DOI:
10.1039/C7RA05258H
(Paper)
RSC Adv., 2017,
7, 29645-29655
Surface modification using crosslinking of diamine and a bis(diarylcarbene): synthesis, characterization, and antibacterial activity via binding hydrogen peroxide†
Received
9th May 2017
, Accepted 31st May 2017
First published on 7th June 2017
Abstract
A bis(diaryldiazomethane) substituted with amino groups was synthesized and used for the surface modification of diverse materials, such as polystyrene, polyethylene, titanium dioxide, and aluminum plate, giving amino-functionalized materials. These were subsequently reacted with phenyl isocyanate to give urea-functionalized materials, so that hydrogen peroxide could be reversibly bound, giving in turn antibacterial materials with high activity. These functionalized materials were characterized using a combination of XPS, IR, and NMR spectroscopy, and clear evidence for the crosslinking on the surface of materials was achieved. The crosslinking reaction in the deposited layer contributed to higher loadings of hydrogen peroxide, and hence the antibacterial activity of urea-functionalized materials significantly increased. This approach gives a new protocol to prepare high loading antiseptic/antibacterial materials and surfaces.
1. Introduction
Antibacterial materials and surfaces have been widely used1,2 and have become increasingly important to avoid nosocomial infections because of the rise of antibiotic-resistant and virulent bacteria.3,4 A great deal of research has created diverse antiseptic/antibacterial materials and surfaces by the incorporation of biostats/biocides, such as nanosilvers,5,6 zinc oxides,7,8 tetralkyl ammoniums,9,10 chitins/chitosans,11,12 and N-halamines.13,14 The action of hydrogen peroxide has been known in food chemistry for some time,15 and antibodies are known to produce hydrogen peroxide when irradiated in the presence of oxygen,16,17 so that reactive oxygen species are capable of exerting an immediate and effective bactericidal effect.18,19 However, hydrogen peroxide is often overlooked for the preparation of antibacterial materials and surfaces, probably due to its instability, which cannot easily give long-term antibacterial activity to materials or surfaces. In fact, ureas are capable of binding hydrogen peroxide, and a stabilized form, urea hydrogen peroxide (UHP), is a very common type of catalyst for oxidation,20,21 which also been used for fuel cells.22,23 The stability of UHP lies in its extensive network of hydrogen bonding and if a similar immobilization of hydrogen peroxide might be achieved on surfaces, it could be used to prepare antibacterial materials.24,25 We have reported that it is possible to incorporate hydrogen peroxide binding and release behaviour into polystyrenes (PS) by a post-polymerization modification.26 However, it was less successful with very inert surfaces with low surface energy, such as polyethylene (PE) and polytetrafluoroethylene (PTFE), since only an incomplete monolayer coverage was achieved on those surfaces, which leads to low binding of hydrogen peroxide and therefore very poor antibacterial activity.26
In this work, a new bis(diaryldiazomethane) with two terminal amino groups is reported, along with its application for the modification of various substrates, including PS bead, PE powder, titanium dioxide powder, and aluminum plate. An unexpected crosslinking between those diamines and the dicarbene intermediate was detected, giving a structurally robust surface which improved the loading of hydrogen peroxide and greatly increased the antibacterial activity of materials or surfaces. Moreover, this crosslinking expands the surface modification approach, which has already been applied in colour,27 fluorescence,28 biocompatibility,29 protein binding30 and adsorption,31 hydrophobicity,32 and payload delivery.33,34
2. Results and discussion
2.1 Synthesis and characterization of bis(diazomethane)
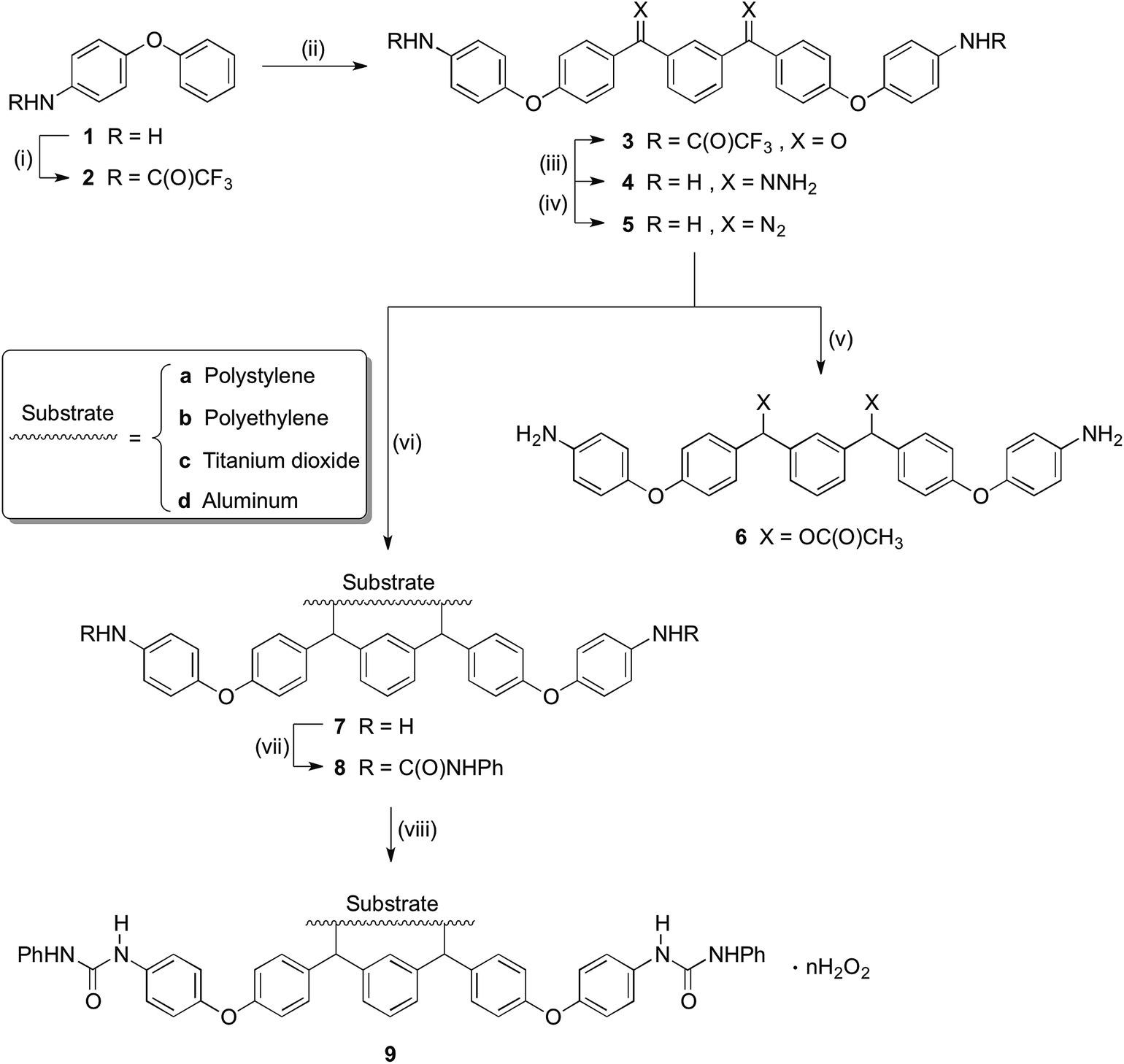
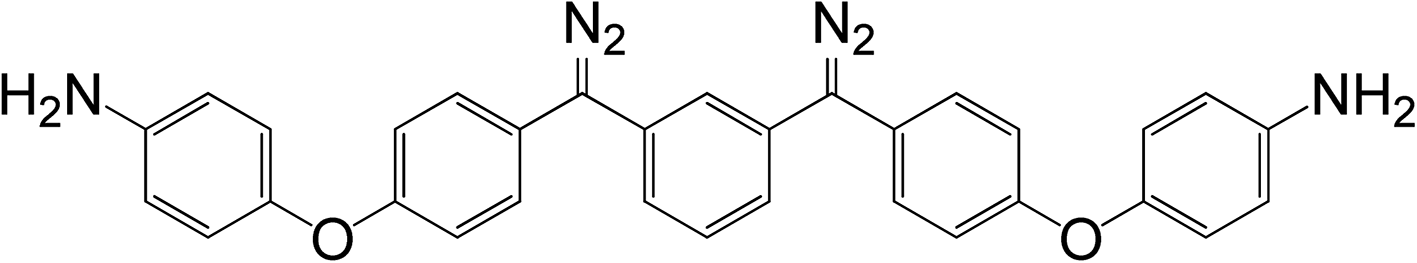
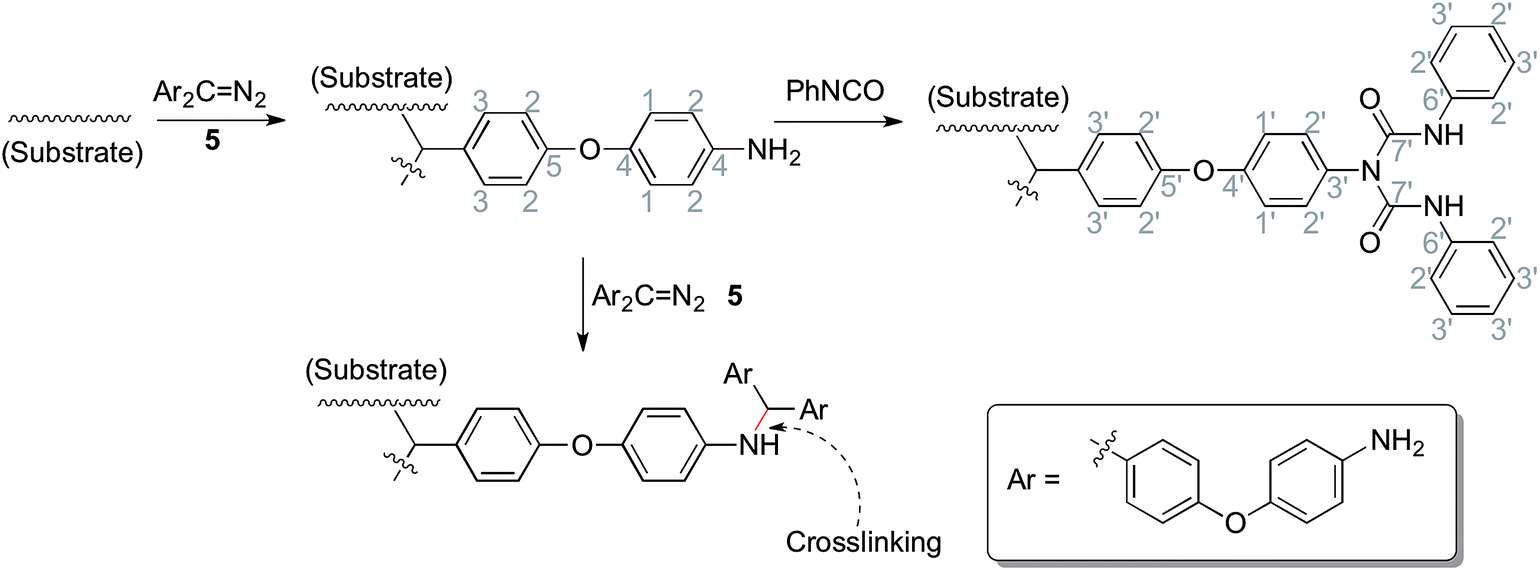
As shown in Scheme 1, trifluoroacetic anhydride was used to protect the amino group in 4-phenoxyaniline 1 to give amide 2, which was subsequently converted by Friedel–Crafts reaction to bis(arylketone) 3 in relatively high yield.36 The bis(arylketone) 3 was treated with hydrazine monohydrate to generate bis(hydrazone) 4, which was subsequently oxidized with manganese dioxide to obtain bis(diaryldiazomethane) 5. This protocol is noteworthy for two reasons. Firstly, it has been reported that amines are not active enough to react with diaryldiazomethanes at room temperature,37 but easily react with carbenes through the N–H insertion reaction at higher temperature; this reactivity difference permits the synthesis and storage of amino-substituted diazomethane 5 at room temperature, which is required for its convenient application as a surface modifying agent, but also for its participation in carbene-amine crosslinking reaction upon heating. Thus, unlike many diaryldiazomethanes, which are not very stable, the characterization of diazomethane 5 by 13C NMR spectra was possible. Secondly, bis(hydrazone) 4 contains unprotected terminal amino groups, but the use of manganese dioxide as oxidant permits a very mild, chemoselective oxidation of the hydrazone to the diazo compound 5,38–40 even in the presence of the free amine. The greater stability of these systems compared to the diazo compound derived from Michler's ketone is noteworthy.26
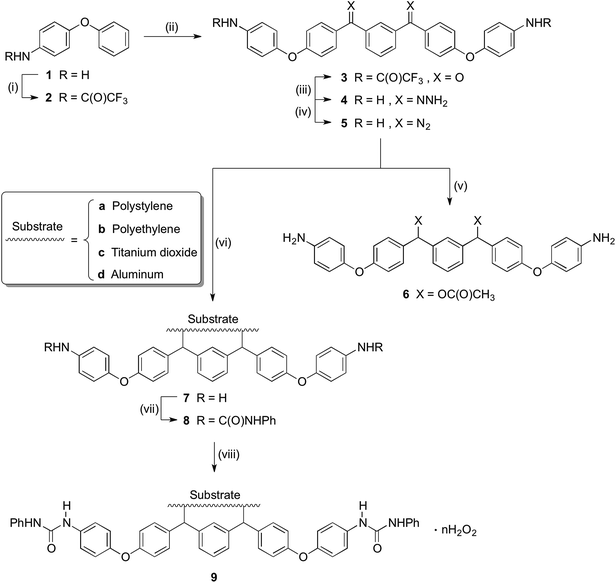 |
| | Scheme 1 Synthesis of precursor and modification of materials. Conditions: (i) TFAA, pyridine, DCM, 5 °C-r.t., overnight (96%); (ii) isophthaloyl chloride, AlCl3, DCM, 5 °C-r.t., 2h (89%); (iii) NH2NH2, H2O, HAc, EtOH, reflux, 40h (87%); (iv) MnO2, Na2SO4, KOH, DCM, r.t., 5 h (89%); (v) HOAc, DCM, r.t., 20 minutes (68%); (vi) Substrate, 120 °C; (vii) Triethylamine, PhNCO, acetone, 56 °C, 2 h (or r.t., overnight); (viii) 30%(m/m) H2O2, r.t., overnight. | |
2.2 Kinetic studies
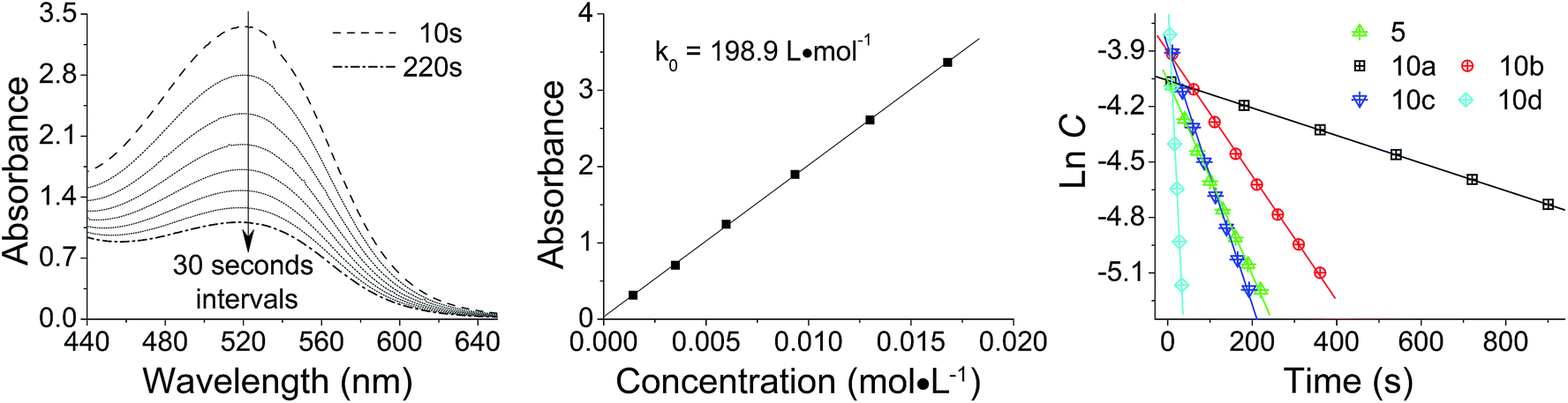
The reactivity of diazomethane 5 was assessed before being used for surface modification, in order to confirm that both diazo functions were capable of reacting at a suitable rate. Therefore, acetic acid was reacted with 5 to form ester 6, which was isolated and characterized as the diester by IR, NMR, and MS techniques. Moreover, the reaction rate of 5 with acetic acid was compared with several other diazomethanes. In Fig. 1(1), clear changes of UV spectra at 527 nm appeared after acetic acid was added, and that peak was used for kinetic studies. The Beer–Lambert law was used to determine the linear relationship between absorbance (A) and concentration (C) in UV spectra (Fig. 1(2)), giving a value of k0 (=198.9 L mol−1) in eqn (1).
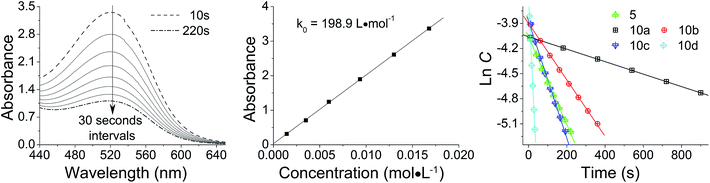 |
| | Fig. 1 Reaction kinetics of various diazomethanes with acetic acid: (1) absorbance (A) versus reaction time (t) curve for diazomethane 5 at 527 nm; (2) calibration from absorbance (A) to concentration (C); (3) ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) C versus t plot for various diazomethanes. C versus t plot for various diazomethanes. | |
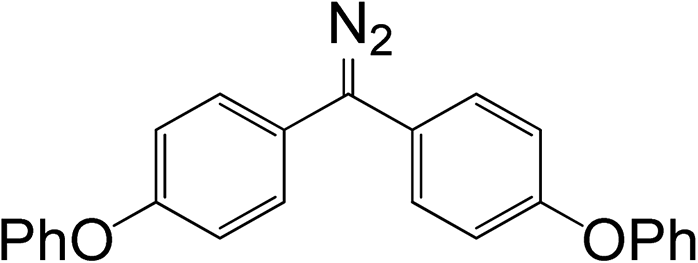
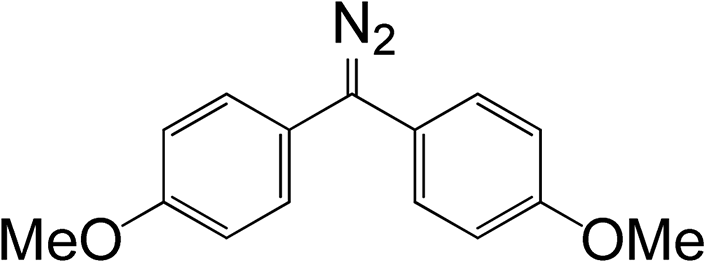
Because the amount of acetic acid was in large excess over that of diazomethane, the conversion of the diazo group could be given by the pseudo first-order expression. Thus, the relationship between lnC and t for different diazomethanes was plotted to obtain the rate constants (Fig. 1(3)). Detailed data are shown in Table 1, as well as the peak position of the diazo group in UV and IR spectra. From this data, it is clearly evident that reaction rates can differ by as much as 200-fold, and that the more reactive diazomethanes have the larger wavelength of diazo group in both UV and IR spectra. Significantly, compound 5 is intermediate in reactivity, giving a good balance of stability and reactivity, which is desirable for a reagent to be used “off the shelf” as a surface modifying agent.
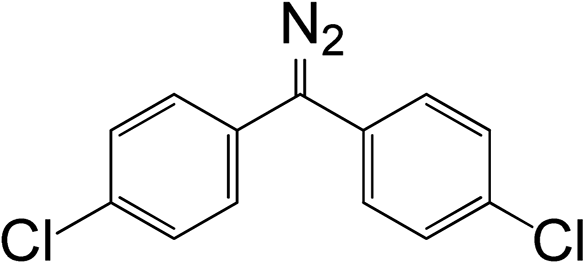
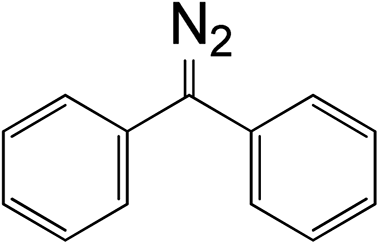
Table 1 Relationships between chemical structure, rate constant, and diazo group peak position in UV and IR spectra
| Compound |
Structure |
Rate constant (s−1) |
UV wavelength (nm) |
IR wavenumber (cm−1) |
|
10a
|
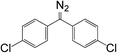
|
7.45 × 10−4 |
521 |
2039.2 |
|
10b
|
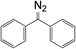
|
3.37 × 10−3 |
523 |
2036.5 |
|
5
|
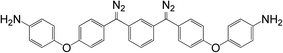
|
5.25 × 10−3 |
527 |
2030.5 |
|
10c
|
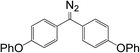
|
6.98 × 10−3 |
535 |
2028.6 |
|
10d
|
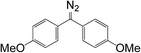
|
4.28 × 10−2 |
556 |
2028.1 |
2.3 Preparation of materials
In order to allow rapid access to functionalized materials, we adapted our previously published approach for surface modification using carbenes.41 This is a convenient and efficient method to introduce diverse chemical functionality onto a solid substrate (Scheme 1). Firstly, each of polystyrene (XAD4), polyethylene or titanium dioxide was carefully mixed with bis(diazomethane) 5 and heated at 120 °C, which formed amino-functionalized polystyrene 7a, polyethylene 7b or titanium dioxide 7c. The amino-functionalized aluminium plate 7d was made by directly heating a plate which had been covered by a solution of bis(diazomethane) 5 before careful solvent removal. In this step, carbenes are generated from the precursor diazomethanes, and their insertion reactions with C–H bonds (on the surface of polystyrene and polyethylene) or O–H bonds (on the surface of titanium dioxide and aluminum) give the modified materials 7a–d. It is interesting that materials 7a–d were a yellow colour after being modified by the diazomethane, consistent with the introduction of aromatic residues, which was very different from the unmodified materials. This immediately suggested that the modification had been successful, but in order to provide additional confirmation, ninhydrin was used to dye materials 7a–d, giving the expected change of color occurred from yellow to brown as a result of the well-known chromogenic reaction which is used as a qualitative test for amino groups.42 The color changes of polyethylene after modification and dyeing are shown in Fig. 2.
 |
| | Fig. 2 Color changes of modified and dyed polyethylenes: (1) blank; (2) amino – functionalized; (3) ninhydrin – dyed. | |
Phenyl isocyanate was reacted with these amino-functionalized materials 7a–d to give urea-functionalized materials 8a–d, and these materials were immersed into hydrogen peroxide aq. solution to give H2O2-loaded materials 9a–d.
2.4 XPS analysis
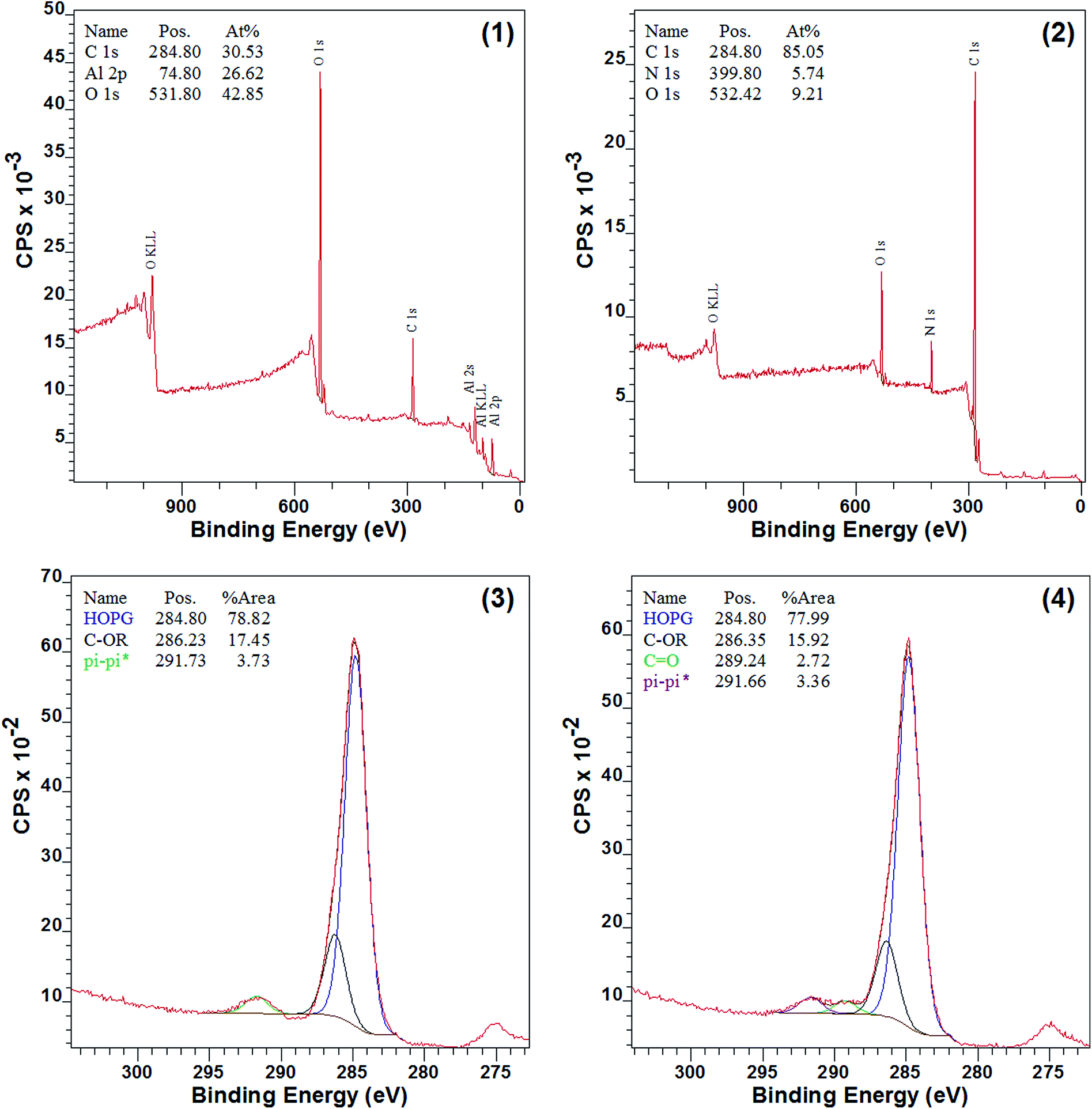
The surface chemical composition of 7a–d and 8a–d should be very different before and after the modification, since nitrogen was introduced, and XPS spectra of Al sheet, 7d, and 8d were obtained (Fig. 3). As shown in Fig. 3(2), nitrogen appeared and aluminum disappeared in 7d when compared to the pure Al sheet (Fig. 3(1)), which indicated that the surface modification by diazomethane 5 was successful. Moreover, as shown in Fig. 3(3), there were three types of carbon on the surface of 7d: HO–C (285 eV), C-OR (286 eV), and π–π* (292 eV), and this is consistent with surface modification by compound 5. The carbon of C-OR confirmed the reaction of carbene with hydroxyls on the surface of aluminum, and the carbon of π–π* showed the appearance of benzene ring, both of which indicated the success of modification. After 7d reacted with phenyl isocyanate, another type of carbon, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (289 eV), appeared on the surface of 8d (Fig. 3(4)). This carbon of CO belonged to the urea groups and indicated the success of reaction with isocyanate.
O (289 eV), appeared on the surface of 8d (Fig. 3(4)). This carbon of CO belonged to the urea groups and indicated the success of reaction with isocyanate.
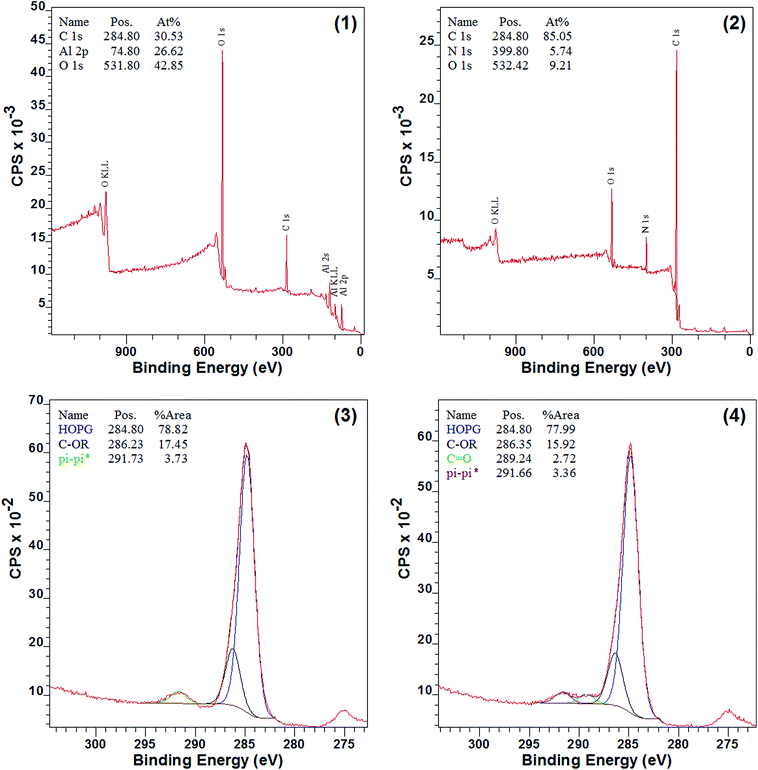 |
| | Fig. 3 XPS spectra before and after modification: (1) survey spectra of pure Al sheet; (2) survey spectra of 7d; (3) C 1s spectra of 7d; (4) C 1s spectra of 8d. | |
Detailed results are shown in Table 2, and these suggest that the modified surface layer of materials 7a–c is likely to be very thin because, for example, the titanium(IV) in TiO2 was found both before and after modification. However, the element aluminum in Al sheet could not be found after modification, consistent with a thicker surface loading, and this suggests that the size and shape of materials might influence the modification process. In addition, the detected nitrogen data for the polyethylene was lower than other materials, and it indicates that the polyethylene is intrinsically less reactive, and is a difficult substrate for modification; possible reasons for this have been previously discussed.26
Table 2 Chemical composition determined by XPS before and after modification
| Materials |
Detected dataa (%) |
Materials |
Detected dataa (%) |
Materials |
Detected dataa (%) |
|
Ratio taken from nitrogen, carbon, oxygen, titanium and aluminum counts in a survey spectrum.
(—) not determined.
Aluminum cannot be detected.
|
| XAD4 beads |
N 6.3, C 80.5, O 13.19 |
7a
|
N 7.0, C 79.6, O 13.4 |
8a
|
N 7.7, C 78.3, O 14.1 |
| PE powder |
—b |
7b
|
N 0.9, C 91.6, O 7.6 |
8b
|
N 2.3, C 90.0, O 7.6 |
| TiO2 powder |
—b |
7c
|
N 2.4, C 76.1, O 18.9 Ti 2.6 |
8c
|
N 2.8, C 76.9, O 19.0 Ti 1.3 |
| Al sheet |
C 30.5, O 42.9, Al 26.6 |
7d
|
N 5.7, C 85.1, O 9.2c |
8d
|
N 8.8, C 85.3, O 8.9c |
2.5 IR analysis
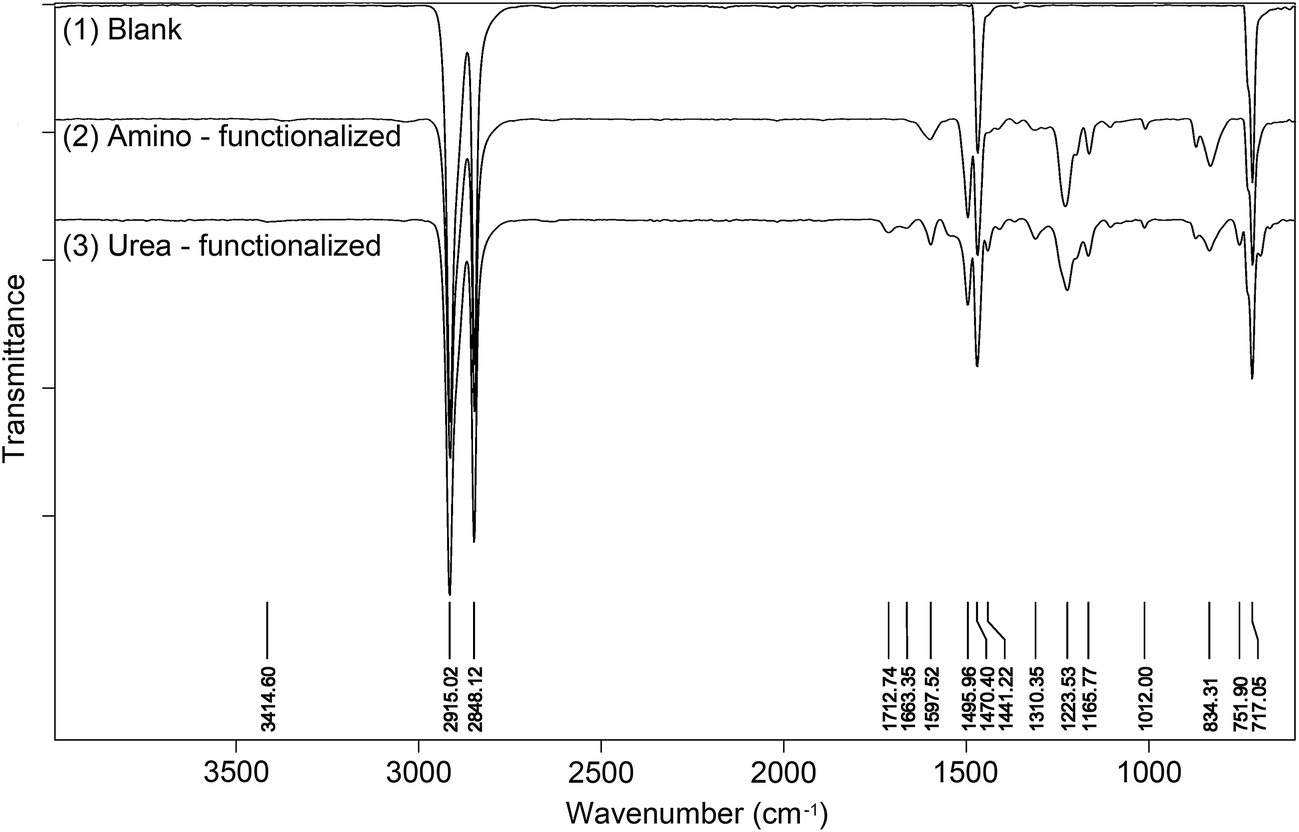
The surface modification with bis(diaryldiazomethanes) introduced aromatic rings onto the surface of materials, which should be detectable by ATR-IR spectroscopy. Fig. 4(1) is the unmodified polyethylene, from which the spectra only display four peaks attributed to the alkyl chain. Fig. 4(2) is the amino-functionalized polyethylene 7b, in which it can be seen that there are several new peaks which have appeared after polyethylene was modified by the bisdiazomethane. The characteristic peaks in the regions of 1598 and 1496 cm−1 belong to the CC double bond in the aromatic ring, and the peak in the region of 1224 cm−1 belongs to the C–O bond in the aryl ether, all of which indicate the successful modification of polyethylene. Subsequent reaction of amine-modified surface with phenyl isocyanate gave new urea groups, which would be expected to show a characteristic vibration band of CO double bonds in the region of 1710–1650 cm−1. In addition, the subsequent reaction of the generated urea groups with excess phenyl isocyanate would give carbamyl urea groups and generate another peak in the region of 1720–1690 cm−1.43,44Fig. 3(3) shows the IR spectra of urea-functionalized polyethylene, and as is expected, two clear peaks in the regions of 1713 and 1663 cm−1 appear (Fig. 4(3)). In fact, other materials show the same changes (see the spectra in ESI†).
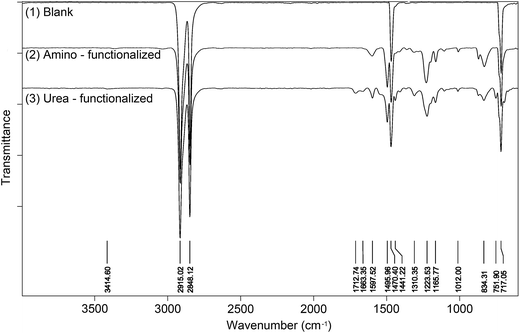 |
| | Fig. 4 IR spectra of blank, amino – functionalized and urea – functionalized polyethylenes. | |
2.6 Solid state NMR analysis
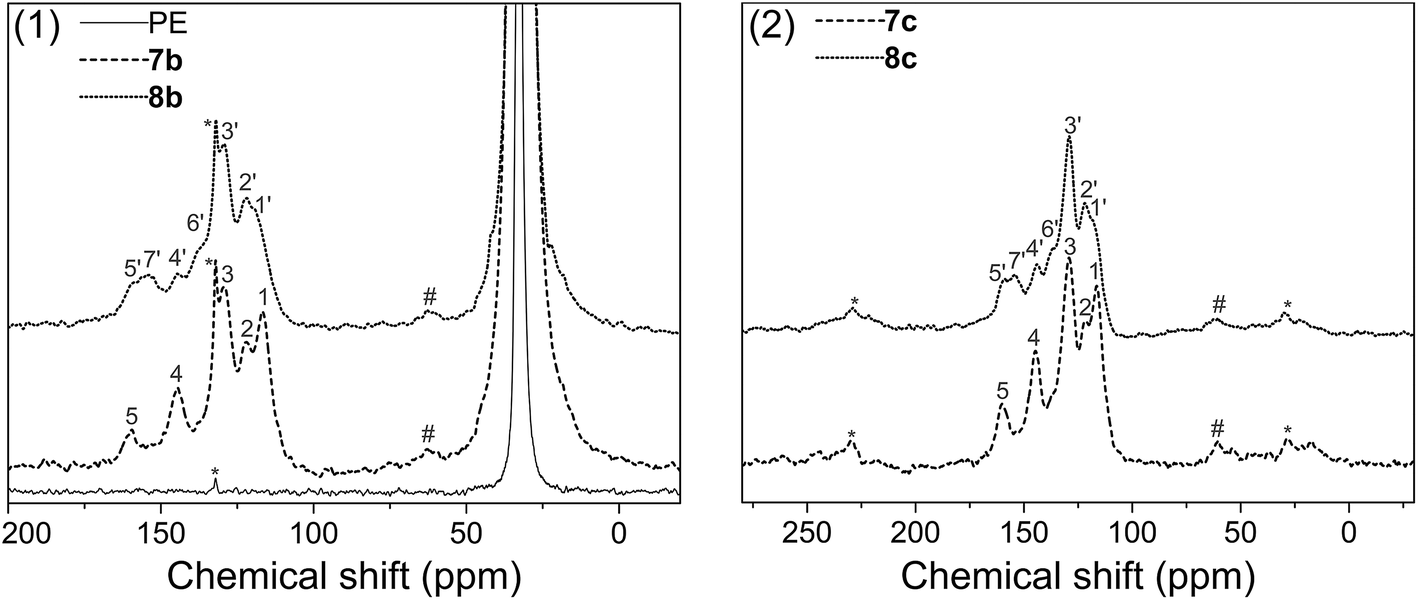
Solid state 13C NMR with CP/MAS was used to further assess the surface structure, since the materials could not be dissolved in any deuterated solvent before or after modification. Fig. 5(1) gives the spectra of pure polyethylene (PE), amino-functionalized polyethylene 7b and urea-functionalized polyethylene 8b, and Fig. 5(2) gives the spectra of amino-functionalized titanium dioxide 7c and urea-functionalized titanium dioxide 8c, respectively (for carbon assignments of Fig. 5, see Scheme 2). After the materials were modified by the bis(diazomethane) 5, aryl rings and amino groups were introduced onto their surface. As a result, five peaks appeared clearly in the spectra of 7b and 7c, which were attributed to the carbons 1–5 (Scheme 2). After 7b or 7c reacted with phenyl isocyanate to give 8b or 8c, additional peaks appeared, which were attributed to the carbons 1′–7′ (Scheme 2). A clear peak (7′) arose in the region of 154 ppm and another peak (4) diminished in size in the region of 145 ppm in Fig. 4, which belonged to the carbon in the urea group and the carbon in the benzene ring that linked with the nitrogen directly. All this evidence confirms successful modification.
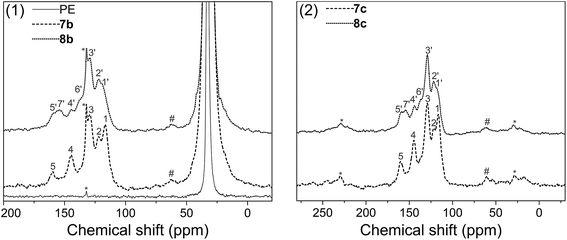 |
| | Fig. 5 Solid state spectra of blank, amino – functionalized and urea – functionalized polyethylenes: (1) polyethylene; (2) titanium dioxide. | |
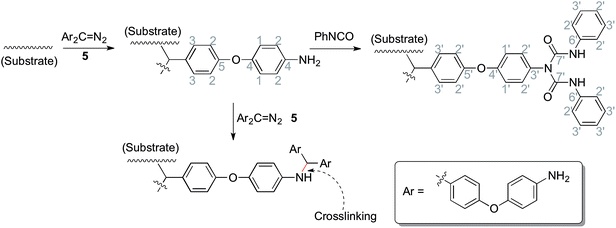 |
| | Scheme 2 The chemical reactions during the preparation of materials (related to Fig. 5). | |
More importantly, there are two diazo groups and two amino groups in each molecule of 5, so there is the possibility of crosslinking inside the surface modification layer. In the NMR spectra, it was noticeable that a small peak (#) in the region of 62.9 ppm (7b) or 60.4 ppm (7c) could be observed, which was likely to belong to the product of the crosslinking reaction between amines and carbenes (Scheme 2), namely, the N–H insertion reaction; this confirms that the modified layer crosslinked on the surface of materials. The chemical shift of this peak is very similar to the result given for a diarylmethylene–N bond by simulation (65.8 ppm).
2.7 Loading of hydrogen peroxide
Since the crosslinking of amines with carbenes forms a very dense crosslinked layer, and the subsequent reaction with isocyanates generates a large number of urea groups, this was expected to enhance the loadings of hydrogen peroxide on the surface of various materials which might be achieved. In order to compare with earlier data,26 a titration method with KI-Na2S2O3 was used to determine the loadings of hydrogen peroxide, and the results are shown in Table 3. Compared with that earlier data, the loading of hydrogen peroxide on the surface of XAD4 and polyethylene increased by about 5 times. In particular, the modification approach was so successful that it led to a large loading of hydrogen peroxide not only for polymers but also for inorganic materials, such as TiO2 and Al. The reason is likely to be that there are large numbers of hydroxyl groups on the surface of metallic oxides, as well as most metals, so that the original carbene modification gives a surface layer that in turn allows the introduction of large numbers of urea groups, which could easily bind hydrogen peroxide with higher loading levels.
Table 3 The loadings of hydrogen peroxide and the antibacterial activity of materials
| Materials |
Polymers |
Detected dataa |
Data from referenceb |
| Loadings of H2O2 (mmol g−1) |
Activity against S. aureus (mm) |
Activity against E. coli (mm) |
Loadings of H2O2 (mmol g−1) |
Activity against S. aureuse (mm) |
Activity against E. colie (mm) |
|
Blank samples of polystyrene, polyethylene, TiO2, and Al sheet all gave no measurable value for hydrogen peroxide loading and no observable bioactivity.
Data from the reference: Langmuir, 2010, 26, 14142.
Zone of inhibition results (diameter for the circle) using material (10 mg) in 10 mm wells in a hole-plate bioassay (against Ceph C. standard).
Zone of inhibition results (length for the square) using 10 × 10 mm sheet on the surface of a plate bioassay (against Ceph C. standard).
Zone of inhibition results using material (50 mg) in 10 mm wells in a hole-plate bioassay (against Ceph C. standard).
(—) not determined.
|
|
9a
|
XAD4 |
0.423 |
23c |
24c |
0.083 |
25 |
17 |
|
9b
|
Polyethylene |
0.108 |
14c |
16c |
0.025 |
Inactive |
—f |
|
9c
|
TiO2 |
0.063 |
12c |
12c |
—f |
—f |
—f |
|
9d
|
Al sheet |
—f |
22d |
22d |
—f |
—f |
—f |
2.8 Antibacterial activity
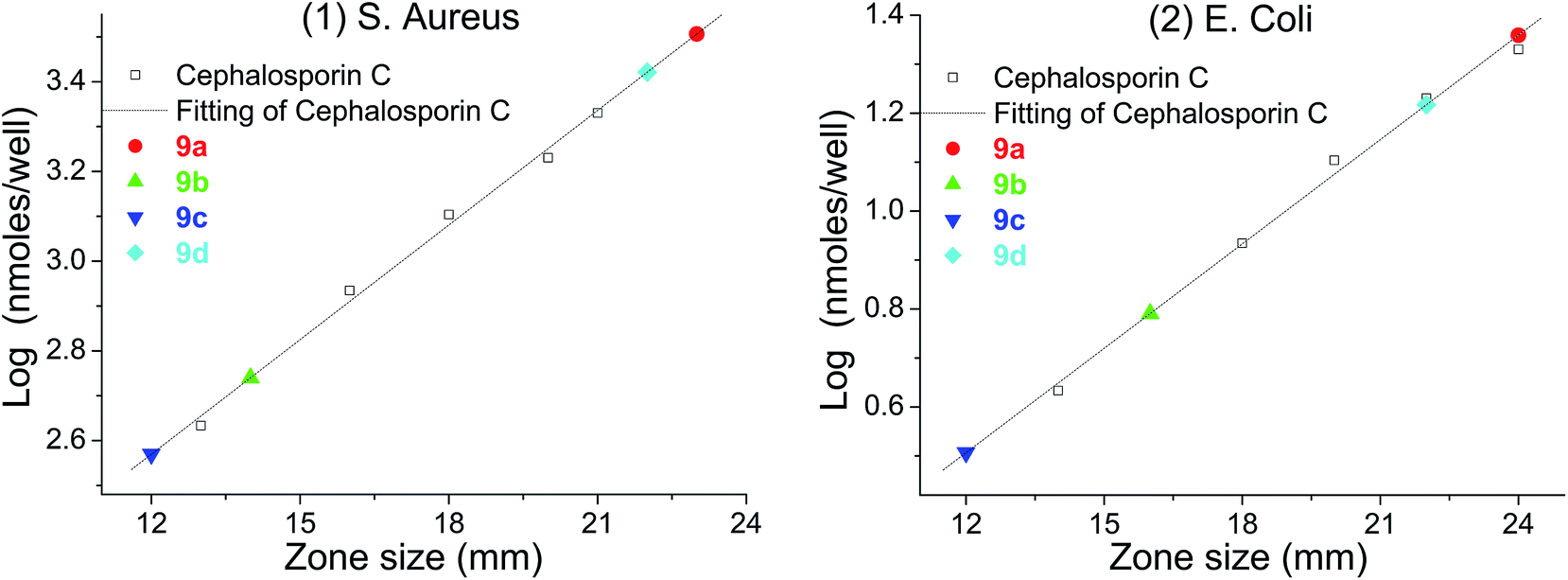
The crosslinking increased the density and the thickness of the modified layer, improved the loading of urea functions and therefore hydrogen peroxide. Upon incubation with bacteria (S. aureus and E. coli), large zones of inhibition were found for each material (Table 3). The most prominent of these materials was polyethylene 9b, which showed clear antibacterial activity; this could be achieved using up to only 1/5 of the amounts which had previously been reported (50 mg).26 The inorganic materials, such as TiO29c and Al sheet 9d, also showed favorable antibacterial activity, which greatly expands the application of the carbene modification approach. In addition, the Al sheet 9d showed better antibacterial activity than that of TiO29c, which might derive from the difference of the morphology and the modification protocol of the substrate. In order to calibrate the antibacterial activity, cephalosporin C in aqueous solution was used as a standard, giving log(nmole per well) versus diameter of inhibition zone as a straight line.45 Using this calibration, the equivalent concentration of cephalosporin C for materials 9a–d (Fig. 6) can be determined from the diameter of their inhibition zones (from Table 3), and compared with other antiseptic treatments.46,47
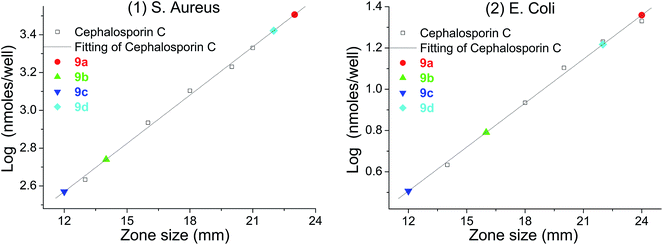 |
| | Fig. 6 log(nmole per well)–diameter plot for cephalosporin C and the equivalent of cephalosporin C for materials: (1) S. aureus; (2) E. coli. | |
3. Experimental section
3.1 Materials and characterization
All commercial reagents were purchased from Sigma-Aldrich Co. XAD4 (composed of polystyrene that is crosslinked with divinylbenzene, and is hereinafter referred to as polystyrene), polyethylene, and titanium dioxide were purchased from Sigma-Aldrich Co. and washed with methanol, distilled water, and acetone sequentially. Aluminum plate was purchased from Merck KGaA as a TLC plate, and immersed into water/methanol (1:1) solution to remove the silica from the surface before use. NMR spectra were recorded using an AVANCE III spectrometer, and ATR-IR spectra were recorded by a Tensor 27 spectrometer, both of which were manufactured by Bruker Co. XPS analysis was performed with a VG – Escalab X-ray Photo-electron Spectrometer VGX900 using Al Kα radiation with an energy of 1486.6 eV at an operating pressure less than 1 × 10−8 mBar. The Pass Energy was set at 50 eV for the survey scan of the sample and 20 eV for scans of specific areas: C 1s, N 1s, O 1s, Ti 2p, and Al 2p. Casa XPS peak fitting software was used to analyze spectra and a reference charge correction of 284.8 eV for C 1s was used. The area under the elemental peaks and standard sensitivity factors (C, 1.00; O, 2.93; N, 1.80; Ti, 7.91; Al, 0.537) were used to calculate relative chemical composition within the surface and near-surface sampling depth of the XPS.
3.2 Synthetic procedures
3.2.1 2,2,2-Trifluoro-N-(4-phenoxyphenyl)acetamide (2).
4-Phenoxyaniline 1 (3.0 g, 16.2 mmol) and pyridine (3.93 mL, 48.6 mmol) were dissolved in DCM (50 mL) with magnetic stirring. After the mixture was cooled to 5 °C in an ice/water bath, a solution of trifluoroacetic anhydride (3.38 mL, 24.3 mmol) in DCM (15 mL) was added dropwise. After the addition, the solution continued stirring overnight at room temperature before evaporation under vacuum. Acetone (10 mL) was added to dissolve the residue and the solution was poured into cold water (300 mL) and stirred for 10 minutes. The mixture was then filtered under vacuum and the solid was collected and dried in an oven at 80 °C (96%). mp: 118–120 °C; δH (400.3 MHz, CDCl3): 6.83–6.95 (m, 4H), 7.02 (t, 1H), 7.24 (t, 2H), 7.40 (d, 2H), 8.27 (s, 1H); δC (100.7 MHz, CDCl3): 115.9(q), 119.1, 119.3, 122.6, 123.8, 129.9, 130.1, 155.2(q), 155.6, 156.8; δF (376.6 MHz, CDCl3): 75.6; IR νmax (cm−1): 3293, 1704, 1593, 1540, 1488, 1229, 1151, 916, 875, 845, 749, 714, 691; HRMS: m/z calcd for C14H11O2NF3: 282.07364; found: 282.07376 [M + H]+.
3.2.2
N,N′-(((Isophthaloylbis(4,1-phenylene))bis(oxy)bis(4,1-phenylene))bis(2,2,2-trifluoroacetamide)) (3).
Aluminum trichloride (0.49 g, 3.70 mmol) was added to DCM (20 mL) and the solution was cooled to 5 °C with a ice/water bath. To that mixture was added 2 (0.42 g, 1.48 mmol) and isophthaloyl chloride (0.15 g, 0.74 mmol) sequentially. The mixture was stirred at room temperature for 2 hours. The reaction mixture was evaporated under vacuum before acetone (10 mL) was added. The mixture in acetone was poured into cold water to yield a light pink precipitate, which was filtered and dried in an oven (80 °C). After that, the solid was dispersed in DCM (10 mL) and filtered again, and the precipitate was dried under room temperature to obtain a light pink solid (89%). mp: 236–238 °C; δH (500.3 MHz, acetone-d6): 7.14 (d, 4H), 7.23 (d, 4H), 7.77 (t, 1H), 7.85 (d, 4H), 7.91 (d, 4H), 8.05 (d, 2H), 8.11 (t, 1H), 10.4 (s, 2H); δC (125.8 MHz, acetone-d6): 116.1(q), 117.2, 120.7, 122.8, 128.8, 130.4, 131.8, 132.4, 132.8, 133.2, 138.1, 152.9, 154.7(q), 161.7, 193.7; δF (376.6 MHz, acetone-d6): 76.1; IR νmax (cm−1): 3285, 1706, 1650, 1598, 1542, 1504, 1238, 1179, 1152, 999, 950, 880, 834, 739, 701; HRMS: m/z calcd for C36H21O6N2F6: 691.13093; found: 691.12988 [M − H]−.
3.2.3 4,4′-((((1E,1′E)-1,3-Phenylenebis(hydrazonomethylene))bis(4,1-phenylene))bis(oxy))dianiline (4).
A suspension of 3 (0.36 g, 0.51 mmol) in ethanol (7 mL) was treated with hydrazine monohydrate (0.51 mL, 10.2 mmol). A few drops of acetic acid (about 0.2 mL) were added as catalyst. The resulting mixture was heated to a gentle reflux for 40 h. After cooling, the solution was evaporated under vacuum. The residue was dissolved in DCM (50 mL), washed with water (50 mL × 4), dried with MgSO4, and evaporated under vacuum. The crude product 4 (87%) was a white solid as a mixture of diastereoisomers and was used without further purification. mp: 92–96 °C; δH (500.3 MHz, CDCl3): 3.52 (s, 4H), 5.43 (s, 4H), 6.52–6.64 (m, 4H), 6.70–6.85 (m, 6H), 6.89–6.96 (m, 2H), 7.05–7.13 (m, 3H), 7.25–7.60 (m, 5H); δC (125.8 MHz, CDCl3): 116.2, 116.3, 116.8, 117.5, 117.6, 121.2, 121.3, 121.6, 121.7, 124.7, 125.5, 126.0, 126.4, 126.5, 127.1, 127.8, 127.9, 128.3, 129.3, 129.4, 130.3, 132.3, 132.5, 133.0, 134.4, 138.7, 140.0, 142.9, 143.0, 143.3, 143.4, 147.4, 147.6, 147.8, 148.1, 148.3, 148.8, 149.1, 159.1, 159.3, 159.5, 159.7; IR νmax (cm−1): 3353, 3210, 3038, 1603, 1495, 1228, 1198, 1165, 871, 834, 701; HRMS: m/z calcd for C32H29O2N6: 529.23465; found: 529.23431 [M + H]+.
3.2.4 4,4′-(((1,3-Phenylenebis(diazomethylene))bis(4,1-phenylene))bis(oxy))dianiline (5).
A mixture of manganese dioxide (0.22 g, 2.48 mmol), sodium sulfate (0.19 g, 1.34 mmol), and potassium hydroxide (0.06 g, 1.35 mmol) was added to a solution of 5 (0.24 g, 0.46 mmol) in dichloromethane (15 mL). The mixture was vigorously stirred for 5 hours in the dark, and then filtered through a pad of Celite™. The filtrate was concentrated under vacuum to yield 5 (89%) as a purple solid and then stored at sub-ambient temperature to avoid decomposition. mp: 115 °C (decomposed); δH (500.3 MHz, CDCl3): 3.52 (s, 4H), 6.50–7.34 (m, 20H); δC (125.8 MHz, CDCl3): 61.8, 116.3, 118.1, 119.9, 121.1, 121.1, 122.1, 127.4, 129.8, 131.3, 142.9, 148.4, 157.5; IR νmax (cm−1): 3446, 3366, 3038, 2031, 1590, 1495, 1228, 870, 828.
3.2.5 1,3-Phenylenebis((4-(4-aminophenoxy)phenyl)methylene) diacetate (6).
Acetic acid (2.86 mL, 50.0 mmol) was added to a solution of 5 (0.27 g, 0.5 mmol) in DCM (10 mL). The mixture was stirred in the dark at room temperature until the color changed from purple to light yellow (about 20 min). The mixture was washed with 2 M Na2CO3 aq. solution (60 mL), water (20 mL × 3), dried with MgSO4 and concentrated under reduced pressure. The crude product was purified by chromatography on a silica gel column eluting with DCM/acetone (10/1) to give the product 6 as a light yellow solid (68%). mp: 76–80 °C; δH (500.3 MHz, CDCl3): 2.03 (d, 6H), 3.51 (s, 4H), 6.51–6.58 (m, 4H), 6.70–6.82 (m, 10H), 7.08–7.18 (m, 6H), 7.18–7.25 (m, 2H); δC (125.9 MHz, CDCl3): 21.3, 76.3, 116.3, 117.0, 121.3, 125.4(d), 126.4(d), 128.8, 133.5, 140.6, 143.1, 148.1, 158.9, 170.1; IR νmax (cm−1): 3453, 3368, 3040, 2980, 1733, 1611, 1498, 1370, 1223, 1167, 1022, 872, 833; HRMS: m/z calcd for C36H32O6N2Na: 611.21526; found: 611.21453 [M + Na]+.
3.3 Kinetic studies
Bis-4-(chlorophenyl)diazomethane 10a, diphenyldiazomethane 10b, bis-4-(phenoxyphenyl)diazomethane 10c, and bis-4-(methoxyphenyl)diazomethane 10d were synthesized according to the literature.35 Firstly, UV spectra were obtained for all compounds. Secondly, a series of diazomethane solutions with different concentrations were prepared. The relationship between UV absorbance and concentration was recorded and fitted linearly to calculate the molar absorption coefficient. This allowed a calibration curve from absorbance to concentration to be determined, which was necessary for kinetic studies. Finally, to the cuvette with diazomethane solutions were added two drops of acetic acid (about 20 times excess). The mixture was immediately scanned by the UV spectrometer at regular intervals until adequate spectra were recorded for calculation of the reaction rate (25 °C, in CH2Cl2).
3.4 Materials functionalization
3.4.1 Preparation of amino-functionalized beads and powders (7a–c).
To a solution of diazomethane 5 (1.0 g) in dichloromethane (50 mL) was added the required material (4.0 g), and the mixture was concentrated under vacuum. The polymer was collected and heated (120 °C for 30 minutes) in a flask with a nitrogen balloon. The resulting solid was washed three times with acetone and dried to yield the amino – functionalized polystyrene 7a, polyethylene 7b, and titanium dioxide 7c. A portion of materials 7a–c were mixed with ninhydrin in a solution of water/ethanol (1:1) and refluxed for 1 h until the color changed from yellow to brown.
3.4.2 Preparation of amino-functionalized sheet (7d).
To a solution of diazomethane 5 in dichloromethane (2 mL) was added the required aluminum sheet (10 × 10 mm). After being immersed into the solution for some time, the aluminum sheet was taken out and dried under N2 flow. Subsequently, it was heated with a heat gun for some time until the pink color faded. The resulting sheet was washed with acetone for 3 times and dried to yield the amino – functionalized Al sheet 7d.
3.4.3 Preparation of urea-functionalized beads and powders (8a–c).
To a stirred suspension of required materials 7a–c (0.6 g) in acetone (25 mL) was added triethylamine (5 mL) and phenyl isocyanate (2 mL). The mixture was heated to reflux for 2 hours. The modified materials were collected by filtration, washed with acetone (25 mL) for 3 times, and dried in an oven (80 °C, 4 h) to obtain urea-functionalized polystyrene 8a, polyethylene 8b, and titanium dioxide 8c.
3.4.4 Preparation of urea-functionalized sheet (8d).
To a one-neck flask with amino – functionalized Al sheet (10 × 10 mm) was added triethylamine (5 mL), acetone (25 mL) and phenyl isocyanate (2 mL). The mixture was stirred overnight at room temperature. The Al sheet were collected, washed with acetone (25 mL) for 3 times, and dried at room temperature to obtain urea-functionalized Al sheet 8d.
3.4.5 Preparation of hydrogen peroxide-loaded materials (9a–d).
The required materials 8a–d were immersed in hydrogen peroxide solution (30% in water) overnight. The materials were collected, washed with water (100 mL), acetone (5 mL), and dried at room temperature to obtain hydrogen peroxide-loaded polystyrene 9a, polyethylene 9b, titanium dioxide 9c, and Al sheet 9d.
3.5 Hydrogen peroxide loading determination
A sample of the required material (10 mg for 9a, 30 mg for 9b or 9c) was added to a mixture of potassium iodide aq. solution (10% (m m−1), 20 mL) and acetic acid (10 mL). After standing for 5 minutes, aqueous starch solution (1% (m m−1), 2 mL) was added and the suspension was left to stand for 1 h. The resulting dark-blue solution was titrated with 0.001 M sodium thiosulfate to a colorless end-point. Typically it required 5.0–10.0 mL of solution. Blank material samples without loading H2O2 were taken as a reference.
3.6 Bioassay
The specific bacterial solution was diluted to 1:50 with sterile water and was added to cooled molten agar in a 1:100 ratio (bacterial solution (1:50): agar, v/v). The inoculated agar (17 mL) was pipetted into empty Petri dishes (90 mm), which were then swirled to ensure even thickness of agar. The Petri dishes were allowed to set and were refrigerated until needed.
Using a sterile method, 10 mm-diameter circles were punched in the bacteria-seeded agar plate, and the inner agar was removed to produce empty wells. The required material 9a–c (10 mg) and DMSO (30 μL) were added to pre-punched wells of the seeded agar plates. The well was then sealed with another 70 μL of molten agar so that a uniform layer of agar was produced. For the material 9d, the sheet was placed on the agar plate seeded with bacteria and sealed with some agar on the surface. The agar plates were covered and incubated for 18 h to encourage bacterial growth. The diameter (for 9a–c) or the length (for 9d) of the antimicrobial clear zones around each material was measured and recorded. Blank material samples without loading H2O2 were taken as a reference.
An aq. solution of cephalosporin C was used to calibrate the antibacterial activity. For Staphylococcus aureus, the concentration of cephalosporin C was 1.0 mg mL−1 and the dosage was 20 μL, 40 μL, 60 μL, 80 μL, and 100 μL. For Escherischia coli, the concentration of cephalosporin C was 10 μg mL−1 and the dosage was 20 μL, 40 μL, 60 μL, 80 μL, and 100 μL. After the solution was added to pre-punched wells of the seeded agar plates, the well was fulfilled to 100 μL with distilled water. The agar plates were covered and incubated for 18 h to encourage bacterial growth. The diameter of the antimicrobial clear zones around each well was measured and recorded.
4. Conclusions
An amino-functionalized bis(aryldiazomethane) was synthesized, and kinetic studies with acetic acid by UV spectroscopy confirmed its reactivity. This was used to functionalize polystyrene, polyethylene, titanium dioxide, and Al plate by a bis(arylcarbene) insertion reaction. These were subsequently reacted with phenyl isocyanate to give urea-functionalized materials. The signals of nitrogen in the XPS spectra showed that the protocol is successful for both organic and inorganic materials. The peaks of 1713 and 1663 cm−1 in their IR spectra confirmed their surface chemical structure, which were shown to be comprised of urea and carbamyl urea groups. The difference of urea-functionalized materials and amino-functionalized materials to unmodified substrates was shown by solid state NMR spectra, which confirmed the reaction of amino groups with isocyanate groups. In particular, an unexpected crosslinking between amines and carbenes was discovered by the solid state NMR spectroscopy in the modified layer. The surface urea layer was used for binding hydrogen peroxide to give antibacterial activity. Compared with the earlier data, this crosslinking layer with the urea groups increases the loading of hydrogen peroxide by 5 times, and hence increases the antibacterial activity greatly. The discovery of this crosslinked modification followed reaction with isocyanates opens a new horizon for loading hydrogen peroxide, which is expected to be general for not only organic but also inorganic materials and surfaces to give excellent antibacterial activity. This approach neatly complements other approaches recently reported which introduce antibacterial activity using silver nanoparticles.48,49
Acknowledgements
This work is financially supported by the National Natural Science Foundation of China (No. 21406121) and the Distinguished Middle-Aged and Young Scientist Encourage and Reward Foundation of Shandong Province (No. BS2013CL037).
References
- E. D. Brown and G. D. Wright, Nature, 2016, 529, 336–343 CrossRef CAS PubMed.
- E. R. Kenawy, S. D. Worley and R. Broughton, Biomacromolecules, 2007, 8, 1359–1384 CrossRef CAS PubMed.
- R. O. Darouiche, N. Engl. J. Med., 2004, 350, 1422–1429 CrossRef CAS PubMed.
- D. Campoccia, L. Montanaro and C. R. Arciola, Biomaterials, 2013, 34, 8533–8554 CrossRef CAS PubMed.
- K. Chaloupka, Y. Malam and A. M. Seifalian, Trends Biotechnol., 2010, 28, 580–588 CrossRef CAS PubMed.
- B. L. Ouay and F. Stellacci, Nano Today, 2015, 10, 339–354 CrossRef.
- Y. Liu, Z. Lin, W. Lin, K. S. Moon and C. P. Wong, ACS Appl. Mater. Interfaces, 2012, 4, 3959–3964 CAS.
- A. Sirelkhatim, S. Mahmud, A. Seeni, N. H. M. Kaus, L. C. Ann, S. K. M. Bakhori, H. Hasan and D. Mohamad, Nano-Micro Lett., 2015, 7, 219–242 CrossRef CAS.
- L. A. T. W. Asri, M. Crismaru, S. Roest, Y. Chen, O. Ivashenko, P. Rudolf, J. C. Tiller, H. C. Mei, T. J. A. Loontjens and H. J. Busscher, Adv. Funct. Mater., 2014, 24, 346–355 CrossRef CAS.
- S. Buffet-Bataillon, P. Tattevin, M. Bonnaure-Mallet and A. Jolivet-Gougeon, Int. J. Antimicrob. Agents, 2012, 39, 381–389 CrossRef CAS PubMed.
- A. Regiel-Futyra, M. Kus-Liskiewicz, V. Sebastian, S. Irusta, M. Arruebo, G. Stochel and A. Kyziol, ACS Appl. Mater. Interfaces, 2015, 7, 1087–1099 CAS.
- R. L. Kumar, A. K. Narayan, S. Dhivya, A. Chawla, S. Saravanan and N. Selvamurugan, Carbohydr. Polym., 2016, 151, 172–188 CrossRef PubMed.
- H. B. Kocer, I. Cerkez, S. D. Worley, R. M. Broughton and T. S. Huang, ACS Appl. Mater. Interfaces, 2011, 3, 2845–2850 CAS.
- F. Hui and C. Debiemme-Chouvy, Biomacromolecules, 2013, 14, 585–601 CrossRef CAS PubMed.
- K. Brudzynski, Can. J. Microbiol., 2006, 52, 1228–1237 CrossRef CAS PubMed.
- A. D. Wentworth, L. H. Jones, P. Wentworth, K. D. Janda and R. A. Lerner, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 10930–10935 CrossRef CAS.
- P. Wentworth, L. H. Jones, A. D. Wentworth, X. Zhu, N. A. Larsen, I. A. Wilson, X. Xu, W. A. Goddard, K. D. Janda and A. Eschenmoser, Science, 2001, 293, 1806–1811 CrossRef CAS PubMed.
- L. M. Wolfson and S. S. Sumner, J. Food Prot., 1993, 56, 887–892 CrossRef CAS.
- B. J. Juven and M. D. Pierson, J. Food Prot., 1996, 59, 1233–1241 CrossRef CAS.
- G. Soldaini, F. Cardona and A. Goti, Org. Lett., 2007, 9, 473–476 CrossRef CAS PubMed.
- X. Liu and K. F. Jensen, Green Chem., 2013, 15, 1538–1541 RSC.
- F. Guo, D. Cao, M. Du, K. Ye, G. Wang, W. Zhang, Y. Gao and K. Cheng, J. Power Sources, 2016, 307, 697–704 CrossRef CAS.
- F. Guo, K. Cheng, K. Ye, G. Wang and D. Cao, Electrochim. Acta, 2016, 199, 290–296 CrossRef CAS.
- E. F. Panarin, K. K. Kalninsh and D. V. Pestov, Eur. Polym. J., 2001, 37, 375–379 CrossRef CAS.
- E. F. Panarin, K. K. Kalninsh and V. V. Azanova, Polym. Sci., Ser. A, 2007, 49, 275–283 CrossRef.
- J. P. Griffiths, B. Maliha, M. G. Moloney, A. L. Thompson and I. Hussain, Langmuir, 2010, 26, 14142–14153 CrossRef CAS PubMed.
- K. M. Awenat, P. J. Davis, M. G. Moloney and W. Ebenezer, Chem. Commun., 2005, 990–992 RSC.
- H. Wang, J. P. Griffiths, R. G. Egdell, M. G. Moloney and J. S. Foord, Langmuir, 2008, 24, 862–868 CrossRef CAS PubMed.
- C. Choong, J. P. Griffiths, M. G. Moloney, J. Triffitt and D. Swallow, React. Funct. Polym., 2009, 69, 77–85 CrossRef CAS.
- C. Choong, J. S. Foord, J. P. Griffiths, E. M. Parker, L. Baiwen, M. Bora and M. G. Moloney, New J. Chem., 2012, 36, 1187–1200 RSC.
- G. W. Nelson, E. M. Parker, K. Singh, C. F. Blanford, M. G. Moloney and J. S. Foord, Langmuir, 2015, 31, 11086–11096 CrossRef CAS PubMed.
- J. P. GriffithS, D. M. L. Leonard, M. G. Moloney and N. J. Stratton, J. Mol. Eng. Mater., 2013, 1, 1250002 CrossRef.
- P. Luksirikul, B. Ballesteros, G. Tobias, M. G. Moloney and M. L. H. Green, Carbon, 2010, 48, 1912–1917 CrossRef CAS.
- A. Aphaiwong, M. G. Moloney and M. Christlieb, J. Mater. Chem., 2012, 22, 24627–24636 RSC.
- P. J. Davis, L. Harris, A. Karim, A. L. Thompson, M. Gilpin, M. G. Moloney, M. J. Pound and C. Thompson, Tetrahedron Lett., 2011, 52, 1553–1556 CrossRef CAS.
-
G. Sartori, R. Maggi and V. Santacroce, in Arene Chemistry: Reaction Mechanisms and Methods for Aromatic Compounds, ed. J. Mortier, John Wiley & Sons, Inc, Hoboken, 2015, ch. 3, pp. 59–82 Search PubMed.
- W. Sander, G. Bucher and S. Wierlacher, Chem. Rev., 1993, 93, 1583–1621 CrossRef CAS.
- S. M. Nicolle and C. J. Moody, Chem.–Eur. J., 2014, 20, 4420–4425 CrossRef CAS PubMed.
- S. M. Nicolle, C. J. Hayes and C. J. Moody, Chem.–Eur. J., 2015, 21, 4576–4579 CrossRef CAS PubMed.
- H. Liu, Y. Wei and C. Cai, New J. Chem., 2016, 40, 674–678 RSC.
- D. Leonard, M. G. Moloney and C. Thompson, Tetrahedron Lett., 2009, 50, 3499–3502 CrossRef CAS.
- M. Weigele, S. DeBernardo, J. Tengi and W. Leimgruber, J. Am. Chem. Soc., 1972, 94, 5927–5928 CrossRef CAS.
- U. Toyozo, M. Katsunosuke and S. Yutaka, Bull. Chem. Soc. Jpn., 1969, 42, 1539–1545 CrossRef.
- B. A. Shainyan, N. N. Chipanina, T. N. Aksamentova, L. P. Oznobikhina, G. N. Rosentsveig and I. B. Rosentsveig, Tetrahedron, 2010, 66, 8551–8556 CrossRef CAS.
- S. W. B. Tan, C. L. L. Chai, M. G. Moloney and A. L. Thompson, J. Org. Chem., 2015, 80, 2661–2675 CrossRef CAS PubMed.
- D. Radulescu, G. Voicu, A. E. Oprea, E. Andronescu, V. Grumezescu, A. M. Holban, B. S. Vasile, A. V. Surdu, A. M. Grumezescu, G. Socol, L. Mogoanta, G. D. Mogosanu, P. C. Balaure, R. Radulescu and M. C. Chifiriuc, Appl. Surf. Sci., 2016, 374, 165–171 CrossRef CAS.
- L. Shi, S. Santhanakrishnan, Y. S. Cheah, M. Li, C. L. L. Chai and K. G. Neoh, ACS Appl. Mater. Interfaces, 2016, 8, 33131–33138 CAS.
- Z. C. Xiong, Y. J. Zhu, F. F. Chen, T. W. Sun and Y. Q. Shen, Chem.–Eur. J., 2016, 22, 11224–11231 CrossRef CAS PubMed.
- L. Liu, P. Yang, J. Li, Z. Zhang, X. Yu and L. Lu, Appl. Surf. Sci., 2017, 403, 435–440 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra05258h |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  ab and
Mark G.
Moloney
ab and
Mark G.
Moloney