
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A theoretical study about the excited state intermolecular proton transfer mechanisms for 2-phenylimidazo[4,5-b]pyridine in methanol solvent†
Dapeng Yang‡
 *ab,
Jinfeng Zhao‡b,
Min Jiaa and
Xiaoyan Songa
*ab,
Jinfeng Zhao‡b,
Min Jiaa and
Xiaoyan Songa
aCollege of Mathematics and Information Science, North China University of Water Resources and Electric Power, Zhengzhou 450046, China. E-mail: dpyang_ncwu@163.com
bState Key Laboratory of Molecular Reaction Dynamics, Theoretical and Computational Chemistry, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China
First published on 6th July 2017
Abstract
In this study, within the framework of density functional theory (DFT) and time-dependent DFT (TDDFT) methods, we theoretically investigated the novel system 2-phenylimidazo[4,5-b]pyridine (PIP) with respect to the dynamical behavior of its excited state in methanol (MeOH) solvents. Herein, two hydrogen-bonded networks have been discussed between PIP and MeOH, and it has been found that two MeOH connected to PIP (PIP–2MeOH) should be the best arrangement in both S0 and S1 states. Investigations on the electronic spectra of PIP–2MeOH have verified this point. Via analysis of hydrogen bond wires and corresponding infrared (IR) vibrational spectra, we have found that N1–H2⋯O3 of PIP–2MeOH undergoes the biggest change upon photoexcitation that reflects the tendency of the excited state intermolecular proton transfer (ESIPT) process. According to the results of our theoretical potential energy curves along different coordinates, we confirmed that ESIPT reaction should occur along the hydrogen bond wire N1–H2⋯O3 first. After the ESIPT reaction, proton transfer of PIP–2MeOH-PT* could proceed via intersystem crossing (ISC) process from S1 state to T1 state with a negligible energy gap 0.031 eV. Due to this non-radiation process, the fluorescence peak of PIP–2MeOH-PT* could be quenched. Our study not only explains previous successful experiment, but also proposes a new excited state dynamical mechanism for the PIP system.
1. Introduction
The excited state intermolecular proton transfer (ESIPT) reaction is the initial event of numerous photophysical and photochemical processes existing in nature, and it is crucial in chemistry.1–5 In addition, because of the transient property of the ground state, molecules undergoing ESIPT have been used in several applications. As is well-known, the basic photochemical and photophysical principle of ESIPT is that ESIPT molecules possessing proton donors (O–H or N–H) and acceptors (carbonyl oxygen or aromatic nitrogen) may undergo the ESIPT process upon electronic excitation.6–8 Generally, ESIPT reactions could result in a corresponding balance between enol and keto tautomers, stimulating a dual emission and large Stokes shifts.9–12 Moreover, two fluorescence bands are observed and a broader range of the steady-state emission can be covered, making these ESPT molecules suitable for optical chemosensors, white-emitting OLEDs, material chemistry, and UV filters, among other applications.13–26A molecule containing a proton donor and acceptor in close proximity might undergo ESIPT. On the other hand, if the groups involved in the proton transfer are far from each other or do not have appropriate geometry to form an intramolecular hydrogen bond, these molecules undergo ESIPT process with the assistance of other molecules (generally solvent molecules) with both hydrogen-bond accepting and donating abilities. This kind of reaction occurs because solvent molecules act as bridges between proton acceptors and proton donors.27–31 In addition, microenvironment is a key factor for maintaining normal cell metabolism and abnormal changes in the microenvironment might lead to cytopathy.32,33 Recently, as a kind of biologically active system, 2-phenylimidazo[4,5-b]pyridine (PIP) has been tested to be an inhibitor for Aurora-A, Aurora-B, and Aurora-C kinases34–36 that have been certified to be good probes for microenvironments.33 Krishnamoorthy and co-workers experimentally synthesized and investigated PIP and its analogues. They found that a single-emission phenomenon could be found just in a polar protic methanol (MeOH) solvent, which is different from that of PIP analogues (2-(4′-N,N-dimethylaminophenyl)imidazo[4,5-b]pyridine (DMAPIP-b) and 2-(4′-N,N-dimethylaminophenyl)imidazo[4,5-c]pyridine (DMAPIP-c)).34,35 Therefore, the excited state dynamical mechanism of PIP is different from that of the abovementioned two molecules. Krishnamoorthy and co-workers mainly focused on PIP analogues, and they lost sight of PIP in their previous study. In effect, detailed study of PIP is very meaningful not only in biological aspects, but also in photochemical and photophysical fields. Moreover, since PIP is similar to DMAPIP-b and DMAPIP-c, can the proton transfer process also exist in the excited state? If this is the case, why the second fluorescence could not be detected? Furthermore, it is well known that spectroscopic techniques, such as steady state absorption and fluorescence spectroscopy and time-resolved fluorescence spectroscopy, can only provide indirect information about some photochemical and photophysical properties.37–45 The specific mechanism still depends on quantum chemical calculations.
To further understand the excited state reaction mechanism of PIP in a MeOH solvent, herein, a detailed quantum chemical computational investigation has been adopted to study the PIP systems. As mentioned in ref. 34, the structures of PIP in MeOH are shown in Fig. 1. Based on the fundamental chemical structure stability, we speculated that hydrogen bond wire for PIP with one MeOH or two MeOH molecules might be stable; moreover, three or more components could be too soft to retain the stability of the structures. Thus, two types of compound modes have been considered in this study (i.e. PIP–MeOH and PIP–2MeOH). We put forward a new excited state mechanism for PIP molecules based on the density functional theory (DFT) and time-dependent density functional theory (TDDFT) methods. The rest of our study has been organized as follows: the next section describes the theoretical method; Section 3 shows the results and discussions of PIP calculations including geometry analyses, infrared (IR) vibrational analyses, electronic spectra, charge redistribution, and mechanism analysis; and finally, the last section summarizes and provides the conclusion of this study.
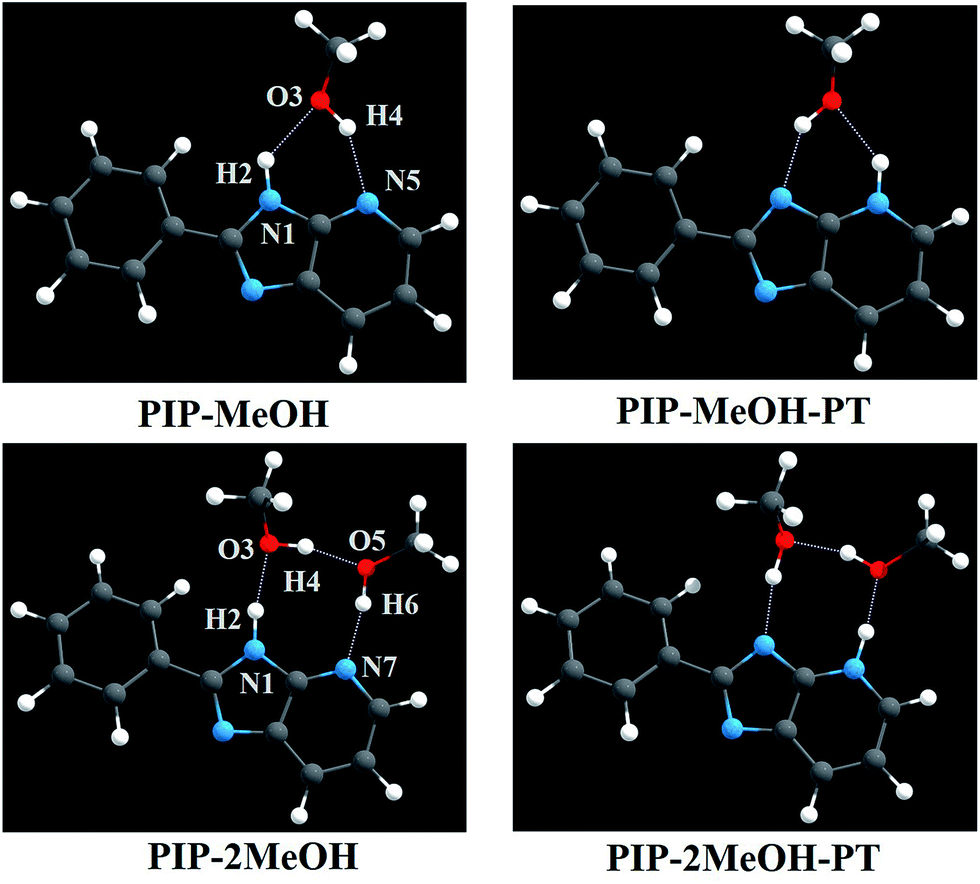 | ||
| Fig. 1 The optimized structures for PIP–MeOH (PIP combined with one MeOH molecule) and PIP–2MeOH (PIP combined with two MeOH molecules) systems in methanol (MeOH) solvents. For the sake of description, we have labelled sequence numbers on relevant atoms involved in hydrogen bond wires. Dark grey: C; white: H; red: O; and sapphire: N. | ||
2. Theoretical method
In this study, all the quantum chemical calculations have been performed via density functional theory (DFT) and time-dependent density functional theory (TDDFT) methods using the Becke's three-parameter hybrid exchange function with the Lee–Yang–Parr gradient-corrected correlation functional (B3LYP)46–48 in combination with the triple-ζ valence quality with one set of polarization functions (TZVP)49 basis set in the Gaussian 09 program.50 MeOH has been selected as a solvent in our calculations based on the polarizable continuum model (PCM) using the integral equation formalism variant (IEFPCM)51,52 that considers a solute in a cavity of overlapping solvents (with an average area of 0.4 Å) with apparent charges to reproduce the electrostatic potential caused by the polarized dielectric within the cavity. All initial geometries for PIP–MeOH and PIP–2MeOH complexes were generated from the standard geometrical parameters by minimization, without any constraints for symmetry, bond length, bond angle, and dihedral angle, to obtain the true minima based on DFT and TDDFT. IR vibrational frequencies were calculated at the same level to confirm the absence of imaginary modes. The calculations of vertical excitation energies were also performed from the ground-optimized structures based on TDDFT with IEFPCM (MeOH), and our theoretical calculations predicted six low-lying absorbing transitions. To illustrate the excited state mechanism for the PIP system, all stationary points along the reaction coordinate were scanned by constraining optimizations to obtain thermodynamic corrections in the corresponding electronic states.53–57 Zero-point energy corrections and thermal corrections to Gibbs free energy were also carried out according to harmonic vibrational frequencies. The energies of intermolecular hydrogen bonds were calculated using the counterpoise method (basis set superposition error (BSSE)) of Boys and Bernardi.583. Results and discussion
3.1. Analysis of the structures
As abovementioned, all structural optimizations of corresponding molecular configurations were obtained based on DFT and TDDFT/B3LYP/TZVP theoretical levels in both S0 and S1 states. Atomic coordinates of all optimized structures involved in this study are provided in ESI.† For the sake of presentation, we have labelled the order numbers on corresponding atoms involved in the hydrogen bond wires (see Fig. 1). In addition, some staple parameters of these intermolecular hydrogen bonds for the PIP–MeOH and PIP–2MeOH systems are listed in Table 1 and Table 2, respectively. Herein, according to the AIM theory (mainly based on the analysis of electron density at the specific point (ρ(r))), identification of a critical point (CP) and the existence of a bond path in equilibrium geometry are necessary and sufficient conditions for assigning an interaction between two primary atoms.59,60 Moreover, the AIM analysis of the title compounds ensures the presence of an appreciable interaction between the concerned atoms. The relevant AIM topological parameters involved in the optimized geometries demonstrate that the ρ(r) values at the bond critical point (BCP) of PIP–MeOH and PIP–2MeOH systems are close to 0.04 a.u. (the maximum threshold value proposed by Popelier to confirm the presence of hydrogen bonds59,60). In addition, the corresponding ∇2 ρc values are in the range 0.02–0.15 a.u.59,60 Therefore, we can confirm that all these intermolecular hydrogen bonds should be formed for both PIP–MeOH and PIP–2MeOH.| Electronic state | PIP–MeOH | PIP–MeOH-PT | ||
|---|---|---|---|---|
| S0 | S1 | S0 | S1 | |
| N1–H2 | 1.016 | 1.018 | 1.955 | 1.961 |
| H2–O3 | 2.088 | 2.054 | 0.980 | 0.978 |
| O3–H4 | 0.978 | 0.978 | 1.984 | 2.050 |
| H4–N5 | 1.985 | 1.987 | 1.026 | 1.020 |
| δ(N1–H2–O3) | 132.7 | 134.3 | 146.8 | 148.8 |
| δ(O3–H4–N5) | 149.5 | 148.4 | 139.0 | 137.7 |
| Electronic state | PIP–2MeOH | PIP–2MeOH-PT | |||
|---|---|---|---|---|---|
| S0 | S1 | S0 | S1 | T1 | |
| N1–H2 | 1.031 | 1.032 | 1.810 | 1.828 | 1.923 |
| H2–O3 | 1.813 | 1.806 | 0.992 | 0.989 | 0.981 |
| O3–H4 | 0.986 | 0.986 | 1.753 | 1.770 | 1.774 |
| H4–O5 | 1.759 | 1.756 | 0.987 | 0.985 | 0.984 |
| O5–H6 | 0.989 | 0.989 | 1.744 | 1.790 | 1.765 |
| H6–N7 | 1.840 | 1.837 | 1.042 | 1.035 | 1.037 |
| δ(N1–H2–O3) | 170.4 | 171.0 | 170.2 | 171.1 | 168.9 |
| δ(O3–H4–O5) | 165.0 | 165.1 | 163.2 | 163.1 | 162.2 |
| δ(O5–H6–N7) | 170.4 | 170.2 | 174.1 | 173.8 | 176.7 |
For the PIP–MeOH complex, note that the length of the hydrogen bond (H2⋯O3) is shortened from S0 state (2.088 Å) to S1 state (2.054 Å), with the bond angle δ(N1–H2⋯O3) increasing from 132.7° to 134.3°. It demonstrates that the hydrogen bond (N1–H2⋯O3) should be strengthened upon photoexcitation. On the contrary, H4⋯N5 is elongated from 1.985 Å to 1.987 Å together with the decrease of δ(O3–H4⋯N5) from 149.5° to 148.4°, which reveals that this hydrogen bond is weakened. That is to say, if ESIPT occurs in the PIP–MeOH system, the proton should transfer along N1–H2⋯O3 first.27–30 Moreover, turning to the PIP–2MeOH system, on similar analysis, we found that the hydrogen bond N1–H2⋯O3 undergoes the greatest change among the three hydrogen bond wires. Thus, the ESIPT reaction should occurs first along this hydrogen bond wire.
It is well known that IR vibrational modes of molecules connected by hydrogen bonds could be considered as another determinant to detect variations in the excited state hydrogen bonds.37–43 Therefore, theoretical IR vibrational spectra were used in this study to reveal the effects originating from photoexcitation (Fig. 2). Note that our theoretical stretching vibrational frequency of N1–H2 for PIP–MeOH shows a red-shift of 22 cm−1 from the S0 to S1 states, whereas that of O3–H4 shows little change. For the PIP–2MeOH complex, IR vibration of N1–H2 shows the biggest red-shift from the S0 state (3197.9 cm−1) to the S1 state (3169.1 cm−1) among the three hydrogen bond wires. All these variations in hydrogen bonds for PIP–MeOH and PIP–2MeOH confirm that protons should first transfer along the N1–H2⋯O3 hydrogen bond in both PIP–MeOH and PIP–2MeOH in the event of an ESIPT reaction. In addition, based on the calculation of hydrogen bond binding energy via basis set superposition error (BSSE),28 we have found that the binding energy (23.61 kcal mol−1) for PIP–2MeOH in the S0 state is more stable than that for PIP–MeOH (10.58 kcal mol−1). Thus, we hypothesize that two MeOH molecules will be more suitable for PIP since two MeOH molecules render the PIP complex to be more compact.
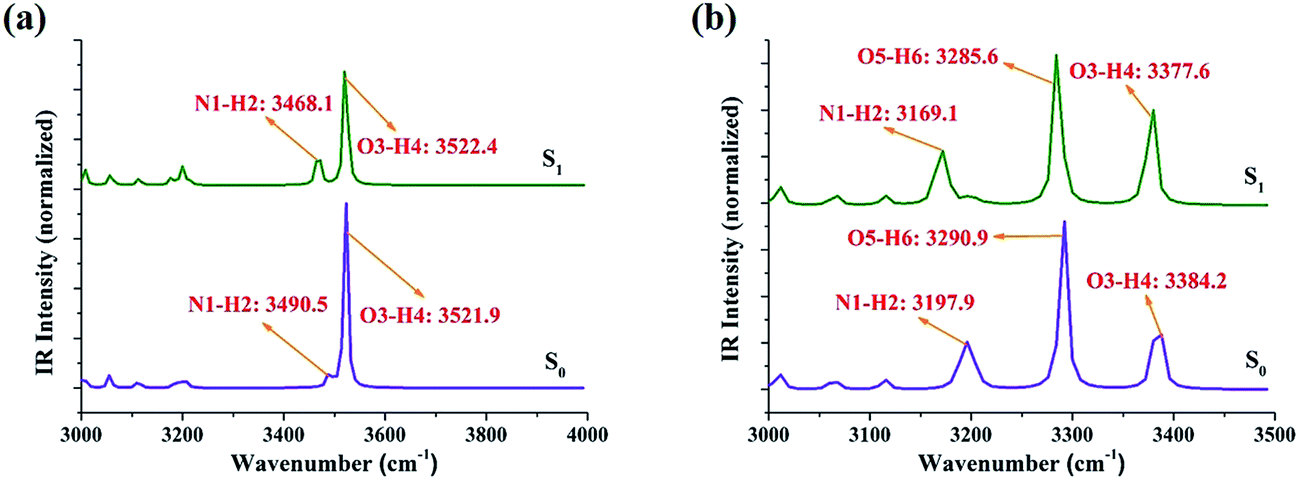 | ||
| Fig. 2 The theoretical IR spectra of PIP–MeOH (a) and PIP–2MeOH (b) structures in MeOH solvents in the spectral regions of corresponding chemical bonds in both S0 and S1 states. | ||
3.2. Electronic spectra and charge redistribution
To further inspect the influence of the photoexcitation process, the corresponding theoretical electronic spectra are displayed in Fig. 3. Note that our calculated absorption peaks for PIP–MeOH and PIP–2MeOH are obtained at 302.7 nm and 301.9 nm, respectively, which are in line with the experimental results.34 Moreover, our simulated fluorescence peaks for PIP–MeOH and PIP–2MeOH are obtained at 377.1 nm and 376.9 nm, respectively, which are also in agreement with the experimentally obtained 378 nm.34 Thus, we not only conclude that the theoretical level used in this study is reasonable and feasible for the system, but also confirm that both these structures are reasonable. Which structure is better has been discussed in the Section 3.3.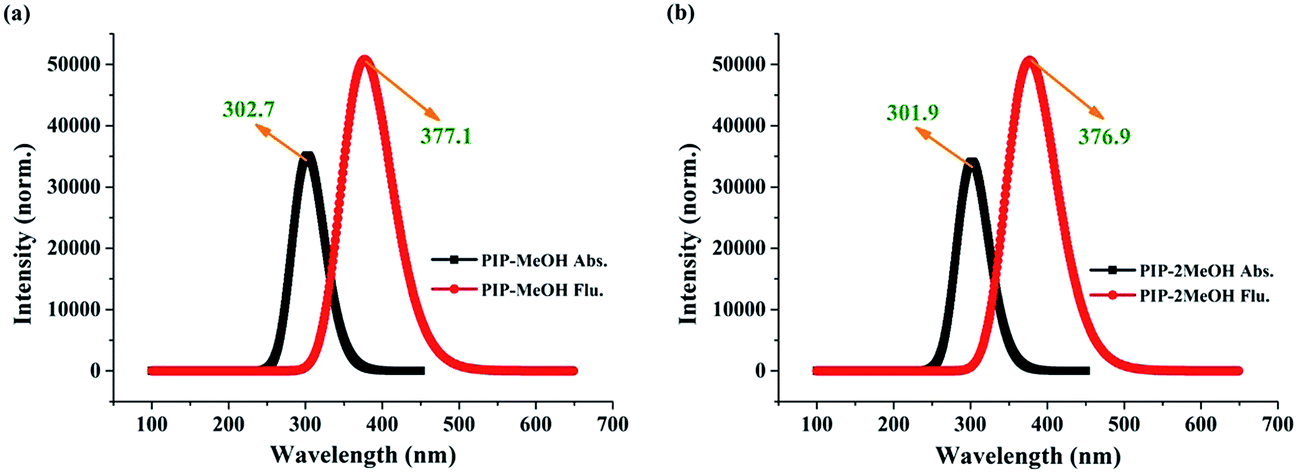 | ||
| Fig. 3 The calculated absorption and emission peaks for both PIP–MeOH (a) and PIP–2MeOH (b) complex in MeOH solvents based on TDDFT/B3LYP/TZVP theoretical levels. | ||
In addition, to qualitatively further study charge redistribution upon photo-excitation, the frontier molecular orbitals (MOs) have been discussed in this section. Herein, based on our calculated results, the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) are shown in Fig. 4 since these two orbitals are mainly associated with the transition from S0 to S1 states. It can be clearly seen that the distributions of PIP–MeOH and PIP–2MeOH are almost the same. Undoubtedly, the S1 state involves a dominant π–π*-type transition from HOMO to LUMO, which is in agreement with previous results.34 In fact, note that there is almost no charge located around N5 of PIP–MeOH and N7 of PIP–2MeOH in HOMO orbital, while charge is observed at these sites in the LUMO orbital. It demonstrates that the S1 state is the charge-transfer state. The change in electron density at N5 of PIP–MeOH and N7 of PIP–2MeOH could directly affect intermolecular hydrogen bonds. This kind of redistribution of charge transfer indicates a tendency for the ESIPT reaction.37–43
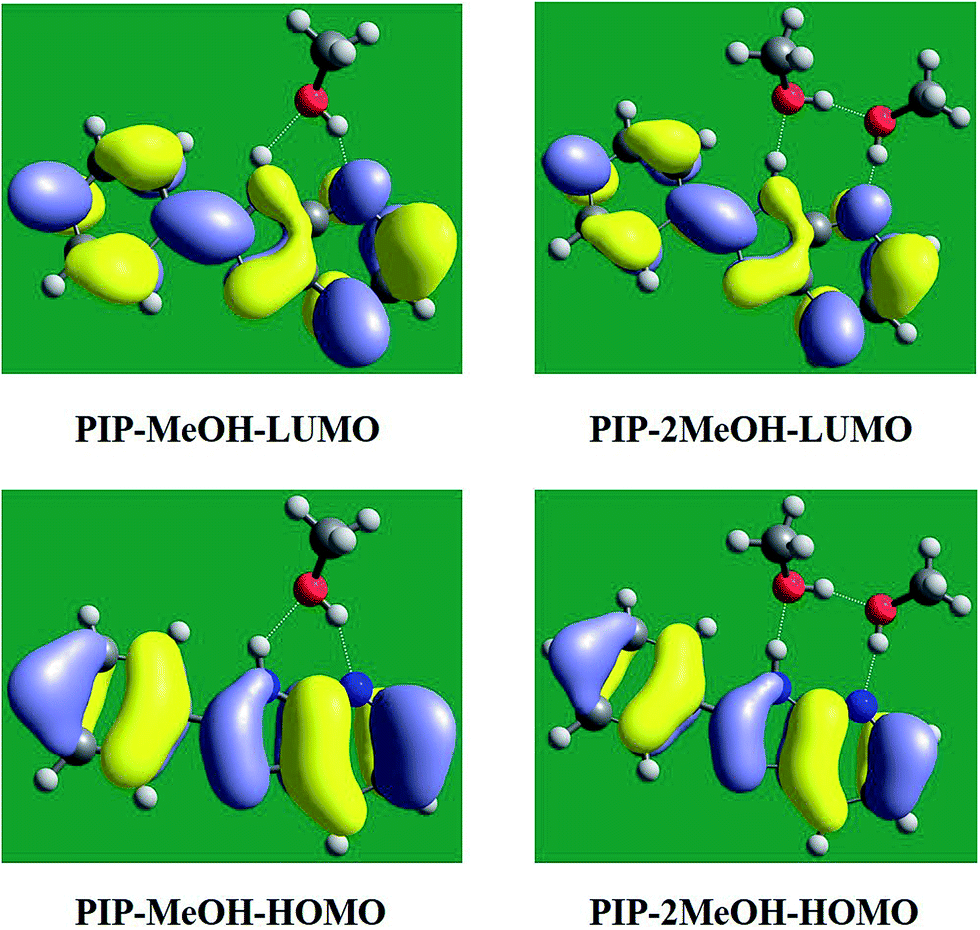 | ||
| Fig. 4 View of frontier molecular orbitals (HOMO and LUMO) for both PIP–MeOH and PIP–2MeOH systems. | ||
3.3. Mechanism analyses
It is well known that potential energy curves should be a kind of conventional method to solve problems associated with excited state behavior;27–31 therefore, we theoretically constructed potential energy curves to further study in detail the excited state mechanism for the PIP system in MeOH solvent (Fig. 5). The constructed potential energy curves of both S0 and S1 states are at the step of 0.05 Å along with corresponding bond lengths. Herein, it is noted that the correct ordering of the closely spaced excited state might not be expected to be accurately yielded based on the TDDFT method, whereas some previous studies show that this method could be reliable to provide qualitative energetic pathways for studying the ESIPT processes.61,62 In Fig. 5, three kinds of possible ESIPT conditions for both PIP–MeOH and PIP–2MeOH complexes have been considered (i.e., stepwise proton-transfer manner along with ambilateral hydrogen bond wires and synchronous pathways). Obviously, for PIP–MeOH, the ESIPT reaction could not occur along the O3–H4⋯N5 hydrogen bond wire and the so-called synchronous pathway along both N1–H2⋯O3 and O3–H4⋯N5. The best path for ESIPT is along the hydrogen bond N1–H2⋯O3 with a moderate potential energy barrier of 11.72 kcal mol−1 in the S1 state. By contrast, a lower potential energy barrier (9.97 kcal mol−1) exists in the S1 state for PIP–2MeOH to complete the ESIPT process. Moreover, under the other two manners, the potential energy barriers are much lower than that for PIP–MeOH. Combined with the hydrogen bond energies, we confirmed that two MeOH solvent molecules are more reasonable for connecting with the PIP system rather than one MeOH molecule, as mentioned in a previous study.34 | ||
| Fig. 5 View of three kinds of potential energy curves for PIP–MeOH and PIP–2MeOH systems in both S0 and S1 states. (a) Shortening the distance of H2⋯O3 bond length (Å) along N1–H2⋯O3 for PIP–MeOH; (b) shortening the distance of H4⋯N5 bond length (Å) along O3–H4⋯N5 for PIP–MeOH; (c) synchronous shortening of H2⋯O3 and H4⋯N5 bond lengths along N1–H2⋯O3 and O3–H4⋯N5 for PIP–MeOH; (d) shortening the distance of H2⋯O3 bond length (Å) along N1–H2⋯O3 for PIP–2MeOH; (e) shortening the distance of H6⋯N7 bond length (Å) along O5–H6⋯N7 for PIP–2MeOH; (f) synchronous shortening of H2⋯O3 and H6⋯N7 bond lengths along N1–H2⋯O3 and O5–H6⋯N7 for PIP–2MeOH. | ||
It is, however, difficult to judge whether ESIPT occurs at a barrier of 9.97 kcal mol−1 although according to thermodynamic Boltzmann distribution, ESIPT reactions can occur under this condition. In this case, we theoretically optimized the proton-transfer in PIP–2MeOH-PT* with an energy of −858.63014221 hartree in the S1 state. Unexpectedly, the emission peak at about 392.1 nm was calculated using a large oscillator strength of around 1.174 based on the TDDFT/B3LYP/TZVP theoretical level. Why was this second fluorescence not mentioned in previous experiments? We speculated that it must involve a non-radiation path. While further investigating this, we surprisingly found that the triple (T1) state of PIP–2MeOH-PT was optimized to be −858.63150605 hartree. As shown in Fig. 6, the gap between S1 and T1 states is just 0.031 eV (0.855 kcal mol−1), and this low energy gap might result in the intersystem crossing process from S1 to T1.28 Then, the undetected phosphorescence with a 0.0016 oscillator strength in the previous experiment should be emitted back from T1 back to S0 state. Therefore, to date, we have successfully explained previous experiments and concluded that excited state dynamical mechanism for PIP coupled with MeOH is as follows: two MeOH molecules combine with a PIP molecule, forming PIP–2MeOH in the S0 state. Upon photoexcitation, PIP–2MeOH is excited to the S1 state with a fluorescence peak at 377 nm; subsequently, the ESIPT reaction occurs first along the hydrogen bond wire N1–H2⋯O3, forming PIP–2MeOH-PT* in the S1 state. Then, due to the intersystem crossing process, S1-state PIP–2MeOH-PT* turns to the T1 state in a non-radiation way back to the S0 state.
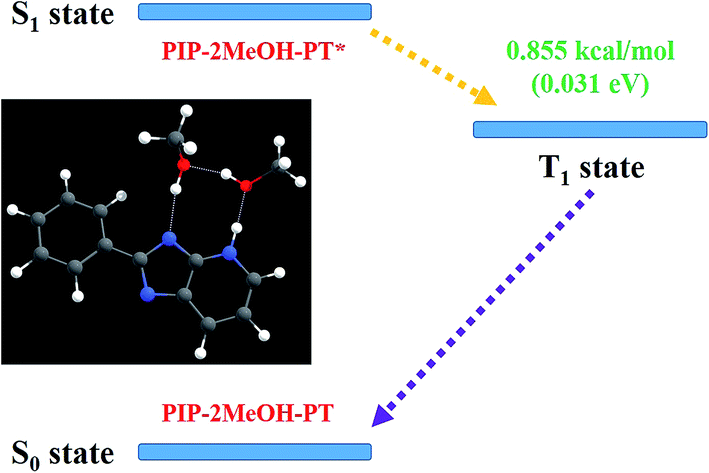 | ||
| Fig. 6 The probable excited state non-radiative mechanism for the PIP–2MeOH system. | ||
4. Conclusions
In this study, we theoretically proposed a new excited state mechanism for the PIP system in MeOH solvents based on DFT and TDDFT methods. We confirmed that two MeOH solvent molecules should connect with PIP forming PIP–2MeOH rather than one MeOH molecule combining to form PIP–MeOH. The theoretical electronic spectra of PIP–2MeOH confirmed the previous experimental results. The strengthened intermolecular hydrogen bond wire and corresponding charge redistribution provide the possibility for ESIPT reaction. By constructing potential energy curves, we confirmed that the ESIPT process should first occur along the hydrogen bond wire N1–H2⋯O3. After the ESIPT process, the S1-state PIP–2MeOH-PT* proceeds the intersystem crossing to the T1 state, with a small energy gap 0.031 eV. Hence, the fluorescence of PIP–2MeOH-PT* is quenched, which should be the reason that only one emission peak has been reported previously.34Acknowledgements
This work was supported by the National Natural Science Foundation of China (grant no. 11604333) and the Key Scientific Research Project of Colleges and Universities of Henan Province of China (grant no. 18A140023).References
- K. C. Tang, C. L. Chen, H. H. Chuang, J. L. Chen, Y. J. Chen, Y. C. Lin, J. Y. Shen, W. P. Hu and P. T. Chou, J. Phys. Chem. Lett., 2011, 2, 3063 CrossRef CAS.
- K. Y. Chen, C. C. Hsieh, Y. M. Cheng, C. H. Lai and P. T. Chou, Chem. Commun., 2006, 42, 4395 RSC.
- L. Liu, D. P. Yang and P. Li, J. Phys. Chem. B, 2014, 118, 11707 CrossRef CAS PubMed.
- D. P. Yang, Y. G. Yang and Y. F. Liu, Commun. Comput. Chem., 2013, 1, 205 Search PubMed.
- J. Zhao, J. Chen, Y. Cui, J. Wang, L. Xia, Y. Dai, P. Song and F. Ma, Phys. Chem. Chem. Phys., 2015, 17, 1142 RSC.
- J. Zhao, H. Yao, J. Liu and M. R. Hoffmann, J. Phys. Chem. A, 2015, 119, 681 CrossRef CAS PubMed.
- Y. H. Liu and T. S. Chu, Chem. Phys. Lett., 2011, 505, 117 CrossRef CAS.
- G. Y. Li, G. J. Zhao, K. L. Han and G. Z. He, J. Comput. Chem., 2011, 32, 668 CrossRef CAS PubMed.
- Y. Liu and S. C. Lan, Commun. Comput. Chem., 2013, 1, 235 Search PubMed.
- J. Zhao, P. Song and F. Ma, Commun. Comput. Chem., 2014, 2, 117 Search PubMed.
- H. Ma and J. D. Huang, RSC Adv., 2016, 6, 96147 RSC.
- P. Song and F. Ma, Int. Rev. Phys. Chem., 2013, 32, 589 CrossRef CAS.
- J. Zhao and P. Li, RSC Adv., 2015, 5, 73619 RSC.
- Y. H. Liu and P. Li, J. Lumin., 2011, 131, 2116 CrossRef CAS.
- F. B. Yu, P. Li, G. Y. Li, G. J. Zhao, T. S. Chu and K. L. Han, J. Am. Chem. Soc., 2011, 133, 11030 CrossRef CAS PubMed.
- Y. Cui, P. Li, J. Wang, P. Song and L. Xia, J. At. Mol. Sci., 2015, 6, 23 Search PubMed.
- M. W. Chung, J. L. Liao, K. C. Tang, C. C. Hsieh, T. Y. Lin, C. Liu, G. H. Lee, Y. Chi and P. T. Chou, Phys. Chem. Chem. Phys., 2012, 14, 9006 RSC.
- D. D. Wang, R. Lu, M. H. Yuan, A. P. Fu and T. S. Chu, Spectrochim. Acta, Part A, 2014, 125, 131 CrossRef CAS PubMed.
- J. Zhao and Y. Yang, Commun. Comput. Chem., 2016, 4, 1 CrossRef.
- P. Song and J. Zhao, Commun. Comput. Chem., 2017, 5, 1 CrossRef.
- Y. H. Chen, K. C. Tang, Y. T. Chen, J. Y. Shen, Y. S. Wu, S. H. Liu, C. S. Lee, C. H. Chen, T. Y. Lai, S. H. Tung, R. J. Jeng, W. Y. Hung, M. Jiao, C. C. Wu and P. T. Chou, Chem. Sci., 2016, 7, 3556 RSC.
- S. Chai, G. J. Zhao, P. Song, S. Q. Yang, J. Y. Liu and K. L. Han, Phys. Chem. Chem. Phys., 2009, 11, 4385 RSC.
- A. P. Demchenko, K. C. Tang and P. T. Chou, Chem. Soc. Rev., 2013, 42, 1379 RSC.
- W. T. Hsieh, C. C. Hsieh, C. H. Lai, L. M. Cheng, M. L. Ho, K. K. Wang, G. H. Lee and P. T. Chou, ChemPhysChem, 2008, 9, 293 CrossRef CAS PubMed.
- T. S. Chu and J. Xu, J. Mol. Model., 2016, 22, 200 CrossRef PubMed.
- Y. Li, J. S. Chen and T. S. Chu, J. Lumin., 2016, 179, 203 CrossRef CAS.
- Y. H. Liu, S. M. Wang, C. W. Wang, C. Y. Zhu, K. L. Han and S. H. Lin, J. Chem. Phys., 2016, 145, 164314 CrossRef PubMed.
- Y. H. Liu, S. C. Lan, C. Y. Zhu and S. H. Lin, J. Phys. Chem. A, 2015, 119, 6269 CrossRef CAS PubMed.
- Y. H. Liu, M. S. Mehata and J. Y. Liu, J. Phys. Chem. A, 2011, 115, 19 CrossRef CAS PubMed.
- Y. Wang, H. Yin, Y. Shi, M. X. Jin and D. J. Ding, New J. Chem., 2014, 38, 4458 RSC.
- H. Yin, H. Li, G. Xia, C. Ruan, Y. Shi, H. Wang, M. Jin and D. Ding, Sci. Rep., 2016, 6, 19774 CrossRef CAS PubMed.
- S. Spranger, R. Bao and T. F. Gajewski, Nature, 2015, 523, 231 CrossRef CAS PubMed.
- D. W. Hein, R. J. Alheim and J. J. Leavitt, J. Am. Chem. Soc., 1957, 79, 427 CrossRef CAS.
- A. Mishra, S. Sahu, N. Dash, S. K. Behera and G. Krishnamoorthy, J. Phys. Chem. B, 2013, 117, 9469 CrossRef CAS PubMed.
- S. K. Behera and G. Krishnamoorthy, Photochem. Photobiol. Sci., 2015, 14, 2225 CAS.
- V. Bavatsias, C. Sun, N. Bouloc, J. Reynisson, P. Workman, S. Linardopoulos and E. Mcdonald, Bioorg. Med. Chem. Lett., 2007, 17, 6567 CrossRef PubMed.
- G. J. Zhao and K. L. Han, Acc. Chem. Res., 2012, 45, 404 CrossRef CAS PubMed.
- G. J. Zhao, B. H. Northrop, P. J. Stang and K. L. Han, J. Phys. Chem. A, 2010, 114, 3418 CrossRef CAS PubMed.
- G. J. Zhao and K. L. Han, Phys. Chem. Chem. Phys., 2010, 12, 8914 RSC.
- G. J. Zhao and K. L. Han, ChemPhysChem, 2008, 9, 1842 CrossRef CAS PubMed.
- G. J. Zhao and K. L. Han, J. Phys. Chem. A, 2007, 111, 9218 CrossRef CAS PubMed.
- J. Zhao, J. Chen, J. Liu and M. R. Hoffmann, Phys. Chem. Chem. Phys., 2015, 17, 11990 RSC.
- J. Zhao, P. Song and F. Ma, Commun. Comput. Chem., 2015, 3, 44 Search PubMed.
- L. Cong, H. Yin, Y. Shi, M. Jin and D. Ding, RSC Adv., 2015, 5, 1205 RSC.
- H. Li, H. Yin, X. Liu and Y. Shi, J. At. Mol. Sci., 2016, 7, 115 Search PubMed.
- C. T. Lee, W. T. Wang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS.
- B. Mennucci, E. Cances and J. Tomasi, J. Phys. Chem. B, 1997, 101, 10506 CrossRef CAS.
- B. Miehlich, A. Savin, H. Stoll and H. Preuss, Chem. Phys. Lett., 1989, 157, 200 CrossRef CAS.
- D. Feller, J. Comput. Chem., 1996, 17, 1571 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, revision C.01, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- R. Cammi and J. Tomasi, J. Comput. Chem., 1995, 16, 1449 CrossRef CAS.
- S. Miertus, E. Scrocco and J. Tomasi, Chem. Phys., 1981, 55, 117 CrossRef CAS.
- H. W. Tseng, J. Q. Liu, Y. A. Chen, C. M. Chao, K. M. Liu, C. L. Chen, T. C. Lin, C. H. Hung, Y. L. Chou, T. C. Lin, T. L. Wang and P. T. Chou, J. Phys. Chem. Lett., 2015, 6, 1477 CrossRef CAS PubMed.
- C. Y. Peng, J. Y. Shen, Y. T. Chen, P. J. Wu, W. Y. Hung, W. P. Hu and P. T. Chou, J. Am. Chem. Soc., 2015, 137, 14349 CrossRef CAS PubMed.
- J. Zhao, P. Song and F. Ma, Commun. Comput. Chem., 2015, 2, 146 Search PubMed.
- Z. Zhang, Y. H. Hsu, Y. A. Chen, C. L. Chen, T. C. Lin, J. Y. Shen and P. T. Chou, Chem. Commun., 2014, 50, 15026 RSC.
- H. W. Tseng, T. C. Lin, C. L. Chen, T. C. Lin, Y. A. Chen, J. Q. Liu, C. H. Hung, C. M. Chao, K. M. Liu and P. T. Chou, Chem. Commun., 2015, 51, 16099 RSC.
- S. F. Boys and F. Bernardi, Mol. Phys., 2002, 100, 65 CrossRef.
- P. Hobza and Z. Havlas, Chem. Rev., 2000, 100, 4253 CrossRef CAS PubMed.
- P. L. A. Popelier, J. Phys. Chem. A, 1998, 102, 1873 CrossRef CAS.
- Y. Saga, Y. Shibata and H. Tamiaki, J. Photochem. Photobiol., C, 2010, 11, 15 CrossRef CAS.
- A. L. Serrano and M. Merchan, J. Photochem. Photobiol., C, 2009, 10, 21 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra05976k |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2017 |