1) |
3.771 ± 1.852 |
0.578 ± 0.489 |
0.203 ± 0.203 |
| Fa (%) |
11.68 |
— |
31.30 |
| Fr (%) |
— |
— |
267 |
The plasma concentration of the first group and the third group were determined in 48 h, while the second group could not be determined after 12 h. The maximum concentration (Cmax) of the three groups was 110.3 ± 0.274, 337.12 ± 66.28 and 56 ± 0.0233 ng mL−1, respectively. Tmax of n-HA was shorter than that of PLGA/HA NP after oral administration group (1.03 ± 0.274 h and 2.822 ± 1.083 h), respectively. The delayed Tmax demonstrated a relatively sustained release of HA in PLGA NPs. The Vd of the three groups was 141.87 ± 37.92, 17.01 ± 4.413 and 182.31 ± 13.475 L, respectively, implying that oral administration of n-HA and PLGA/HA NPs increased the volume of distribution compared to n-HA after i.v. administration. The CL of three groups was 3.771 ± 1.852, 0.203 ± 0.203 and 0.578 ± 0.489 L h−1, respectively. These results implied that the elimination rate of HA was low. The CL of the oral administration group was 18.5-fold lower than that of the i.v. administration group.
Recently, PLGA drug delivery systems were developed to offer important advantages in the bioavailability of drugs. In our study, the terminal elimination half-life (t1/2β) and bioavailability of the three groups differed significantly. The t1/2β of the three groups was 6.649 ± 0.0058, 2.564 ± 0.0943 and 18 ± 0.303 h, respectively. The results showed that PLGA/HA NPs had low clearance and long half-life compared with n-HA in rat. PLGA/HA NPs prolonged the absorption phase and decreased the distribution, metabolism and obvious controlled release of HA from the nanoparticles. However, this finding was not consistent with previous results. Gu et al. reported HA-liposome t1/2β was 9.85 h.1 Pharmacokinetic parameters were altered depending upon the carriers and animal models. In addition, PLGA is frequently used for drug carriers because it may increase the residency time of the sustained release formulation and enhance the drug absorption due to intimacy of contact with the epithelium cells.21 AUC0−t values of the three groups were 1158.2 ± 171.01, 991.67 ± 219.4 and 969.69 ± 230.24 ng h mL−1, respectively. The absolute bioavailability of HA was significantly increased from 11.68 to 31.30%. The relative bioavailability of PLGA/HA NPs was approximately 267% that of HA. The bioavailability of PLGA/HA NPs increased 2.67-fold compared to n-HA.
To date, few studies have reported the bioavailability of HA; only the pharmacokinetic parameters of hypericin, which has an analogue structure of HA, was reported. A study by Schulz et al. showed the poor solubility and bioavailability of hypericin.32 In our study, the results demonstrated that PLGA used for HA delivery by oral administration increased HA bioavailability. Therefore, PLGA could potentially be applied to increase the bioavailability of other hydrophobic compounds.
According to experiment results, PLGA contributed to the improved solubility of HA and facilitated HA transport in vivo. In addition, PLGA protected HA from degradation under physiological conditions and enhanced its stability. All of these advantages favoured improved HA bioavailability. Because of the strong attraction of the particles to the negatively charged mucosa of the intestine responsible for a longer residence time in the intestine, mucoadhesive nanoparticles improve the bioavailability of poorly absorbed drugs.33 Nanoparticles have a plethora of advantages over other delivery systems, including the ability to protect drugs from gastrointestinal degradation, prolong systemic circulation, reduce fed/fasted variable absorption, and control drug release.34 Therefore, it is expected that with all the aforementioned advantages, the bioavailability of orally delivered drugs in nano form will be improved. However, the mechanism by which PLGA/HA NPs improved HA bioavailability was not detected, and further research is necessary.
4. Conclusions
In our study, PLGA/HA NPs have been successfully prepared. PLGA/HA NPs had superior solubility and stability compared with n-HA in physiological conditions. In pharmacokinetic study, a rapid, specific, selective, and reproducible LC-MS/MS method was first developed and validated for the determination of HA in rat plasma. The method was successfully applied to analyse plasma drug concentrations following oral administration of n-HA, PLGA/HA NPs and i.v. administration of n-HA. The pharmacokinetic parameters revealed that PLGA/HA NPs enhanced bioavailability of HA and prolonged HA systemic circulation. Moreover, the improved water solubility of HA was the main reason for its increased bioavailability. Conclusively, encapsulating hydrophobic HA on PLGA polymer will become a promising method for enhanced HA solubility, stability and improved bioavailability.
Conflicts of interest
There are no conflicts to declare. The authors alone are responsible for the content and writing of this article.
Acknowledgements
This study was supported by the National Natural Science Foundation of China (Project No. 81673214) the National Key Technology Research and Development Program of the Ministry of Science and Technology of China (Project No. 2012BAD36B0502) and the Priority Academic Program Development of Jiangsu Higher Educational Institutions.
Notes and references
- Z. Wang, Y. He, C. Huang, J. Huang, Y. Huang, J. An and Y. Gu, J. Photochem. Photobiol., B, 1999, 70, 773–780 CrossRef CAS.
- J. Zhang, E. Cao, J. Li, T. Zhang and W. Ma, J. Photochem. Photobiol., B, 1998, 43, 106–111 CrossRef CAS.
- A. P. Castano, T. N. Demidova and M. R. Hamblin, Photodiagn. Photodyn. Ther., 2005, 2, 91–106 CrossRef CAS PubMed.
- S. Qi, X. Lin, M. Zhang, S. Yan, S. Yu and S. Chen, RSC Adv., 2014, 4, 40085 RSC.
- Z. Diwu, Photochem. Photobiol. Sci., 1995, 61, 529–539 CrossRef CAS.
- M. J. Fehr, S. L. Carpenter, Y. Wannemuehler and J. W. Petrich, Biochemistry, 1996, 34, 15845–15848 CrossRef.
- Y. Su, J. Sun, S. Rao, Y. Cai and Y. Yang, J. Photochem. Photobiol., B, 2011, 103, 29–34 CrossRef CAS PubMed.
- G. Ma, S. I. Khan, M. R. Jacob, B. L. Tekwani, Z. Li, D. S. Pasco, L. A. Walker and I. A. Khan, Antimicrob. Agents Chemother., 2004, 48, 4450–4452 CrossRef CAS PubMed.
- S. Xia, Chin. Sci. Bull., 1981, 26, 1040 Search PubMed.
- L. Zhou, J. Zhou, C. Dong, F. Ma, S. Wei and J. Shen, Dyes Pigm., 2009, 82, 90–94 CrossRef CAS.
- M. Thommes, D. R. Ely, M. T. Carvajal and R. Pinal, Mol. Pharmaceutics, 2011, 8, 727–735 CrossRef CAS PubMed.
- Y. Hu, J. An and L. Jiang, J. Photochem. Photobiol., B, 1993, 17, 195–201 CrossRef CAS.
- Y. Hu, J. An and L. Jiang, J. Photochem. Photobiol., B, 1994, 22, 219–227 CrossRef CAS.
- J. Zhou, J. Liu, S. Xia, X. Wang and B. Zhang, J. Phys. Chem. B, 2005, 109, 19529–19535 CrossRef CAS PubMed.
- J. Zhou, S. Xia, J. Chen, X. Wang and B. Zhang, Chem. Commun., 2003, 12, 1372–1373 RSC.
- Z. Wei, J. An and L. Jiang, J. Photochem. Photobiol., B, 1996, 33, 73–78 CrossRef.
- Z. Wang, H. Ying, L. Cui, J. Zhang, W. Wang and L. Liu, Chin. J. Clin., 2012, 6, 1443–1446 CAS.
- Y. Qiu, L. U. An and P. Sun, China Surfactant Deterg. Cosmet., 2011, 41, 422–425 CAS.
- N. M. Khalil, T. C. do Nascimento, D. M. Casa, L. F. Dalmolin, A. C. de Mattos, I. Hoss, M. A. Romano and R. M. Mainardes, Colloids Surf., B, 2013, 101, 353–360 CrossRef CAS PubMed.
- J. L. Italia, D. K. Bhatt, V. Bhardwaj, K. Tikoo and M. N. Kumar, J. Controlled Release, 2007, 119, 197–206 CrossRef CAS PubMed.
- Z. Panagi, A. Beletsi, G. Evangelatos, E. Livaniou, D. S. Ithakissios and K. Avgoustakis, Int. J. Pharm., 2001, 221, 143–152 CrossRef CAS PubMed.
- X. Xie, Q. Tao, Y. Zou, F. Zhang, M. Guo, Y. Wang, H. Wang, Q. Zhou and S. Yu, J. Agric. Food Chem., 2011, 59, 9280–9289 CrossRef CAS PubMed.
- X. Gu, J. Zhou, Y. Feng, S. Wei, X. Wang and B. Zhang, J. Instrum. Anal., 2005, 24, 130–132 CAS.
- C. Wischke and S. P. Schwendeman, Int. J. Pharm., 2008, 364, 298–327 CrossRef CAS PubMed.
- D. A. Norris, N. Puri and P. J. Sinko, Adv. Drug Delivery Rev., 1998, 34, 135–154 CrossRef CAS PubMed.
- S. Hariharan, V. Bhardwaj, I. Bala, J. Sitterberg, U. Bakowsky and M. N. V. R. Kumar, Pharm. Res., 2006, 23, 184 CrossRef CAS PubMed.
- M. Sugimoto, T. Okagaki, S. Narisawa, Y. Koida and K. Nakajima, Int. J. Pharm., 1998, 160, 11–19 CrossRef CAS.
- I. Bala, S. Hariharan and M. N. Kumar, Crit. Rev. Ther. Drug Carrier Syst., 2004, 21(5), 387–422 CrossRef CAS PubMed.
- C. Mohanty and S. K. Sahoo, Biomaterials, 2010, 31, 6597–6611 CrossRef CAS PubMed.
- E. Fattal, S. Pecquet, P. Couvreur and A. Andremont, Int. J. Pharm., 2002, 242, 15–24 CrossRef CAS PubMed.
- W. He, S. W. Horn and M. D. Hussain, Int. J. Pharm., 2007, 334, 173–178 CrossRef CAS PubMed.
- H. U. Schulz, M. Schürer, D. Bässler and D. Weiser, Arzneim. Forsch., 2005, 55, 15–22 CAS.
- L. C. Simon, R. W. Stout and C. Sabliov, Nanobiomedicine, 2016, 3, 1–10 CrossRef.
- L. Plapied, N. Duhem, A. D. Rieux and V. Préat, Curr. Opin. Colloid Interface Sci., 2011, 16, 228–237 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Shu-Zhen Yan
a,
Shu-Zhen Yan