
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
New endeavours involving the cooperative behaviour of TMAO and urea towards the globular state of poly(N-isopropylacrylamide)†
Payal Narang and
Pannuru Venkatesu *
*
Department of Chemistry, University of Delhi, Delhi – 110007, India. E-mail: venkatesup@hotmail.com; pvenkatesu@chemistry.du.ac.in; Fax: +91-11-2766-6605; Tel: +91-11-2766-6646 ext. 142
First published on 5th July 2017
Abstract
Studies have provided evidence for the destruction of the hydrogen bonds of poly(N-isopropylacrylamide) (PNIPAM) in the presence of osmolytes such as trimethylamine N-oxide (TMAO) and urea. Herein, we explore the effect of a mixed osmolytic environment on PNIPAM, which has thus far been an unanswered question in the literature, and we show the ease with which the addition of co-solvents like urea and TMAO individually, and as a mixture, result in the quick formation of the globular state of PNIPAM. The investigation of the globular or collapsed state of aqueous PNIPAM solution in the presence of co-solvents is exploited using various biophysical techniques like UV-visible spectroscopy, fluorescence spectroscopy, dynamic light scattering studies (DLS) and Fourier transform infrared spectroscopy (FTIR). The decrease in the lower critical solution temperature (LCST) with the addition of osmolytes and their mixed environment may be due to the different types of interactions (direct or indirect) with the hydrated, extended, coiled and globular PNIPAM states. The extent of hydrogen bonding and polarization of these additives is the major driving force for the collapsed conformation of PNIPAM. TMAO is a better inducer for the collapsed state of PNIPAM than urea, and their mixture shows a drastic decrease in the LCST of PNIPAM, because of the LCST decrease in both osmolytes individually, but the limited number of binding sites of PNIPAM is the main reason the mixture of urea and TMAO is not able to show an additive effect on PNIPAM agglomeration. Varying ratios of TMAO and urea are compared for a clear understanding of their behavior toward PNIPAM. Their mixed environment is a very good inducer of the globular form, compared to individual urea at higher concentration. The collapsed state of the polymer may provide sites or domains for drug encapsulation and release at the target by changing its conformation.
Introduction
In the modern era, the reports of infectious diseases have been on the increase. These diseases are clinical disorders resulting from the presence of pathogenic agents such as viruses, bacteria, fungi or parasites.1 Their ability to get transferred from one person or species to another is the reason they are called communicable diseases.1,2 More than half of the deaths worldwide can be attributed to these diseases, particularly in developing countries, and it is very important to find a cure. Thermoresponsive polymers (TRPs) play an integral role in the advancement of drug delivery technology by providing mechanisms for the controlled release of therapeutic agents and the tunable release of both hydrophilic and hydrophobic drugs.3 The pharmaceutical and therapeutic properties of drugs can be modulated by the use of polymeric materials and their arrangement in drug delivery systems.4,5 The huge medicinal applications of TRPs may control the aforementioned diseases. Moreover, the polymeric materials can also be used for targeted delivery in tissues and different cellular systems.6Among all polymers, stimuli responsive polymers, which respond to the external stimuli such as temperature, pH and certain other chemicals, are of particular interest with respect to the polymer–protein and polymer–drug conjugates.3 These smart polymers have wide applications in drug delivery and many other research areas such as matrix preparation for bioseparation, biomedical applications, actuators for chemical valves, lubricants and tissue engineering, etc.7–10 The TRPs undergo conformational changes with temperature, attributed to the changes in the interactions among the polymer chains and water molecules.11 Poly(N-isopropylacrylamide) (PNIPAM) is a well-known “intelligent” amphiphilic polymer that shows phase transition from the coiled to the globular state at around 32–33 °C, which is very close to body temperature and is promising for various biomedical applications. The collapsed form of PNIPAM leads to turbidity at higher temperatures because it is not water soluble. The temperature below which the equilibrium shifts from coil to collapsed or globular form is said to be its lower critical solution temperature (LCST).6,12–16
The globular form of PNIPAM expels water while undergoing transition and forms hydrophobic nanopockets or domains that are stabilized by the hydrophobic interactions of isopropyl groups.17,18 The collapsed state of the polymers have been used in the preparations of nanoparticles and nanoreactors.19,20 These domains may or may not be long range order, depending on the process by which they are undergoing collapse.21–24 The permeability of various molecules can be altered by varying the size of the domains in the collapsed network of polymers, resulting in the formation of switchable valves and semi-interpenetrating polymeric networks.25,26 Moreover, the swelling and deswelling of hydrogels formed from the PNIPAM chain or with its copolymer may lead to a better understanding of hydrophobicity/hydrophilicity for biomolecular engineering.27–30
Osmolytes are low molecular weight co-solutes known to protect cells against osmotic stress; these are molecules, like proteins, polymers, etc., that can alter macromolecular function by varying the intra or intermolecular interactions.31–34 Trimethylamine N-oxide (TMAO) and urea are two well-known osmolytes that have the ability to change the conformation of macromolecules, resulting in different functions.32,35 TMAO is a kind of amphiphilic osmolyte containing a small hydrophilic group (N+O−) and a bulky hydrophobic part (three methyl groups),32 while urea is a small non-protecting osmolyte consisting of polar moieties that can easily form H-bonds with other structural moieties. Earlier studies revealed that osmolytes increase the thermodynamic stability of many folded proteins by providing protection against stresses that lead to the loss of its native structure.36–38 Many researchers have focused on the biomolecular interaction studies of TMAO and urea with proteins. A literature survey has shown that urea is a denaturating co-solvent for proteins, but it is still a matter of debate whether it is through direct interaction or indirect interactions with respect to concentration.13,14 TMAO molecules are known to show greater thermodynamic stability towards the folded protein, while there is a reverse effect for urea.27 TMAO also has a counteracting effect against the denaturation induced by urea and guanidinium hydrochloride for many proteins like α-chymotrypsin, etc.32,39–46
Interestingly, PNIPAM is a well-accepted model protein containing amide bonds and hydrophobic isopropyl groups as a branched moiety.8 Various studies have already demonstrated the individual effect of urea and TMAO on the PNIPAM aqueous solution; however, the molecular mechanism for the interaction of urea is still not completely understood and remains an open question, despite many studies. The mixture effect of TMAO + urea on PNIPAM is absent in the literature and the mixed osmolytic environment study is essentially important to tune the properties of PNIPAM hydrogels and target specific drug achievements. It has been reported by many research groups,6,8,47 using various spectroscopic and molecular dynamics studies, that the direct interaction between amide moieties of PNIPAM with urea is thought to act as a cross linker among various amide moieties in the PNIPAM chain.48 The propensity of urea for the collapsed state is less prominent at lower concentration and greater at higher concentrations.10,15,31 Furthermore, simulation studies have shown that the accumulation of urea in the first solvation shell and the globular state of PNIPAM is stabilized via strong interactions between urea and PNIPAM, which lead to the exclusion of water molecules from the PNIPAM surface to the bulk solution.49,50 Moreover, the effect of methylated urea and alcoholic mixtures on the phase transition studies of PNIPAM have been studied using different techniques.47,51
Schroer et al.52 reported that the effect of TMAO on the transition studies of PNIPAM is more significant than that of urea. Subsequently, Reddy et al.53 showed the destruction of the hydrogen bonds of PNIPAM in the presence of TMAO, resulting in faster agglomeration (i.e. lower LCST). The destruction of hydrogen bonds between water and polymer is attributed to the sharp phase transition. Binding studies also corroborate the above-mentioned result and show that there is a negligible number of PNIPAM–TMAO hydrogen bonds. The effect of TMAO and urea, individually, on the hydrophobic homopolymer polystyrene, was investigated using atomic force spectroscopy and molecular dynamics simulations. The studies on polystyrene show the reverse effect, since TMAO has a high binding affinity for the collapsed state, but there is a low affinity for urea molecules.27,53 PNIPAM and its copolymer were also studied with various additives like DMSO/water, methanol/water mixtures and buffers to tune the transition temperature, which could lead to applications in biomedical sciences, polymeric nanoparticle composites and separation studies.16,54–57 Additionally, the thermo-responsive properties of PNIPAM can also be modulated by replacing the end groups of the chain with urea or any other group containing hydrogen bonding properties.58 Fig. 1 displays the chemical structure of PNIPAM, urea and TMAO.
 | ||
| Fig. 1 Chemical structure of poly(N-isopropylacrylamide), urea and trimethylamine-N-oxide. | ||
Since the molecular interactions with PNIPAM in a mixed osmolytic environment are absent in the literature. We made an attempt to study the ease of formation of the globular conformation of PNIPAM to explore the knowledge regarding PNIPAM applications in the formation of hydrogels with varying cross linking domains, biomedical applications and other systems. Nevertheless, only a handful of simulation studies on hydrophobic macromolecules with mixed osmolytes have been reported in the literature.59
In this work, we have employed UV-visible spectroscopy, fluorescence spectroscopy and dynamic light scattering (DLS) and Fourier transform infrared (FTIR) techniques to investigate the effect of TMAO and urea osmolytes on the aggregation of PNIPAM polymer individually, as well as in the mixed osmolytic environment in varying ratios. By studying the effect of co-solvent on the globular state, an attempt has been made to closely explore the changes in molecular interaction (direct or indirect), as there are virtually no studies of this sort in the literature. This study provides a better insight to further explore knowledge pertinent to engineering drug delivery systems, surface modification, biosensors and bioseparation, etc.
Experimental section
Materials and sample preparation
PNIPAM (Mn = 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–40000), 8-anilino-1-naphthalenesulfonic acid (ANS) and trimethylamine N-oxide (TMAO) were purchased from Sigma-Aldrich (USA). The polydispersity index value of the PNIPAM aqueous sample was 0.258 at room temperature, evaluated by dynamic light scattering studies. Urea was purchased from Sisco Research Laboratory Pvt. Ltd. These chemicals were used directly, without further purification. All samples were prepared by dissolving the appropriate amount of polymer gravimetrically in double distilled deionized water (Ultra 370 series, Rions India, India) with 18.3 MΩ resistivity. The required amounts of chemicals were weighed using an analytical balance (Mettler Toledo) with a precision of ±0.0001 g in screw-capped vials. The polymer concentration was maintained at 3 mg mL−1 for UV-visible and fluorescence measurements. For DLS and FTIR measurements, we maintained the concentration of polymer at 10 mg mL−1. Varied concentrations of urea (0.5, 1.0, 1.5, 2.0, 2.5, 3.0, 4.0, 5.0 and 6.0 M) were used for one set of experiments, and different concentrations of TMAO were used (0.4, 0.5, 0.6, 0.7, 0.8, 0.9 and 1.0 M) for another set of experiments and mixtures with varying ratios were used for the comparison study. The DLS experiments were carried out using 0.22 μm disposable filters (Millipore, Millex-GS) to filter the resulting sample solution through a medical syringe. The sample solution was maintained at room temperature prior to the measurements.
000–40000), 8-anilino-1-naphthalenesulfonic acid (ANS) and trimethylamine N-oxide (TMAO) were purchased from Sigma-Aldrich (USA). The polydispersity index value of the PNIPAM aqueous sample was 0.258 at room temperature, evaluated by dynamic light scattering studies. Urea was purchased from Sisco Research Laboratory Pvt. Ltd. These chemicals were used directly, without further purification. All samples were prepared by dissolving the appropriate amount of polymer gravimetrically in double distilled deionized water (Ultra 370 series, Rions India, India) with 18.3 MΩ resistivity. The required amounts of chemicals were weighed using an analytical balance (Mettler Toledo) with a precision of ±0.0001 g in screw-capped vials. The polymer concentration was maintained at 3 mg mL−1 for UV-visible and fluorescence measurements. For DLS and FTIR measurements, we maintained the concentration of polymer at 10 mg mL−1. Varied concentrations of urea (0.5, 1.0, 1.5, 2.0, 2.5, 3.0, 4.0, 5.0 and 6.0 M) were used for one set of experiments, and different concentrations of TMAO were used (0.4, 0.5, 0.6, 0.7, 0.8, 0.9 and 1.0 M) for another set of experiments and mixtures with varying ratios were used for the comparison study. The DLS experiments were carried out using 0.22 μm disposable filters (Millipore, Millex-GS) to filter the resulting sample solution through a medical syringe. The sample solution was maintained at room temperature prior to the measurements.
UV-visible absorption measurements
The absorption spectra of PNIPAM with various osmolytes were recorded using ultraviolet-visible (UV-visible) spectroscopy from 190 to 700 nm, using a double beam UV-visible spectrophotometer (UV-1800, Shimadzu Co., Japan) at room temperature. The required quantity of sample solution was transferred uniformly into the quartz cell of 1 cm path length. The spectrophotometer had a quartz cuvette with a spectral band width of 1 nm and wavelength accuracy of 0.3 nm, with automatic wavelength correction. We used ANS aqueous solution as the blank or reference in all UV-visible measurements.Fluorescence intensity measurements
A Cary Eclipse fluorescence spectrophotometer (Varian optical spectroscopy instruments, Mulgrave, Victoria, Australia) with an intense xenon flash lamp as the light source was used to acquire fluorescence spectra. A multicell holder, which was electrothermally controlled at precise temperature, with precision of 0.05 °C, regulated by a Peltier device, was used with the instrument. The emission spectra were recorded using the PMT voltage of 720 V and slit width of 5/5 nm. Similarly, thermal fluorescence spectra were collected within a specific temperature range with the interval of 1 °C and with an accuracy of 0.1 °C. The scan speed for all measurements was kept constant at 1200 nm min−1. For the sake of better accuracy in the thermal measurements, the samples were equilibrated for at least 20 min at each temperature before measurement. The excitation wavelength at 360 nm was taken in fluorescence experiments and the emission intensity was collected at the emission wavelength of 510 nm as the function of temperature. All the values reported were an average of three measurements.Dynamic light scattering (DLS) measurements
The hydrodynamic diameter (dH) was determined by means of a Zetasizer Nano ZS90 dynamic light scattering (DLS) instrument (Malvern Instruments Ltd., UK), which measured the time dependent fluctuation in the intensity of light scattered from particles in solution at a fixed scattering angle of 90°, followed by analysis using Malvern Zetasizer software version 7.01. The instrument was equipped with a 4 mW He–Ne laser at a fixed wavelength (λ) of 633 nm as the light source. All DLS experiments were performed as a function of temperature, with intervals of 1 °C, in a thermostatic sample chamber at desired temperatures in the range 23–38 °C. The temperature of the measurements was controlled with an accuracy of ±0.1 °C. A filtered bubble-free sample of approximately 1.5 mL was transferred into a quartz sample cell, which was sealed with a Teflon-coated screw cap to protect from dust. Then, the air-tight sample was introduced into the sample chamber holder of the DLS instrument. The Brownian motion of particles was detected by DLS and correlated to the particle size. The relationship between the size of a particle and its speed due to Brownian motion is defined by the Stokes–Einstein equation| dH = kT/(3πηD) | (1) |
Fourier transform infrared (FTIR) spectroscopy measurements
An iS 50 FT-IR (Thermo-Fisher scientific) spectrometer was used for recording the Fourier transform infrared spectrum in the wavenumber range of 600 cm−1 to 4000 cm−1. The sample must be bubble-free in an IR cell assembled with two ZnSe windows. The continuous monitoring of the temperature inside the sample chamber was performed by a chromel–alumel K-type thermocouple. Each IR spectrum reported was an average of 240 scans, using a spectral resolution of 4 cm−1. The IR spectra were recorded and stored using spectroscopic software (Varian Resolutions, Version 4.10). A background spectrum was obtained directly before the sample spectra. Specifically, for each sample containing PNIPAM and different co-solvents in varying concentrations, the deuterium oxide (D2O) spectrum was used as the background.Results and discussion
Absorption spectroscopy analysis of PNIPAM in the presence of TMAO, urea and their mixture
To investigate the individual effects of urea and TMAO and their mixture, UV-visible spectroscopy studies were carried out over a particular range of wavelengths at room temperature. For the UV-visible studies of PNIPAM, ANS was used as an extensive probe60 in very low concentration (2 × 10−5 M) in order to avoid interferences and to investigate the changes happening in the presence of varying concentrations of osmolytes. Fig. 2 displays the variation of the absorbance spectra of PNIPAM in the presence of urea, TMAO and their mixture as a function of wavelength. The ANS in the presence of PNIPAM shows its band at around 270 nm, which is consistent with the literature value.61 In Fig. 2(a), the band absorbance at 270 nm continuously decreases with the increase in the concentration of urea. Evidently, a new band observed at ∼288 nm may be due to the direct hydrogen bonding interaction between urea and the PNIPAM polymer. The interactions are more significant at higher concentrations as the intensity keeps on increasing with the concentration of urea,8 since urea is a polar molecule that binds to the chain of PNIPAM through favorable hydrogen bonds; Sagle et al.8 reported its ability to form hydrogen bonds with several carbonyl groups as bridging ligands. Interestingly, to observe the effect of urea on the absorbance spectra of ANS in the absence of PNIPAM, we performed the experiment and found that the absorbance is very small, or negligible, which is similar to that in the case of ANS absorbance in water (data shown in the Fig. S1 of ESI†). It can be concluded that absorbance at ∼288 nm might resemble the bonding interactions between PNIPAM and urea. Similarly, TMAO is an amphiphilic osmolyte, which is not able to provide a hydrophobic environment for ANS spectra, as in the hydrated PNIPAM case that leads to the decrease in absorbance at 270 nm in a concentration dependent manner (Fig. 2(b)). However, no significant band was observed at ∼288 nm, which clearly shows that there is no direct bonding between TMAO and PNIPAM to an appreciable extent.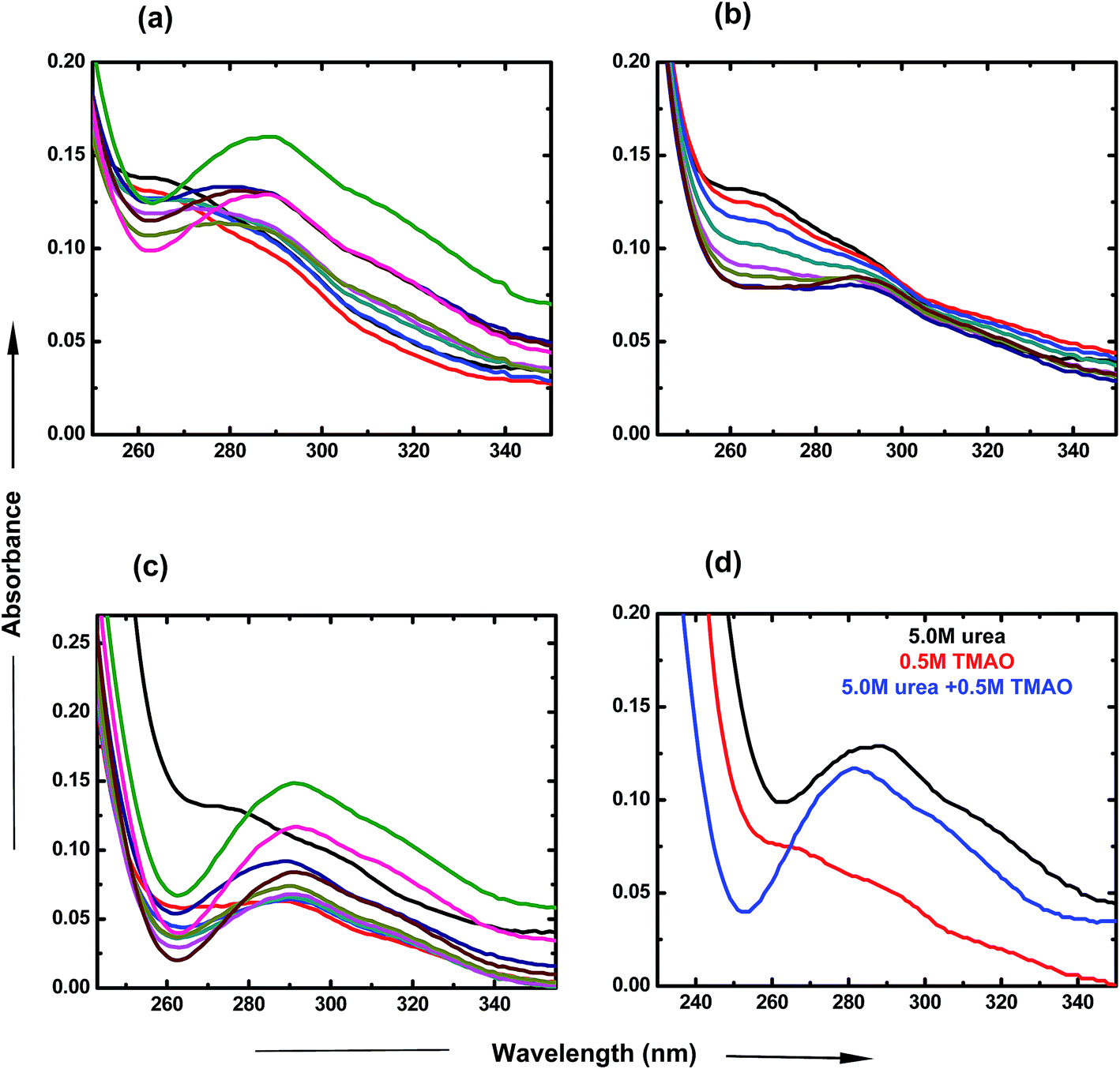 | ||
| Fig. 2 UV visible spectra of ANS in PNIPAM aqueous solution containing (a) urea, PNIPAM in aqueous solution (black), 0.5 M (red), 1.0 M (blue), 1.5 M (dark cyan), 2.0 M (pink), 2.5 M (dark yellow), 3.0 M (dark blue), 4.0 M (brown), 5.0 M (magenta) and 6.0 M (green). (b) TMAO, PNIPAM in aqueous solution (black), 0.4 M (red), 0.5 M (blue), 0.6 M (dark cyan), 0.7 M (pink), 0.8 M (dark yellow), 0.9 M (dark blue), 1.0 M (brown). (c) Mixture of TMAO + urea, PNIPAM in aqueous solution (black), (0.5 + 0.5) M (red), (0.5 + 1.0) M (blue), (0.5 + 1.5) M (dark cyan), (0.5 + 2.0) M (pink), (0.5 + 2.5) M (dark yellow), (0.5 + 3.0) M (dark blue), (0.5 + 4.0) M (brown), (0.5 + 5.0) M (magenta) and (0.5 + 5.0) M (green). (d) 1:10 ratio of TMAO:urea, 5.0 M urea (black), 0.5 M TMAO (red), (0.5 + 5.0) M (blue) under atmospheric conditions. | ||
Fig. 2(c) represents the UV-visible spectra of the mixture of osmolytes by varying the concentration of urea and keeping the TMAO concentration constant. The figure clearly shows that the decrease in absorbance at around 270 nm is more prominent as compared to the effect due to the individual osmolytic environment. Clearly, on increasing the urea concentration (0.5 to 6.0 M) in fixed (0.5 M) TMAO amounts, the band absorbance at 288 nm increases as more urea favours more direct binding with PNIPAM.8 Moreover, different ratios of TMAO and urea are shown to clearly depict the role of osmolyte in the mixed environment. The lower molar ratios (0.5:0.5 and 0.5:1.0) of TMAO and urea show the dominating role of TMAO as it is a better inducer for the collapsed state than urea at lower concentrations. In Fig. 2(d), the 0.5:5.0 molar ratio of TMAO:urea shows a prominent band at 288 nm, but has less absorbance as is the case of individual urea at 5.0 M concentration. This might be due to the lower probability for direct hydrogen bonding, or less binding sites of PNIPAM available for urea in the presence of TMAO. Individually, TMAO is a better inducer of the globular state than urea, even at low concentrations. Specifically, it was observed that a mixture of TMAO and urea had an even higher propensity for the globular form of PNIPAM.
Fluorescence spectroscopy analysis of PNIPAM in the presence of TMAO, urea and their mixture
:5.0 molar ratio of TMAO and urea is clearly represented in Fig. 3(d), which shows an increase in intensity, up to 650 a.u. The spectra show that lower ratios are less likely for the formation of the globular state, but more likely for higher ratios of osmolyte concentrations.
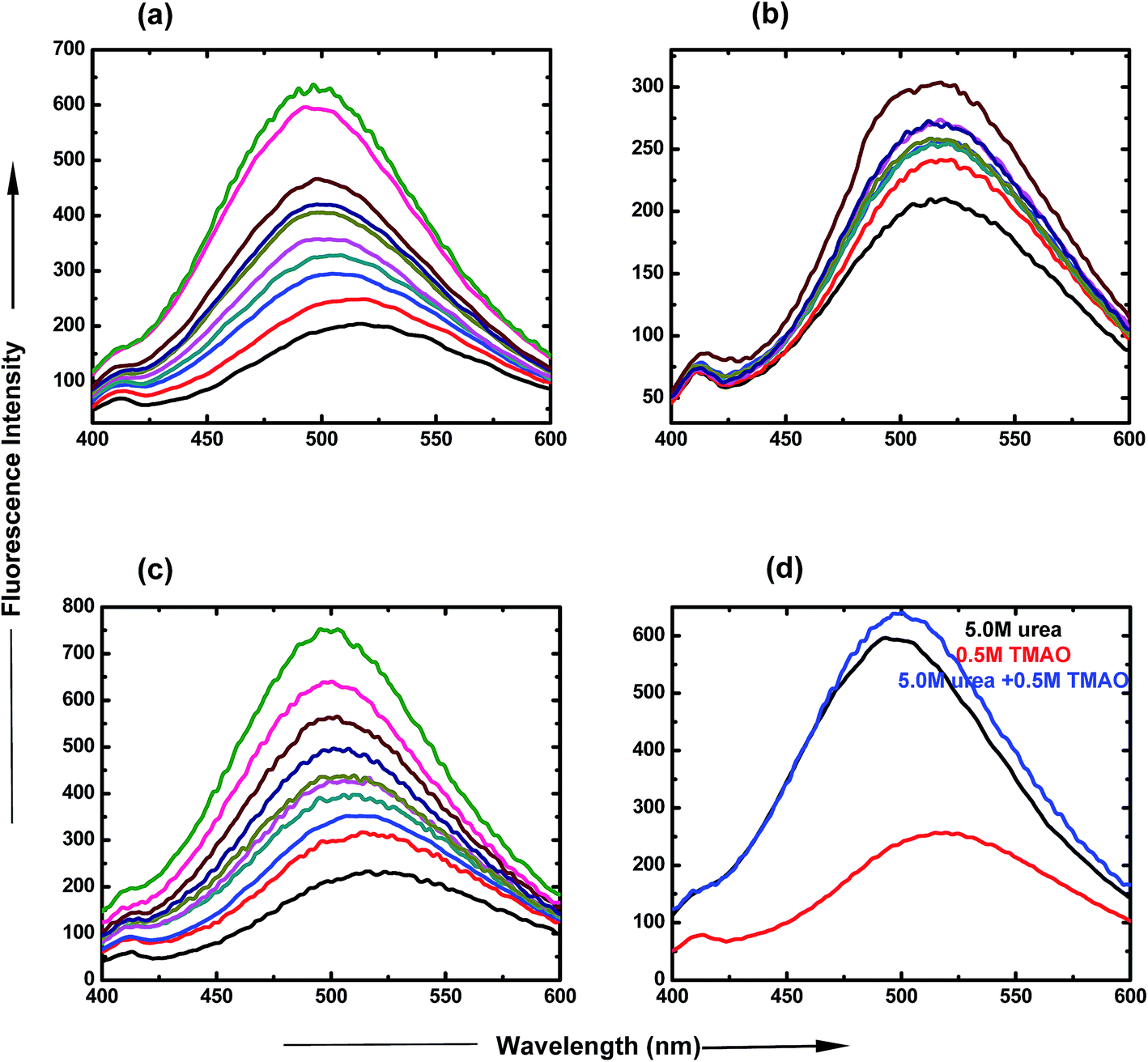 | ||
| Fig. 3 Steady state fluorescence spectra of ANS in PNIPAM aqueous solution containing (a) urea, PNIPAM in aqueous solution (black), 0.5 M (red), 1.0 M (blue), 1.5 M (dark cyan), 2.0 M (pink), 2.5 M (dark yellow), 3.0 M (dark blue), 4.0 M (brown), 5.0 M (magenta) and 6.0 M (green). (b) TMAO, PNIPAM in aqueous solution (black), 0.4 M (red), 0.5 M (blue), 0.6 M (dark cyan), 0.7 M (pink), 0.8 M (dark yellow), 0.9 M (dark blue), 1.0 M (brown). (c) TMAO + urea mixture, PNIPAM in aqueous solution (black), (0.5 + 0.5) M (red), (0.5 + 1.0) M (blue), (0.5 + 1.5) M (dark cyan), (0.5 + 2.0) M (pink), (0.5 + 2.5) M (dark yellow), (0.5 + 3.0) M (dark blue), (0.5 + 4.0) M (brown), (0.5 + 5.0) M (magenta) and (0.5 + 5.0) M (green). (d) 1:10 ratio of TMAO:urea, 5.0 M urea (black), 0.5 M TMAO (red), (0.5 + 5.0) M (blue) under atmospheric conditions. | ||
:5.0 ratio of TMAO and urea as a function of temperature. The LCST of PNIPAM decreased to a lesser extent (30.9 °C and 29.9 °C) for lower ratios (0.5:0.5 and 0.5:1.0) of TMAO and urea, and shifted to 25.5 °C in the case of the 0.5:5.0 molar ratio. Therefore, the 0.5:5.0 molar ratio of TMAO and urea facilitated the globular conformation to the greater extent, while the 0.5:0.5 molar ratio to the lesser extent.
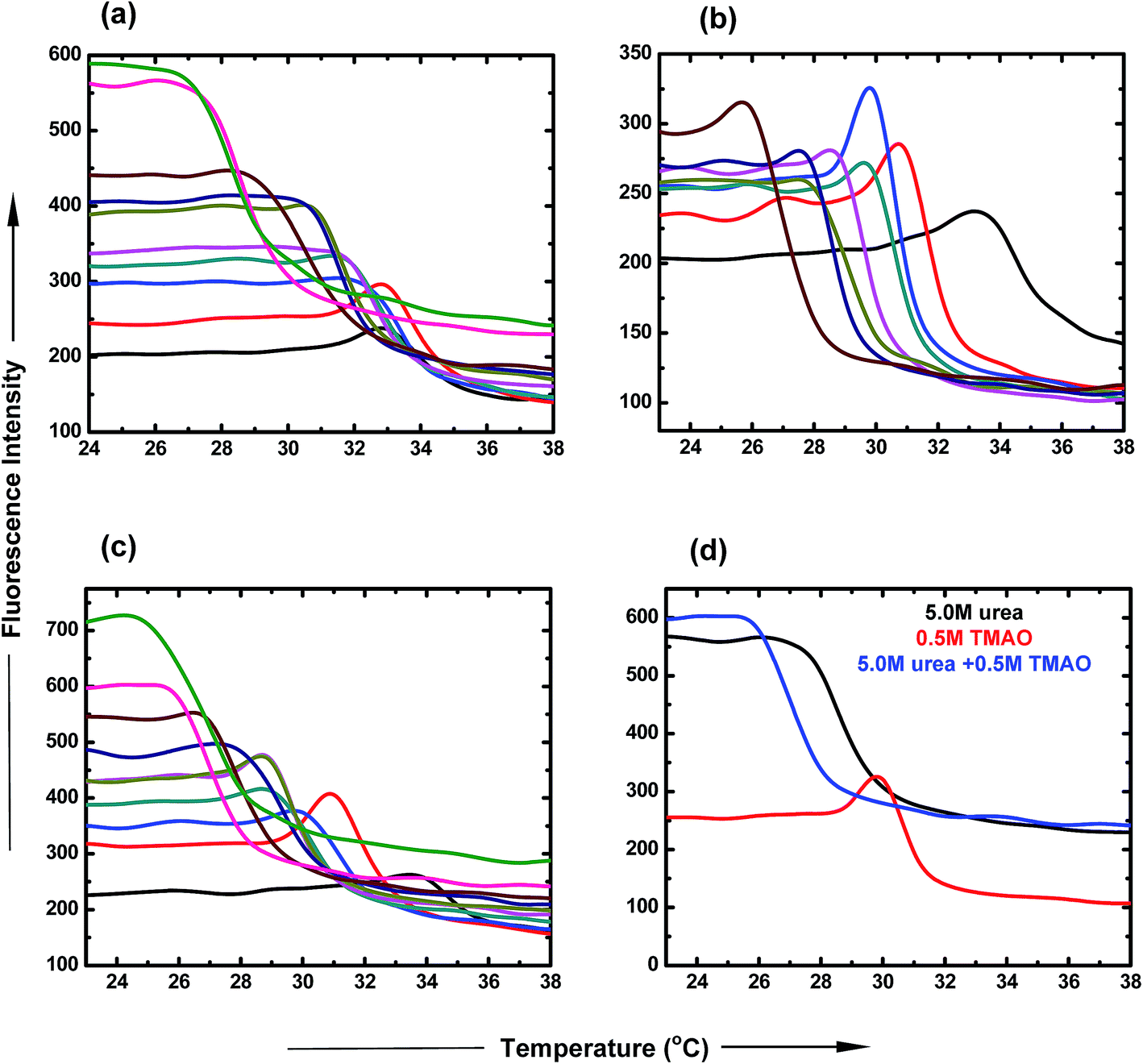 | ||
| Fig. 4 Thermal fluorescence spectra of ANS in PNIPAM aqueous solution containing (a) urea, PNIPAM in aqueous (black), 0.5 M (red), 1.0 M (blue), 1.5 M (dark cyan), 2.0 M (pink), 2.5 M (dark yellow), 3.0 M (dark blue), 4.0 M (brown), 5.0 M (magenta) and 6.0 M (green). (b) TMAO, PNIPAM in aqueous (black), 0.4 M (red), 0.5 M (blue), 0.6 M (dark cyan), 0.7 M (pink), 0.8 M (dark yellow), 0.9 M (dark blue), 1.0 M (brown). (c) TMAO + urea mixture, PNIPAM in aqueous solution (black), (0.5 + 0.5) M (red), (0.5 + 1.0) M (blue), (0.5 + 1.5) M (dark cyan), (0.5 + 2.0) M (pink), (0.5 + 2.5) M (dark yellow), (0.5 + 3.0) M (dark blue), (0.5 + 4.0) M (brown), (0.5 + 5.0) M (magenta) and (0.5 + 5.0) M (green). (d) 1:10 ratio of TMAO:urea, 5.0 M urea (black), 0.5 M TMAO (red), (0.5 + 5.0) M (blue) under atmospheric conditions. | ||
| Concentration of urea [M] | Tm (°C) | Concentration of TMAO [M] | Tm (°C) | Mixed ratios TMAO:urea [M] |
Tm (°C) |
|---|---|---|---|---|---|
| 0.0 | 33.0 | 0.0 | 33.0 | 0.0 | 33.0 |
| 0.5 | 32.8 | 0.4 | 30.7 | 0.5: 0.5 | 30.9 |
| 1.0 | 32.3 | 0.5 | 29.8 | 0.5: 1.0 | 29.9 |
| 1.5 | 31.5 | 0.6 | 29.5 | 0.5: 1.5 | 28.8 |
| 2.0 | 31.6 | 0.7 | 28.5 | 0.5: 2.0 | 28.7 |
| 2.5 | 30.7 | 0.8 | 27.5 | 0.5: 2.5 | 28.6 |
| 3.0 | 30.5 | 0.9 | 27.5 | 0.5: 3.0 | 27.7 |
| 4.0 | 28.7 | 1.0 | 25.6 | 0.5: 4.0 | 26.6 |
| 5.0 | 27.4 | 0.5: 5.0 | 25.5 | ||
| 6.0 | 26.9 | 0.5: 6.0 | 24.3 |
Dynamic light scattering studies of PNIPAM in the presence of TMAO, urea and their mixture
DLS is a very important tool to study the agglomeration of PNIPAM in different co-solvent environments. The clear size distribution of the macromolecule can be estimated using this biophysical technique. The hydrophobic collapse of PNIPAM is directly proportional to the hydrodynamic diameter of the aggregates. The typical LCST of PNIPAM in water was found to be at 32 °C.53 At the transition temperature, the dH increases drastically, which is due to the large sizes of the polymer agglomerates. Further, a decrease in the scattering intensities was observed in all samples, which may be due to two main reasons. First, the solution is highly turbid and thus, does not allow the laser to pass through it. Secondly, due to high aggregate formation and increasing temperature, the polymer aggregates settle down and are not able to scatter light further. Fig. 5 represents the hydrodynamic diameter of the polymer with temperature in varying concentrations of osmolyte and their mixtures. The presence of urea induces the hydrophobic collapse of PNIPAM with the faster rate at lower temperature than the typical LCST of PNIPAM in aqueous solution (Fig. 5(a)). The decrease in LCST, up to ∼27.4 °C, is maximum in the case of 6.0 M urea. The LCST change is not appreciable at lower concentrations; however, there is a more significant effect at higher concentrations. The dH of agglomerates increases in a concentration dependent manner. Fig. 5(b) shows that the decrease in LCST is more prominent in the presence of TMAO, even at low concentrations. After 1.0 M concentration, the LCST reached very near to room temperature, resulting in the formation of a turbid solution. Furthermore, as the LCST decreases with concentration, the size of agglomerates also keeps on increasing.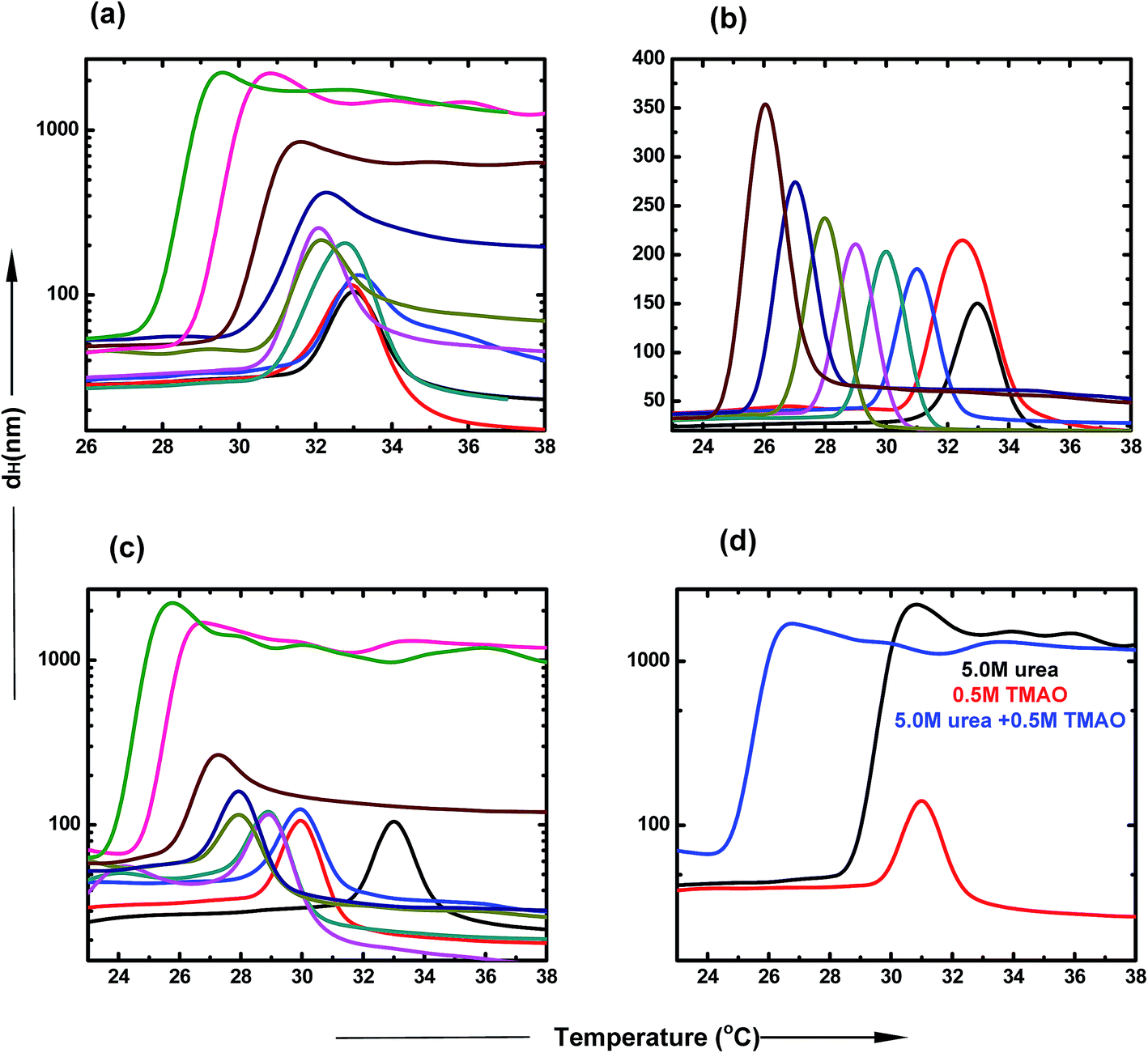 | ||
| Fig. 5 Dynamic light scattering studies of PNIPAM aqueous solution containing (a) urea, PNIPAM in aqueous solution (black), 0.5 M (red), 1.0 M (blue), 1.5 M (dark cyan), 2.0 M (pink), 2.5 M (dark yellow), 3.0 M (dark blue), 4.0 M (brown), 5.0 M (magenta) and 6.0 M (green). (b) TMAO, PNIPAM in aqueous solution (black), 0.4 M (red), 0.5 M (blue), 0.6 M (dark cyan), 0.7 M (pink), 0.8 M (dark yellow), 0.9 M (dark blue), 1.0 M (brown). (c) Mixture TMAO + urea, PNIPAM in aqueous solution (black), (0.5 + 0.5) M (red), (0.5 + 1.0) M (blue), (0.5 + 1.5) M (dark cyan), (0.5 + 2.0) M (pink), (0.5 + 2.5) M (dark yellow), (0.5 + 3.0) M (dark blue), (0.5 + 4.0) M (brown), (0.5 + 5.0) M (magenta) and (0.5 + 5.0) M (green). (d) 1:10 ratio of TMAO:urea, 5.0 M urea (black), 0.5 M TMAO (red), (0.5 + 5.0) M (blue) under atmospheric conditions. | ||
In Fig. 5(c), the different ratios of TMAO and urea were taken as osmolytes and the dH was assessed with the change in temperature. The LCST decreased to 23.5 °C with the addition of 6.0 M urea and 0.5 M TMAO in the aqueous solution of PNIPAM. The size of agglomerates increased to a much higher value at 5.0 and 6.0 M urea, keeping the TMAO concentration constant at 0.5 M. The extent to which LCST decreases is more pronounced in the case of the mixed osmotic environment, in comparison to the individual osmolyte effect. The effect of formation of the collapsed state occurs more easily in a mixed environment and at lower temperature, and is basically attributed to the direct hydrogen bond capacity of urea and some hydrophobic interactions within the PNIPAM chains induced by TMAO. The dH of the 0.5:5.0 ratio of TMAO and urea was studied with respect to temperature, as shown in Fig. 4(d). The LCST change and size is maximum in the case of the 0.5:5.0 molar ratio of TMAO:urea, relative to that of lower ratios (0.5:0.5 and 0.5:1.0). The mixed osmolytic environment was observed to decrease the LCST to a greater extent than individual urea or TMAO osmolytes; however, they did not show an additive effect because of the limited number of binding sites of PNIPAM available.
Fourier transform infrared spectroscopy studies of PNIPAM in the presence of TMAO, urea and their mixture
FTIR spectroscopy studies further confirmed the types of interactions between the co-solvent and amide moiety of PNIPAM. FTIR spectra were obtained in D2O instead of water in order to avoid the interference of the OH band at around 1640 cm−1 with the carbonyl band. PNIPAM in D2O is observed to show two bands named amide I and amide II at 1624 cm−1 62,63 and 1460 cm−1 respectively. The amide I band corresponds to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O⋯D–O–D hydrogen bonding interactions and the amide II band is attributed to the N–D deformation prompted by the D2O molecules. Fig. 6(a) shows that in the case of urea, the peak at around 1610 cm−1 and 1490 cm−1 64,65 was observed up to 2.0 M, predominantly due to the deuterated urea, which itself contains the amide bond, and its direct interactions with the amide moiety of PNIPAM are not significant at lower concentrations. Moreover, at higher concentrations of urea, the direct interactions between urea and PNIPAM are not pronounced and lead to the faster hydrophobic collapse, clearly shown from the peak at 1650 cm−1. The peak keeps on shifting towards 1650 cm−1 and gets broadened at higher concentration, which is mainly due to both hydrophobic collapse and deuterated urea. In the case of TMAO, depicted in Fig. 6(b), the absorbance at 1624 cm−1 keeps on decreasing with concentration as more and more TMAO concentration leads to faster intramolecular interactions, but the peak at 1650 cm−1 attributed to the CO⋯N–D is not very prominent, since a decrease in absorbance is greater, due to the indirect interactions for the hydrophobic collapse of the polymer. Very similar to urea in Fig. 6(c), the mixture of urea and TMAO in varying ratios showed the peak at 1610 cm−1, again due to the presence of deuterated urea.63,64 Since both osmolytes induce the hydrophobic collapse, the peak keeps on shifting towards 1650 cm−1 faster than in the case of urea, as both osmolytes facilitate intramolecular interactions within the polymer, and therefore enhance the hydrophobic collapse. The aggregation of PNIPAM is more pronounced at higher ratios (0.5:5.0 and 0.5:6.0), as compared to that at lower ratios (0.5:0.5 and 0.5:1.0) of TMAO and urea.
O⋯D–O–D hydrogen bonding interactions and the amide II band is attributed to the N–D deformation prompted by the D2O molecules. Fig. 6(a) shows that in the case of urea, the peak at around 1610 cm−1 and 1490 cm−1 64,65 was observed up to 2.0 M, predominantly due to the deuterated urea, which itself contains the amide bond, and its direct interactions with the amide moiety of PNIPAM are not significant at lower concentrations. Moreover, at higher concentrations of urea, the direct interactions between urea and PNIPAM are not pronounced and lead to the faster hydrophobic collapse, clearly shown from the peak at 1650 cm−1. The peak keeps on shifting towards 1650 cm−1 and gets broadened at higher concentration, which is mainly due to both hydrophobic collapse and deuterated urea. In the case of TMAO, depicted in Fig. 6(b), the absorbance at 1624 cm−1 keeps on decreasing with concentration as more and more TMAO concentration leads to faster intramolecular interactions, but the peak at 1650 cm−1 attributed to the CO⋯N–D is not very prominent, since a decrease in absorbance is greater, due to the indirect interactions for the hydrophobic collapse of the polymer. Very similar to urea in Fig. 6(c), the mixture of urea and TMAO in varying ratios showed the peak at 1610 cm−1, again due to the presence of deuterated urea.63,64 Since both osmolytes induce the hydrophobic collapse, the peak keeps on shifting towards 1650 cm−1 faster than in the case of urea, as both osmolytes facilitate intramolecular interactions within the polymer, and therefore enhance the hydrophobic collapse. The aggregation of PNIPAM is more pronounced at higher ratios (0.5:5.0 and 0.5:6.0), as compared to that at lower ratios (0.5:0.5 and 0.5:1.0) of TMAO and urea.
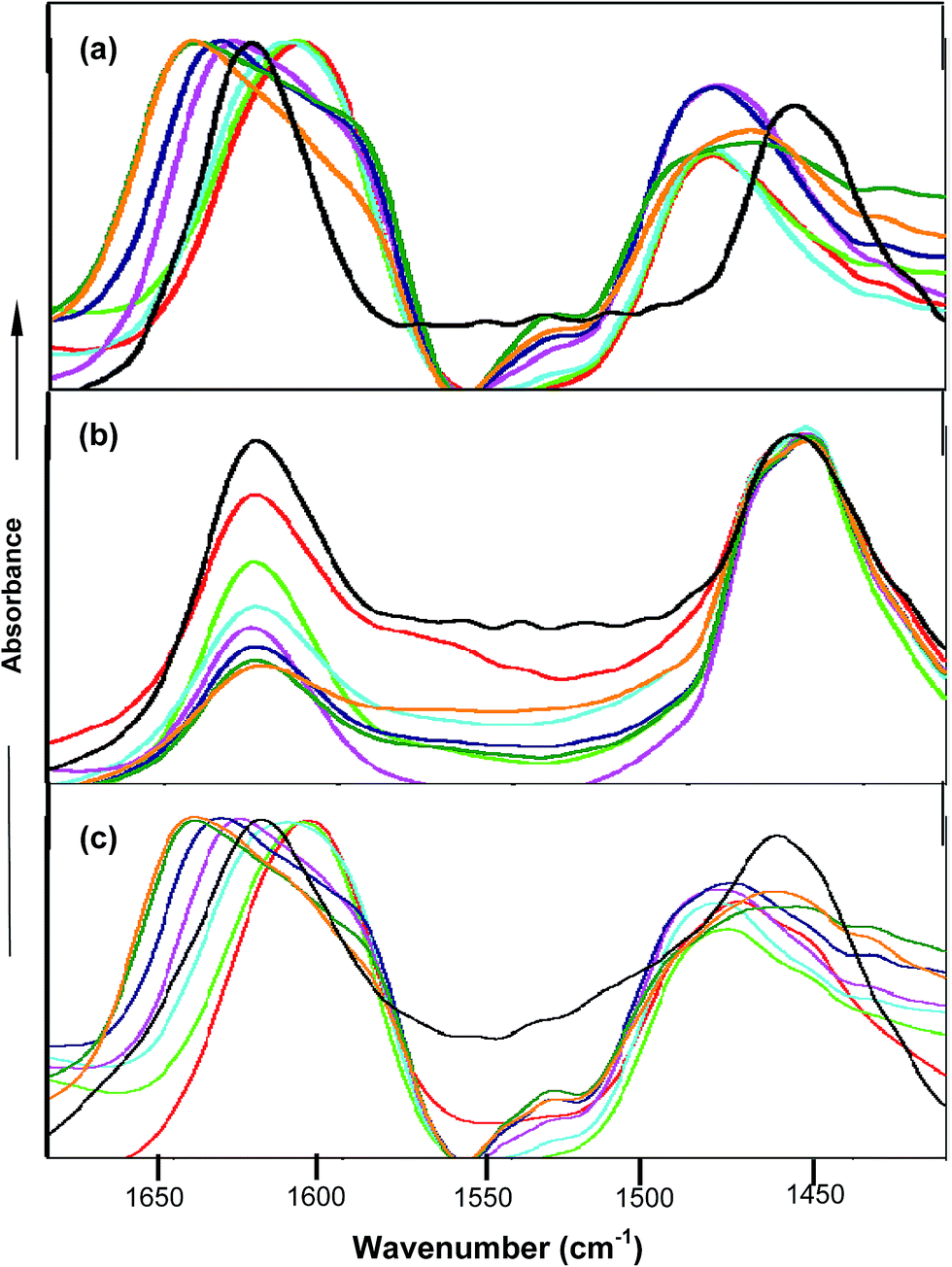 | ||
| Fig. 6 FTIR spectra of PNIPAM in D2O containing (a) urea, PNIPAM in D2O (black), 0.5 M (red), 1.0 M (green), 2.0 M (cyan), 3.0 M (pink), 4.0 M (blue), 5.0 M (dark green) and 6.0 M (yellow). (b) TMAO, PNIPAM in D2O (black), 0.4 M (red), 0.5 M (green), 0.6 M (cyan), 0.7 M (pink), 0.8 M (blue), 0.9 M (dark green), 1.0 M (yellow). (c) TMAO + urea mixture, PNIPAM in D2O (black), (0.5 + 0.5) M (red), (0.5 + 1.0) M (green), (0.5 + 2.0) M (cyan), (0.5 + 3.0) M (pink), (0.5 + 4.0) M (blue), (5.0 + 0.5) M (dark green) and (6.0 + 0.5) M (yellow). | ||
Our results for the individual osmolyte study are consistent with the prior existing results of TMAO27,52,53 and urea,8,48–50 since they both reduce the LCST of PNIPAM to lower temperatures. As mentioned earlier, a new band was observed at ∼288 nm in the UV-absorption spectra, as the addition of urea alters the bonding interaction between PNIPAM and water, which may be because of its ability to form hydrogen bonds and intercalate between several carbonyl groups. Zhou et al.48 also reported that it acts as a cross-linker and has the ability to replace water molecules that were earlier bound to the amide moiety of the PNIPAM chain. This kind of study has also been reported by many researchers, where there is a stabilizing effect of urea on the collapsed state of PNIPAM via its accumulation in the first solvation layer of PNIPAM.8,48–50 TMAO was found to stabilize the collapsed state of PNIPAM to a higher extent than urea, which also matched the previous studies. The stabilizing action of TMAO towards the globular state is induced by its extent of polarization towards water molecules. Polarization with respect to PNIPAM is related to the polar separation of charges in the water molecules that lead to changes in the microenvironment around the macromolecule in the presence of TMAO. Several research groups52,53 have revealed that the driving force for the concomitant sharp configurational transition has been attributed to the rupture of hydrogen bonds between water and polymer and to the hydrophobic association of the polymer. FTIR results obtained for PNIPAM (performed in D2O) reflect results in accordance with those of Cheng et al.,66 indicating that with an increase in the temperature, there is a corresponding decrease in the absorption peak area attributed to the hydrogen bonding, and a new peak appears that is related to the intramolecular hydrogen bonding. Moreover, the binding study mechanism depicts that there are negligible numbers of PNIPAM–TMAO hydrogen bonds. The mixture effect of these osmolytes have not been studied experimentally, while only one simulation study has been performed with the hydrophobic macromolecule.58 The simulation study depicted the effect of the TMAO:urea mixture on the hydrophobic macromolecule. This can be taken as the reference for studying the effect of TMAO + urea on the globular state of amphiphilic polymer PNIPAM, but since their outer exposed moieties in aqueous solution would be much different, they may or may not give similar stabilizing/destabilizing effects for TMAO and urea. This change in the outer surface for interaction with different co-solvents causes the change in the behavior of osmolyte over the PNIPAM surface.
The main findings of our results are that the hydrophobic collapse of PNIPAM in aqueous mixtures of TMAO and urea is pronounced at much lower temperatures as compared to their individual effects on PNIPAM. In order to see the effect of even lower concentrations of TMAO (0.5 M) over high concentrations of urea (5.0 M), we compared our results at 0.5:5.0 molar ratio of TMAO:urea. The mixture of both showed a higher propensity for the collapsed state than their individual effect. Fig. 7 shows the overall change in Tm values with the concentration of TMAO, urea and their various ratios. The graph shows the decrease in LCST on addition of any osmolytes. The slope is most negative for TMAO and least negative for urea. However, the 0.5:5.0 molar ratio is found to decrease the LCST up to 25.5 °C, which is greater as compared to 27.4 and 29.8 °C at 5.0 M urea and 0.5 M TMAO, respectively. The LCST values of PNIPAM decreased for 0.5 M TMAO and urea at various molar concentrations (from 0.5 to 6.0 M). However, they were not able to show a complete additive effect, as ideally expected, due to a limited number of PNIPAM sites available for binding and greater steric hindrance of the osmolyte molecules to interact directly or indirectly with the PNIPAM chain. It can be easily concluded that the 0.5:5.0 molar ratio of TMAO:urea is more facilitating of the globular state of PNIPAM than the extended coiled state.
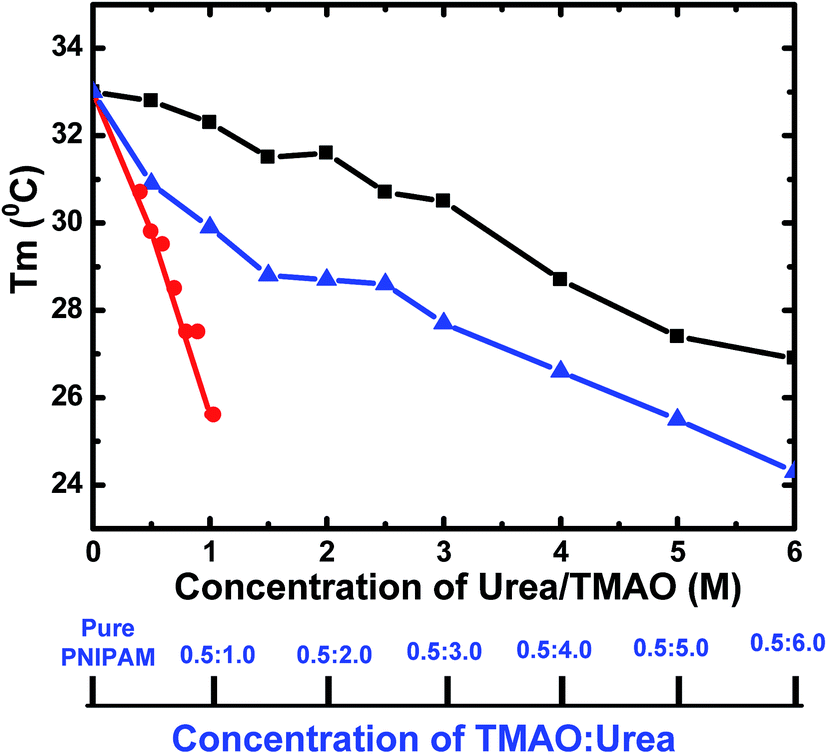 | ||
| Fig. 7 Variation of Tm values as a function of different concentrations of urea (black), TMAO (red) and varying ratios of the mixture TMAO:urea (blue). | ||
Conclusion
The effects of two different osmolytes (TMAO + urea) in a mixed environment were studied using various biophysical techniques, and the results observed from each technique showed that the mixing of TMAO and urea in varying molar ratios led to the stabilization of the collapsed state of PNIPAM through various bonding interactions induced by TMAO and urea molecules. The types of interactions may be direct, or via polarization that leads to the decrease in LCST of PNIPAM in a concentration dependent manner. The greater affinity of urea for direct hydrogen bonding with the amide moiety of PNIPAM makes the extended coiled form more soluble, up to 2.0 M concentration, unlike TMAO, as it is soluble up to a maximum of 1.0 M concentration at room temperature. Our findings reveal that both the osmolytes show their effect in the mixture, resulting in the formation of agglomerates of PNIPAM at very low temperatures, compared to the individual. Moreover, thermal fluorescence studies and DLS data clearly show the aggregated state of PNIPAM at lower temperature, as compared to the typical LCST of PNIPAM in aqueous solution. In a mixed osmolytic environment, the TMAO effect is appreciable even at 0.5 M concentration, in order to decrease the LCST of PNIPAM more towards lower temperature. The 0.5:5.0 molar ratio is observed to stabilize the collapsed form of PNIPAM, compared to lower molar ratios. This tuning of LCST may help in the exploration of the applications of the intelligent PNIPAM polymer in target specific applications in scientific areas.
Acknowledgements
We acknowledge the financial support from the Department of Science and Technology (DST), New Delhi, India (Grant No. SB/SI/PC-109/2012), P. N. is grateful to UGC, New Delhi for providing JRF (Junior Research Fellowship).References
- E. Haladjova, N. T. Moncheva, M. D. Apostolova, B. Trzebicka, A. Dworak, P. Petrov, I. Dimitrov, S. Rangelov and C. B. Trsvetanov, Biomacromolecules, 2014, 15, 4377–4395 CrossRef CAS PubMed.
- M. Salouti, A. Ahangari, InTech, 2014, DOI:10.5772/58423.
- W. B. Liechty, D. R. Kryscio, B. V. Slaughter and N. A. Peppas, Annu. Rev. Chem. Biomol. Eng., 2010, 1, 149–173 CrossRef CAS PubMed.
- A. P. Nikalje, Med. Chem., 2015, 5, 81–89 Search PubMed.
- O. Sedláček, P. Černoch, J. Kučka, R. Konefal, P. Štěpánek, M. Vetrík, T. P. Lodge and M. Hruby, Langmuir, 2016, 32, 6115–6122 CrossRef PubMed.
- R. Singh and J. W. Lillard, Exp. Mol. Pathol., 2009, 86, 215–223 CrossRef CAS PubMed.
- Y. Guan and Y. Zhang, Soft Matter, 2011, 7, 6375–6384 RSC.
- L. B. Sagle, Y. Zhang, V. A. Litosh, X. Chen, Y. Cho and P. S. Cremer, J. Am. Chem. Soc., 2009, 131, 9304–9310 CrossRef CAS PubMed.
- L. W. Xia, R. Xie, X. J. Ju, W. Wang, Q. Chen and L. Y. Chu, Nat. Commun., 2013, 4, 1–11 Search PubMed.
- Y. Yu, B. D. Kieviet, F. Liu, I. Siretanu, E. Kutnyánszky, G. J. Vancso and S. D. Beer, Soft Matter, 2015, 11, 8508–8516 RSC.
- T. Patel, G. Ghosh, S. I. Yusa and P. Bahadur, J. Dispersion Sci. Technol., 2011, 32, 1111–1118 CrossRef CAS.
- A. Halperin, M. Kröger and F. M. Winnik, Angew. Chem., Int. Ed., 2015, 54, 15342–15367 CrossRef CAS PubMed.
- Y. Fang, J. C. Qiang, D. D. Hu, M. Z. Wang and Y. L. Cui, Colloid Polym. Sci., 2001, 279, 14–21 CAS.
- Y. Gao, J. Yang, Y. Ding and X. Ye, J. Phys. Chem. B, 2014, 118, 9460–9466 CrossRef CAS PubMed.
- J. Wang, B. Liu, G. Ru, J. Bai and J. Feng, Macromolecules, 2016, 49, 234–243 CrossRef CAS.
- S. T. Jones, Z. W. Korb, S. J. Barrow, S. L. Henderson, J. D. Barrio and O. A. Scherman, ACS Nano, 2016, 10, 3158–3165 CrossRef CAS PubMed.
- A. Burmistrova, M. Richter, M. Eisele, C. Üzüm and R. V. Klitzing, Polymers, 2011, 3, 1575–1590 CrossRef CAS.
- Z. Ahmed, E. A. Gooding, K. V. Pimenov, L. Wang and S. A. Asher, J. Phys. Chem. B, 2009, 113, 4248–4256 CrossRef CAS PubMed.
- H. Jia, R. Roa, S. Angioletti-Uberti, K. Henzler, A. Ott, X. Lin, J. Möser, Z. Kochovski, A. Schnegg, J. Dzubiella, M. Ballauff and Y. Lu, J. Mater. Chem. A, 2016, 4, 9677–9684 CAS.
- B. Sanz, C. von Bilderling, J. S. Tuninetti, L. Pietrasanta, C. Mijangos, G. S. Longo, O. Azzaronic and J. M. Giussi, Soft Matter, 2017, 13, 2453–2464 RSC.
- Z. Huang, Y. Liu, Q. Zhang, X. Chang, A. Li, L. Deng, C. Yi, Y. Yang, N. M. Khashab, J. Gong and Z. Nie, Nat. Commun., 2016, 7, 1–8 Search PubMed.
- D. Hu, J. Yu, K. Wong, B. Bagchi, P. J. Rossky and P. F. Barbara, Nature, 2000, 405, 1030–1033 CrossRef CAS PubMed.
- E. Mah and R. Ghosh, Processes, 2013, 1, 238–262 CrossRef CAS.
- R. C. Caceres, A. S. Iglesias, M. Karg, I. P. Santos, J. P. Juste, J. Pacifico, T. Hellweg, A. F. Barbero and M. L. L. Marzan, Adv. Mater., 2008, 20, 1666–1670 CrossRef.
- Y. S. Park, Y. Ito and Y. Imanishi, Langmuir, 1998, 14, 910–914 CrossRef CAS.
- L. Verestiuc, C. Ivanov, E. Barbu and J. Tsibouklis, Int. J. Pharm., 2004, 269, 185–194 CrossRef CAS PubMed.
- J. Mondal, D. Halverson, I. T. S. Li, G. Stirnemann, G. C. Walker and B. J. Berne, Proc. Natl. Acad. Sci. (USA), 2015, 112, 9270–9275 CrossRef CAS PubMed.
- H. Wang and S. C. Heilshorn, Adv. Mater., 2015, 27, 3717–3736 CrossRef CAS PubMed.
- S. Shekhar, M. Mukherjee and A. K. Sen, Adv. Mater. Res., 2012, 1, 269–284 CrossRef.
- M. Yang, C. Liu, Y. Lian, K. Zhao, D. Zhub and J. Zhou, Soft Matter, 2017, 13, 2663–2676 RSC.
- S. Micciulla, J. Michalowsky, M. A. Schroer, C. Holm, R. V. Klitzing and J. Smiatek, Phys. Chem. Chem. Phys., 2016, 18, 5324–5335 RSC.
- J. Mondal, G. Stirneman and B. J. Berne, J. Phys. Chem. B, 2013, 117, 8723–8732 CrossRef CAS PubMed.
- G. A. Spitz, C. M. Furtado, M. S. Penna and P. Zancan, Biochem. Pharmacol., 2009, 77, 46–53 CrossRef CAS PubMed.
- S. O. Costa, M. M. Sorenson and M. S. Penna, FEBS J., 2008, 275, 3388–3396 CrossRef PubMed.
- P. J. Rossky, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 16825–16826 CrossRef CAS PubMed.
- M. V. Athawale, J. S. Dordick and S. Garde, Biophys. J., 2005, 89, 858–866 CrossRef CAS PubMed.
- A. Kumar and P. Venkatesu, Chem. Rev., 2012, 112, 4283–4307 CrossRef CAS PubMed.
- P. Venkatesu, M. J. Lee and H. M. Lin, J. Phys. Chem. B, 2007, 111, 9045–9056 CrossRef CAS PubMed.
- A. Kumar, P. Attri and P. Venkatesu, Int. J. Biol. Macromol., 2010, 47, 540–545 CrossRef CAS PubMed.
- J. Ma, I. M. Pazos and F. Gai, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 8476–8481 CrossRef CAS PubMed.
- S. O. Costa, M. M. Sorenson and M. S. Penna, FEBS J., 2005, 272, 2760–2772 CrossRef PubMed.
- P. Attri and P. Venkatesu, Thermochim. Acta, 2011, 526, 143–150 CrossRef CAS.
- P. Venkatesu, H. M. Lin and M. J. Lee, Thermochim. Acta, 2009, 491, 20–28 CrossRef CAS.
- P. Venkatesu, M. J. Lee and H. M. Lin, J. Phys. Chem. B, 2009, 113, 5327–5338 CrossRef CAS PubMed.
- P. Attri, P. Venkatesu and M. J. Lee, J. Phys. Chem. B, 2010, 114, 1471–1478 CrossRef CAS PubMed.
- P. Venkatesu, M. J. Lee and H. M. Lin, Arch. Biochem. Biophys., 2007, 466, 106–115 CrossRef CAS PubMed.
- Y. Gao, J. Yang, H. Fan, Y. Ding and X. Ye, J. Polym. Sci., Part B: Polym. Phys., 2016, 54, 1145–1151 CrossRef CAS.
- Y. Lu, X. Ye, K. Zhou and W. Shi, J. Phys. Chem. B, 2013, 117, 7481–7488 CrossRef CAS PubMed.
- F. Rodríguez-Ropero and N. F. A. van der Vegt, Phys. Chem. Chem. Phys., 2015, 17, 8491–8498 RSC.
- A. Picaa and G. Graziano, Phys. Chem. Chem. Phys., 2016, 18, 14426–14433 RSC.
- I. Bischofberger, D. C. E. Calzolari and V. Trappe, Soft Matter, 2014, 10, 8288–8295 RSC.
- M. A. Schroer, J. Michalowsky, B. Fischer, J. Smiatek and G. Grürel, Phys. Chem. Chem. Phys., 2016, 18, 31459–31470 RSC.
- P. M. Reddy, M. Taha, P. Venkatesu, A. Kumar and M. J. Lee, J. Chem. Phys., 2012, 136, 234904 CrossRef PubMed.
- D. Mukherji, C. M. Marques and K. Kremer, Nat. Commun., 2014, 5, 1–6 Search PubMed.
- M. Taha, B. S. Gupta, I. Khoiroh and M. J. Lee, Macromolecules, 2011, 44, 8575–8589 CrossRef CAS.
- H. Yamauchi and Y. Maeda, J. Phys. Chem. B, 2007, 111, 12964–12968 CrossRef CAS PubMed.
- M. Wang, Y. Fang and D. Hu, React. Funct. Polym., 2001, 48, 215–221 CrossRef CAS.
- K. Fuchise, R. KakuchiI, S. T. Lin, R. Sakai, S. I. Sato, T. Satoh, W. C. Chen and T. Kakuchi, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 6259–6268 CrossRef CAS.
- I. Tah and J. Mondal, J. Phys. Chem. B, 2016, 120, 10969–10978 CrossRef CAS PubMed.
- P. M. Reddy, R. Umapathi and P. Venkatesu, Phys. Chem. Chem. Phys., 2014, 16, 10708–10718 RSC.
- P. M. Reddy, M. Taha, A. Kumar, P. Venkatesu and M. J. Lee, Polymer, 2013, 54, 791–797 CrossRef CAS.
- B. Sun, Y. Lin, P. Wu and H. W. Siesler, Macromolecules, 2008, 41, 1512–1520 CrossRef CAS.
- E. Kesselman, O. Ramon, R. Berkovici and Y. Paz, Polym. Adv. Technol., 2002, 13, 982–991 CrossRef CAS.
- M. Salman, Int. J. Life Sci. Pharma Res., 2012, 2, 128–138 CAS.
- M. M. Al-Majthoub and M. Salman, J. Chem. Pharm. Res., 2012, 4(3), 1856–1863 CAS.
- H. Cheng, L. Shen and C. Wu, Macromolecules, 2006, 39, 2325–2329 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra05120d |
| This journal is © The Royal Society of Chemistry 2017 |