DOI:
10.1039/C7RA01306J
(Paper)
RSC Adv., 2017,
7, 29656-29664
Selenadiazole derivatives antagonize glucocorticoid-induced osteoblasts cells apoptosis by blocking ROS-mediated signaling, a new anti-osteoporosis strategy
Received
31st January 2017
, Accepted 22nd May 2017
First published on 7th June 2017
Abstract
Long-term use of glucocorticoids (GC), especially dexamethasone (Dex), could result in osteoporosis through induction of oxidative stress-mediated apoptosis of osteoblasts and increased differentiation of osteoclasts, finally leading to bone loss. Therefore, searching for new agents that could block Dex-induced cytotoxicity would be a good way to treat osteoporosis. In this study, we show that, synthetic benzo[1,2,5]selenadiazole derivatives (SeDs) could be used as effective inhibitors of Dex-induced osteoblasts apoptosis. This protective effect was correlated with their lipophilicity, cellular uptake and antioxidant activities. Furthermore, mechanistic studies reveal that, treatment of osteoblast cells with Dex resulted in overproduction of intracellular reactive oxygen species (ROS), DNA fragmentation, activation of caspase-3/-9, mitochondria fragmentation, phosphorylation of p53, and activation of MAPKs and AKT pathways. However, pre-treatment of the cells with the synthetic SeDs effectively blocked these intracellular events, which suggest that SeDs could protect osteoblast cells against Dex-induced cell apoptosis via attenuating oxidative stress and downstream signalling pathways. Therefore, this study demonstrates a new therapeutic application of SeDs to antagonize GC-induced osteoporosis.
Introduction
Glucocorticoid (GC) therapy is widely used in the treatment of chronic inflammatory and autoimmune diseases such as rheumatic, asthma disease, cancer and transplantation.1 Nowadays, hundreds of thousands of people receive GC treatment. However, long-term using of GCs could bring severe side effects, such as reduction in bone density.2 Bone, a dynamic remodeling tissue, formation is a coupled process including bone resorption by osteoclasts and bone formation by osteoblasts.3 Osteocyte, the most abundant bone cell, is the terminally differentiated osteoblasts.4 Increasing osteocyte cell death is also considered to be highly associated with accumulation of bone micro damage which is caused by loss of ability to sense micro damage and repair by other bone cells.5
GC-induced osteoblasts cells apoptosis is the most common reason of drug-related osteoporosis, and ranks third of osteoporosis following the postmenopausal and senile varieties.6 Early studies have showed that GC mainly led to bone loss through the influence on osteocyte, osteoblasts and osteoclasts.7 Excess GCs have prosurvival effect on osteoclasts differentiation, inhibit the activity of osteoblasts, and induce apoptosis of osteoblasts and osteocytes, leading to reduced bone formation and increased bone resorption.8 Nowadays, a variety of pharmacological treatments, including bisphosphonates, hormone replacement therapy, and calcium supplementation have been developed for osteoporosis therapy. Fluoride, testosterone, parathyroid hormone, calcitonin and anabolic steroids have been proposed to prevent GC-induced bone lose.9,10 However, looking for efficient and low toxicity protectants still kindled great interest of scientists.
Studies have confirmed that many diseases were closely correlated with the generation of reactive oxygen species (ROS).11 Multiple cellular molecules could be oxidized by ROS such as unsaturated fatty acid, and the oxidative substances are harmful to human health. Moreover, oxidative stress has been reported to induce osteoblasts apoptosis in MC3T3 osteoblast cells.12 Therefore, drugs that possess antioxidants are potential pharmacologic and therapeutic agents. Selenium (Se) is an important trace element, and antioxidant activity is an important biological function. Studies have showed that, inside the biological systems, selenocompounds could effectively remove ROS.13 Animal experiments confirmed that, organic selenocompounds showed significant protective effects at the range of safe dose against cell damage.14 Se could significantly activate SOD, GSH-PX activity, increase the GSH content, and thus enhance the intracellular anti-oxidative capacity.15 Allophycocyanin isolated from Se-rich algae could protect the blood cell from AAPH-induced oxidative stress damage by inhibiting ROS generation.10 Sodium selenite could reverse the glutamate-induced cytotoxicity associated with ROS production and enhanced oxygen consumption.16 In addition, supplementation of Se could bring the gospel to many neurodegenerative diseases by restoring the activity of antioxidant enzymes.17 Comparing with inorganic Se, organic selenocompounds show advantages of higher absorptivity and lower toxicity.18 Therefore, more and more attentions have been focused on the rational design and discovery of organoselenium in cancer and chemotherapy.
During the past decade, a large number of potent organoselenium compounds, including selenocyanate, selenobetaine and seleno-amino acids, have been designed to achieve greater chemopreventive efficacy. In our previous studies, synthetic selenadiazole derivatives have been found be able to induce cancer cell death through caspase- and p53-dependent apoptosis.19 Selenadiazole derivatives could also antagonize hyperglycemia-induced drug resistance in cancer cells through the AMPK-mTORC1 and p53 signaling pathways.20,21 Another series of selenadiazole derivatives were also identified as thioredoxin reductase inhibitors that induced cancer cell apoptosis.22,23 Recently, a novel selenium-containing ruthenium complex was found to enhance radiation-induced DNA damage, which led to G2/M arrest and apoptosis in cancer cells by activating ROS-mediated pathways.24 All of our previous works has suggested that Se-containing compounds exhibited potency in cancer therapy. However, little information on their ability to antagonize GC-induced osteoporosis is available. Interestingly, herein a series of benzo[1,2,5]selenadiazole derivatives have been screened and identified as effective inhibitors of Dex-induced osteoblasts apoptosis. This protective effect was correlated with their lipophilicity, cellular uptake and antioxidant activities. Furthermore, we demonstrated that, treatment of MC3T3 osteoblast cells with Dex resulted in overproduction of intracellular reactive oxygen species (ROS), DNA fragmentation, activation of caspase-3/-9, mitochondria fragmentation, phosphorylation of p53, and activation of MAPKs and AKT pathways. However, pre-treatment of the cells with the synthetic SeDs effectively blocked these intracellular events induced by Dex, which suggest that SeDs could protect osteoblast cells against Dex-induced cell apoptosis via attenuating oxidative stress and downstream signalling pathways. Therefore, this study demonstrates a new therapeutic application of SeDs to antagonize GC-induced osteoporosis.
Results and discussion
Relationship among the lipophilicity, cellular uptake and antioxidant activities of SeDs
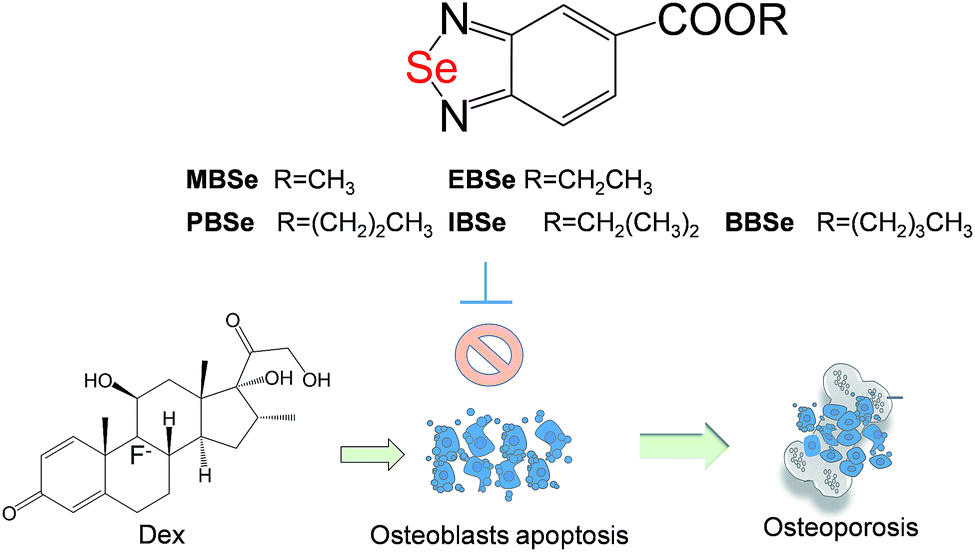
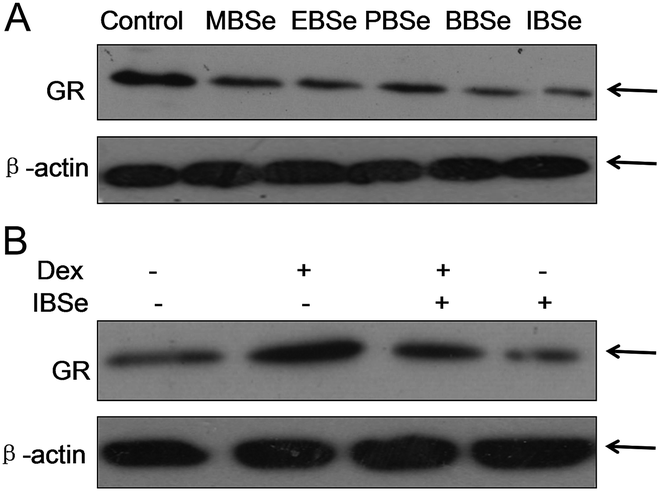
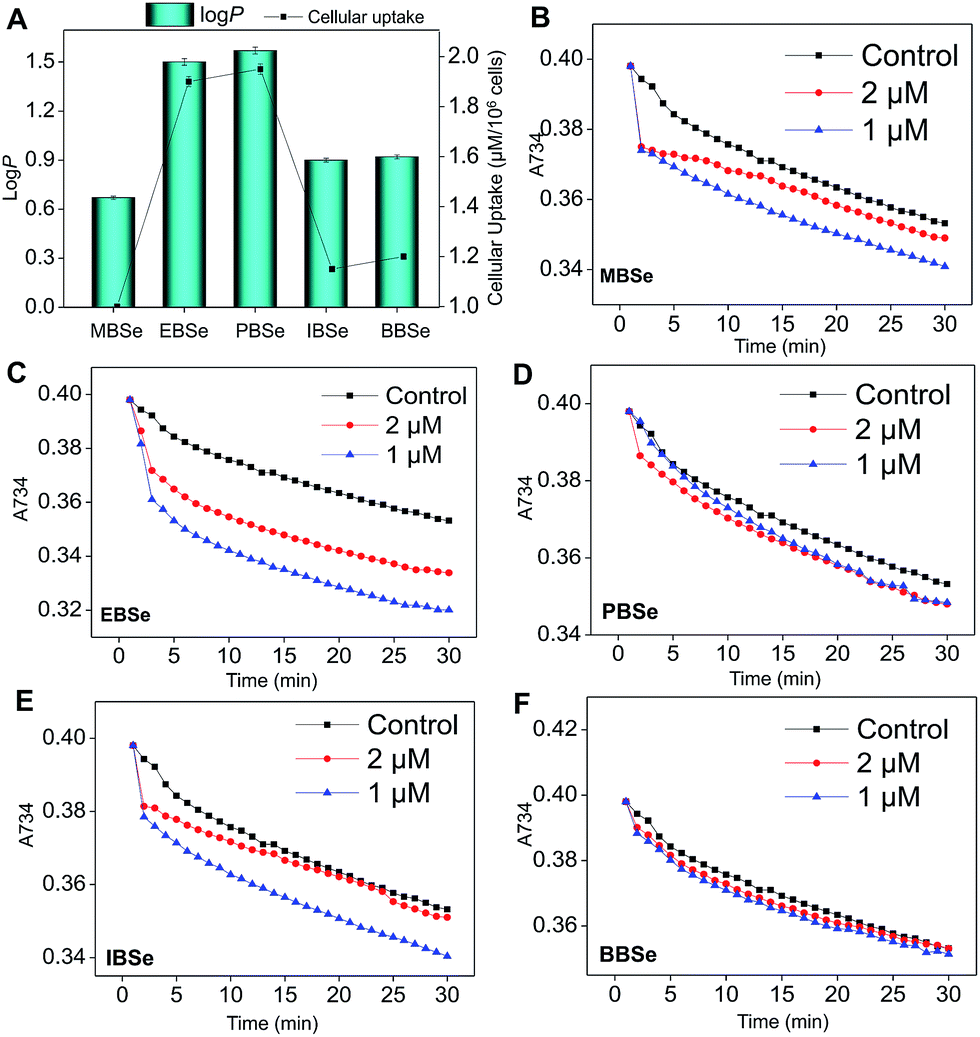
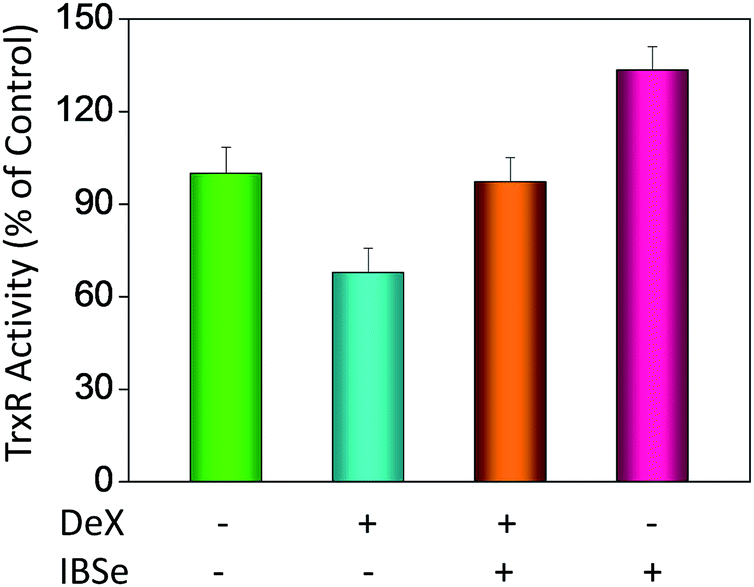
Our previous work has showed that organoselenium compounds with selenadiazole moiety had good anticancer and antioxidantactivities.14,20–23,25–32 Therefore we tried to make some decoration on the benzo[1,2,5]selenadiazole by introduction of various alcohol to enhance the solubility and antioxidant activities. A series of 5-alkylbenzenecarboxyl[c][1,2,5]selenadiazoles with different alkyls, such as methyl, ethyl, propyl, isopropyl, butyl groups, were synthesized according to previous described.21,33 These SeDs were expected to antagonize Dex-induced apoptosis and anti-osteoporosis activities in MC3T3 osteoblast cells (Fig. 1). Glucocorticoid receptor (GR) transactivation induced by Glucocorticoid (GC) has been confirmed to highly correlate with GC-induced osteoblasts cells apoptosis. In order to investigate whether selenadiazole derivatives (SeDs) have anti-glucocorticoid receptor (GR) effect, we firstly carried out experiments to examine the inhibition activities of SeDs on GR transactivation. As shown in Fig. 2A, the SeDs alone could effectively inhibit the expression of GR. Especially, IBSe demonstrated the highest inhibitory effects. Furthermore, we tried to test the effects of Dex and the inhibitory effect of IBSe. As shown in Fig. 2B, Dex alone significantly increased the expression level of GR. In contrast, the combined treatment with IBSe effectively reversed this effect. Taken together, these results suggest that, GR may be involved in the antagonizing activity of SeDs against Dex-induced osteoblasts cells apoptosis. Evidence have revealed the correlationship between oxidative stress and osteoblasts apoptosis,12 therefore, these SeDs were then screened for their antioxidant and anti-osteoporosis activities. As we all known, the lipophilicity of a compound is well known to have significant influence on its biological activity.34 Therefore, we have detected the relationship between lipo-hydro partition coefficient (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P) and cellular uptake of the synthetic compounds. As shown in Fig. 3A, the increase in the length and molecular weight of alkyl groups didn't linearly increased the lipophilicity of the synthetic selenadiazole derivatives, with higher logP values detected in EBSe and PBSe. Consistently, EBSe and PBSe demonstrated significant higher cellular uptake in MC3T3 cells and the five compounds were well lipophilic. These results demonstrate the positive relationship between the lipophilicity and cellular uptake of the synthetic compounds. We then examined the antioxidant activities of SeDs by using ABTS assay. As showed in Fig. 3B–F, all of the compounds could effectively scavenge the free radicals in a time-dependent manner, which demonstrating the potent antioxidant activity of these compounds. It is well known that TrxR is homodimeric selenoenzyme and plays crucial role in antioxidant defense through regulating the activation and reduction of Trx as well as other oxidized cellular components by counteracting excess reactive oxygen species (ROS). Therefore, in order to further investigate whether SeDs antioxidant activity was due to the upregulation of selenoproteins. We next examined the effects of IBSe and Dex on the activity of selenoenzyme, TrxR. As shown in Fig. 4, Dex alone significantly inhibited the activity, IBSe effectively elevated TrxR activity. In contrast, the combined treatment with IBSe effectively reversed this effect. Taken together, these results suggest that, IBSe may antagonize Dex-induced osteoblasts cells apoptosis by increasing selenoenzyme activity which supports the application potential of the compounds against oxidative stress-related diseases.
P) and cellular uptake of the synthetic compounds. As shown in Fig. 3A, the increase in the length and molecular weight of alkyl groups didn't linearly increased the lipophilicity of the synthetic selenadiazole derivatives, with higher logP values detected in EBSe and PBSe. Consistently, EBSe and PBSe demonstrated significant higher cellular uptake in MC3T3 cells and the five compounds were well lipophilic. These results demonstrate the positive relationship between the lipophilicity and cellular uptake of the synthetic compounds. We then examined the antioxidant activities of SeDs by using ABTS assay. As showed in Fig. 3B–F, all of the compounds could effectively scavenge the free radicals in a time-dependent manner, which demonstrating the potent antioxidant activity of these compounds. It is well known that TrxR is homodimeric selenoenzyme and plays crucial role in antioxidant defense through regulating the activation and reduction of Trx as well as other oxidized cellular components by counteracting excess reactive oxygen species (ROS). Therefore, in order to further investigate whether SeDs antioxidant activity was due to the upregulation of selenoproteins. We next examined the effects of IBSe and Dex on the activity of selenoenzyme, TrxR. As shown in Fig. 4, Dex alone significantly inhibited the activity, IBSe effectively elevated TrxR activity. In contrast, the combined treatment with IBSe effectively reversed this effect. Taken together, these results suggest that, IBSe may antagonize Dex-induced osteoblasts cells apoptosis by increasing selenoenzyme activity which supports the application potential of the compounds against oxidative stress-related diseases.
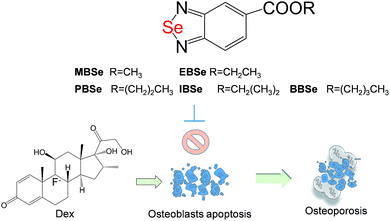 |
| | Fig. 1 Antagonization of Dex-induced apoptosis by selenadiazole derivatives. | |
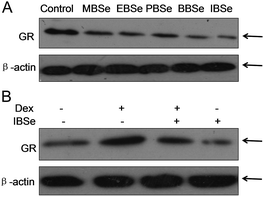 |
| | Fig. 2 GR expression inhibition of selenadiazole derivatives. (A) SeDs inhibited GR transactivation. (B) IBSe reversed GR transactivation induced by DeX. | |
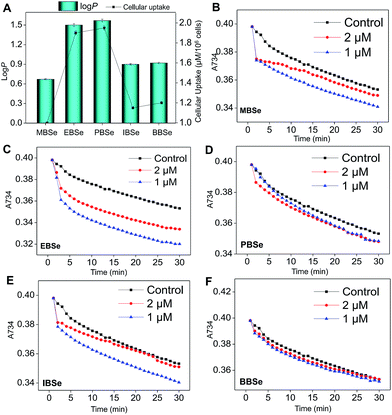 |
| | Fig. 3 Lipophilicity, cellular uptake and antioxidant activities of selenadiazole derivatives. (A) The lipophilicity and cellular uptake of SeDs. The intracellular Se concentration in cells exposed to 0.2 μM SeDs was determined by ICP-AES analysis. Antioxidant activities of MBSe (B), EBSe (C), PBSe (D), IBSe (E) and BBSe (F) by ABTS assay. | |
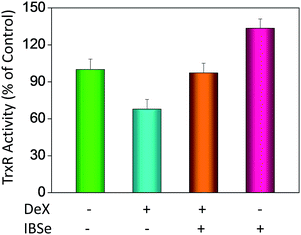 |
| | Fig. 4 Selenadiazole derivatives reverse Dex-inhibited intracellular TrxR activity in MC3T3 cells. | |
SeDs reduce Dex-induced cytotoxicity through blocking cell apoptosis
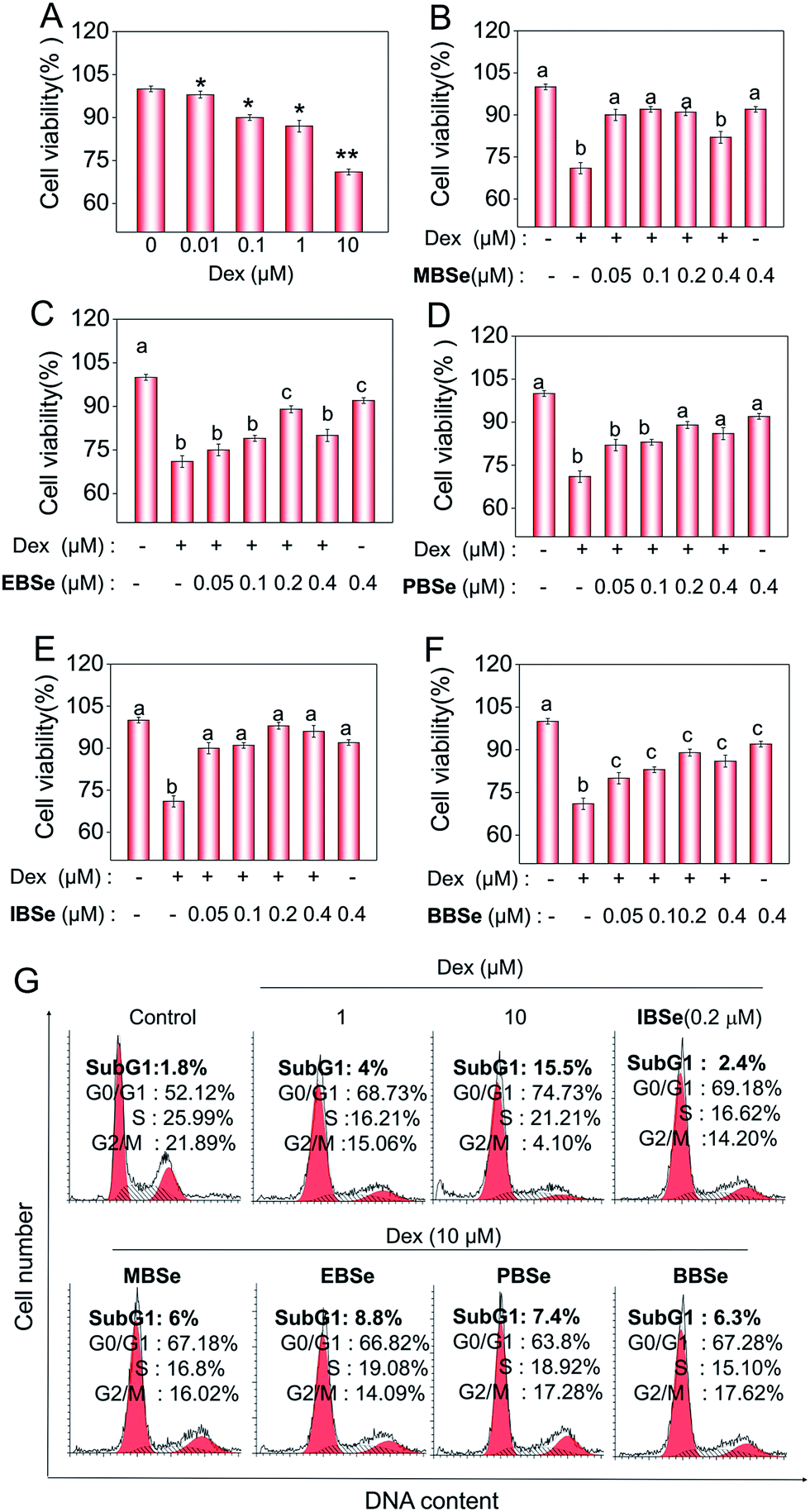
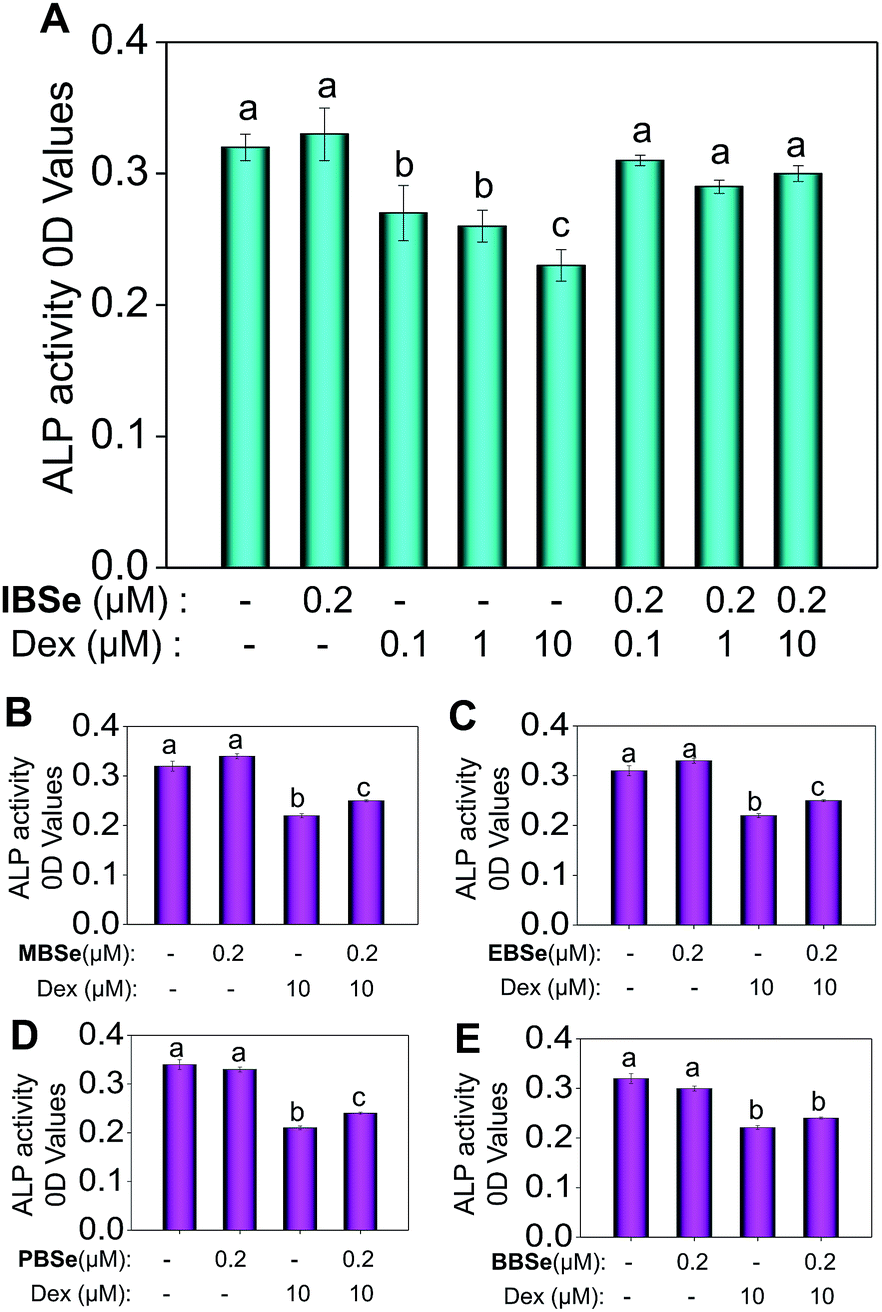
Studies were also carried out to evaluate the protective effects of the synthetic compounds on Dex-induced cytotoxicity in MC3T3 osteoblast cells. As shown in Fig. 5A, treatment of the cells with Dex at 0.1 to 10 μM for 48 h reduced the cell viability in a dose-dependent manner. However, pre-treatments of the cells with all the SeDs significantly increased the cell viability (Fig. 5B–F), while the compounds alone showed slightly inhibition effects in the cell growth. Among them, IBSe exhibited better protective effect on Dex-induced cytotoxicity, which may be due to its strong antioxidant activity. Studies were also conducted to examine the action mechanisms of Dex and the synthetic SeDs. Apoptosis is a highly programmed cell death, which has been identified as the major mode of cell death induced by Dex in MC3T3 osteoblast cells.35 Therefore, in this study, firstly, we have analyzed the effects of Dex and selenadiazole derivatives on the cell cycle distribution by flow cytometric analysis. As shown in Fig. 5G, Dex (10 μM) caused 15.7% of the cell apoptosis and 22.61% G0/G1 cycle arrest. However pre-treated the cells with MBSe, EBSe, PBSe, IBSe and BBSe (0.2 μM) for 2 h, obviously reduced the apoptosis from 15.7% to 6%, 8.8%, 7.4%, 2.4%, 6.3% and G0/G1 cycle arrest from 22.6% to 15.06%, 12.7%, 11.68%, 15.05%, 15.16%. Among them, preventive effect of IBSe was the most obvious. Alkaline phosphatase (ALP) is the earliest marker of osteoblasts MC3T3 osteoblast cells differentiation.36,37 Then we detected the activity of ALP to assess the protective effect of the five SeDs on osteoblasts differentiation. As shown in Fig. 6A, incubation of cells with Dex (0.1, 1, 10 μM) for 72 h visibly reduced the activity of ALP. However, pretreatment of IBSe for 2 h significantly abolished these effects induced by Dex. Additionally, we found that MBSe, EBSe, PBSe and BBSe displayed neglectable effects towards ALP activity (Fig. 6B–E) Taken together, these results suggest that IBSe is capable of reversing the apoptosis effects induced by Dex.
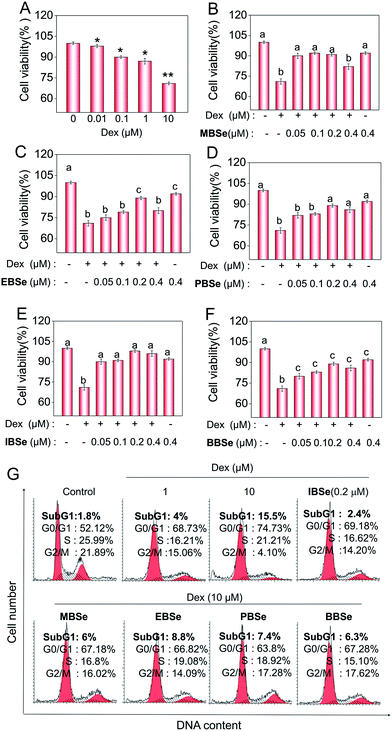 |
| | Fig. 5 SeDs reverse the apoptosis-inducing effects of Dex. Cytotoxicity of Dex and SeD toward MC3T3 cells (A–F). (G) SeDs attenuate Dex-induced apoptosis in MC3T3 cells. Quantitative analysis of apoptotic cell death induced by SeDs and Dex were analyzed by flow cytometric analysis. All data here are expressed as means ± SD of triplicates. Bars with different characters (a–c) are statistically different at P < 0.05 level. *P < 0.05 vs. untreated control. **P < 0.01 vs. untreated control. | |
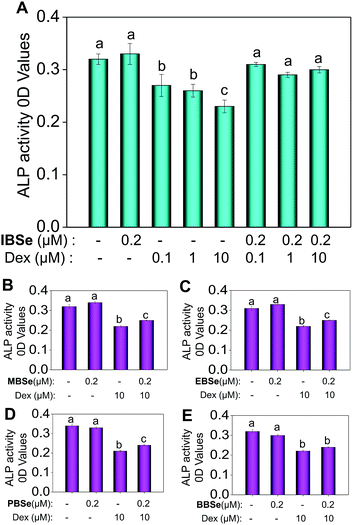 |
| | Fig. 6 Effects of DEX and SeDs on the ALP activity of osteoblast. Osteoblast was used to be treated with Dex or SeDs with different concentration, (A) IBSe, (B) MBSe, (C) EBSe, (D) PBSe, (E) BBSe. Bars with different characters (a–c) are statistically different at P < 0.05 level. | |
IBSe alleviates Dex-induced apoptosis through inhibition of caspase activation and mitochondrial dysfunction
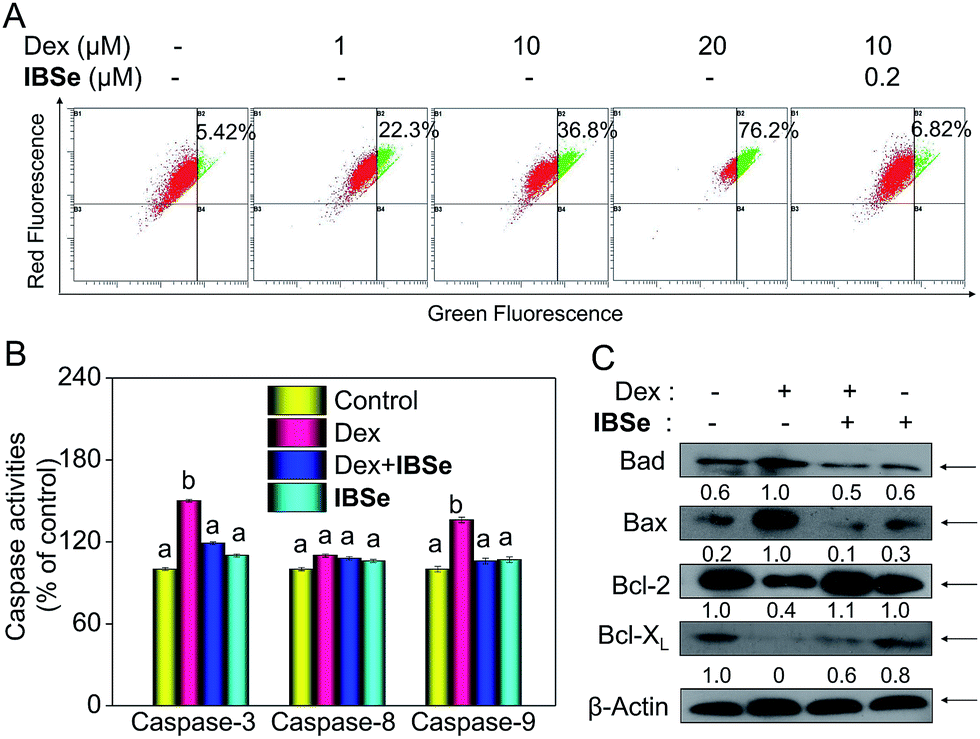
Caspase, a family of cysteine acid proteases, is known as important mediators of cell apoptosis and contributed to the apoptotic morphology through cleavage of various cellular substrates.38 Generally upon stimulation, apoptosis occurs through mitochondria-mediated (intrinsic) pathway and death receptor-mediated (extrinsic) pathway.39,40 These two pathways exert their roles by activating various caspases. Therefore, caspases activation is an important indicator of apoptosis. Consequently, we conducted fluorometric assay to detect whether the activation of caspase-3/-8/-9 was involved in this process. As depicted in Fig. 7B, IBSe induced slightly upregulation of actived caspase-3/-8/-9, while Dex triggered significant activation of caspase-3/-9 in MC3T3 cells. In contrast, pretreatment of the cells with 0.2 μM IBSe remarkably restrained the caspases activation induced by Dex, which indicated the protective effects of IBSe against the caspases activation inducing capacity of Dex towards MC3T3 osteoblast cells. Both the extrinsic and intrinsic apoptotic signals have been shown to integrated in mitochondria. And deleption of mitochondria membrane potential (Δψm) is considered to be a highly relevant event upon apoptosis stimulation.41 Therefore, we conducted the following study, cells were pretreated with IBSe for 2 h, then co-cultured with Dex for 48 h, after that, cells were stained with the mitochondria-selective JC-1 dye and analyzed by flow cytometry. The red fluorescence represented normal mitochondrial membranes potential, while intensity of green fluorescence represented the cells that lost mitochondrial membrane potential as a result of the JC-1 monomers. As shown in Fig. 7A, Dex treatment dose-dependently induced deleption of Δψm from 5.42% to 22.3%, 36.8% and 76.2%. However, pretreatment of 0.2 μM IBSe effectively blocked the mitochondria depolarization induced by Dex, as demonstrated by the decreased green fluorescence intensity.
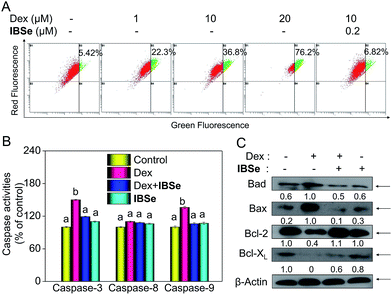 |
| | Fig. 7 IBS reduces caspase mediated mitochondrial membrane potential induced by Dex. (A) Cells were pre-treated with IBSe for 2 h, then co-cultured with Dex for another 48 h and analyzed by JC-1 flow cytometry. (B) Caspase activities were measured by specific fluorescent substrates for caspase-3/8/9. (C) The expression of Bcl-2 protein family. Significant difference between treatment and control groups in indicated at P < 0.05(*) and P < 0.01(**) levels. | |
The Bcl-2 family members have been known as the key regulators of mitochondrial permeability. Here we measured the expression level of Bcl-2 family members by immunoblotting assay. Dex incubation up-regulated the expression of pro-apoptosis protein, such as Bax and Bad, but the down-regulated the expression level of pro-survival proteins, such as Bcl-2 and Bcl-xL. However, noticeable reversed effects were observed in Bcl-2 family proteins expression triggered by Dex after the pretreatment with IBSe for 2 h (Fig. 7C). Together, these results confirm that IBSe protects MC3T3 cells against Dex-induced apoptosis through inhibition of caspases activation and mitochondria dysfunction.
IBSe prevents Dex-induced mitochondria fragmentation
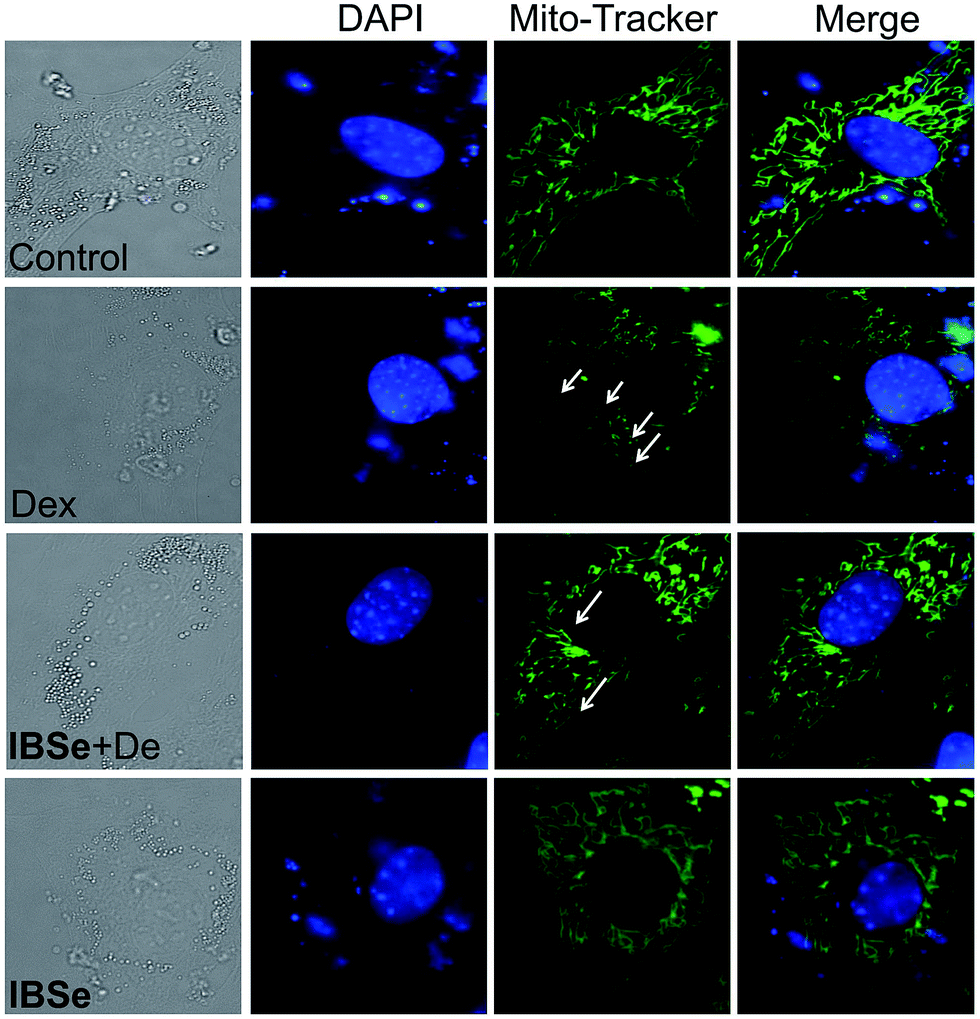
Since IBSe obtains protective effects against Dex destruction on mitochondria, we next conducted morphological studies of mitochondria by using Mito Tracker Green CMXRos as a marker through employing fluorescence microscopy.42 As illustrated in Fig. 8, 0.2 μM IBSe incubation induced neglectable disruption in mitochondria structure, while treatment of the MC3T3 cells with 10 μM Dex resulted in remarkable fragmentation of mitochondria compared with the control groups. Significant reduction in these changes was observed in groups that pretreated with IBSe for 2 h and then cotreated with Dex for 48 h. The morphological improvements demonstrated that IBSe could restrain the Dex-induced damage to mitochondria in MC3T3 cells.
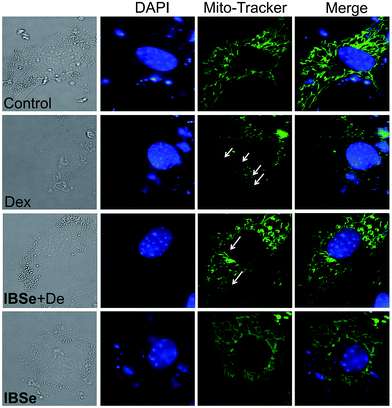 |
| | Fig. 8 IBSe reduces mitochondria fragmentation induced by Dex. Cells were pre-treated with or without IBSe (0.2 μM) for 2 h, then cultured with Dex (10 μM) for 48 h, after that cells were Stained by Green Mito-Tracker for 2 h and stained by DAPI for 30 min. | |
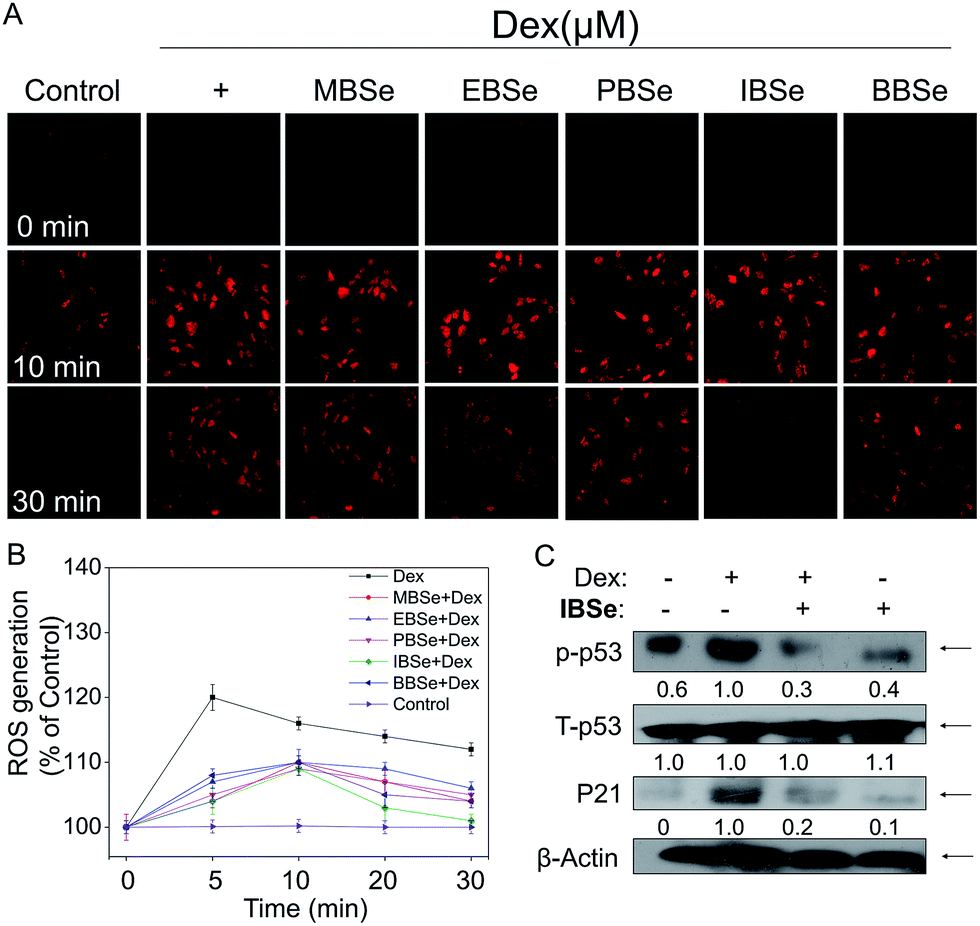
IBSe decreases Dex-induced p53 phosphorylation through eliminating ROS accumulation in MC3T3 cells
ROS, including superoxide, hydrogen peroxide, and hydroxyl radical, accumulation has been confirmed to be the results of mitochondria dysfunction. And ROS generation is shown to be the important mechanisms of osteoblasts apoptosis induced by high dose GCs. Thus, we examined whether attenuation of ROS was implicated in IBSe protective effects towards Dex-induced apoptosis by using a specific probe DHE.43 As shown in Fig. 9A, significant increase of ROS was observed in cells that treated with Dex (10 μM), which induced the apoptosis of MC3T3 osteoblast cells. However, pretreatment of the cells with MBSe, EBSe, PBSe, IBSe and BBSe significantly reduced the Dex-induced ROS generation a time-dependent manner, Moreover, IBSe was found to potently inhibited Dex triggered ROS generation in osteoblast cells (Fig. 9B). Evidences have demonstrated that overproduction of ROS may result in DNA damage and thus activated p53, which takes part in apoptosis modulation through regulating various downstream proteins expression, such as p21, Bax, Bcl-2 and Bcl-XL. Therefore, p53 and p21 proteins expression level was evaluated by western blotting assay to confirm the protective effects. The results revealed that IBSe strongly blocked Dex-induced p53 activation and p21 expression (Fig. 9C). Together, these results demonstrate that IBSe effectively inhibits Dex-induced p53 phosphorylation through decreasing ROS accumulation in MC3T3 cells.
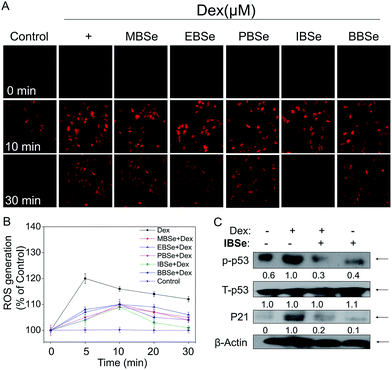 |
| | Fig. 9 IBSe prevents Dex induced ROS generation in MC3T3 cells. (A) Cells pretreatment with SeDs for 2 h and then co-incubated with Dex. After treatment cells were stained with DHE-DA for 30 min, and then imaged under the fluorescence microscope (magnification, 10×). (B) Dex and SeDs induced ROS accumulation in MC3T3 cells. Cells were treated with indicated concentrations of drugs for various periods of time. The levels of intracellular ROS were determined by DHE-DA assay. (C) Protein expression of p53 and p21. | |
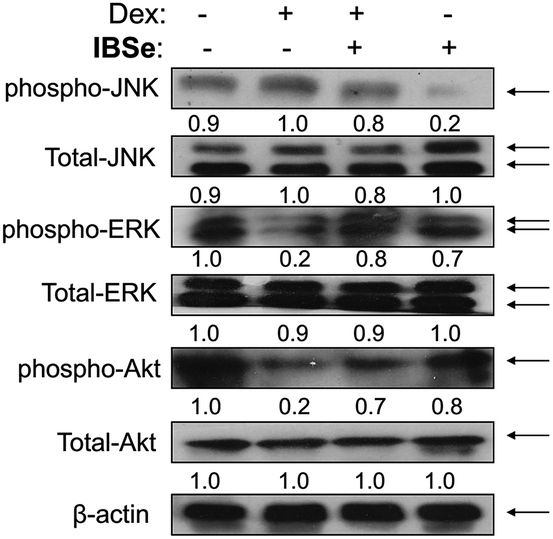
IBSe attenuates Dex-induced apoptosis through regulating AKT and MAPKs pathways
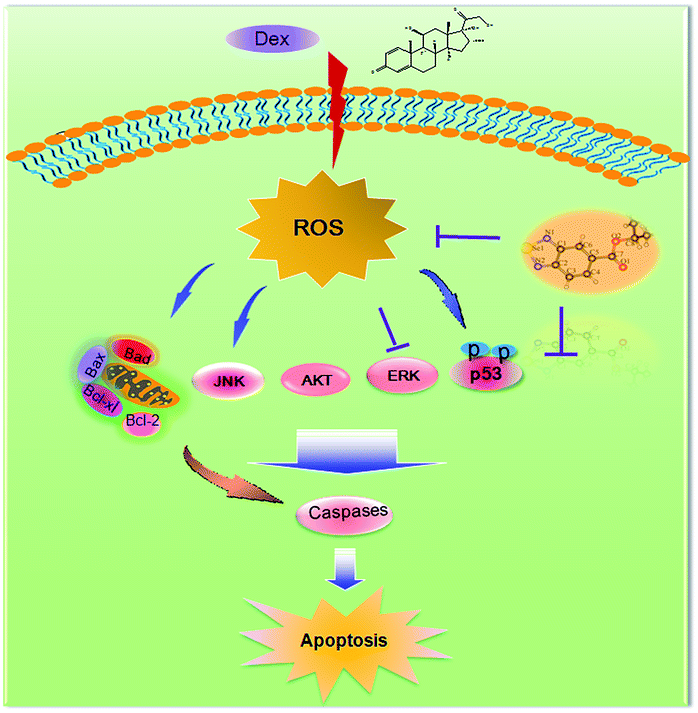
ERK and Akt signaling pathways have been confirmed to play important role in cell apoptosis and cell proliferation.44 Akt enhances the survival of cells by inhibiting the expression of pro-apoptotic proteins, such as caspase-9 and p53.45 Similar to Akt, ERK are involved in many kinds of cellular processes, including apoptosis, cell proliferation and differentiation.46 It could also prevent cell apoptosis by blocking the activation of caspase.47 Contrary to Akt and ERK, MAPK has been reported to induce cell apoptosis in response to external stimuli.48 Oxidative stress has been identified as a key modulator of MAPK signaling cascades.49 In this study, ROS overproduction induced by Dex up-regulated the phosphorylation of JNK, and down-regulated the phosphorylation of ERK and AKT (Fig. 10). In contrast, pretreatment of the cells with IBSe effectively reversed these effects induced by Dex. These results indicate that, SeDs prevented Dex-induced osteoblast cells apoptosis by suppression of pro-apoptotic JNK kinase and activation of pro-survival ERK and AKT kinases. Based on the above results, we proposed a signaling network for the antagonism action of selenadiazole derivative on Dex-induced apoptosis (Fig. 11).
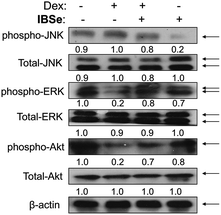 |
| | Fig. 10 IBSe and Dex impact on Akt and MAPK signaling pathway. Cells were pre-treated with IBSe (0.2 μM) for 2 h, then cultured with Dex (10 μM) for 48 h. The phosphorylation status and expression levels of Akt and MAPK after treatment of IBSe and Dex. Equal protein loading was confirmed by Western analysis of β-actin in the protein extracts. | |
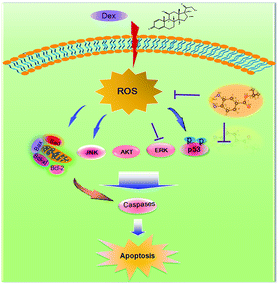 |
| | Fig. 11 The proposed protective signaling pathway of IBSe againsts Dex-induced apoptosis in MC3T3 cells. | |
Conclusions
In this study, a series of benzo[1,2,5]selenadiazole derivatives has been screened and identified as effective inhibitors of Dex-induced osteoblasts apoptosis. The protective effects were correlated with their lipophilicity, cellular uptake and antioxidant activities against Dex-induced oxidative damage. Further mechanic studies suggest that the synthetic selenadiazole derivatives displayed noticeable protective effects against Dex-induced cell apoptosis through attenuating oxidative damage mediated p53 phosphorylation and regulating AKT and MAPKs signaling pathways. Taken together, this study demonstrates a new therapeutic application of SeDs to antagonize GC-induced osteoporosis.
Experimental section
Materials
Dexamethasones (Dex), Propidium Iodide (PI), thiazolyl blue tetrazolium bromide (MTT), solid JC-1, dihydroethidium (DHE), bicinchoninic acid (BCA) kit were purchased from Sigma (sigma-Aldrich, St. Louis, MO). All antibodies were purchased from Cell Signaling Technology (Beverly, MA). Caspase-8, caspase-9 and caspase-3 substrates were purchased from Calbiochem.
Synthesis and characterization of selenadiazole derivatives
The synthesis of SeDs was conducted according to our previous methods.20,21
Cell culture, drugs treatment and measurement of cell viability
MC3T3 cell line was obtained from American Type Culture Collection (ATCC, Manassas, Virginia). The cells were cultured with α-MEM medium, which supplemented with fetal bovine serum (10%), penicillin (100 units per mL) and streptomycin (50 units per mL) at 37 °C in a humidified (5% CO2) atmosphere. The cells were seeded in culture plates for 24 h, and then pre-treated with five benzo[1,2,5]selenadiazole derivatives for 2 h and co-incubated with Dex for another 48 h to examine the synergistic effects on MC3T3 osteoblast cells. Cell viability was determined by using MTT assay as previously described.50
Flow cytometric analysis
The cell cycle distribution was analyzed by flow cytometry as previously described.50 Apoptotic cells were measured by quantifying the sub-G1 peak. For each experiment, 10000 events per sample were recorded.
Determination of lipophilicity and cellular uptake
The lipophilicity of MBSe, EBSe, PBSe, IBSe and BBSe was determined by using the “shake-flask” method previously reported.51 The content of Se in each phase was determined by ICP-MS analysis. logP was calculated as the logarithmic ratio of the concentrations of the drug in the n-octanol phases and aqueous phases. The intracellular Se concentration was determined by ICP-MS method, as previously described.52 MC3T3 osteoblast cells were harvested after incubation with 0.2 μM five compounds for 2 h, and then washed with PBS buffer for three times. Cells were digested for 3 h in mixture of perchloric acid and nitric acid at the ratio of 3:1. Then digestive was volume to 5 mL with pure water.
Caspase activity assay
The specific caspase substrates (Ac-DEVD-AMC for caspase-3, Ac-IETD-AMC for caspase-8 and Ac-LEHD-AMC for caspase-9) were placed in 96-well plates and then total cell lysates (50 μg per well) were added.50 Plates were incubated for 2 h at 37 °C in darkness and the caspase activity was determined by fluorescence intensity with the excitation and emission wavelengths set at 380 and 460 nm respectively.
Evaluation of mitochondrial membrane potential Δψm
After treatment, cells were in incubated with 0.5 mL of PBS buffer containing 10 μg per mL of JC-1 for 20 min. Stained cells were immediately centrifuged to remove the supernatant, and added 0.5 mL PBS, at last analyzed by flow cytometry.53 The percentage of green fluorescence was used to represent the cells that lost Δψm.
Mitochondria fragmentation
The cells were pretreated with IBSe for 2 h, and then co-cultured with Dex for another 48 h; cell mitochondria were stained with 100 nM MitoTracker Green CMXRos for 90 min and nucleuses stained with 1 μg per mL DAPI for another 20 min. After that, the cells were washed with PBS twice, and cultured in fresh medium on a thermo-cell culture FCS2 chamber of Carl Zeiss Cell Observer (Jena, Germany). Cell images were captured with a monochromatic CoolSNAPFX camera (Roper Scientific, USA) and analyzed by using Axio Vision 4.2 software (Carl Zeiss).
Measurement of intracellular reactive oxygen species (ROS) generation
The intracellular ROS generation was detected by DHE probe fluorescence assay.54 The DHE fluorescence intensity of treated cells was expressed as percentage of control (as 100%)
ABTS free radical scavenging assay
ABTS free radical scavenging activity of MBSe, EBSe, PBSe, IBSe and BBSe were measured as previously described.38 To determine the antioxidant activity, 1 mL ABTS reagent and 50 μL of sample or positive controls was mixed. Absorbance was detected at 734 nm for 2 h.
Alkaline phosphatase (ALP) activity assay
The induction of ALP is an unequivocal marker for bone cell differentiation. To measure the ALP activity, cells were seeded in 48-well plate at 1.5 × 104 per well. After 24 h cells were pretreated with/without MBSe, EBSe, PBSe, IBSe and BBSe (0.2 μM), and then cultured with Dex (0.1, 1, 10 μM). After culture for 3 days, the medium was collected. 20 μL medium were taken on the 96-wells, then 100 μL reaction liquid (0.5 M Tris, 10 mM p-nitro phenyl phosphate, 2 mM MgCl2, 1 mM ZnCl2, pH = 9.0) were added. After incubation for 30 min at 37 °C in the incubator, the reaction was terminated by 1 M NaOH (80 μL). The absorbance at 409 nm was measured using a spectrophotometric plate reader.
Determination of TrxR inhibition
The inhibition of TrxR in MC3T3 cells induced by SeDs at different concentrations within 1 h was determined by the thioredoxin reductase assay kit (Cayman) as previously described.25
Western blot analysis
The effects of SeDs and Dex on the proteins expression level of glucocorticoid receptor (GR) and other different signaling pathways were detected by Western blot analysis.55 β-actin was used to confirm the equal loading and transfer of proteins.
Statistical analysis
Experiments were carried out at least in triplicate and repeated three times. All data were expressed as mean ± S.D. Statistical analysis was performed using SPSS statistical package (SPSS 13.0 for Windows; SPSS, Inc. Chicago, IL). The difference between two groups was analyzed by two-tailed Student's t-test. The difference between three or more groups was analyzed by one-way analysis of variance multiple comparisons. Differences with P < 0.05 or P < 0.01 was considered statistically significant.
Acknowledgements
This work was supported by Natural Science Foundation of China (21371076), National High-level personnel of special support program, Science Foundation for Distinguished Young Scholars of Guangdong Province (S2013050014667), YangFan Innovative & Entrepreneurial Research Team Project (201312H05), Guangdong Special Support Program and Guangdong Frontier Key Technological Innovation Special Funds (2014B050505012) and Fundamental Research Funds for the Central Universities.
Notes and references
- H. Xie, S.-Y. Tang, H. Li, X.-H. Luo, L.-Q. Yuan, D. Wang and E.-Y. Liao, Amino Acids, 2008, 35, 419–423 CrossRef CAS PubMed.
- D. Patschan, K. Loddenkemper and F. Buttgereit, Bone, 2001, 29, 498–505 CrossRef CAS PubMed.
- D. Zhang, J. Zhang, C. Fong, X. Yao and M. Yang, Biochimie, 2012, 94, 2514–2522 CrossRef CAS PubMed.
- L. F. Bonewald and M. L. Johnson, Bone, 2008, 42, 606–615 CrossRef CAS PubMed.
- S. C. Manolagas, J. Bone Miner. Res., 2000, 15, 1001–1005 CrossRef CAS PubMed.
- G. Gu, T. Hentunen, M. Nars, P. Härkönen and H. Väänänen, Apoptosis, 2005, 10, 583–595 CrossRef CAS PubMed.
- S. C. Manolagas, Endocr. Rev., 2000, 21, 115–137 CAS.
- P. Moutsatsou, E. Kassi and A. G. Papavassiliou, Trends Mol. Med., 2012, 18, 348–359 CrossRef CAS PubMed.
- J.-F. Zhang, G. Li, C.-L. Meng, Q. Dong, C.-Y. Chan, M.-L. He, P.-C. Leung, Y.-O. Zhang and H.-F. Kung, Phytomedicine, 2009, 16, 521–529 CrossRef CAS PubMed.
- H. Zhang, T. Chen, J. Jiang, Y.-S. Wong, F. Yang and W. Zheng, J. Agric. Food Chem., 2011, 59, 8683–8690 CrossRef CAS PubMed.
- H. Kawagishi and T. Finkel, Nat. Med., 2014, 20, 711–713 CrossRef CAS PubMed.
- M. Arai, Y. Shibata, K. Pugdee, Y. Abiko and Y. Ogata, IUBMB Life, 2007, 59, 27–33 CrossRef CAS PubMed.
- G. K. Azad and R. S. Tomar, Mol. Biol. Rep., 2014, 41, 4865–4879 CrossRef CAS PubMed.
- Y.-F. Li, Z. Dong, C. Chen, B. Li, Y. Gao, L. Qu, T. Wang, X. Fu, Y. Zhao and Z. Chai, Environ. Sci. Technol., 2012, 46, 11313–11318 CrossRef CAS PubMed.
- E. Song, C. Su, J. Fu, X. Xia, S. Yang, C. Xiao, B. Lu, H. Chen, Z. Sun and S. Wu, Life Sci., 2014, 109, 37–43 CrossRef CAS PubMed.
- S. Kumari, S. L. Mehta and P. A. Li, PLoS One, 2012, 7, e39382 CAS.
- K. Venardos, G. Harrison, J. Headrick and A. Perkins, J. Trace Elem. Med. Biol., 2004, 18, 81–88 CAS.
- B. Laffon, V. Valdiglesias, E. Pásaro and J. Méndez, Biol. Trace Elem. Res., 2010, 133, 12–19 CrossRef CAS PubMed.
- T. Chen and Y.-S. Wong, Int. J. Biochem. Cell Biol., 2009, 41, 666–676 CrossRef CAS PubMed.
- Y. Yang, S. Deng, Q. Zeng, W. Hu and T. Chen, Dalton Trans., 2016, 45, 18465–18475 RSC.
- Y. Liu, Y. Luo, X. Li, W. Zheng and T. Chen, Chem. –Asian J., 2015, 10, 642–652 CrossRef CAS PubMed.
- Q. Xie, Y. L. Zhou, G. Q. Lan, L. Y. Yang, W. J. Zheng, Y. W. Liang and T. F. Chen, Biochem. Biophys. Res. Commun., 2014, 449, 88–93 CrossRef CAS PubMed.
- Y. Liu, Y. Luo, X. Li, W. Zheng and T. Chen, Chem. –Asian J., 2015, 10, 642–652 CrossRef CAS PubMed.
- S. Movva, L. Rodriguez, H. Arias-Pulido and C. Verschraegen, Cancer, 2009, 115, 3166–3180 CrossRef CAS PubMed.
- L. He, S. Ji, H. Lai and T. Chen, J. Mater. Chem. B, 2015, 3, 8383–8393 RSC.
- T. Chen, Y. S. Wong, W. Zheng and J. Liu, Chem.-Biol. Interact., 2009, 180, 54–60 CrossRef CAS PubMed.
- T. Chen and Y. S. Wong, J. Agric. Food Chem., 2008, 56, 10574–10581 CrossRef CAS PubMed.
- T. Chen, W. Zheng, Y. S. Wong and F. Yang, J. Steroid Biochem., 2008, 62, 77–84 CAS.
- Q. Xie, Y. Zhou, G. Lan, L. Yang, W. Zheng, Y. Liang and T. Chen, Biochem. Biophys. Res. Commun., 2014, 449, 88–93 CrossRef CAS PubMed.
- Y. W. Liang, J. Zheng, X. Li, W. Zheng and T. Chen, Eur. J. Med. Chem., 2014, 84, 335–342 CrossRef CAS PubMed.
- Q. Xie, L. He, H. Lai, W. Zheng and T. Chen, RSC Adv., 2014, 4, 34210–34216 RSC.
- M. Zhou, S. Ji, Z. Wu, Y. Li, W. Zheng, H. Zhou and T. Chen, Eur. J. Med. Chem., 2015, 96, 92–97 CrossRef CAS PubMed.
- Y. Yang, S. Deng, Q. Zeng, W. Hu and T. Chen, Dalton Trans., 2016, 45, 18465–18475 RSC.
- L. Messori, F. Scaletti, L. Massai, M. A. Cinellu, C. Gabbiani, A. Vergara and A. Merlino, Chem. Commun., 2013, 49, 10100–10102 RSC.
- L. He, Y. Huang, H. Zhu, G. Pang, W. Zheng, Y. S. Wong and T. Chen, Adv. Funct. Mater., 2014, 24(19), 2754–2763 CrossRef CAS.
- Z.-S. Xu, X.-Y. Wang, D.-M. Xiao, L.-F. Hu, M. Lu, Z.-Y. Wu and J.-S. Bian, Free Radical Biol. Med., 2011, 50, 1314–1323 CrossRef CAS PubMed.
- L. Jin, Y. Nonaka, S. Miyakawa, M. Fujiwara and Y. Nakamura, Mol. Ther., 2016, 24, 1974–1986 CrossRef CAS PubMed.
- T. Chen and Y.-S. Wong, J. Agric. Food Chem., 2008, 56, 4352–4358 CrossRef CAS PubMed.
- M. R. de Oliveira, Toxicol. Lett., 2016, 258, 185–191 CrossRef CAS PubMed.
- W.-X. Zong, J. D. Rabinowitz and E. White, Mol. Cell, 2016, 61, 667–676 CrossRef CAS PubMed.
- S. Pierotti, L. Gandini, A. Lenzi and A. M. Isidori, J. Steroid Biochem., 2008, 108, 292–299 CrossRef CAS PubMed.
- M. G. Scioli, V. Cervelli, G. Arcuri, P. Gentile, E. Doldo, A. Bielli, E. Bonanno and A. Orlandi, J. Cell. Physiol., 2014, 229, 2077–2087 CrossRef CAS PubMed.
- M. Almeida, L. Han, E. Ambrogini, R. S. Weinstein and S. C. Manolagas, J. Biol. Chem., 2011, 286, 44326–44335 CrossRef CAS PubMed.
- J. A. McCubrey, L. S. Steelman, W. H. Chappell, S. L. Abrams, G. Montalto, M. Cervello, F. Nicoletti, P. Fagone, G. Malaponte and M. C. Mazzarino, Oncotarget, 2012, 3, 954–987 CrossRef PubMed.
- B. D. Manning and L. C. Cantley, Cell, 2007, 129, 1261–1274 CrossRef CAS PubMed.
- A. Dhanasekaran, S. Kotamraju, C. Karunakaran, S. V. Kalivendi, S. Thomas, J. Joseph and B. Kalyanaraman, Free Radical Biol. Med., 2005, 39, 567–583 CrossRef CAS PubMed.
- J.-Y. Yang, D. Michod, J. Walicki and C. Widmann, Biochem. Pharmacol., 2004, 68, 1027–1031 CrossRef CAS PubMed.
- G. Makin and C. Dive, Breast Cancer Res. Treat., 2001, 3, 150–153 CrossRef CAS.
- T. Thalhamer, M. McGrath and M. Harnett, Rheumatology, 2008, 47, 409–414 CrossRef CAS PubMed.
- Y. Zhang, X. Li, Z. Huang, W. Zheng, C. Fan and T. Chen, J. Nanomed. Nanotechnol., 2013, 9, 74–84 CrossRef CAS PubMed.
- V. Pierroz, T. Joshi, A. Leonidova, C. Mari, J. Schur, I. Ott, L. Spiccia, S. Ferrari and G. Gasser, J. Am. Chem. Soc., 2012, 134, 20376–20387 CrossRef CAS PubMed.
- W. Chen, N. Xu, L. Xu, L. Wang, Z. Li, W. Ma, Y. Zhu, C. Xu and N. A. Kotov, Macromol. Rapid Commun., 2010, 31, 228–236 CAS.
- L. Li, Y.-S. Wong, T. Chen, C. Fan and W. Zheng, Dalton Trans., 2012, 41, 1138–1141 RSC.
- J. Su, H. Lai, J. Chen, L. Li, Y.-S. Wong, T. Chen and X. Li, PLoS One, 2013, 8, e63502 CAS.
- J. Chen, Z. Luo, Z. Zhao, L. Xie, W. Zheng and T. Chen, Biomaterials, 2015, 71, 168–177 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *b
*b