DOI:
10.1039/C7RA05161A
(Paper)
RSC Adv., 2017,
7, 33812-33821
Rapid synthesis and properties of segmented block copolymers based on monodisperse aromatic poly(N-methyl benzamide) and poly(propylene oxide)†
Received
8th May 2017
, Accepted 28th June 2017
First published on 4th July 2017
Abstract
Monodisperse aromatic N-methyl benzamide-based molecules (repeat number: x = 5–10, molar mass distribution Mw/Mn = 1.02–1.04) connected with terephthalic acid at both sides (MABx–x) were rapidly prepared by a step-wise condensation reaction using the reported one-pot dendrimer synthetic method. These were copolymerized with NH2-terminated poly(propylene oxide) (PPOy) using a condensation reagent at ambient temperature, and the relationship between the structure and the properties was evaluated. The copolymers, poly(MABx–x-b-PPOy), had Mn values up to 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 g mol−1 (Mw/Mn = 1.6), with only a single Tg at −70 °C by DSC, assignable to the pure PPO domain. Only for the copolymer films consisting of a monodisperse MABx–x segment, did the dynamic mechanical analysis (DMA) show two Tg values at −70 and above 50 °C, indicating the clear phase segregation of the copolymers. The mechanical properties of the copolymer films were mostly dependent on the fraction of the monodisperse hard segment, and the tensile modulus and elongation at break varied from 3.3 to 32.5 MPa and 150 to 540%, respectively.
500 g mol−1 (Mw/Mn = 1.6), with only a single Tg at −70 °C by DSC, assignable to the pure PPO domain. Only for the copolymer films consisting of a monodisperse MABx–x segment, did the dynamic mechanical analysis (DMA) show two Tg values at −70 and above 50 °C, indicating the clear phase segregation of the copolymers. The mechanical properties of the copolymer films were mostly dependent on the fraction of the monodisperse hard segment, and the tensile modulus and elongation at break varied from 3.3 to 32.5 MPa and 150 to 540%, respectively.
Introduction
A material prepared of a block copolymer having hard and soft segments (HSs and SSs) in the same chain shows unique properties due to the immiscible polymer segments, and is thus applicable in various fields such as membranes,1–7 photonics,8–12 lithography,12–16 nanocarriers,17,18 and thermoplastic elastomers (TPEs).19 TPEs are currently receiving much attention since they are recyclable and do not require additional chemical cross-linking reactions. The hard domains are strongly governed by physical packing, rather than chemical crosslinking, and are melted or softened at high temperatures. TPEs exhibit elastomeric properties that are comparable to those of chemically cross-linked rubbers, and like conventional thermoplastics, show good processability. Poly(ether-b-amide) multiblock copolymers (PEBA) are a class of important TPE materials due to the tuneable properties with variable polyether SSs and the highly thermostable aliphatic amide segment that realizes the reliable mechanical strength even at high temperatures.20–25 One of the most important application of PEBA can be the gas separation membrane,26–31 in which the crystalline amide HS increases the separation selectivity while the soft poly(ether) segment functions as high gas permeability. The properties of TPE materials are, of course, dependent on the types, length, and the molar mass dispersity of the individual segments. However, in generally speaking, polymers like PEBA are prepared via condensation polymerization, and thus, the lengths of the HSs are not well aligned (broad molar-mass distribution (Mw/Mn) of around 2). On the condensation polymers, Harrell first prepared well-defined non-hydrogen-bonding segmented poly(urethane)s via step-wise reactions.32 The mechanical properties of these polymers were strongly affected by the length and size distribution of the HSs not by SSs. He also found that an HS with one repeat unit only cocrystallized with those containing two repeat units and not with those containing more repeat units. Samuels and Wilkes studied poly(urethane)s using WAXS, where an increasing HS length resulted in higher crystallinity with a thicker lamellar structure.33 Hydrogen-bondable segmented poly(urethane)s based on 4,4′-methylenebis(phenyl isocyanate) (MDI), poly(tetramethylene oxide) (PTMO), and butanediol were then reported by Miller and his co-workers, and found that the polymers having a low Mw/Mn in the HSs showed a smaller degree of phase mixing, and thus, higher strain in the polymers.34 Despite the controllable toughness and the elongation ability by choosing an isocyanate molecule as a proper HS unit, the flow temperature of these polymers is typically from 50 to 150 °C, and they tend to degrade on heating over the melting temperature. Therefore, thermally more stable amide groups can be favored, especially for the high temperature TPE usage. As a novel type PEBA, Gaymans and Van Hutten reported a series of hydrogen-bondable segmented copolymers based on a monodisperse aliphatic- and aromatic-mixed bis(ester amide) as a HS unit. For example, a HS composed of two terephthaloyl with one 1,4-butylene diamino centre, called (T4T) was copolymerized with aliphatic diols. The T4T units (4 indicates the C number in aliphtatic chain) crystallized very fast and almost completely, resulting in a low diamide concentration in the polyether phase.35 The uniform T4T segments formed perfect and stable crystalline lamellae, resulting in a temperature-independent rubbery plateau. They also prepared monodisperse aliphatic- and aromatic-mixed diamide HSs such as T2T,36 T6T,37 and oligoamide HSs of T6T6T and T6T6T6T.38 The tuneable Tm from 111 to 225 °C with the monodisperse HS structure be useful as high-performance TPE materials, and also the materials were applied to gas separation membrane with the excellent CO2 permeability.39 Despite such many useful properties, the synthesis of the hydrogen-bondable HSs with uniform length requires multi-step tedious procedures, which may constrict the industrial application.
Aromatic polyamides, such as poly(p-phenylene terephthalamide) (Kevlar) and poly(benzamide), have attracted extensive attention due to their inherently extended rigid-rod structure, which is caused by the para-linked benzene rings with the double bond character of the trans-amide linkage.40–42 A structural study based on X-ray crystallography, with conformational energy calculations of hydrogen-bondable terephthalamide model compounds, was performed by Flory and his coworker.43 On the other hands, nonhydrogen-bondable N-methylbenzamide was prepared, and the stereo chemistry of the amide bond structure was determined to be cis by X-ray analysis, whereas that of benzanilide was trans.44–47 They also prepared a 1,2-bis(N-benzoyl-N-methylamino)benzene molecule, from which optically active crystals were obtained. The amide linkage was again cis, but the molecule had C2 symmetry.48 Therefore, the circular dichroic spectra of the crystal solutions show positive and negative Cotton effects. Yokozawa and his co-workers prepared non hydrogen-bondable aromatic poly(benzamide) bearing a chiral side chain via condensative chain polymerization (thus, the Mw/Mn was around 1.1) and investigated the structure in solution and in the solid state, where the polymer had a unique one-handed helical structure depending on the chirality.49 We have prepared a series of non hydrogen-bondable poly(N-alkyl benzamide)s [PABAn (n indicates the C number of CnH2n+1 at the side group)] by conventional direct condensation polymerization of p-(N-alkylamino)benzoic acids and elucidated the structures and packing of the PABAn polymers using IR spectroscopy, elemental analysis, DSC, WAXD, and SAXS. The prepared PABA1 and PABAn (where n = 2–5) samples were insoluble in all organic solvents, and the tight packing was assignable to a monoclinic and orthorhombic structure between the main-chain dipolar interaction of the cis amide function. In contrast, while PABA17 also possessed crystalline structures, it crystalized in hexagonal lattices through weak van der Waals interactions of the alkyl side chains.50 Various higher-ordered layered structures of PABAn were also manufactured by the Langmuir Blodgett method, and the color of the interference structure was observed from the differences in the layer thickness of the stepwise multiparticle layers.51–53 For the synthesis of aromatic polyamide dendrimers, we have demonstrated a simple and highly efficient convergent approach without protection and deprotection steps, using thionyl chloride.54 Therefore, utilizing this reliable step-wise technique, we expected that aromatic PABAn molecules with a “perfectly” defined molecular weight can be rapidly prepared and applied to the synthesis of a novel type aromatic-based-PEBA with poly(propylene oxide) (PPO). The nonhydrogen-bondable monodisperse aromatic amide HS would show higher thermostability compared to the aliphatic-based-PEBA material. Moreover, the use of N-methylated aromatic amide HS with a cis configuration would show higher solubility, so that the design and the synthesis of the HS with the desired length be easily attainable. Here, we demonstrate the synthesis and properties of a block copolymer based on oligo(4-N-methyl benzamide) (crystalline molecule) having an almost uniform length with PPO (typical amorphous polymer), using the facile dendrimer synthetic approach.
Results and discussion
Synthesis of aramid molecules
We performed a step-by-step reaction to construct the rigid-rod aramid molecules; the two carboxylic acids at the terminals of the compound were activated in situ with thionyl chloride, followed by condensation with the building block, 4-(methylamino)benzoic acid (MAB), according to the procedure for our previously reported dendrimer synthetic method,54 as shown in Scheme 1.
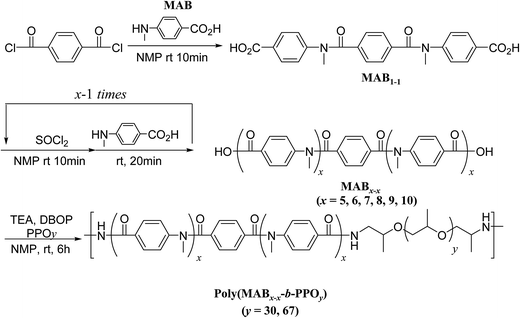 |
| | Scheme 1 Synthesis of the N-methyl poly(p-benzamide)s (MABx–x) and the block copolymerization with PPOy. | |
The highly pure targeted aramid compounds with the desired number of repeating units (MABx–x, where x = 5, 6, 7, 8, 9, and 10) were readily obtained in good yields (77–83%). The aramid molecules are crystalline compounds, and thus, were recrystallized with methanol/water mixed solvent, giving pale brown fine powders. Fig. 1 depicts the 1H NMR spectra of the MABx–x molecules. Along with the protons of the main chain, a–d, the protons at the terminal units, c(t), were observed at 7.85 ppm to have reasonable integration values, which supported the successful formation of the highly pure targeted HS, MABx–x molecule. The 1H NMR spectra also showed that the prepared molecules had cis geometry at the amide function.44
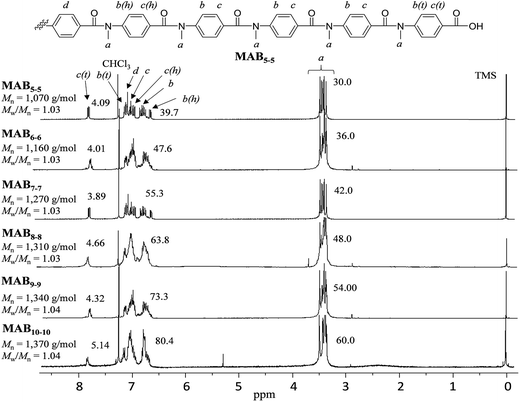 |
| | Fig. 1 1H NMR spectra (CDCl3) of the N-methyl poly(p-benzamide)s (MABx–x) and the molecular-weight data by GPC. | |
The N-methylated aramide molecules were then subjected to the GPC analysis. Fig. 2 depicts typical MABx–x molecules prepared (a) by the step-wise and (b) by the conventional polycondensation methods using terephthalic acid in one-step reaction in NMP. The former MAB5–5 molecule shows the reasonable Mn of 1071 g mol−1 (theoretically 1500 g mol−1) along with the quite narrow Mw/Mn value of 1.03. In contrast, the conventional one-step polycondensation polymer (bMAB5–5) shows the Mn of 1470 g mol−1 with the Mw/Mn of 1.45. However, the MALDI-TOF mass spectrum of the monodisperse MAB5–5 molecule shows that the molecule is not a single one, but still the mixture of MABx–x compounds as shown in Fig. 3. Although the MAB5–5 by the step-wise reaction has some molar-mass distribution, we thought that this still can be useful for the copolymerization with PPO to elucidate the phase segregation and the mechanical properties under the effect of the molar-mass distribution.
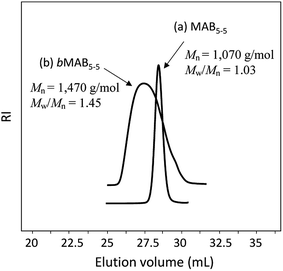 |
| | Fig. 2 GPC profiles of (a) MAB5–5 (Mn = 1070 g mol−1, Mw/Mn = 1.03) and (b) bMAB5–5 (Mn = 1470 g mol−1, Mw/Mn = 1.45). | |
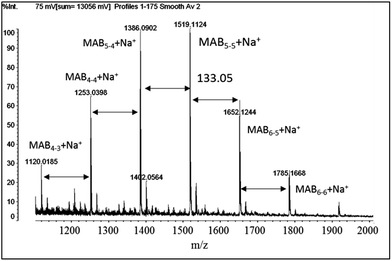 |
| | Fig. 3 MALDI-TOF MS spectrum of MAB5–5 (Mn = 1110 g mol−1, Mw/Mn = 1.03). | |
To investigate the thermal behaviour, we then performed a DSC experiment. Fig. 4 depicts the DSC profiles of MABx–x molecules prepared via step-wise process. In a previous paper, we reported that PABAn (n = 1–5) were all crystalline polymers, whose crystal structures were well assigned by WAXD experiments. However, the melting temperature (Tm) of poly(N-methyl benzamide) (poly(MAB)) was too high to measure with DSC, and only Tg was observed at 200 °C.35 The MABx–x molecules prepared in this study showed a clear thermal transition at around 170–178 °C, and thus, can be assigned as the Tg of the molecules. As expected, these values increased with the length of the MABx–x molecules.
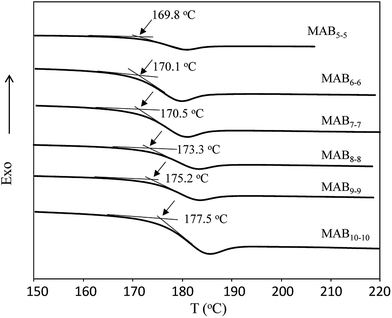 |
| | Fig. 4 DSC profiles of MABx–x samples (heating rate 20 °C min−1, N2). | |
Synthesis of block copolymers of MABx–x with PPOy
As described in Introduction, the mechanical properties of TPEs were primarily affected by the length and size distribution of the HSs, not of the SSs. Therefore, we then copolymerized monodisperse MABx–x with commercially available PPOy, whose Mw/Mn was not well controlled, as shown in Scheme 1. The resultant mixture was poured into water to precipitate the polymer. The viscous pale brown coloured polymer was washed with hot water several times to remove low-molecular-weight impurities.
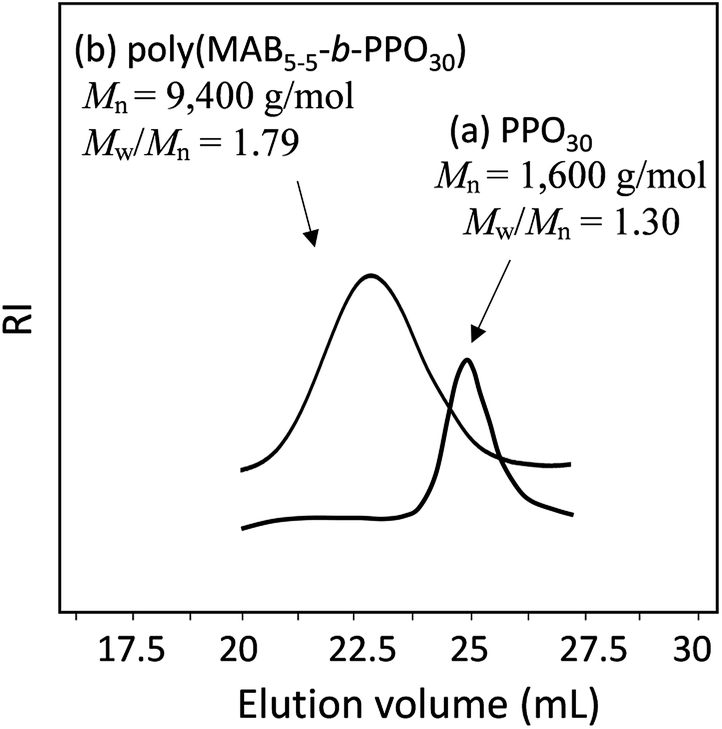
Fig. 5 shows the GPC profiles of (a) PPO30 and the block copolymer (b) poly(MAB5–5-b-PPO30). The unimodal peak of PPO30 was completely shifted into the higher-molecular-weight region, with a reasonable Mw/Mn of 1.79 as a condensation polymer. Fig. 6 depicts the 1H NMR spectra of (a) MAB5–5, (b) PPO30, and (c) poly(MAB5–5-b-PPO30) in CDCl3. In the spectrum of the finally obtained polymer, the signals assignable to MAB5–5 segments a′, b′, c′, and d′ were observed at 3.35–3.72 and 6.75–7.21 ppm in addition to the signals assignable to PPO30 segments f′, g′, and h′ at 3.45–3.75, 3.45–3.20, and 0.93–1.20 ppm, respectively. The signal e′ corresponding to the two-terminal CH3 protons of PPO30 was originally observed at 1.01 ppm, and this was shifted to 1.30 ppm after the formation of the amide bond from the amine functions. The integrations of the terminal e′ protons should not change to be 6.00, and in this case, the integration at around 3.5 ppm became 124.6, in which about 91.6 integration values could be assigned to PPO30 segment f′, g′, and h′ protons, so the 124.6–91.6 = 33.0 integration is assignable to the CH3 protons of the MAB5–5 segment. From these calculations, we can estimate the compositions of MAB5–5 and PPO30 in the copolymer to be 1.1:1.0, which is in good agreement with the feed of the molecules (1.0:1.0). Along with the GPC results, it was obvious that the copolymer, poly(MAB5–5-b-PPO30), was satisfactorily formed with the expected structure by this process.
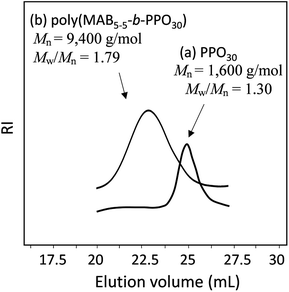 |
| | Fig. 5 GPC profiles of (a) PPO30 (Mn = 1600 g mol−1, Mw/Mn = 1.30) and (b) poly(MAB5–5-b-PPO30) (Mn = 9400 g mol−1, Mw/Mn = 1.79). | |
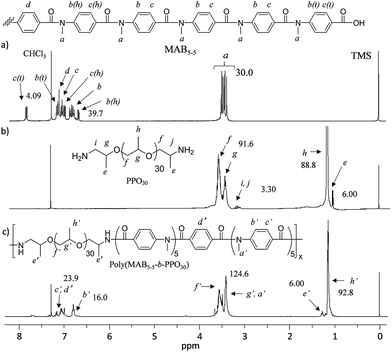 |
| | Fig. 6 1H NMR spectra (CDCl3) of (a) MAB5–5, (b) PPO30, and (c) poly(MAB5–5-b-PPO30). | |
Table 1 summarizes the results of the copolymerization. All polymers were obtained in good yield with Mn values of up to 12500 g mol−1. The morphology of the resulting polymers ranged from viscous sticky materials to hard solids, depending on the weight fraction (fHS) of the aramid molecules. The resulting polymers showed excellent solubility to typical common organic solvents including methanol, acetone, chloroform, tetrahydrofuran, and polar aprotic solvent, N,N-dimethylformamide. Fig. 7 depicts the DSC profiles of the copolymers. All polymers have a clear thermal transition at −70 °C, assignable to the PPO segments, but no other transitions were observed over the temperature range measured.
Table 1 Synthesis of copolymers poly(MABx–x-b-PPOy)a
| Run |
x/y |
Yield (%) |
Mnb (g mol−1) |
Mw/Mnb |
fHSc (%) |
| Conditions; DBOP 1.2 equiv. in 2 mL NMP at 20 °C for 6 h. Determined by GPC (NMP containing 0.01 M LiBr, PSt standards). Weight fraction of HS. DBOP 1.0 equiv. DBOP 1.1 equiv. |
| 1d |
5/30 |
85.1 |
8400 |
1.4 |
44.8 |
| 2e |
5/30 |
84.3 |
8800 |
1.5 |
44.8 |
| 3 |
5/30 |
84.2 |
9400 |
1.8 |
44.8 |
| 4 |
6/30 |
88.8 |
9100 |
2.1 |
48.8 |
| 5 |
6/67 |
83.2 |
11600 |
2.0 |
30.7 |
| 6 |
7/30 |
80.6 |
9400 |
1.8 |
52.2 |
| 7 |
7/67 |
86.0 |
11000 |
1.8 |
33.8 |
| 8 |
8/30 |
70.4 |
8800 |
2.3 |
55.3 |
| 9 |
8/67 |
65.6 |
9000 |
2.2 |
36.5 |
| 10 |
9/30 |
77.0 |
10000 |
2.3 |
57.9 |
| 11 |
9/67 |
73.1 |
9800 |
2.3 |
39.1 |
| 12 |
10/30 |
74.0 |
11000 |
1.6 |
60.3 |
| 13 |
10/67 |
76.0 |
12500 |
1.6 |
41.5 |
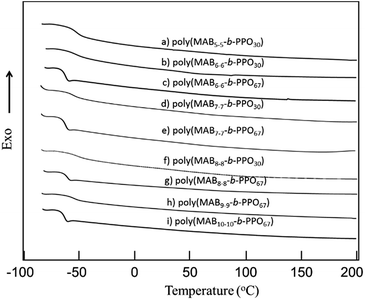 |
| | Fig. 7 DSC profiles (second scan) of (a) poly(MAB5–5-b-PPO30), (b) poly(MAB6–6-b-PPO30), (c) poly(MAB6–6-b-PPO67), (d) poly(MAB7–7-b-PPO30), (e) poly(MAB7–7-b-PPO67), (f) poly(MAB8–8-b-PPO30), (g) poly(MAB8–8-b-PPO67), (h) poly(MAB9–9-b-PPO67), and (i) poly(MAB10–10-b-PPO67) (20 °C min−1, N2). | |
Properties of block copolymers [poly(MABx–x-b-PPOy)] and the influence of Mw/Mn
Fig. 8 depicts the DMA and the tanδ curves of poly(MAB5–5-b-PPO67) (Mn = 8800 g mol−1, Mw/Mn = 2.2) and poly(bMAB5–5-b-PPO67) (Mn = 9400 g mol−1, Mw/Mn = 1.8) prepared with monodisperse (Mw/Mn = 1.03) and polydisperse (Mw/Mn = 1.45) MAB5–5 molecules as HSs, respectively. Even though these finally obtained polymers have similar Mn and Mw/Mn values, the initial storage modulus E′ 4.84 GPa of poly(MAB5–5-b-PPO67) consisting of monodisperse HS was higher than that of poly(bMAB5–5-b-PPO67) film (E′ = 4.15 GPa). The peak intensity of tanδ at −50.7 °C of poly(MAB5–5-b-PPO67) consisting of monodisperse HS was obviously lower than the other sample, and also it has two distinct peaks at −50.7 and 47.9 °C in tanδ, which indicated the existence of more clear phase segregation of the HS block in the copolymer. Indeed, the appearance of poly(MAB5–5-b-PPO67) consisting of monodisperse HS film was opaque and tough, while that of the other one [poly(bMAB5–5-b-PPO67) film] was transparent and soft. These results indicated that in order to make the clear phase-segregated structure in the polymer film consisting of non-hydrogen-bondable N-methylated aromatic poly(p-benzamide), the use of the monodisperse HS block is essential.
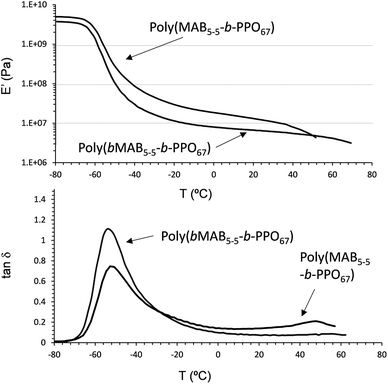 |
| | Fig. 8 DMA analysis of poly(MAB5–5-b-PPO67) having monodisperse HS (Mw/Mn = 1.03) and poly(bMAB5–5-b-PPO67) having polydisperse HS (Mw/Mn = 1.45) (E′ vs. Temp and tanδ vs. Temp plots). | |
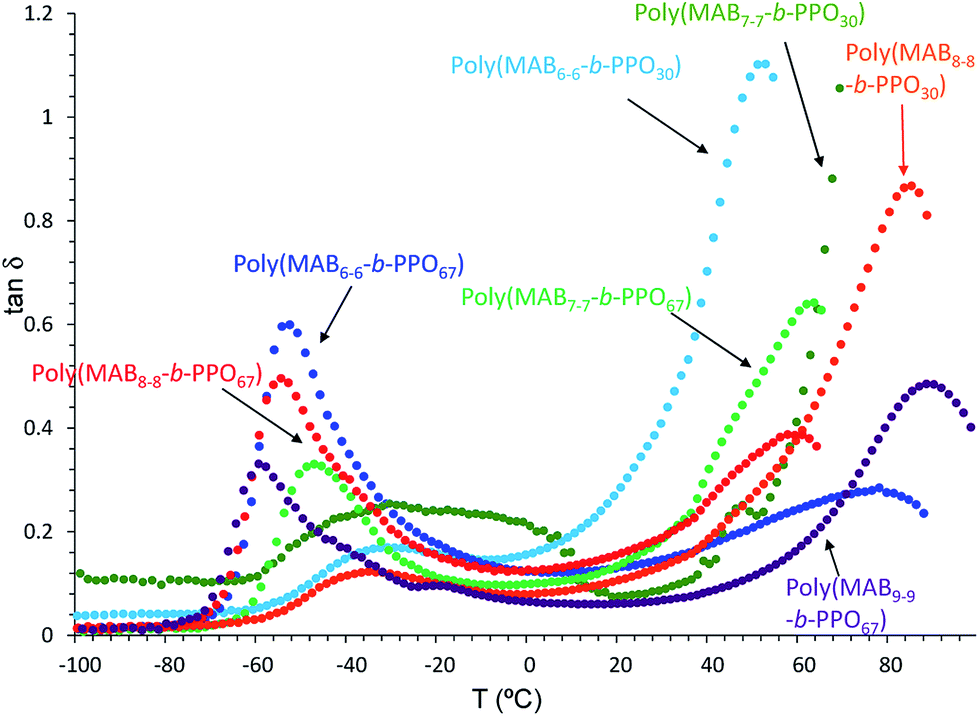
Fig. 9 depicts the relationship between tanδ and the measured temperature of the copolymer films. In contrast to the weak intensity of tanδ peak observed in Fig. 8, more clear peaks were observed above 50 °C for the poly(MABx–x-b-PPOy) (x ≥ 6) samples. For example, poly(MAB6–6-b-PPO30) shows a small and large peak at −30.5 and 53.0 °C, respectively, suggesting the stronger effect of the HS molecules. However, as seen in the DMA curve (Fig. S3†), there is no plateau region. For the polymer having a longer HS, poly(MAB8–8-b-PPO30), the tanδ peak was shifted into higher region. For the polymers having a longer SS, such as poly(MAB6–6-b-PPO67), poly(MAB7–7-b-PPO67), poly(MAB8–8-b-PPO67), and poly(MAB9–9-b-PPO67), all showed a clearer drop in E′ at a lower temperature of −60 °C. In particular, for a poly(MAB9–9-b-PPO67) film, the tanδ peak intensity at −57.7 and 90.3 °C became similar. In the DMA curve, E′ started to decrease at around −71.2 °C and the second drop was observed at from 68.1 °C having clear plateau region between them.
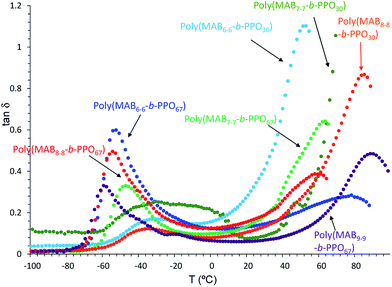 |
| | Fig. 9 Relationship between tanδ and Temp obtained by DMA analysis of poly(MABx–x-b-PPOy) block copolymers. | |
From the data obtained above, we can conclude that the phase-separation conditions of the films are as follows. (A) For the polymers having a short SS, there can be a phase-separated region consisting of SSs significantly contaminated with HSs and HSs containing some quantities of SSs. (B) For the polymers having long SSs with long HSs, the two phases can be separated, and the phase separation should be better for longer HS samples.
To investigate the polymer microstructures in the films, we then performed a WAXD experiment (Fig. 10). In our previous study, we prepared poly(MAB) via conventional polycondensation of MAB with a small amount of terephthalic acid, and the crystal structure was assigned to be monoclinic with parameters a = 7.7 Å, b = 5.7 Å, c = 7.9 Å, α = γ = 90°, and β = 67.5°. The WAXD profile of the polymer powder of poly(MAB5–5-b-PPO30) is depicted in Fig. 10(b), from which the crystal structure was assigned as triclinic with parameters a = 4.6 Å, b = 6.5 Å, c = 7.9 Å, α = 86°, β = 67.5°, and γ = 100°. Note that the crystallized MAB5–5 from a methanol/water mixed solvent did not show a clear diffraction pattern in the WAXD experiment. Thus, we assumed that the HSs cannot align well alone. However, they are placed in a regular manner in the solid-state block copolymer, where the PPO segment (Tg = −68 °C) acted as the solvent even at ambient temperature. In contrast, for the poly(MABx–x-b-PPOy), whose x values range from 6 to 9, all samples showed only amorphous hollows in the profiles. However, the packing was recovered for the poly(MAB10–10-b-PPOy) sample, returning to the crystal structure to be original monoclinic lattice. This can be explained by the polymers having relatively short HSs being unable to crystalize into the stable monoclinic lattice because of the inadequate length of the HSs.
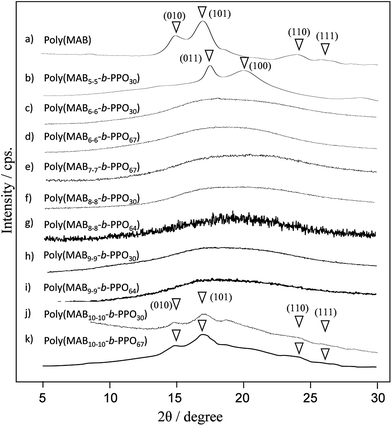 |
| | Fig. 10 Powder WAXD profiles of (a) the MAB homopolymer poly(MABx–y), and (b)-(k) poly(MABx–x-b-PPOy). | |
In order to evaluate the phase segregated structure in more detail, we measured AFM of the poly(MAB5–5-b-PPO67), poly(MAB9–9-b-PPO67) films (Fig. S11†). Unfortunately, not very clear phase-separation was observed in the images. The former sample seems the rough surface with large phase separation in topography, and the other one shows more precise separation. In the prepared films, the phase separation mainly occurred within the films, and was not appeared on the film surface. Since the interaction between the HS units in our system (non-hydrogen bondable) is rather weak compared with those of hydrogen-bondable PEBA, the existence of the longer HS must be critical to form clear phase segregation structure. This resulted in the mechanical properties of the films. Namely, the longer HS unit in the copolymer is the better for the mechanical properties. In other words, the mechanical properties can be slowly changed, and thus we have more opportunity to tune the properties.
Fig. 11 depicts the stress–strain curves of the block copolymer films, and the mechanical properties are summarized in Table 2. Polymer films having short SSs were hard, but they showed an adequate elongation at break (EB) of over 150%. In contrast, the films having long SSs showed much better ductility, and thus, the EB values were over 500%. The tensile modulus (TM) of the films depended on the fraction and length of the HSs; TM values were found to be in the following order: poly(MAB8–8-b-PPO30) (fHS = 55.3%) > poly(MAB6–6-b-PPO30) (fHS = 48.8%) > poly(MAB8–8-b-PPO67) (fHS = 36.5%) > poly(MAB7–7-b-PPO67) (fHS = 33.8%) > poly(MAB6–6-b-PPO67) (fHS = 30.7%). The mechanical strength data in Table 2 are all superior to those of hyper-branched type poly(MABx–x-b-PPOy) in our previous report,55 so the packing of the HS molecules be favored in the linear polymer. The modulus against fHS is plotted in Fig. 12. The hardness of the film is related to the fHS value, but the slope of the fitted line seems different between PPO30- and PPO67-based copolymers. And these plots are not on the same fitted line. As described in Introduction, the use of monodisperse N-methylated aromatic poly(p-benzamide) segment as a novel PEBA material could allow us a fine tuning of the film properties, the results in Fig. 12 supported this assumption. In addition, the HS of the poly(MABx–x-b-PPOy) shows an excellent thermal stability of 5% weight loss temperature in N2 Td5 = 408 °C, while aliphatic amide, nylon 6 shows the value of at 320 °C. The ease of preparation and higher thermostability can be useful of the application of this monodisperse N-methylated aromatic poly(p-benzamide) compound.
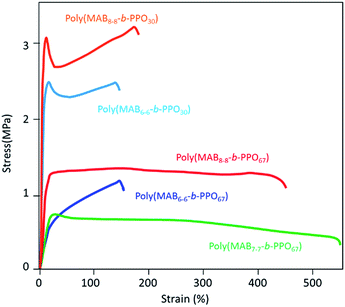 |
| | Fig. 11 Strain–stress curves of multiblock copolymers (poly(MABx–x-b-PPOy)). | |
Table 2 Mechanical properties of poly(MABx–x-b-PPOy)
| Polymer |
TMa (MPa) |
TSb (MPa) |
EBc(%) |
fHS (%) |
| Young's modulus. Tensile strength. Elongation at break. |
| Poly(MAB6–6-b-PPO30) |
28.0 |
2.2 |
150 |
48.8 |
| Poly(MAB6–6-b-PPO67) |
3.3 |
1.7 |
155 |
30.7 |
| Poly(MAB7–7-b-PPO67) |
4.8 |
0.75 |
540 |
33.8 |
| Poly(MAB8–8-b-PPO30) |
32.5 |
3.5 |
185 |
55.3 |
| Poly(MAB8–8-b-PPO67) |
6.0 |
1.6 |
455 |
36.5 |
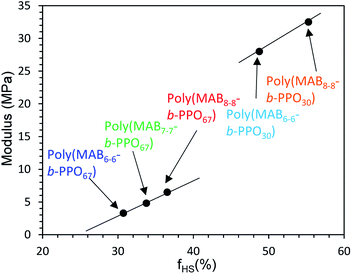 |
| | Fig. 12 Relationship between modulus and strain of poly(MABx–x-b-PPOy). | |
Experimental
Materials
4-(N-Methylamino)benzoic acid (MBA) was purchased from Wako Pure Chemical Industries, Ltd., and purified by sublimation. Poly(propylene glycol) bis(2-aminopropyl ether)s (PPOy) were purchased from Sigma-Aldrich Co. LLC., and the number-average molecular weight (Mn) and degree of polymerization (y) were estimated by 1H NMR measurement. Two samples of PPOy (Mn = 1875 g mol−1, y = 30 and Mn = 4025 g mol−1, y = 67) were used. N-Methylpyrrolidone (NMP) was dried over calcium hydride (CaH2) and distilled under reduced pressure. Thionyl chloride was distilled prior to use. Diphenyl (2,3-dihydro-2-thioxo-3-benzoxazolyl)phosphonate (DBOP) was prepared according to the literature.56 The other reagents and solvents were used without further purification.
Synthesis of low molar mass distribution 4-(N-methyl)benzamide molecules (MABx–x)
Typical procedure (MAB5–5). Terephthalic acid chloride (0.406 g, 2.00 mmol) and NMP (2 mL) were added to a 30 mL two-necked flask equipped with a three-way stopcock and a nitrogen inlet, at 20 °C. The building block, MAB (0.604 g, 4.00 mmol), was added to the mixture, and the reaction was performed at 20 °C for 30 min to give the condensation product MAB1–1. Thionyl chloride (0.604 g, 4.00 mmol) was then added and stirred for 30 min at that temperature (activation). Again, MBA (0.604 g, 4.00 mmol) was added to the mixture to perform the condensation reaction, resulting in the condensation product MAB2–2. This activation and condensation reaction process was repeated another three times, and the resulting solution was poured into distilled water to precipitate the product. The collected light brown powder was recrystallized with methanol/water mixed solvent and dried at 100 °C for 8 h to afford the title compound.
MAB5–5. Yield: 1.24 g (82.8%). 1H NMR (CDCl3, 400 MHz, ppm): δ 3.31–3.72 (m, 30H, –CH3), 6.71–7.36 (m, 40H, –ArH), 7.85 (m, 4H, –ArH). 13C NMR (CDCl3, 100 MHz, ppm): δ 35.5, 120.5, 126.2, 127.5, 129.1, 130.3, 136.9, 141.6, 143.3, 163.7, 172.0. IR (KBr, cm−1): ν 3466 (COOH), 3048 (ArH), 2938 (CH3), 1650 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1602 (CC), 1354 (amide C–N), 1102 (CH3–N). Anal. calcd for (C88H76N10O14): C, 70.58; H, 5.12; N, 9.35%. Found: C, 67.71; H, 5.07; N, 8.95%.
O), 1602 (CC), 1354 (amide C–N), 1102 (CH3–N). Anal. calcd for (C88H76N10O14): C, 70.58; H, 5.12; N, 9.35%. Found: C, 67.71; H, 5.07; N, 8.95%.
MAB6–6. Yield: 82.5%. 1H NMR (CDCl3, 400 MHz, ppm): δ 3.29–3.68 (m, 36H, –CH3), 6.73–7.35 (m, 48H, –ArH), 7.76 (m, 4H, –ArH). 13C NMR (CDCl3, 100 MHz, ppm): δ 34.7, 120.3, 126.0, 127.3, 128.6, 130.0, 137.3, 140.9, 142.8, 163.4, 172.0. IR (KBr, cm−1): ν 3494 (COOH), 3051 (ArH), 2937 (CH3), 1651 (CO), 1603 (CC), 1356 (amide C–N), 1109 (CH3–N).
MAB7–7. Yield: 81.1%. 1H NMR (CDCl3, 400 MHz, ppm): δ 3.31–3.72 (m, 42H, –CH3), 6.71–7.36 (m, 56H, –ArH), 7.75 (m, 4H, –ArH). 13C NMR (CDCl3, 100 MHz, ppm): δ 35.7, 120.5, 126.2, 127.3, 129.0, 130.1, 137.1, 141.6, 143.1, 163.8, 172.1. IR (KBr): ν 3051 (N–H), 2923 (aliphatic C–H), 1645 (CO), 1368 (Ar–N), 1175 (amide C–N) cm−1. Anal. calcd for (C120H104N14O18): C, 70.99; H, 5.16; N, 9.66%. Found: C, 68.51; H, 5.17; N, 9.04%.
MAB8–8. Yield: 78.5%. 1H NMR (CDCl3, 400 MHz, ppm): δ 3.28–3.69 (m, 48H, –CH3), 6.70–7.40 (m, 64H, –ArH), 7.87 (m, 4H, –ArH). 13C NMR (CDCl3, 100 MHz, ppm): δ 35.9, 120.5, 126.2, 126.7, 127.1, 129.2, 130.3, 136.8, 141.6, 142.8, 162.5, 171.8.
MAB9–9. Yield: 76.7%. 1H NMR (CDCl3, 400 MHz, ppm): δ 3.25–3.72 (m, 54H, –CH3), 6.66–7.38 (m, 72H, –ArH), 7.86 (m, 4H, –ArH). 13C NMR (CDCl3, 100 MHz, ppm): δ 35.7, 121.5, 126.5, 127.8, 132.4, 138.1, 143.6, 145.6, 163.8, 171.1, 175.4. IR (KBr, cm−1): ν 3478 (COOH), 3053 (ArH), 2940 (CH3), 1646 (CO), 1602 (CC), 1353 (amide C–N), 1102 (CH3–N).
MAB10–10. Yield: 77.5%. 1H NMR (CDCl3, 400 MHz, ppm): δ 3.22–3.73 (m, 60H, –CH3), 6.61–7.45 (m, 80H, –ArH), 7.75 (m, 4H, –ArH). 13C NMR (CDCl3, 100 MHz, ppm): δ 34.7, 120.3, 127.2, 128.5, 130.1, 132.4, 136.0, 137.1, 141.6, 143.1, 163.8, 172.1, 175.5. IR (KBr, cm−1): ν 3498 (COOH), 3052 (ArH), 2935 (CH3), 1647 (CO), 1602 (CC), 1351 (amide C–N), 1102 (CH3–N). Anal. calcd for (C168H146N20O24): C, 71.32; H, 5.20; N, 9.90%. Found: C, 69.51; H, 5.22; N, 9.36%.
Synthesis of broad molar mass distribution oligo[4-(N-methyl)benzamide] (bMAB5–5)
Terephthalic acid (0.166 g, 1.00 mmol) and MAB (1.510 g, 10.08 mmol) were dissolved with NMP (1 mL) in a 30 mL two-necked flask equipped with a three-way stopcock and a nitrogen inlet at 20 °C. Thionyl chloride (2.32 g, 4.04 mmol) was then added and stirred for 7 h at that temperature. The additional 2 mL NMP was added, and kept stirring for another 7 h. The resulting solution was poured into distilled water to precipitate the product. The collected light brown powder was dried at 100 °C for 10 h to afford the title compound. Yield: 1.335 g (84.2%). 1H NMR (CDCl3, 400 MHz, ppm): δ 3.31–3.72 (m, 30H, –CH3), 4.00–4.90 (br, 9.2H, ArNH(CH3)), 6.71–7.36 (m, 40H, –ArH), 7.91 (m, 4H, –ArH). 13C NMR (CDCl3, 100 MHz, ppm): δ 35.5, 120.5, 126.2, 127.5, 129.1, 130.3, 136.9, 141.6, 143.3, 163.7, 172.0. IR (KBr, cm−1): ν 3466 (COOH), 3048 (ArH), 2938 (CH3), 1650 (CO), 1602 (CC), 1354 (amide C–N), 1102 (CH3–N). Anal. calcd for (C88H76N10O14): C, 70.58; H, 5.12; N, 9.35%. Found: C, 67.71; H, 5.07; N, 8.95%.
Multiblock copolymerization of MABx–x with PPOy (poly(MABx–x-b-PPOy))
The typical procedure for poly(MAB5–5-b-PPO30) was as follows. MAB5–5 (0.375 g, 0.250 mmol), PPO30 (0.469 g, 0.250 mmol), triethylamine (0.0600 g, 0.600 mmol), and NMP (2 mL) were added to a 30 mL two-necked flask equipped with a three-way stopcock and a nitrogen inlet, at 20 °C. DBOP (0.230 g, 0.600 mmol) was added to initiate the polymerization. After stirring at 20 °C for 6 h, the solution was poured into 3 wt% NaHCO3 (aq.). The precipitate was collected and dried under vacuum at 100 °C for 8 h to afford a viscous brown polymeric compound. Yield: 84.2%. 1H NMR (CDCl3, 400 MHz, ppm): δ 1.12–1.26 (m, –CH3), 1.27 (t, –CH3), 3.35–3.72 (m, –CH2, –CH3), 6.75–7.21 (m, –ArH). 13C NMR (CDCl3, 100 MHz, ppm): δ 17.6, 73.0, 73.5, 75.4, 120.4, 123.6, 126.1, 129.3, 129.9. IR (KBr, cm−1): ν 3051 (N–H), 2923 (aliphatic C–H), 1645 (CO), 1368 (Ar–N), 1175 (CH3–N).
Measurements
Fourier transform IR spectra were measured with a Jasco IR-5500 (Jasco Co., Ltd.) by transmittance absorption spectroscopy (KBr tablet method). The molecular mass of polymers [number- and weight-average (Mn and Mw)] was determined with a Tosoh HLC-8120 GPC using a consecutive polystyrene gel column (TSK-GEL GMHHR-M and GMHHR-N) at 40 °C, eluted with NMP containing 0.01 mol L−1 LiBr at a flow rate of 1.0 mL min−1. Nuclear magnetic resonance (NMR) was performed on a Bruker AC-400P spectrometer at 400 MHz for 1H and 100 MHz for 13C measurements. Deuterated chloroform (CDCl3) was used as a solvent with tetramethylsilane as an internal reference. Thermal analyses were performed on a Seiko thermal analyzer (SCC 5200 system) at a heating rate of 10°C min−1 for thermogravimetric analysis (TGA by TG/DTA 320) under air or nitrogen. Differential scanning calorimetry (DSC) was performed on a Shimadzu DSC-60 at a heating rate of 20°C min−1 under nitrogen. Dynamic mechanical analysis (DMA) was performed on a Shimadzu DMS-210 at a heating rate of 20°C min−1 under nitrogen. Polymer films were cut into sizes of 20 × 9 × 0.12 mm3, and a load was applied to the films with a strain amplitude of 5 μm and a frequency of 1 Hz. The modes of several polymers in the crystalline phase were examined by wide-angle X-ray powder diffraction (WAXD) measurements using a Rigaku R-axis Rapid diffractometer equipped with a graphite monochromator, with CuKα radiation at 45 kV and 200 mA. Tapping mode atomic force microscopy (AFM) measurements were performed with a NanoNavi S-image SPM system (Hitachi High-Tech Sci. Co., Tokyo, Japan) using a cantilever with a force constant of approximately 15 N m−1 and a resonance frequency of 120 kHz. The scan size and a tip velocity of the AFM measurements are 10 × 10 μm2 and 200 nm s−1, respectively. Matrix-assisted laser desorption/ionization time-of-flight mass spectra (MALDI-TOF MS) were recorded on an AXIMA Confidence (SHIMADZU Corp., Kyoto, Japan). Dithranol was used as the matrix.
Conclusions
In this study, we prepared N-methyl benzamide-based monodisperse molecules (repeat number: 5–10 × 2) MABx–x for copolymerization with PPOy, characterized the structures, and evaluated the properties. The MABx–x molecules prepared by an authentic step-wise condensation reaction, using the reliable dendrimer synthetic method, exhibited quite narrow molar-mass distribution below 1.04 and the Tg was 170–178 °C, depending on the length of the repeating unit. These rigid-rod molecules were successfully copolymerized with PPOy, giving the copolymer poly(MABx–x-b-PPOy) in good yield with Mn up to 12500 g mol−1 (Mw/Mn = 1.6). As a control, the polydisperse HS (bMAB5–5, Mn 1470, Mw/Mn = 1.45) was prepared, and copolymerized with PPO as well to form poly(bMAB5–5-b-PPO67) (Mn 9400 g mol−1, Mw/Mn = 1.8). Even though these finally obtained block copolymers have similar Mn and Mw/Mn values, the initial storage modulus E′, the peak intensity of tanδ at −50.7 °C, the tanδ peak at 47.9 °C, and the appearance showed large differences. Thus, for the formation of the phase-segregated structure with non-hydrogen-bondable block copolymer system, the use of the monodisperse HS block is essential. The copolymers using monodisperse HS blocks showed a single Tg at −60 °C, assignable to the pure PPO domain by DSC. DMA analysis of the copolymer films clearly showed two Tg values at −60 and above 60 °C, indicating the phase segregation of the copolymers. The mechanical properties of the copolymer films were mainly dependent on the fraction of the hard segment, and the tensile modulus and elongation at break varied from 3.3 to 28.0 MPa and 150 to 540%, respectively.
Acknowledgements
This work was financially supported by the Grant Co-funded by 3NUNT (National Universities of Northern Tohoku), Japan, and by the Iwate University Support Fund for Focused Research and formation of the Centre of Excellence, 2013, and CSTI, SIP (Research and development of innovative manufacture using molecular adhesion technology). We also thank Prof. Hideo Sawada (Hirosaki University) for their valuable discussion on this research. The authors thank Ms Shiduko Nakajo for MALDI-TOF MS measurement.
Notes and references
- A. Asatekin, S. Kang, M. Elimelech and A. M. Mayes, J. Membr. Sci., 2007, 298, 136 CrossRef CAS.
- S. Y. Yang, I. Ryu, H. Y. Kim, J. K. Kim, S. K. Jang and T. P. Russell, Adv. Mater., 2006, 18, 709–712 CrossRef CAS.
- S. Y. Yang, J. Park, J. Yoon, M. Ree, S. K. Jang and J. K. Kim, Adv. Funct. Mater., 2008, 18, 1371–1377 CrossRef CAS.
- W. A. Phillip, M. Amendt, B. O'neill, L. Chen, M. A. Hillmyer and E. L. Cussler, ACS Appl. Mater. Interfaces, 2009, 1, 472–480 CAS.
- E. E. Nuxoll, M. A. Hillmyer, R. Wang, C. Leighton and R. A. Siegel, ACS Appl. Mater. Interfaces, 2009, 1, 888–893 CAS.
- K. Peinemann, V. Abetz and P. F. W. Simon, Nat. Mater., 2007, 6, 992–996 CrossRef CAS PubMed.
- H. Uehara, M. Kakiage, M. Sekiya, D. Sakuma, T. Yamonobe, N. Takano, A. Barraud, E. Meurville and P. Ryser, ACS Nano, 2009, 3, 924–932 CrossRef CAS PubMed.
- A. Urbas, R. Sharp, Y. Fink, E. L. Thomas, M. Xenidou and L. J. Fetters, Adv. Mater., 2000, 12, 812–814 CrossRef CAS.
- M. R. Bockstaller, Y. Lapetnikov, S. Margel and E. L. Thomas, J. Am. Chem. Soc., 2003, 125, 5276–5277 CrossRef CAS PubMed.
- J. Yoon, R. T. Mathers, G. W. Coates and E. L. Thomas, Macromolecules, 2006, 39, 1913–1919 CrossRef CAS.
- C. Osuji, C.-Y. Chao, I. Bita, C. K. Ober and E. L. Thomas, Adv. Funct. Mater., 2002, 12, 753–758 CrossRef CAS.
- J. Yoon, W. Lee and E. L. Thomas, Nano Lett., 2006, 6, 2211–2214 CrossRef CAS PubMed.
- R. A. Segalman, H. Yokoyama and E. J. Kramer, Adv. Mater., 2001, 13, 1152–1155 CrossRef CAS.
- D. Sundrani, S. B. Darling and S. J. Sibener, Nano Lett., 2004, 4, 273–276 CrossRef CAS.
- T. L. Morkved, M. Lu, A. M. Urbas, E. E. Ehrichs, H. M. Jaeger, P. Mansky and T. P. Russell, Science, 1996, 273, 931–933 CAS.
- C. D. Rosa, C. Park, E. L. Thomas and B. Lotz, Nature, 2000, 405, 433–436 CrossRef PubMed.
- K. Kataoka, A. Harada and Y. Nagasaki, Adv. Drug Delivery Rev., 2001, 47, 113–131 CrossRef CAS PubMed.
- A. Rösler, G. W. M. Vandermeulen and H.-A. Klok, Adv. Drug Delivery Rev., 2012, 64, 270–279 CrossRef.
- R. J. Gaymans, Prog. Polym. Sci., 2011, 36, 713–748 CrossRef CAS.
- G. Rabani, G. M. Rosair and A. Kraft, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 1449–1460 CrossRef CAS.
- M. Peyravi, A. A. Babaluo, M. A. Ardestani, M. K. R. Aghjeh, S. R. Pishghadam and P. Hadi, J. Appl. Polym. Sci., 2010, 118, 1211–1218 CAS.
- J. F. Feller and Y. Grohens, Sens. Actuators, B, 2004, 97, 231–242 CrossRef CAS.
- C. Yi. Z. Peng, H. Wang, M. Li and C. Wang, Polym. Int., 2011, 60, 1728–1736 CrossRef.
- I. K. Yang and P. H. Tsai, J. Polym. Sci., Part B: Polym. Phys., 2005, 43, 2557–2567 CrossRef CAS.
- M. Van der Schuur, J. D. Boer and R. Gaymans, Polymer, 2005, 46, 9243–9256 CrossRef CAS.
- C. Y. Yeong and H. J. Won, J. Appl. Polym. Sci., 1994, 54, 585–591 CrossRef.
- C. Y. Yeong and H. J. Won, J. Appl. Polym. Sci., 1995, 56, 895–904 CrossRef.
- L. Liu, A. Chakma and X. Feng, J. Membr. Sci., 2004, 235, 43–52 CrossRef CAS.
- L. Liu, A. Chakma and X. Feng, Chem. Eng. Sci., 2006, 61, 6142–6153 CrossRef CAS.
- A. Gugliuzza and E. Drioli, Polymer, 2003, 44, 2149–2157 CrossRef CAS.
- S. Sridhar, R. Suryamurali, B. Smitha and T. M. Aminabhavi, Colloids Surf., A, 2007, 297, 267–274 CrossRef CAS.
- L. J. Harrell, Macromolecules, 1969, 2, 607–612 CrossRef CAS.
- G. L. Wilkes, S. L. Samuels and R. Crystal, J. Macromol. Sci., Part B: Phys., 1974, 10, 203–229 CrossRef CAS.
- J. A. Miller, S. B. Lin, K. K. S. Hwang, K. S. Wu, P. E. Gibson and S. L. Cooper, Macromolecules, 1985, 18, 32–44 CrossRef CAS.
- R. J. Gaymans and J. L. de Haan, Polymer, 1993, 34, 4360–4364 CrossRef CAS.
- K. Bouma, G. A. Wester and R. J. Gaymans, Polym. Sci. Eng, 2001, 41, 1173–1180 Search PubMed.
- G. Perego, M. Cesari, G. Della Fortuna and J. St, Appl. Polym. Sci., 1984, 29, 1141–1155 CrossRef CAS.
- J. M. Van der Schuur and R. J. Gaymans, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 4769–4781 CrossRef.
- D. Husken, T. Visser, M. Wessling and R. J. Gaymans, J. Membr. Sci., 2010, 346, 194–201 CrossRef CAS.
- P. W. Morgan, Macromolecules, 1977, 10, 1381–1390 CrossRef CAS.
- S. L. Kwolek, P. W. Morgan, J. R. Schaefgen and L. W. Gulrich, Macromolecules, 1977, 10, 1390–1396 CrossRef CAS.
- T. I. Bair, P. W. Morgan and F. L. Killian, Macromolecules, 1977, 10, 1396–1400 CrossRef CAS.
- J. P. Hummel and P. J. Flory, Macromolecules, 1980, 13, 479–484 CrossRef CAS.
- A. Itai, Y. Toriumi, N. Tomioka, H. Kagechika, I. Azumaya and K. Shudo, Tetrahedron Lett., 1989, 30, 6177–6180 CrossRef CAS.
- I. Azumaya, H. Kagechika, Y. Fujiwara, M. Itoh, K. Yamaguchi and K. Shudo, J. Am. Chem. Soc., 1991, 113, 2833–2838 CrossRef CAS.
- K. Yamaguchi, G. Matsumura, H. Kagechika, I. Azumaya, Y. Ito, A. Itai and K. Shudo, J. Am. Chem. Soc., 1991, 113, 5474–5475 CrossRef CAS.
- A. Itai, Y. Toriumi, S. Saito, H. Kagechika and K. Shudo, J. Am. Chem. Soc., 1992, 114, 10649–10650 CrossRef CAS.
- I. Azumaya, I. Yamaguchi, Y. Okamoto, H. Kagechika and K. Shudo, J. Am. Chem. Soc., 1995, 117, 9083–9084 CrossRef CAS.
- A. Tanatani, A. Yokoyama, I. Azumaya, Y. Takakura, C. Mitsui, S. Motoo, M. Uchiyama, A. Muranaka, N. Kobayashi and T. Yokozawa, J. Am. Chem. Soc., 2005, 127, 8553–8561 CrossRef CAS PubMed.
- Y. Shibasaki, Y. Abe, N. Sato, A. Fujimori and Y. Oishi, Polym. J., 2010, 42, 72–80 CrossRef CAS.
- S. Chiba, S. Arai, Y. Kaneko, M. Taguchi, Y. Shibasaki and A. Fujimori, Trans. Mater. Res. Soc. Jpn., 2012, 37, 283–286 CrossRef CAS.
- A. Fujimori, Y. Kaneko, T. Kikkawa, S. Chiba and Y. Shibasaki, J. Colloid Interface Sci., 2014, 418, 338–349 CrossRef CAS PubMed.
- A. Fujimori, S. Miura, T. Kikkawa and Y. Shibasaki, J. Polym. Sci., Part B: Polym. Phys., 2014, 418, 338–349 CAS.
- I. Washio, Y. Shibasaki and M. Ueda, Macromolecules, 2005, 38, 2237–2246 CrossRef CAS.
- S. Masukawa, T. Kikkawa, A. Fujimori, Y. Oishi and Y. Shibasaki, Chem. Lett., 2015, 44, 536–538 CrossRef CAS.
- M. Ueda, M. Katayama, T. Morosumi and R. Sato, Polym. J., 1991, 23, 167–176 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra05161a |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *a
*a