
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mechanistic investigation on radical-induced construction of oxindoles: radical versus electrophilic cyclization†
Zhiqiang Denga,
Lu Jinab,
Yong Wu *a and
Xiang Zhao*a
*a and
Xiang Zhao*a
aDepartment of Chemistry, Institute for Chemical Physics, School of Science, Xi'an Jiaotong University, Xi'an 710049, China. E-mail: xzhao@mail.xjtu.edu.cn; specwy@mail.xjtu.edu.cn
bCollege of Chemical Engineering, Xinjiang Agriculture University, Urumchi, 830025, China
First published on 15th June 2017
Abstract
This work is based on our previous reported experimental results (RSC Advances, 2016, 6, 27000). The mechanism of constructing oxindoles via tandem radical reaction of N-arylacrylamide with tertiary cycloalkanols is theoretically explored by using density functional theory (DFT). A four-step mechanism was put forward for the reaction. Step 1 is related with the ring-opening process of cycloalkanol radical after the oxidation, step 2 corresponds to the intermolecular attack between keto-included alkyl radical and N-methyl-N-phenylmethacrylamide, step 3 is associated with the intramolecular C–C bond formation via the cationic attack, and step 4 is the deprotonation towards the final product. It is found that step 2 is the rate-limiting step. Importantly, step 3 is updated to be a cationic step (channel 2), instead of the previously suggested radical step (channel 1). After the ring-opening processes, comparative study indicates that the intramolecular reactions of β-, γ-, and δ-keto radicals determines the product selectivity. The low selectivity of product in the cyclopentanol-involved reaction would originate from the high competition between intra- and intermolecular reactions, which is in agreement with our previous experimental observations.
1. Introduction
Tandem reactions, which combine multiple reaction steps into a single operation, are powerful and extensively used in organic synthesis.1–4 Due to the advantage of atom and step economy, this strategy has been focused by synthetic chemists and was comprehensively applied in total synthesis.5–9 In recent years, thanks to tandem radical cyclization strategy, the easily accessible N-arylacrylamides have been widely used for the synthesis of 3,3-disubstituted oxindole skeletons10–13 which commonly exist in numerous natural compounds.14–16 It is easy to put a variety of functional groups on the C3-position of oxindoles with different radical substrates.Based on the research interest in the synthesis of oxindoles derivatives,17–21 we have systematically studied a novel transition-metal free tandem radical cyclization of acrylamides with tertiary cycloalkanol.22 To our delight, we achieved the first combination of ring-opening of tertiary cycloalkanols and functionalization of acrylamides via the tandem radical cyclization reaction (Scheme 1a). To gain insight into the mechanism of this reaction, we have also tested two radical scavengers (TEMPO and BHT) under the optimal reaction condition to trap the forming radical in the reaction. In addition, the intra- and intermolecular kinetic isotope effect (KIE) experiments were also performed, respectively. According to the experimental results, we suggested that the reaction might undergo a radical pathway (Scheme 1b).
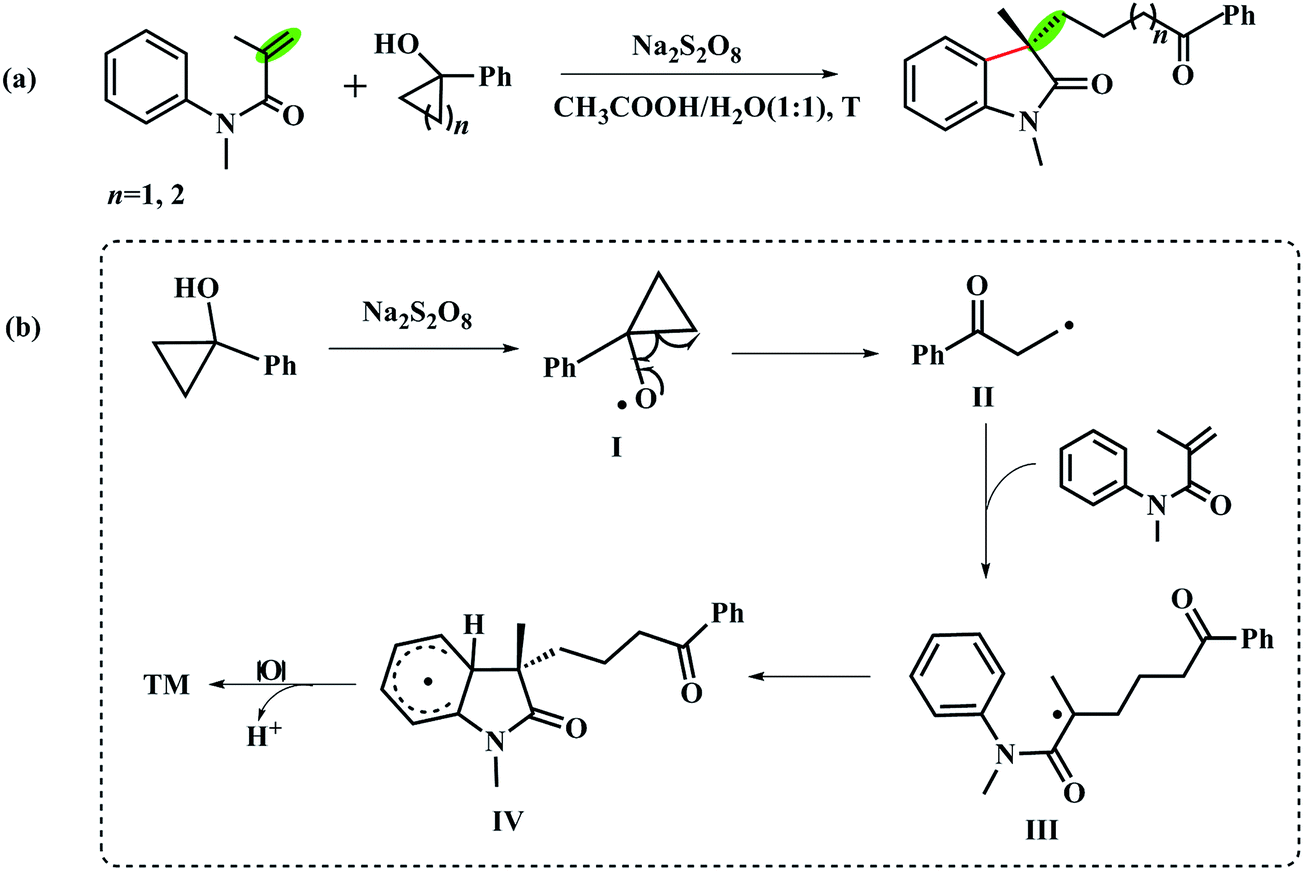 | ||
| Scheme 1 (a) Transition-metal free catalyzed cyclization reaction of acrylamides with the tertiary cycloalkanols. (b) The proposed reaction mechanism in ref. 22. | ||
Although this mechanism could be accepted widely,23–34 some critical issues deserve to take into account. In particular, several details of the reaction mechanism need to be clarified. For example, how to understand the competition between intramolecular cyclization and intermolecular addition? Which step determines the rate of the reaction? And how much is about the redox potential for each reactive intermediates? Generally, it is tough to thoroughly observe and respond these points using experimental methods only. On the other hand, quantum mechanics (QM) computations can practically find the trajectory of reaction processes and provide electronic structure information in detail.
In this work, the reaction mechanism of the experimental reaction (Scheme 1a) is examined by using density functional theory (DFT) method at the M06-2X/6-31+G(d,p) level. Our computations reveal that a four-step mechanism is involved in the radical-induced construction of oxindoles and that the intermolecular attack of cycloalkanol radical on the double bond of acrylamide (II → III, Scheme 1b) is the rate-determining step. Interestingly, except the conventional radical intramolecular ring closure (III → IV, Scheme 1b), we put forward a intramolecular electrophilic ring closure for the formation of oxindole via a cationic step after the oxidization. The evidence of the calculated redox potential supports this cationic step is favorable in the reaction. To the best of our knowledge, it is the first time to demonstrate the favored intramolecular electrophilic cyclization in the radical-induced construction of oxindole.
2. Computational details
All relevant structures were fully optimized at the M06-2X/6-31+G(d,p) level, combined with self-consistent reaction field (SCRF) method. Frequency analysis was used to ensure each optimized stationary point to be a minimum or a saddle point. Intrinsic reaction coordinate (IRC) calculations were also performed to verify that each transition state connects with the two associated minima on the potential energy surface. The solvent effect was evaluated in water (ε = 78.4) with the conductor-like polarized continuum model (C-PCM). To match with the experimental conditions, all thermal energies were corrected at 50 °C and 1 atm. Furthermore, some electronic structures were analyzed by the natural bond orbital (NBO) method. All computations were performed with Gaussian 09 package.35 All 3-D structures were generated by CYLview program.363. Results and discussion
3.1. 1-Phenylcyclopropanol-related reaction mechanism
In this study, 1-phenylcyclopropanol and N-methyl-N-phenylmethacrylamide as the substrates were selected to explore the reaction mechanism. According to our computational results, we divide the overall reaction into four steps to report in this article. Step 1 is related with the ring-opening of 1-phenylcyclopropanol radical after the oxidation, step 2 is corresponding to the intermolecular attack between β-keto radical and N-methyl-N-phenylmethacrylamide, step 3 is associated with intramolecular ring closure to construct a C–C bond, and step 4 is the deprotonation towards the final product.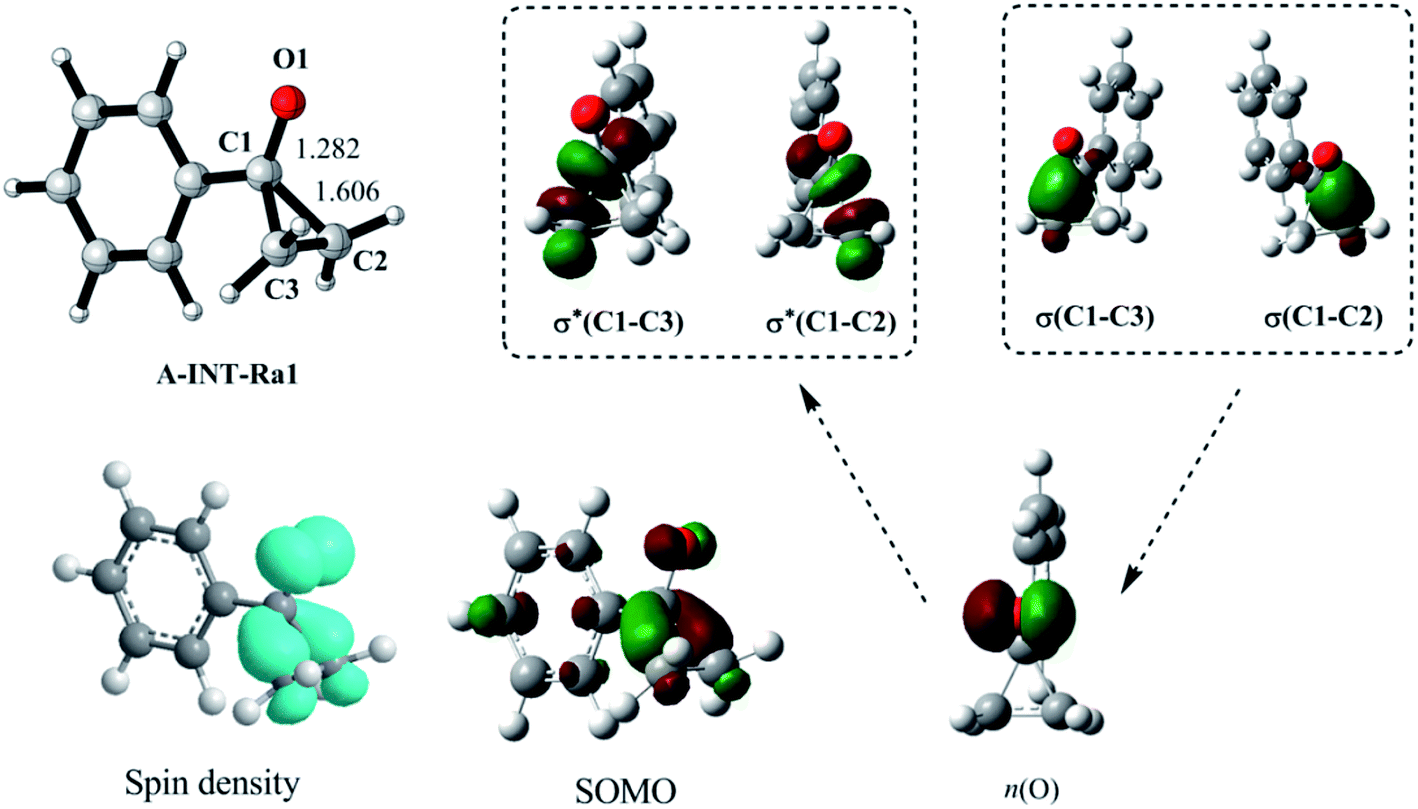 | ||
| Fig. 1 Geometry, spin density, single occupied molecular orbital (SOMO), and correlations of natural orbitals, for the radical A-INT-Ra1. n(O) is an unpaired p orbital. | ||
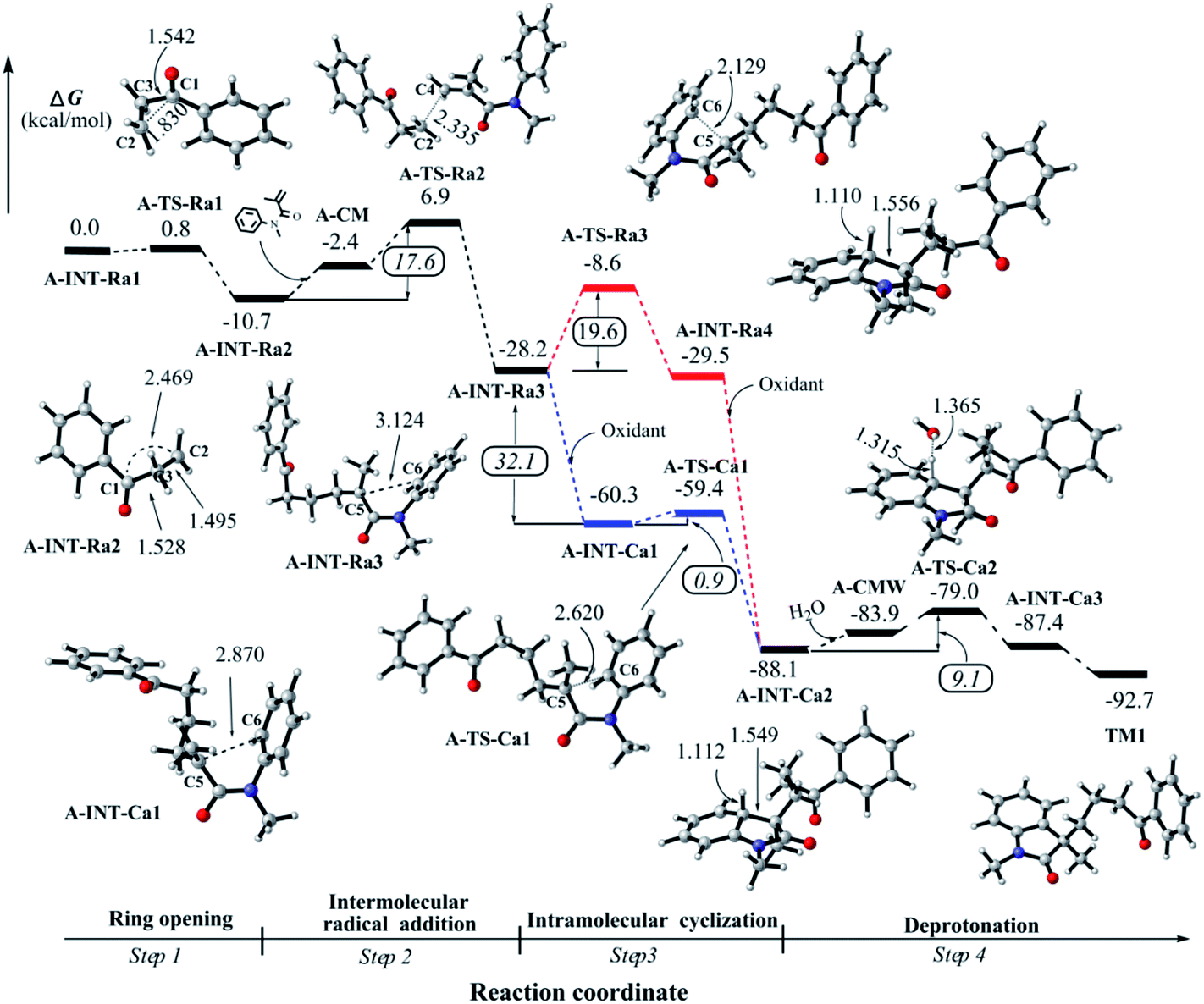 | ||
| Fig. 2 Relative Gibbs free energy profiles for the title reaction. Radical (channel 1, upper) and electrophilic (channel 2, below) cyclizations in step 3 are labeled as red and blue lines, respectively. | ||
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond of N-methyl-N-phenylmethacrylamide; pathway 2 is to construct a five-membered ring by the intramolecular attack on the vicinal aromatic ring. As shown in the ESI† and discussed in the next section, we found that pathway 2 would be unlikely to undergo in this step of the reaction. Herein we firstly report the favored intermolecular attack process.
C double bond of N-methyl-N-phenylmethacrylamide; pathway 2 is to construct a five-membered ring by the intramolecular attack on the vicinal aromatic ring. As shown in the ESI† and discussed in the next section, we found that pathway 2 would be unlikely to undergo in this step of the reaction. Herein we firstly report the favored intermolecular attack process.After the formation of a loose complex A-CM between A-INT-Ra2 and N-methyl-N-phenylmethacrylamide, the corresponding transition state A-TS-Ra2 was optimized (see Fig. 2). The distribution of spin density indicates that the unpaired electron partially moves to N-methyl-N-phenylmethacrylamide in A-TS-Ra2 (see Fig. S4 in the ESI†). The bond distance of C2–C4 is 2.335 Å. The ΔG‡ is 17.6 kcal mol−1 in this step. Then the C2–C4 single bond is formed in the intermediate A-INT-Ra3. It is found that this intermolecular attack step is a strongly irreversible process where the Gibb free energy drops down 17.5 kcal mol−1 from A-INT-Ra2. In A-INT-Ra3, the spin density is expectedly localized on C5 atom. The unpaired electron would be stabilized by the π-conjugation of amide moiety.
Alternatively, the intramolecular electrophilic cyclization (channel 2), namely Friedel–Crafts reaction, was taken into account in this work. We tested the possibility for the oxidation of A-INT-Ra3 by using the external oxidant Na2S2O8. The computational details for the redox potential is provided in the ESI.† Our results indicate that the redox potential is about −32.1 kcal mol−1 (−1.39 eV),37,38 revealing that it would be likely to promote the radical to a cation in the presence of external oxidant Na2S2O8. The high potential and irreversibility would partially benefit from the conjugation effect of the amide moiety. Interestingly, we found that the electrophilic cyclization is very easy to undergo with a barrier of 0.9 kcal mol−1 (Fig. 2). After checking the structural parameters, we found that the transition state A-TS-Ca1 is very close to A-INT-Ca1. The bond distance of C5–C6 is 2.620 in A-TS-Ca1, whereas it is 2.870 Å in A-INT-Ca1. The imaginary vibrational frequency, which is associated with C5–C6 bond stretch mode, is only 73.8 cm−1. Both the reactant-like structure and low imaginary vibrational frequency support this electrophilic cyclization would be a low energy barrier step. That is to say, once the radical A-INT-Ra3 is oxidized to the cation A-INT-Ca1, the process of cyclization would be much easier to take place. After the C–C bond formation, another cation A-INT-Ca2 is offered in the reaction. The corresponding Gibbs free energy continues dramatically dropping down by about 30.0 kcal mol−1.
In all, the mechanistic scenario of the tandem reaction is clarified for 1-phenylcyclopropanol and N-methyl-N-phenylmethacrylamide by using DFT method. The reaction would be triggered by a proton-coupling electron transfer (PCET) process in the presence of the external oxidant, leading to a radical whose spin density is not only localized on the oxygen atom but also distributes in C–C bonds. Next, it would be very easy to undergo the ring-opening of 1-phenylcyclopropanol to afford a β-keto radical whose spin density is mainly localized on the distal carbon atom. After that, the addition of the β-keto radical to the double bond of N-methyl-N-phenylmethacrylamide would happen. Then the intramolecular cyclization would undergo an unexpected cationic process after the oxidation of the formed radical intermediate. Finally, the deprotonation would easily occur to generate the product. Our results indicate that step 2 is the rate-limiting step in the overall reaction, which is consistent with the experimental KIE observations.
3.2. Comparative study on the reactions of tertiary cyclobutanol or cyclopentanol with N-arylacrylamides
Based on the results as mentioned above, we also expand our scope on the reactions of tertiary cyclobutanol or cyclopentanol with N-arylacrylamides. The detailed results are shown in the ESI.† As we expected, the large different exists in the ring-opening step of the cyclobutanol or cyclopentanol radicals after the oxidation (step 1). Due to the smaller ring strain in cyclobutanol and cyclopentanol radicals, the corresponding ΔG‡ of ring-opening slightly increases to 1.2 and 6.2 kcal mol−1, respectively. As for other reaction processes, we found that the trend are very similar to the aforementioned mechanism and that the intramolecular electrophilic cyclization would also be more favorable via the cationic process in step 3.3.3. Intramolecular dearomatization after the ring-opening of cycloalcohol radicals
In our previous experiments, the benzocyclohexanone, an intramolecular addition byproduct, was always detected. Therefore, we attempted to make clear this phenomenon in our study. Starting from keto-included alkyl radicals which generate from the ring-opening of the corresponding cycloalcohol radicals, we explored the intramolecular dearomatization processes (denoted as pathway 2 as mentioned above). Fig. 3 shows the corresponding Gibbs free energy barriers of the intramolecular radical cyclizations in the keto-included alkyl radicals (β-, γ-, and δ-keto radicals). Due to the little ring constrain, the ring-closure ΔG‡ of γ-keto radical would be the lowest via a six-remembered cyclic transition state. Compared with the intermolecular radical addition as stated above, the barriers (ΔG‡) of intramolecular ring-closure processes for β- and δ-keto radicals are somewhat higher. It is very important to note that the ΔG‡ of γ-keto radical in the intramolecular ring-closure is slightly lower than that of intermolecular radical addition process. Therefore, the low selectivity of product in the cyclopentanol-involved reaction would originate from the high competition between intra- and intermolecular reactions, which is in agreement with our experimental observations.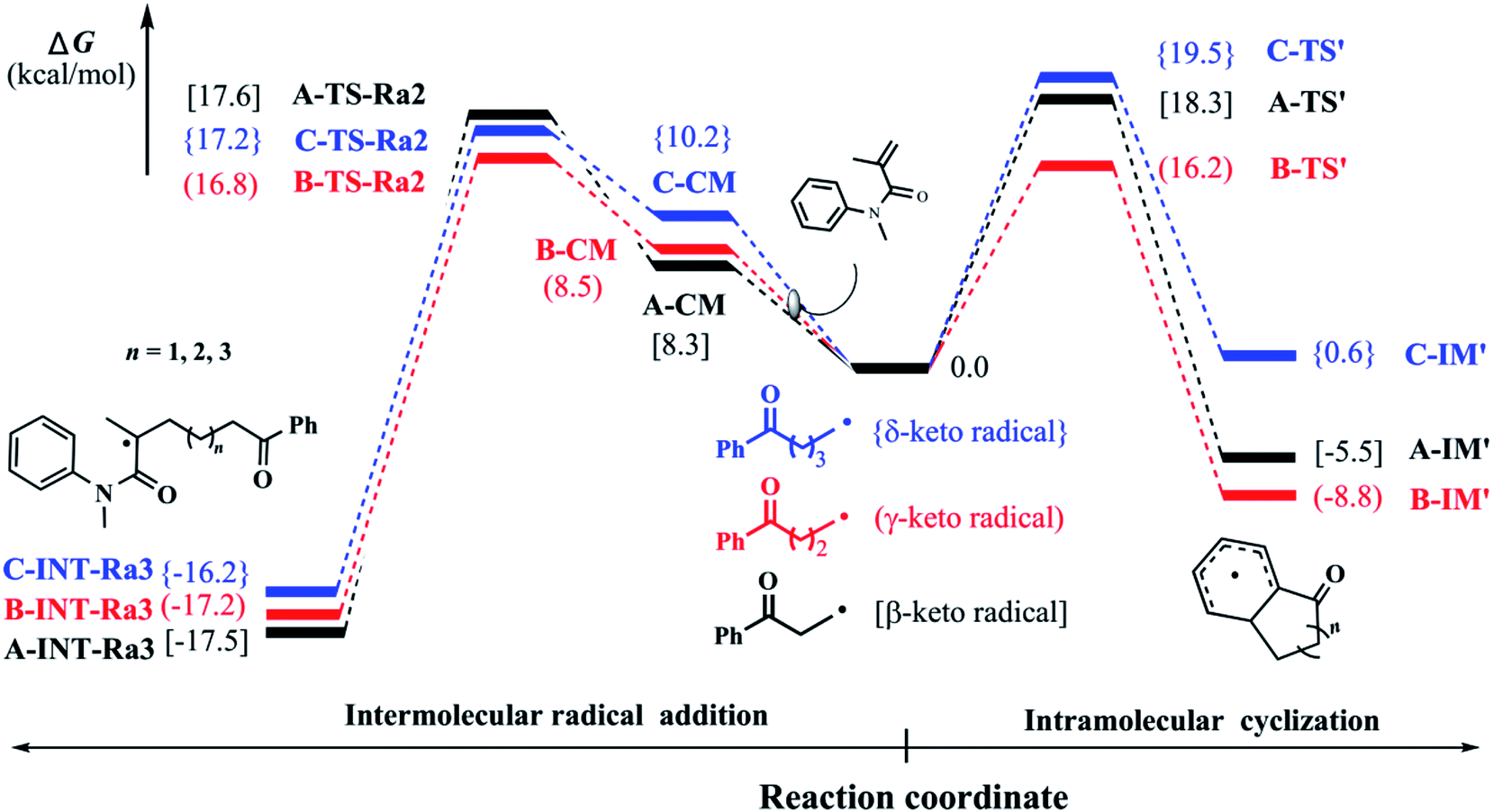 | ||
| Fig. 3 Relative Gibbs free energy (kcal mol−1) profiles of β-, γ-, δ-keto radicals for intramolecular radical cyclization and intermolecular radical addition. | ||
4. Conclusions
The mechanism of constructing oxindoles via tandem radical reaction of N-arylacrylamide with tertiary cycloalkanols was theoretically explored by using density functional theory (DFT) method in this work. Our findings are summarized as follows.(1) A four-step mechanism was put forward for the title reaction. Step 1 is related with the ring-opening process of 1-phenylcyclopropanol radical after the oxidation, step 2 is corresponding to the intermolecular attack between β-keto radical and N-methyl-N-phenylmethacrylamide, step 3 is associated with the intramolecular C–C bond formation via the cationic attack, and step 4 is the deprotonation towards the final product.
(2) Step 2, an intermolecular radical attack, is the rate-limiting step in the overall reaction.
(3) Step 3 was updated to a novel cationic intramolecular ring closure, which is different from the radical process in our previous suggestion.
(4) After the ring-opening processes, comparative study indicates that the intramolecular reactions of β-, γ-, and δ-keto radicals determine the product selectivity. The low selectivity of product in the cyclopentanol-involved reaction would originate from the high competition between intra- and intermolecular reactions.
This novel mechanism of the unexpected cationic intramolecular ring closure enriches the understanding of constructing oxindoles and provides the possibility to control the reaction selectivity. Moreover, it is very crucial for other similar radical reaction to draw a rational mechanism.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21573172) and the National Key Basic Research Program of China (No. 2012CB720904). L. Jin thanks the Postdoctoral Science Foundation of China (No. 2015M582635) and Research Foundation for Advanced Talents in Xinjiang Province. Y. Wu thanks the Fundamental Research Funds for the Central University (No. xjj2015064).References
- S. E. Denmark and A. Thorarensen, Chem. Rev., 1996, 96, 137–165 CrossRef CAS.
- J. H. Kim, Y. O. Ko, J. Bouffard and S. Lee, Chem. Soc. Rev., 2015, 44, 2489–2507 RSC.
- L. F. Tietze, Chem. Rev., 1996, 96, 115–136 CrossRef CAS.
- L. F. Tietze and U. Beifuss, Angew. Chem., Int. Ed., 1993, 32, 131–163 CrossRef.
- R. A. Bunce, Tetrahedron, 1995, 51, 13103–13159 CrossRef CAS.
- K. C. Nicolaou, D. J. Edmonds and P. G. Bulger, Angew. Chem., Int. Ed., 2006, 45, 7134–7186 CrossRef CAS PubMed.
- K. C. Nicolaou, Q. Kang, T. R. Wu, C. S. Lim and D. Y. K. Chen, J. Am. Chem. Soc., 2010, 132, 7540–7548 CrossRef CAS PubMed.
- K. C. Nicolaou and J. Li, Angew. Chem., Int. Ed., 2001, 40, 4264–4268 CrossRef CAS.
- K. C. Nicolaou, T. Montagnon and S. A. Snyder, Chem. Commun., 2003, 551–564 RSC.
- J. Bergman, Advances in Heterocyclic Chemistry, ed. E. F. V. Scriven and C. A. Ramsden, Elsevier Academic Press Inc, San Diego, 2015, vol. 117, pp. 1–81 Search PubMed.
- J. R. Chen, X.-Y. Yu and W.-J. Xiao, Synthesis, 2015, 47, 604–629 CrossRef CAS.
- C. C. Li and S. D. Yang, Org. Biomol. Chem., 2016, 14, 4365–4377 CAS.
- R. J. Song, Y. Liu, Y. X. Xie and J. H. Li, Synthesis, 2015, 47, 1195–1209 CrossRef CAS.
- N. R. Ball-Jones, J. J. Badillo and A. K. Franz, Org. Biomol. Chem., 2012, 10, 5165–5181 CAS.
- C. V. Galliford and K. A. Scheidt, Angew. Chem., Int. Ed., 2007, 46, 8748–8758 CrossRef CAS PubMed.
- H. Lin and S. J. Danishefsky, Angew. Chem., Int. Ed., 2003, 42, 36–51 CrossRef CAS PubMed.
- Y. Meng, L. N. Guo, H. Wang and X.-H. Duan, Chem. Commun., 2013, 49, 7540–7542 RSC.
- H. Wang, L. N. Guo and X.-H. Duan, Chem. Commun., 2013, 49, 10370–10372 RSC.
- H. Wang, L. N. Guo and X.-H. Duan, Adv. Synth. Catal., 2013, 355, 2222–2226 CrossRef CAS.
- H. Wang, L. N. Guo and X.-H. Duan, J. Org. Chem., 2016, 81, 860–867 CrossRef CAS PubMed.
- S. L. Zhou, L. N. Guo, H. Wang and X.-H. Duan, Chem.–Eur. J., 2013, 19, 12970–12973 CrossRef CAS PubMed.
- L. N. Guo, Z. Q. Deng, Y. Wu and J. Hu, RSC Adv., 2016, 6, 27000–27003 RSC.
- W. Fu, F. Xu, Y. Fu, M. Zhu, J. Yu, C. Xu and D. Zou, J. Org. Chem., 2013, 78, 12202–12206 CrossRef CAS PubMed.
- W. J. Fu, F. J. Xu, Y. Q. Fu, M. Zhu, J. Q. Yu, C. Xu and D. P. Zou, J. Org. Chem., 2013, 78, 12202–12206 CrossRef CAS PubMed.
- Y. M. Li, Y. H. Shen, K. J. Chang and S. D. Yang, Tetrahedron, 2014, 70, 1991–1996 CrossRef CAS.
- Y. M. Li, X. H. Wei, X. A. Li and S. D. Yang, Chem. Commun., 2013, 49, 11701–11703 RSC.
- Z. Li, Y. Zhang, L. Zhang and Z. Q. Liu, Org. Lett., 2014, 16, 382–385 CrossRef CAS PubMed.
- M. Z. Lu and T. P. Loh, Org. Lett., 2014, 16, 4698–4701 CrossRef CAS PubMed.
- X. H. Ouyang, R. J. Song and J. H. Li, Eur. J. Org. Chem., 2014, 2014, 3395–3401 CrossRef CAS.
- T. Shen, Y. Yuan, S. Song and N. Jiao, Chem. Commun., 2014, 50, 4115–4118 RSC.
- X. J. Tang, C. S. Thomoson and W. R. Dolbier, Org. Lett., 2014, 16, 4594–4597 CrossRef CAS PubMed.
- J. Y. Wang, X. Zhang, Y. Bao, Y. M. Xu, X. F. Cheng and X.-S. Wang, Org. Biomol. Chem., 2014, 12, 5582–5585 CAS.
- W. T. Wei, M. B. Zhou, J. H. Fan, W. Liu, R. J. Song, Y. Liu, M. Hu, P. Xie and J. H. Li, Angew. Chem., Int. Ed., 2013, 52, 3638–3641 CrossRef CAS PubMed.
- P. Xu, J. Xie, Q. C. Xue, C. D. Pan, Y. X. Cheng and C. J. Zhu, Chem.–Eur. J., 2013, 19, 14039–14042 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- C. Y. Legault, CYLView, 1.0b, Université de Sherbrooke, Canada, 2009, http://www.cylview.org Search PubMed.
- W. S. Chen, Y. C. Jhou and C. P. Huang, Chem. Eng. J., 2014, 252, 166–172 CrossRef CAS.
- C. Liang and H. W. Su, Ind. Eng. Chem. Res., 2009, 48, 5558–5562 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Computational details of reduction potential, calculated energies, Cartesian coordinates of stationary points, and relative energy profiles for comparative studies. See DOI: 10.1039/c7ra04756h |
| This journal is © The Royal Society of Chemistry 2017 |