
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffect of talin1 on apoptosis in hepatoma carcinoma cells via the PI3K/Akt/NF-κB signaling pathway
Zhiqiu Hu†
a,
Kai Jiang†b,
Qimeng Chang†a,
Yuhua Zhangb,
Bing Zhouc,
Ziping Zhang*a and
Ran Tao *bc
*bc
aDepartment of Surgery, Minhang Hospital, Fudan University, 7th Floor, 170 Xinsong Road, Shanghai, 201199, P. R. China. E-mail: zhangzp16@163.com; Tel: +86-18917511598
bDepartment of Hepatobiliary-Pancreatic Surgery, Zhejiang Provincial People's Hospital, 14th Floor, Building 1, 158 Shangtang Road, Hangzhou, 310014, P. R. China. E-mail: taoran898@163.com; Fax: +86-571-85131448; Tel: +86-1330-1919-116
cKey Laboratory of Zhejiang Provincial Institute of Clinical Medicine, Hangzhou, P. R. China
First published on 16th August 2017
Abstract
Talin1 is implicated in many cellular processes, which has been studied in various diseases using molecular biological technology. However, the detailed mechanism of talin1 in hepatocellular carcinoma (HCC) cell apoptosis remains unclear. This study was aimed at exploring the molecular mechanism mediating the effect of talin1 on the apoptosis of human HCC cells. We showed that shRNA-mediated talin1 loss led to a significant increase of apoptosis in HepG2 cells via upregulation of caspase-8, caspase-9, and PARP and downregulation of Bid protein levels. Talin1 knockdown decreased the phosphorylation of Akt and specially counteracted the effect of pCDNA-Akt in the PI3K/Akt pathway and down-regulated p50, p65, and p105 protein levels to inhibit the activation of NF-κB to promote apoptosis in HCC cells. Talin1 knockdown could enhance the inhibitory effects of IκBα on NF-κB and suppress the activation of IKKβ to upregulate caspase-8, caspase-9, and PARP protein levels, thus leading to the increased apoptotic proportion in HCC cells. Moreover, talin1 could attenuate TNF-α and TRAIL-induced apoptosis, which was readily reversed by RIP. Our results demonstrated that inhibition of talin1 suppressed the HCC cell proliferation and promoted apoptosis by decreasing the activity of the PI3K/Akt/NF-κB signaling pathway. This new mechanism provides further understanding of the molecular signaling pathway in HCC cell apoptosis.
1. Introduction
Hepatocellular carcinoma (HCC) is the world's fifth most common cancer, and the third leading cause of cancer death.1 HCC is also the most common primary liver malignancy, which has an incidence of about 780![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cases per year. Many studies have shown that HCV, HBV, alcohol, metabolic, and cholestatic liver diseases are associated with HCC development.2 Most HCCs are diagnosed at the intermediate or advanced disease stages; as a result, they progress rapidly, and curative approaches are often not feasible.3 At present, there are different therapeutic and management options, including resection, transplantation, chemoembolization, and ablation, with varying advantages and disadvantages for HCC therapy.4 However, the long-term prognosis after resection or ablation needs to be improved, and finding a new strategy to prevent recurrence is an important unmet medical need in patients with HCC.
000 cases per year. Many studies have shown that HCV, HBV, alcohol, metabolic, and cholestatic liver diseases are associated with HCC development.2 Most HCCs are diagnosed at the intermediate or advanced disease stages; as a result, they progress rapidly, and curative approaches are often not feasible.3 At present, there are different therapeutic and management options, including resection, transplantation, chemoembolization, and ablation, with varying advantages and disadvantages for HCC therapy.4 However, the long-term prognosis after resection or ablation needs to be improved, and finding a new strategy to prevent recurrence is an important unmet medical need in patients with HCC.
There are two partly interconnected apoptotic mechanisms: classical caspase-dependent apoptosis and the caspase-independent programmed form of cell death, named necroptosis.5,6 The classical, caspase-dependent apoptosis is initiated either by extrinsic or intrinsic factors. The extrinsic pathway is activated by the engagement of transmembrane receptors and eventually leads to the activation of caspase-8, which in turn activates down-stream caspases.7 On the other hand, the intrinsic pathway, also called the mitochondrial pathway, is regulated by the Bcl2 family of proteins, which can form an apoptosome complex to activate caspase-9 that in turn activates down-stream elements of the caspase cascade.8 NF-κB is increasingly recognized as a crucial player in many steps of cancer initiation and progression including the immune response, protection against apoptosis, and inflammation.9 The NF-κB protein family consists of five members: p65 (RelA), RelB, c-Rel (Rel) and the precursor proteins p100 (NF-κB2) and p105 (NF-κB1), and the latter gives rise to p52 and p50, respectively.10 As one of the IKK complex members, IKKβ participates in the regulation of the canonical NF-κB pathway, which typically culminates in translocation of the p50:p65 dimer to the nucleus.11,12 IκB can inhibit the activation of NF-κB through binding to NF-κB dimers and maintaining inactive NF-κB in the cytoplasm. In addition, phosphorylation and activation of Akt has been recognized as an important regulatory factor in the NF-κB signaling pathway.13–15
Akt or protein kinase B is a 57 kDa Ser/Thr kinase. It includes 3 isoforms in mammalian cells: PKBα/Akt1, PKBβ/Akt2, and PKBγ/Akt3.16 Akt is a downstream effector of PI3-kinase and activated by extracellular signals to promote cell proliferation and provide protection from apoptosis in cancer.17 In addition, Akt is the major upstream factor to activate and regulate NF-κB activity via phosphorylation of p65 by IκB kinase (IKK) both directly and indirectly.18 Therefore, Akt may confer some of its pro-survival effects by interacting with other pathways and help to boost the efficacy of new therapeutic agents in cancer.
Talin1, a large 270 kDa cytoskeletal protein that contains 2541 amino acids, is mainly expressed in the kidney, liver, spleen, stomach, lung, and vascular smooth muscle.19,20 Talin1 plays an essential role in integrin activation, which promotes integrins to enhance the functional interaction between cells and the extracellular matrix (ECM); these integrins serve as bidirectional transducers of extracellular and intracellular signals, ultimately regulating cell adhesion, proliferation, anoikis, survival, and tumor progression.21–23 It has been reported that Cdk5-mediated phosphorylation of talin1 leads to β1 integrin activation, resulting in increased metastatic potential of prostate cancer (PCa) cells.24 Many studies have reported that talin1 is upregulated in HCC, and talin1 serum levels in HCC patients are significantly higher.25,26 In this study, we demonstrate that talin1 can protect HCC cells from apoptosis via increasing the activity of the PI3K/Akt/NF-κB signaling pathway. These results provide a novel mechanism via which talin1 activates the PI3K/Akt/NF-κB signaling pathway to inhibit apoptosis and promote the proliferation potential of the HCC cells.
2. Materials and methods
2.1. Cell culture
The HCC cell lines HepG2 and LM-3 and the liver cell line Chang were purchased from Shanghai Institute of Chinese Academy of Sciences (China). All cells were maintained in Dulbecco's modified Eagle's medium (Cat#C11965, Invitrogen) supplemented with 10% v/v inactivated fetal bovine serum (HyClone, Logan, Utah, USA), 1% penicillin (100 U mL−1), and streptomycin (100 μg mL−1) in a humid environment at 37 °C with 95% normal air and 5% CO2.2.2. Vector construction and transfection
To investigate the effect of talin1 overexpression in cells, a pair of primers (forward: 5′-CCGGATCCATGGTTGCACTTTCACT-3′; reverse: 5′-CCGAATTCTAGAAGAGGCTTCTTT-3′) were designed to amplify talin1 as well as AKT and RIP1, cloned into a pCDNA3.1+ expression vector (Invitrogen). Transfection was performed using the Lipofectamine™ 2000 reagent (Invitrogen, Carlsbad, CA). Briefly, cells were inoculated into each well on 6-well plates, and plasmid and liposomal transfection reagent were added. Cells were transfected with plasmids and cloned under G418 selection (Geneticin, Gibco Brl, Grand Island, NY).2.3. Lentivirus-mediated stable talin1 knockdown
The lentiviral expression systems were purchased from System Biosciences (SBI, Mountain View, CA, USA). The shRNA sequence targeting the TLN-1 gene (GenBank. No. NM_006289.3) (5′-GCTCGAGATGGCAAGCTTAAA-3′) and one nonspecific sequence 5′-TTCTCCGAACGTGTCACGTTTC-3′ (scramble shRNA) were designed. After co-transfection, the virus media were harvested. Cells were transduced for 72 h with lentivirus mediated talin1-shRNA and scramble shRNA under puromycin (0.5 mg mL−1; Sigma, USA) selection. Passaging cells into DMEM containing 10% FBS and 0.5 mg mL−1 of puromycin for 12 days (changing the medium every 3 days) and stably transduced cells were obtained.2.4. Caspase activity assay
The activation of caspase-8, -9 was detected via the caspase activity assay. Briefly, cells in 96-well plates were transfected with talin1 and control miRNA as abovementioned. After 24 h, 20 μL of lysis buffer was added to each well. The cell lysate was incubated with 5 μL of chromogenic substrate at room temperature in the dark for 20 min. The optical density of each well was measured by a plate reader at 560 nm.2.5. Quantitative real-time PCR
An RNeasy Mini Kit (QIAGEN, Valencia, CA) was exploited to extract the total RNA samples from cell lines; then, the complementary DNA samples were prepared using a High Capacity cDNA Reverse Transcription Kit (Applied Biosystems Inc.). The following primer pairs were used: talin-1, sense, 5′-TGTAGAGGAGCACGAGACGC-3′; anti-sense, 5′-AAGGAGACAGGGTGGGAGC-3′; GAPDH, sense: 5′-CATGAGAAGTATGACAACAGCCT-3′; and anti-sense, 5′-AGTCCTTCCACGATACCAAAGT-3′. Relative gene expression was quantified by real-time PCR using SYBR Premix Ex Taq™ II (TaKaRa Bio, Dalian, China) via a Lightcycler 480 Real Time PCR System (Roche Diagnostics, Meylan, France). Ct values for talin-1 were normalized to those of GAPDH (ΔCt).2.6. Western blotting
For western blotting, cells were lysed in an M-PER mammalian protein extraction reagent (Thermo, USA), and equal amounts of protein were resolved by SDS-PAGE. Subsequently, the gel-separated proteins were blotted. The following specific primary antibodies were used to visualize proteins: anti-talin1 (ab78291, Abcam, Cambridge, MA, USA) and anti-β-actin (A5441, SIGMA). Bcl2, Bcl-xl, Bax, Bak, PARP, cytochrome oxidase IV (COX IV), cytochrome c (cyt-c), caspase-8, caspase-9, Bid, p-Akt, p50, p65, p105, RIP1, IKKβ, and IκBα antibodies were obtained from Cell Signaling Technology, Inc. (Beverly, MA, USA). Proteins were detected using a Pierce ECL western blotting substrate (Thermo).2.7. Mitochondrial membrane potential (MMP) assay
Mitochondrial membrane potential was measured using the JC-1 fluorescent probe (Invitrogen; Carlsbad, CA, USA). Briefly, talin 1 shRNA-treated HepG2 cells were mixed with 1 JC-1 staining solution and incubated at 37 °C for 15 min. After incubation, cells were washed and analyzed under a fluorescent microscope. Red fluorescence was observed at 525 nm excitation and 590 nm emission, and green fluorescence was observed at 490 nm excitation and 530 nm emission.2.8. Flow cytometry
For measuring apoptosis, transfected cells were dual stained using an annexin V-FITC/7-amino-actinomycin D (7-AAD) kit (Beckman Coulter) according to the manufacturer's protocol. The stained cells were immediately analyzed by flow cytometry using a Cytomics FC500 MPL cytometer, and data were obtained and analyzed using the CXP Version 2.2 software (Beckman Coulter).2.9. Statistical analysis
The data were analyzed using Social Sciences (SPSS) 17.0 software (SPSS Inc., Chicago, IL, USA). Student t test or analysis of variance (ANOVA) was performed to compare the difference between groups if the data were normally distributed. Statistical significance was defined as P < 0.05. P-values have been annotated in the text and figure legends.3. Results
3.1. Effects of talin1 knockdown on apoptosis in HCC cells
Talin1 overexpression has been reported to promote cancer cell adhesion, migration, and invasion.21 To explore whether talin1 was highly expressed in hepatoma cells, we assessed the expression of talin1 in LM-3, HepG2, MHCC-97H, SMMC-7721, and BEL-7402 cell lines through western blot. As shown in Fig. 1a, the HCCs cell lines LM-3, HepG2, and MHCC-97H overexpressed talin1, whereas the expressions of talin1 protein were very low in SMMC-7721 and BEL-7402 cell lines. To assess the effects of talin1 on apoptosis in the HCC cells, we used talin1 shRNA to establish stable talin1 knockdown HepG2 cell lines. Talin1 protein levels in the cells transduced with lentivirus-mediated talin1-shRNA (experimental group) were markedly reduced when compared with those in non-transduced cells (Fig. 1b). The apoptotic potential of talin1 was measured by flow cytometry after staining with annexin V-FITC/7-AAD. After talin1 knockdown, induction of apoptosis in HepG2 cells was dramatically increased when compared with that in normal talin1 expression groups (Fig. 1c). To study the mechanism via which talin1 affected apoptosis, western blot analysis with antibodies against PARP, caspase-8, caspase-9 or Bid was conducted. As shown in Fig. 1d, talin1 knockdown led to the activation of apoptotic proteins including caspase-8 and caspase-9 and its substrate, PARP, whereas it decreased the Bid protein level in the HepG2 cells. Moreover, we assessed the caspase-8, -9 activities through flow cytometry. Fig. 1d shows that caspase-8, -9 activities were markedly increased following talin1 knockdown in the HepG2 cells, which was consistent with western blot results. Then, we pre-treated the cells with the caspase inhibitor z-VAD-fmk (20 μM) before the cells were transduced with talin1 shRNA. While the cell apoptosis proportion in the talin1 shRNA alone group increased, the apoptosis proportion significantly decreased in the talin1 shRNA plus z-VAD group (Fig. 1f); this suggested that z-VAD specifically inhibited the effect of knocked down talin1 on apoptosis in HepG2 cells. In addition, the effects of talin1 shRNA on the levels of Bcl2 family proteins, Bax, Bak, and cytochrome c, as well as the MMP values were examined. As shown in Fig. 2a, the results revealed a marked decrease in mitochondrial cytochrome c and an increase in cytosolic cytochrome c following treatment with talin1 shRNA. The expression levels of proapoptotic Bax, and Bak proteins obviously enhanced, whereas those of antiapoptotic Bcl2 and Bcl2-xl proteins remarkably decreased in response to talin1 shRNA treatment (Fig. 2b). Moreover, talin1 shRNA treatment caused a decrease in MMP as compared to that in the untreated control (Fig. 2c).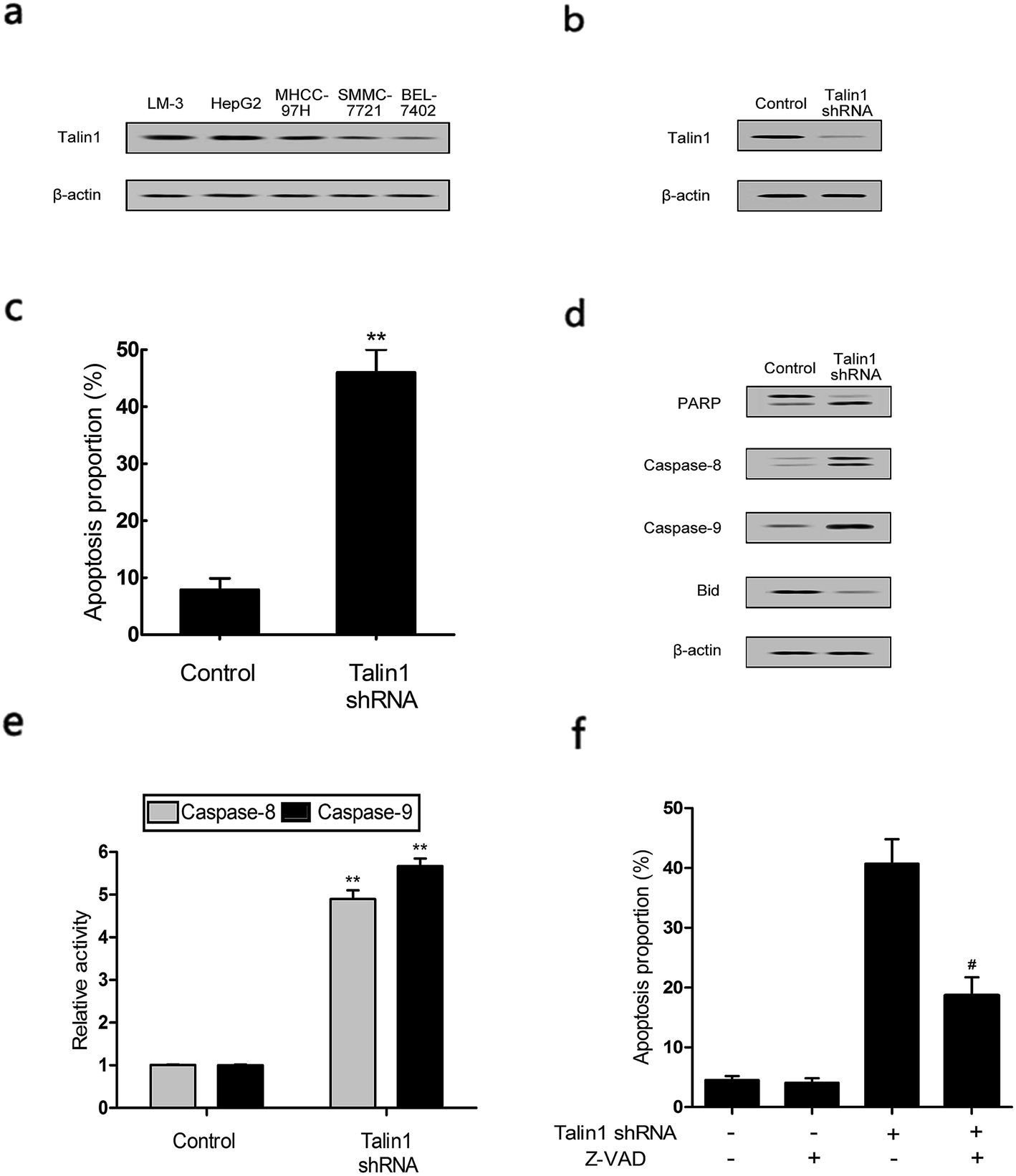 | ||
| Fig. 1 Down-regulation of talin1-induced apoptosis in hepatoma carcinoma cell lines. (a) The expressions of talin1 protein in various HCC cells were determined by western blot. (b) Talin1 protein expression levels were determined by western blot in HepG2 cell lines. (c) The effect of talin1 knockdown on apoptosis in HCC cells. Cells were stained with annexin V-FITC/7-AAD and analyzed by flow cytometry. (d) The protein expression levels of PARP, caspase-8, caspase-9, and Bid were determined by western blot. (e) The effect of talin1 knockdown on caspase-8, -9 activities in HepG2 cells. (f) Cells were treated with or without talin1 shRNA for 48 h after 2 h pre-treatment with the caspase inhibitor z-VAD-fmk (20 μM); then, they were stained with annexin V-FITC/7-AAD and analyzed by flow cytometry. Results were expressed as the mean ± S.D. from three independent experiments. **P < 0.01 versus control. #P < 0.05 versus talin1 shRNA independent treatment. | ||
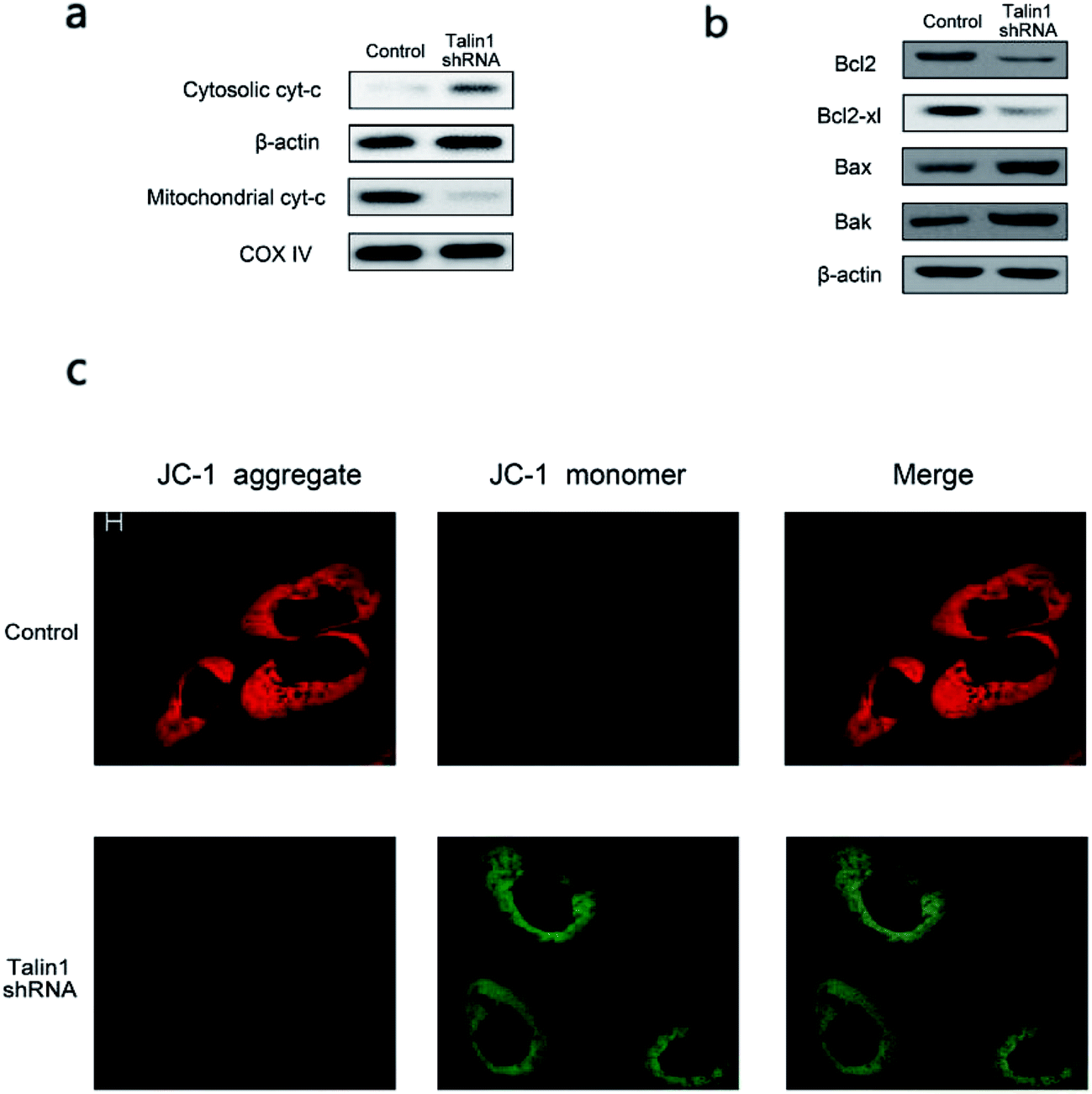 | ||
| Fig. 2 Down-regulation of talin1-induced MMP decline. (a) The protein expression levels of cytochrome c in cytosolic and mitochondrial fractions. (b) The protein expression levels of Bcl2, Bcl-xl, Bax, and Bak in HepG2 cell lines. (c) JC-1 staining revealed down-regulation of talin1-induced MMP decline. In JC-1 stained cells, red fluorescence (JC-1 aggregate) represented a high mitochondrial membrane potential, whereas green fluorescence of the JC-1 monomer depicted the loss of mitochondrial membrane potential. | ||
3.2. Talin1 attenuates the TNF-α and TRAIL-induced apoptosis in HCC cells
TNF-α is a multifunctional cytokine derived from activated macrophages and plays major roles in cell apoptosis, survival, and inflammation.27 To investigate whether talin1 could affect TNF-α-induced apoptosis, we used the talin1 plasmid to establish stable talin1 overexpression in BEL-7402 cell lines. As shown in Fig. 3a, talin1 protein levels were significantly increased after plasmid transfection when compared with those of the control group. According to fluorescence microscopy, the transduction efficiency in BEL-7402 cells (the percentage of GFP-positive cells) was >50% (Fig. 3b). We pre-treated the cells with TNF-α (25 ng mL−1) before they were transduced with talin1 plasmids. As shown in Fig. 3c, TNF-α significantly increased apoptosis in a time-dependent manner, whereas overexpression of talin1 resulted in a marked blockade of the activation of TNF-α-induced apoptosis. TRAIL is a closely related TNF-α and FasL members of the tumor necrosis factor family. TRAIL enhances apoptosis via liaisons such as death receptor 4 (DR4, TRAIL-R1) and death receptor 5 (DR5, TRAIL-R2), leading to the formation of the a death-inducing signaling complex (DISC) with subsequent binding of caspase-8.28 We also pre-treated the cells with TRAIL (25 ng mL−1) before they were transduced with talin1 plasmids and found that the apoptosis of BEL-7402 cells stimulated with TRAIL could be rescued by overexpressing talin1 (Fig. 3d). These results collectively suggested that talin1 could inhibit HCC cell apoptosis through attenuating the TNF-α and TRAIL signaling pathway.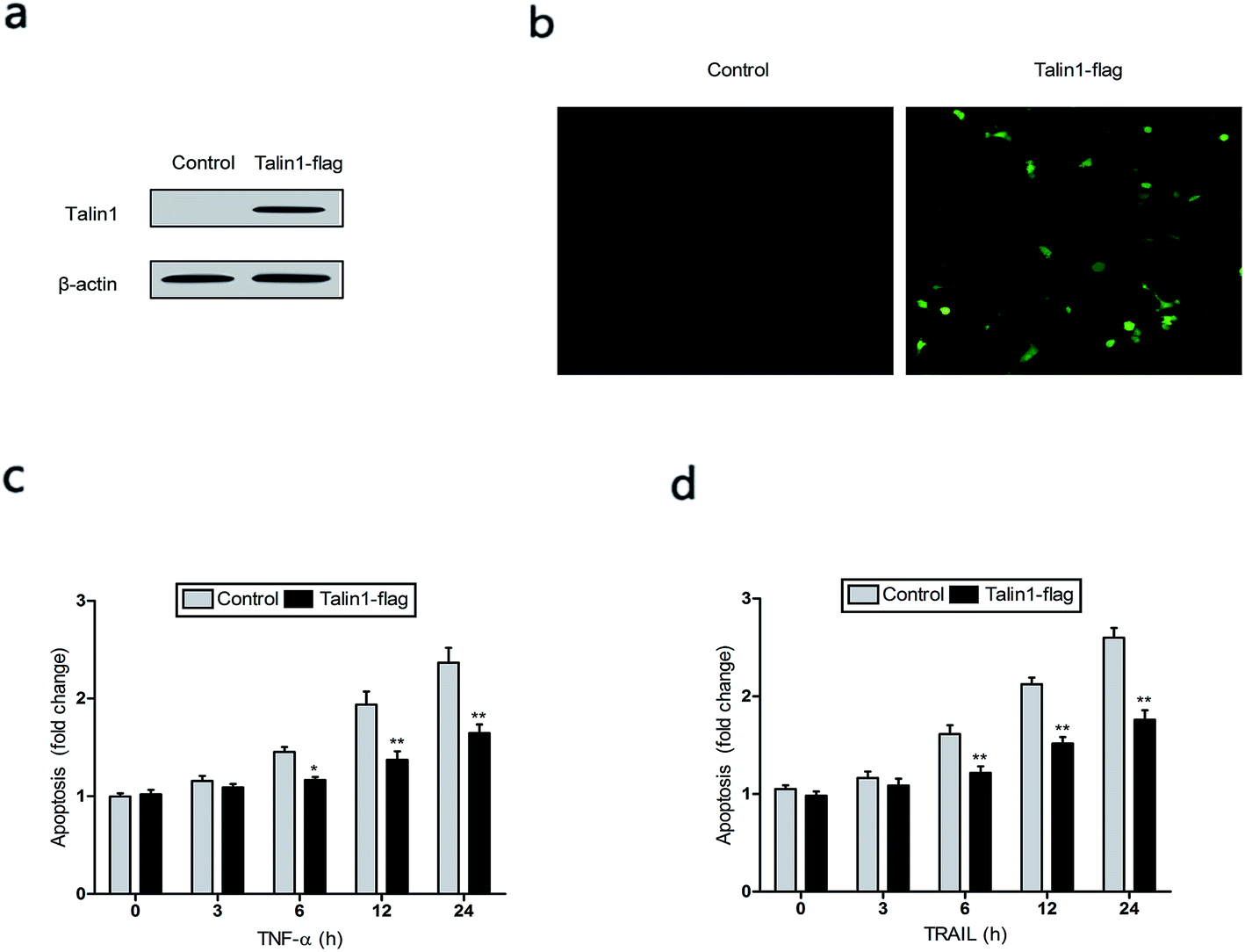 | ||
| Fig. 3 Talin1 attenuated TNF-α and TRAIL-induced apoptosis in hepatoma cells. (a) Talin1 protein expression levels were determined by western blot at 72 h after transduction in BEL-7402 cells. β-Actin was used as an internal control. (b) The transduction efficiency of talin1 was detected by fluorescence microscopy in BEL-7402 cells (magnification: ×200). (c and d) Talin1 attenuated TNF-α and TRAIL-induced apoptosis. Cells were treated with or without talin1 plasmids for 48 h after pre-treatment with TNF-α (25 ng mL−1) and TRAIL (25 ng mL−1) for different amounts of time. Then, cells were stained with annexin V-FITC/7-AAD and analyzed by flow cytometry. Results are expressed as the mean ± S.D. from three independent experiments. *P < 0.05 versus control, **P < 0.01 versus control. | ||
3.3. Talin1 knockdown promotes cell apoptosis via the PI3K/Akt pathway in HCC cells
To determine whether the talin1 knockdown could promote cell apoptosis via the PI3K/Akt pathway, a western blot analysis was performed. As shown in Fig. 4a, talin1 knockdown led to a significant decrease in the expression of phosphorylated Akt signal molecules in HepG2 cells. To further investigate the molecular mechanism mediating the effect of talin1 knockdown on the apoptotic process, cells were transfected with pCDNA-Akt to activate the Akt pathway after transduction. Fig. 4b shows that pCDNA-Akt could readily increase the phosphorylation of Akt protein in the HepG2 cells, whereas cells treated with talin1 shRNA could counteract the effect of pCDNA-Akt (Fig. 4b). Flow cytometry analysis showed that Akt transfection could inhibit the effect of talin1 on apoptosis, whereas cells treated with talin1 shRNA could arrest the Akt pathway and promote apoptosis in the HepG2 cells (Fig. 4c). In addition, we attempted to explore the molecular mechanism of the effect of Akt and talin1 shRNA on apoptosis. As shown in Fig. 4d, Akt inhibited the effects of talin1 knockdown on apoptosis and significantly decreased the caspase-8, caspase-9, and PARP protein levels, whereas it upregulated the Bid protein level. However, treatment of cells with talin1 shRNA plus pCDNA-Akt led to an increase in the expression of caspase-8, caspase-9, and PARP, with a decrease in the expression of Bid. Overall, our data suggested that talin1 could increase the activation of the PI3K/Akt pathway and decrease the expression of pro-apoptotic proteins such as caspase-8, caspase-9, and PARP, whereas it could increase the total Bid; therefore, inhibiting HCC cell apoptosis and resulting in tumor progression.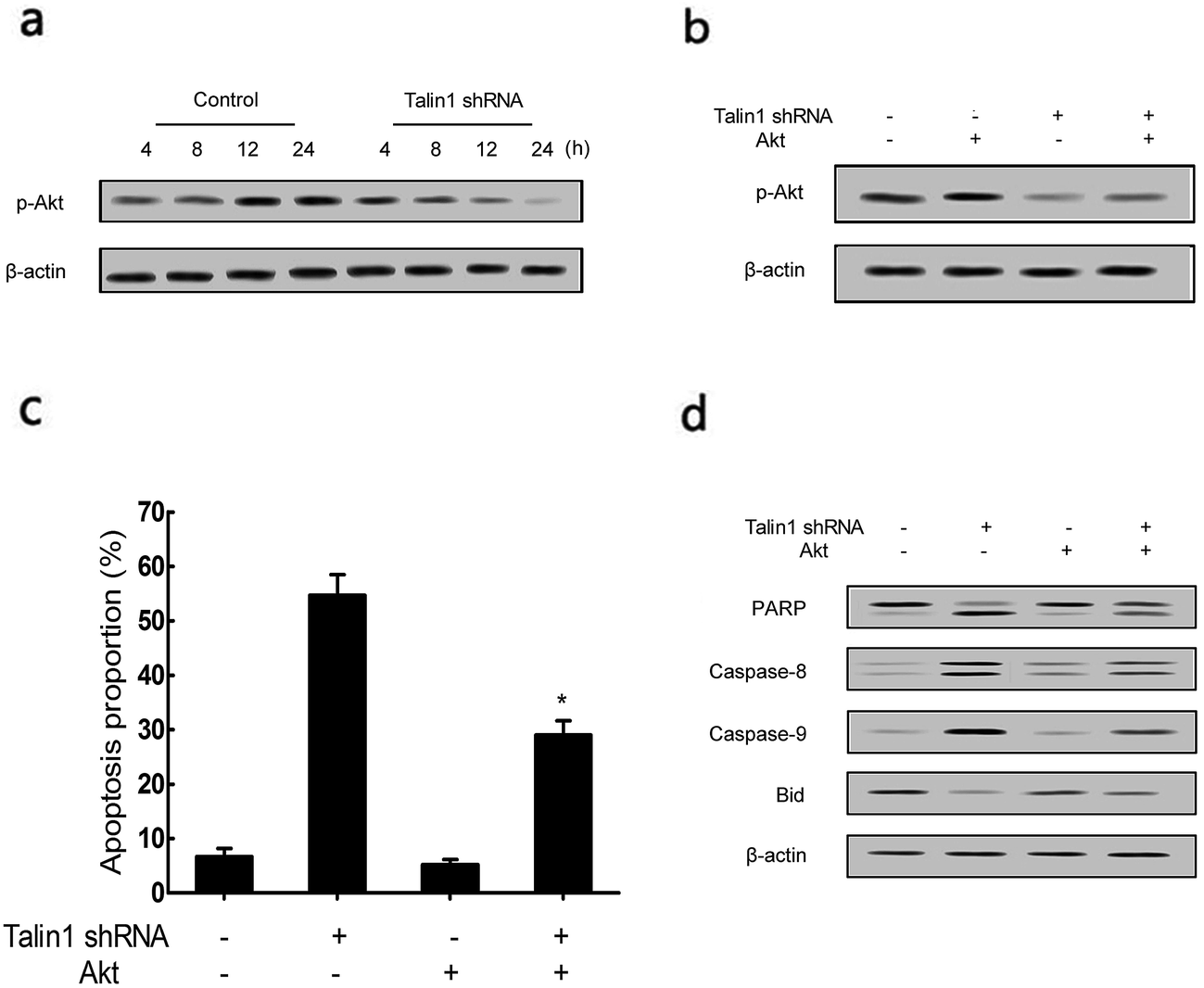 | ||
| Fig. 4 Down-regulation of talin1 inhibited PI3K/Akt signaling in HCC cells. (a) Protein expression of p-Akt was determined by western blot followed by treatment with or without talin1 shRNA for an indicated time period in HepG2 cells. (b) Cells were treated with or without talin1 shRNA following transfection with pCDNA-Akt for 24 h. The expression levels of p-Akt and Akt proteins were determined by western blot. (c) Cells were stained with annexin V-FITC/7-AAD and analyzed by flow cytometry. (d) The expressions of PARP, caspase-8, caspase-9, and Bid were determined by western blot. Results are expressed as mean ± S.D. from three independent experiments. *P < 0.05 versus talin1 shRNA independent treatment. | ||
3.4. Talin1 affects the apoptotic process through the NF-κB pathway
To explore whether talin1 could affect the apoptotic process through the NF-κB pathway, we examined the expression of the NF-κB family members upon the change of talin levels. As shown in Fig. 5a, talin1 knockdown arrested the expression of p50, p65, and p105 proteins, whereas it increased the RIP1 protein level. Then, we used the adenovirus vector to establish stable IKKβ or IκBα overexpressed HepG2 cell lines. Fig. 5b indicated that an upregulated IKKβ level could increase the NF-κB protein level, whereas talin1 knockdown could significantly inhibit the expression of NF-κB. However, IκBα overexpression inhibited NF-κB activation, and talin1 knockdown plus IκBα overexpression enhanced the inhibition of NF-κB activation (Fig. 5c). As shown in Fig. 5d, talin1 knockdown plus the IκBα overexpression group could dramatically promote cell apoptosis as compared to talin1 knockdown plus the IKKβ overexpression group in HepG2 cells. In addition, we showed that talin1 knockdown plus the IκBα overexpression group led to upregulation of caspase-8, caspase-9, and PARP activation and downregulation of the Bid protein level when compared with talin1 knockdown plus the IKKβ overexpression group (Fig. 5e). These results pointed to the fact that talin1 could inhibit the apoptosis of HCCs via activation of the NF-κB pathway.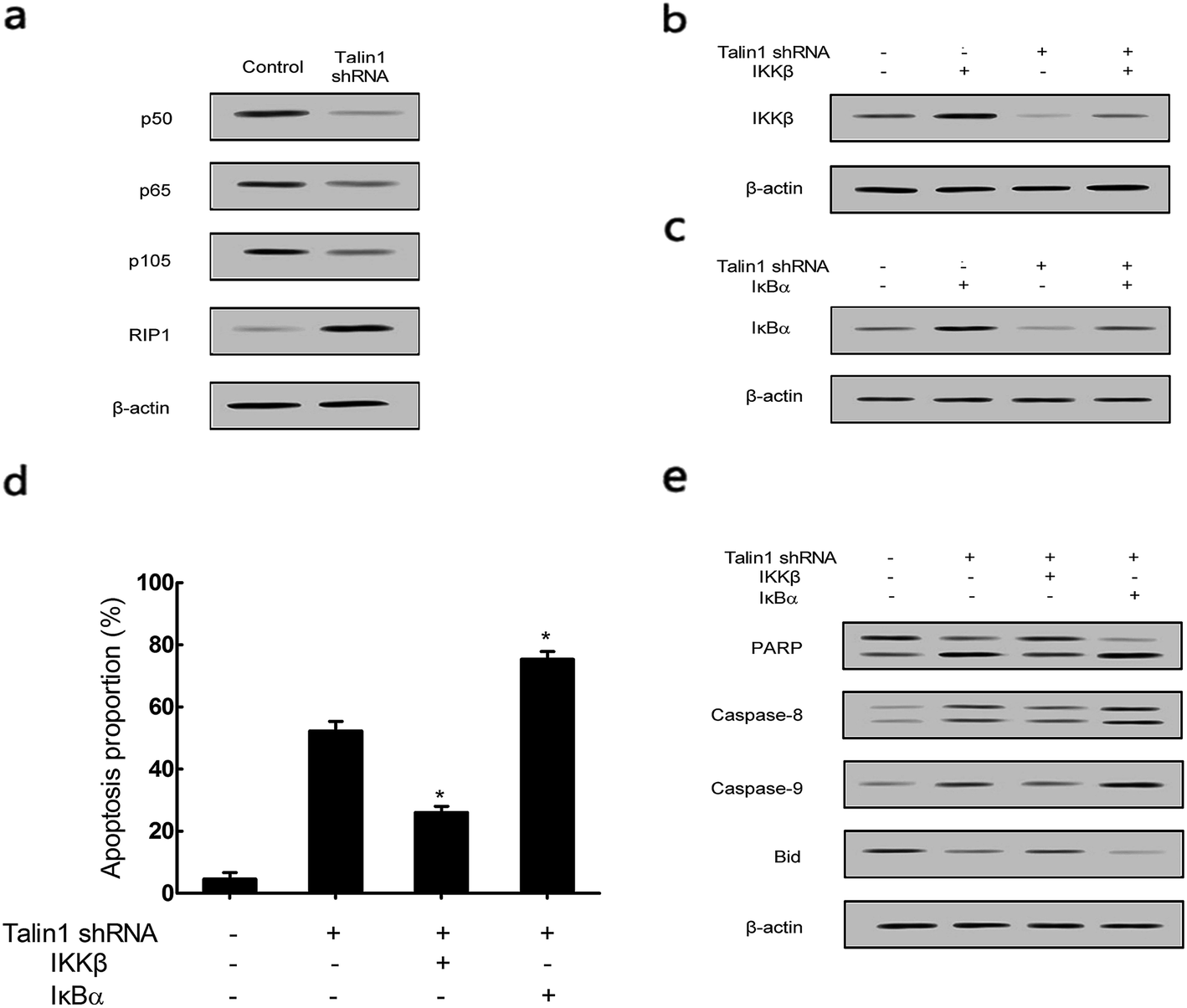 | ||
| Fig. 5 Down-regulation of talin1 inhibited NF-κB signaling in HCC cells. (a) The protein expression levels of p50, p65, p105, and RIP1 were determined by western blot after knocking-down of talin1 in HepG2 cells. (b) The protein expression of IKKβ was determined by western blot after knocking-down of talin1 following treatment with the over-expression of IKKβ. (c) The protein expression of IκBα was determined by western blot after knocking-down of talin1 following treatment with the over-expression of IκBα. (d) Cells were treated with talin1 shRNA following up-regulation of NF-κB by transfection with IKKβ or down-regulation of NF-κB by transfection with IκBα. Cells were stained with annexin V-FITC/7-AAD and analyzed by flow cytometry. (e) The expressions of PARP, caspase-8, caspase-9, and Bid were determined by western blot. Results were expressed as the mean ± S.D. from three independent experiments. *P < 0.05 versus talin1 shRNA independent treatment. | ||
3.5. Attenuation of TNF-α and TRAIL-induced apoptosis by talin1 can be reversed by RIP1
RIP1 has been suggested to be essential for TRAIL-induced activation of the JNK and NF-κB pathways to determine cell death and survival.29 As shown in Fig. 6a, cells pre-treated with TNF-α and TRAIL followed by talin1 overexpression inhibited RIP1 activation when compared with the control group in BEL-7402 cells. Moreover, the mRNA expression of RIP1 also decreased under these conditions (Fig. 6b). pCDNA-RIP1 is a plasmid containing RIP1, which could enhance RIP1 expression after transfection in BEL-7402 cells (Fig. 6c). We next tried to explore whether RIP1 could mediate the effect of talin1 on TNF-α or TRAIL-induced apoptosis using flow cytometry analysis. As indicated in Fig. 6d, talin1 could attenuate TNF-α or TRAIL-induced apoptosis, whereas the apoptotic proportion was markedly increased after transfection with pCDNA-RIP1. Overall, we speculated that RIP1 could reverse the attenuating effect of talin1 on TNF-α or TRAIL-induced apoptosis.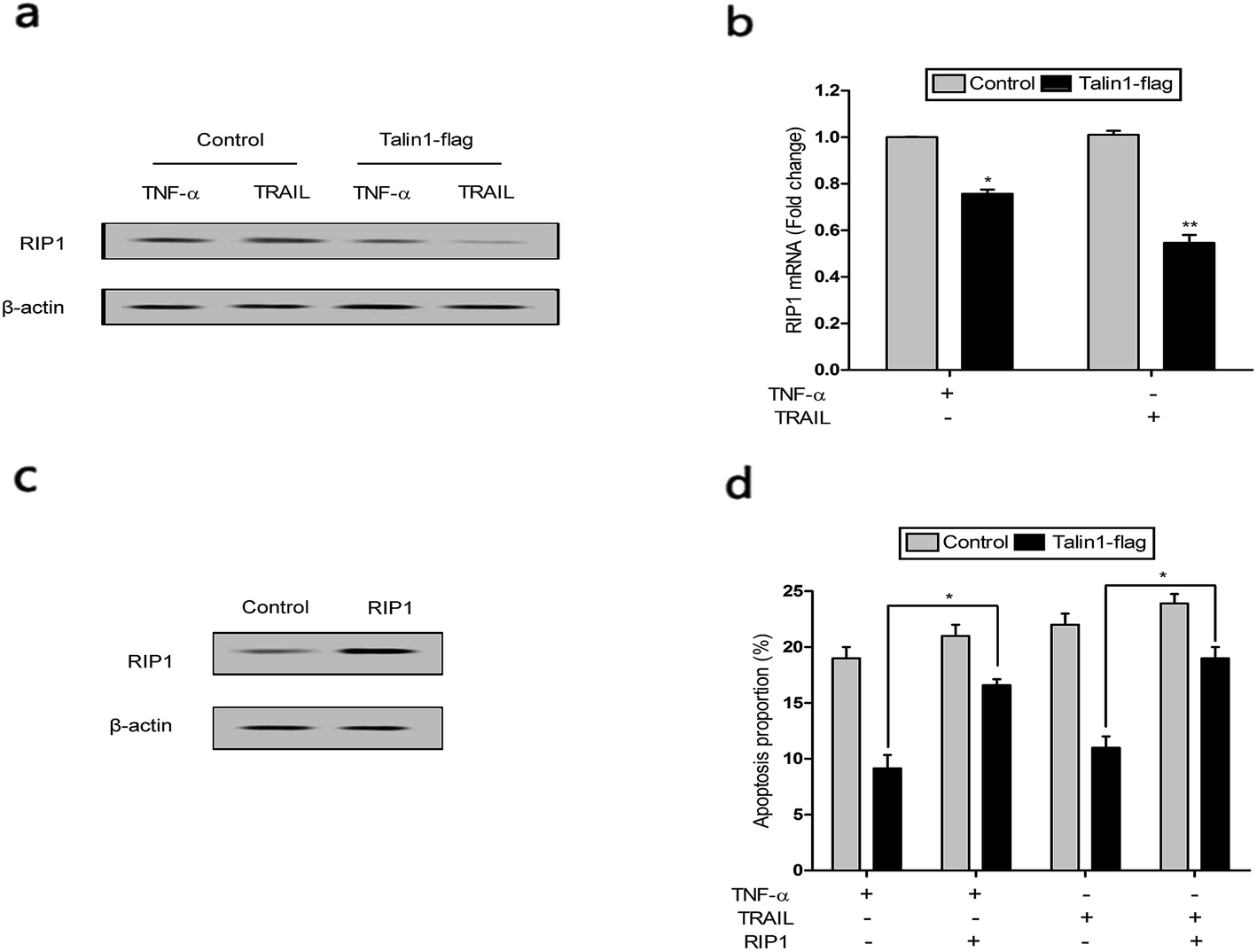 | ||
| Fig. 6 Talin1 attenuated TNF-α or TRAIL-induced apoptosis that could be reversed by RIP1. The expressions of RIP1 protein (a) and mRNA (b) were determined by western blot after treatment with the over-expression of talin1 following pre-incubation with TNF-α and TRAIL in BEL-7402 cells. *P < 0.05 versus control, **P < 0.01 versus control. (c) The expression of RIP1 protein was determined by western blot following transfection with pCDNA-RIP1 in BEL-7402 cells. (d) Cells were treated with or without talin1 shRNA for 24 h following pre-treatment with TNF-α or TRAIL and/or transfection with pCDNA-RIP1. Cell apoptosis was analyzed by flow cytometry using annexin V-FITC/7-AAD staining. Results are expressed as the mean ± S.D. from three independent experiments. *P < 0.05 versus TNF-α or TRAIL group. | ||
4. Discussion
Clinical studies have demonstrated that the talin1 level is up-regulated in poorly differentiated tumors and in cancer cells undergoing angiogenesis or metastasis.30 Fang et al. have shown that the high levels of both talin1 and talin2 correlate with tumorigenicity in human HCC; this indicates that these molecules constitute useful molecular targets in HCC diagnosis and/or treatment.31 Alsebaey et al. found that patients with HCC32 had higher values of serum AFP, serum talin1, AST/ALT ratio, FIB4 score, and fibro-α score than those with cirrhosis, contrary to the fibrosis index score.32 However, the mechanisms underlying the activation and regulation of talin1 in HCC remain poorly understood. The present study demonstrated that talin1 was upregulated in the HCC cell lines LM-3, HepG2, and MHCC-97H, whereas it was lowly expressed in SMMC-7721 and BEL-7402 cell lines. Talin1 knockdown promoted HCC cell apoptosis; this confirmed the involvement of talin1 in HCC progression.Apoptosis is a spontaneous and programmed process of cell death under normal physiological or pathological conditions. Caspases are able to cleave PARP, which is considered a hallmark of apoptosis.33–36 Upon exogenous apoptotic stimulation, caspase-8 is activated and promotes cell apoptosis through the activation of caspase-3, caspase-6, and caspase-7. However, when the amount of activated caspase-8 is not sufficient to initiate cell apoptosis, caspase-8 can cleave Bid (BH3-interacting death agonist) and translate activated Bid.37 The present study showed that talin1 knockdown could promote apoptosis in HCC cells by increasing caspase-8 and caspase-9 activation. Moreover, PARP, caspase-8, and caspase-9 protein levels were increased, whereas the Bid protein level was markedly decreased after treatment with talin1 shRNA. We also found that caspase inhibitor z-VAD-fmk could counteract the regulatory effect of talin1 knockdown on apoptosis in HCC cells. In addition, we found a marked decrease in mitochondrial cytochrome c and an increase in cytosolic cytochrome c following treatment with talin1 shRNA. The expression levels of proapoptotic Bax and Bak proteins were obviously enhanced, whereas those of antiapoptotic Bcl2 and Bcl2-xl proteins remarkably decreased in response to talin1 shRNA treatment. Furthermore, talin1 shRNA treatment caused a marked decrease in MMP as compared to the untreated control. Thus, it is reasonable to speculate that the knockdown of talin1 could induce apoptosis via extrinsic and intrinsic pathways in HCC cells.
TNF-α has been known as an endogenous mediator of apoptosis, which is implicated in the pathogenesis of a wide spectrum of human diseases including cancer.38 RIP1 is a crucial regulator in TNF-α-mediated apoptosis, which acts as a cell-death switch in response to the TNF-α signaling pathway. TNF-α induces apoptosis through the formation of the RIP1-FADD-caspase-8 complex, which triggers caspase-8 activation and subsequent apoptosis.39,40 Our results showed that talin1 overexpression significantly attenuated TNF-α and TRAIL-induced apoptosis and decreased the RIP1 protein level. However, cell transfection with pCDNA-RIP1 could increase the apoptotic proportion and reverse the effects of talin1 on TNF-α and TRAIL-induced apoptosis. These data indicate that talin1 protects the HCC cells from apoptosis by inhibiting the TNF-α pathway.
Several studies have reported that increased PI3K/Akt and ERK activation can induce the proliferation of HCC cells, prevent HCC cell apoptosis,41 and become an independent prognostic index for HCC patients.42 In addition, the Akt signaling pathway also promotes activation of the NF-κB signaling pathway, which inhibits apoptosis by causing up-regulation of key anti-apoptotic proteins such as Bcl-2, Bcl-Xl, XIAP, and survivin.43,44 In this study, we have hypothesized that a functional link might exist between talin1 and PI3K/Akt/NF-κB pathways in the apoptosis process and activation of these pathways may promote proliferation of HCC cells. We found that talin1 knockdown inhibited phosphorylation of Akt in a time-dependent manner. pCDNA-Akt could suppress the apoptosis of HCC cells through activating the PI3K/Akt pathway. However, talin1 knockdown could specially reverse the effects of pCDNA-Akt on apoptosis via increasing the expression of caspase-8, caspase-9, and PARP protein levels and inhibiting Bid activation. In addition, the activations of p50, p65, and p105 proteins in the NF-κB pathway were all down-regulated after treatment with talin1 shRNA in HCC cells. TAB2 or TAB3 promotes phosphorylation of IKKβ through binding with TAK1, resulting in phosphorylated IκB molecules, triggering their K48-linked polyubiquitination via βTrCP followed by proteasomal degradation of IκB and release of active NF-κB.45 The physiological inhibitor of NF-κB, IκBα, is responsible for systematic control of the process of NF-κB signaling, which blocks the nuclear localization and transcriptional activity of NF-κB.46 Herein, we used IKKβ and IκBα to regulate NF-κB activation and found that IKKβ plus talin1 knockdown could inhibit the apoptosis of HCC cells, whereas the NF-κB inhibitor IκBα plus talin1 knockdown promotes the apoptosis of HCC cells through upregulating the activation of caspase-8, caspase-9, and PARP and decreasing the Bid protein level when compared with the talin1 knockdown group. We speculate that talin1 may activate the PI3K/Akt/NF-κB signaling pathway to inhibit apoptosis and promote proliferation of HCC cells.
In summary, the present study may have a variety of implications for understanding the role of talin1 on HCC apoptosis progression via a direct interaction. We found that talin1 could suppress apoptosis via increasing the activation of PI3K/Akt/NF-κB and inhibiting the TNF-α signaling pathway in HCC cells. It is possible that targeting of talin1 may inhibit HCC proliferation and be a potential therapeutic strategy in the management of HCC.
Conflicts of interest
Disclosure of conflict of interest: none.Acknowledgements
This work was supported by the National Nature and Science Foundation (NSF) grant (81001324 and 81373163 to RT) and Zhejiang Provincial Nature and Science Foundation (NSF) grant for “Outstanding Youth” (No. LR15H100001 to RT). The work was also supported by Shanghai Health Bureau scientific research found projects grant (2016MHZ11 to Zhiqiu Hu).References
- H. B. El-Serag and K. L. Rudolph, Hepatocellular carcinoma: epidemiology and molecular carcinogenesis, Gastroenterology, 2007, 132(7), 2557–2576 CrossRef CAS PubMed.
- M. M. Kirstein and A. Vogel, The pathogenesis of hepatocellular carcinoma, J. Dig. Dis., 2014, 32(5), 545–553 CrossRef PubMed.
- S. Torrecilla and J. M. Llovet, New molecular therapies for hepatocellular carcinoma, Clin. Res. Hepatol. Gastroenterol., 2015, 39(Suppl. 1), S80–S85 CrossRef CAS PubMed.
- A. Schlachterman, W. W. Craft Jr, E. Hilgenfeldt, A. Mitra and R. Cabrera, Current and future treatments for hepatocellular carcinoma, World J. Gastroenterol., 2015, 21(28), 8478–8491 CrossRef PubMed.
- K. W. Kinnally, P. M. Peixoto, S. Y. Ryu and L. M. Dejean, Is mPTP the gatekeeper for necrosis, apoptosis, or both?, Biochim. Biophys. Acta, 2011, 1813(4), 616–622 CrossRef CAS PubMed.
- C. C. Smith and D. M. Yellon, Necroptosis, necrostatins and tissue injury, J. Cell. Mol. Med., 2011, 15(9), 1797–1806 CrossRef CAS PubMed.
- S. Elmore, Apoptosis: a review of programmed cell death, Toxicol. Pathol., 2007, 35(4), 495–516 CrossRef CAS PubMed.
- X. Saelens, N. Festjens, L. Vande Walle, M. van Gurp, G. van Loo and P. Vandenabeele, Toxic proteins released from mitochondria in cell death, Oncogene, 2004, 23(16), 2861–2874 CrossRef CAS PubMed.
- N. D. Perkins, Integrating cell-signalling pathways with NF-kappaB and IKK function, Nat. Rev. Mol. Cell Biol., 2007, 8(1), 49–62 CrossRef CAS PubMed.
- M. S. Hayden and S. Ghosh, NF-kappaB, the first quarter-century: remarkable progress and outstanding questions, Genes Dev., 2012, 26(3), 203–234 CrossRef CAS PubMed.
- B. Hoesel and J. A. Schmid, The complexity of NF-kappaB signaling in inflammation and cancer, Mol. Cancer, 2013, 12, 86 CrossRef CAS PubMed.
- J. Zhang, L. Li, A. S. Baldwin Jr, A. D. Friedman and I. Paz-Priel, Loss of IKKbeta but Not NF-kappaB p65 Skews Differentiation towards Myeloid over Erythroid Commitment and Increases Myeloid Progenitor Self-Renewal and Functional Long-Term Hematopoietic Stem Cells, PLoS One, 2015, 10(6), e0130441 Search PubMed.
- T. Tao, C. Cheng, Y. Ji, G. Xu, J. Zhang and L. Zhang, et al., Numbl inhibits glioma cell migration and invasion by suppressing TRAF5-mediated NF-kappaB activation, Mol. Biol. Cell, 2012, 23(14), 2635–2644 CrossRef CAS PubMed.
- L. Jiang, J. Wu, Y. Yang, L. Liu, L. Song and J. Li, et al., Bmi-1 promotes the aggressiveness of glioma via activating the NF-kappaB/MMP-9 signaling pathway, BMC Cancer, 2012, 12, 406 CrossRef CAS PubMed.
- Z. J. Long, J. Xu, M. Yan, J. G. Zhang, Z. Guan and D. Z. Xu, et al., ZM 447439 inhibition of aurora kinase induces Hep2 cancer cell apoptosis in three-dimensional culture, Cell Cycle, 2008, 7(10), 1473–1479 CrossRef CAS PubMed.
- G. Song, G. Ouyang and S. Bao, The activation of Akt/PKB signaling pathway and cell survival, J. Cell. Mol. Med., 2005, 9(1), 59–71 CrossRef CAS PubMed.
- W. Hartmann, J. Kuchler, A. Koch, N. Friedrichs, A. Waha and E. Endl, et al., Activation of phosphatidylinositol-3'-kinase/AKT signaling is essential in hepatoblastoma survival, Clin. Cancer Res., 2009, 15(14), 4538–4545 CrossRef CAS PubMed.
- L. Sun, W. Li, W. Li, L. Xiong, G. Li and R. Ma, Astragaloside IV prevents damage to human mesangial cells through the inhibition of the NADPH oxidase/ROS/Akt/NFkappaB pathway under high glucose conditions, Int. J. Mol. Med., 2014, 34(1), 167–176 CAS.
- F. G. Giancotti and E. Ruoslahti, Integrin signaling, Science, 1999, 285(5430), 1028–1032 CrossRef CAS PubMed.
- M. Moser, K. R. Legate, R. Zent and R. Fassler, The tail of integrins, talin, and kindlins, Science, 2009, 324(5929), 895–899 CrossRef CAS PubMed.
- S. Sakamoto, R. O. McCann, R. Dhir and N. Kyprianou, Talin1 promotes tumor invasion and metastasis via focal adhesion signaling and anoikis resistance, Cancer Res., 2010, 70(5), 1885–1895 CrossRef CAS PubMed.
- D. A. Calderwood, Integrin activation, J. Cell Sci., 2004, 117, 657–666 CrossRef CAS PubMed.
- N. Alam, H. L. Goel, M. J. Zarif, J. E. Butterfield, H. M. Perkins and B. G. Sansoucy, et al., The integrin-growth factor receptor duet, J. Cell. Physiol., 2007, 213(3), 649–653 CrossRef CAS PubMed.
- J. K. Jin, P. C. Tien, C. J. Cheng, J. H. Song, C. Huang and S. H. Lin, et al., Talin1 phosphorylation activates beta1 integrins: a novel mechanism to promote prostate cancer bone metastasis, Oncogene, 2015, 34(14), 1811–1821 CrossRef CAS PubMed.
- H. Kanamori, T. Kawakami, K. Effendi, K. Yamazaki, T. Mori and H. Ebinuma, et al., Identification by differential tissue proteome analysis of talin-1 as a novel molecular marker of progression of hepatocellular carcinoma, J. Oncol., 2011, 80(5–6), 406–415 CrossRef CAS PubMed.
- M. M. Youns, A. H. Abdel Wahab, Z. A. Hassan and M. S. Attia, Serum talin-1 is a potential novel biomarker for diagnosis of hepatocellular carcinoma in Egyptian patients, Asian Pac. J. Cancer Prev., 2013, 14(6), 3819–3823 CrossRef PubMed.
- R. van Horssen, T. L. Ten Hagen and A. M. Eggermont, TNF-alpha in cancer treatment: molecular insights, antitumor effects, and clinical utility, Oncologist, 2006, 11(4), 397–408 CrossRef CAS PubMed.
- M. Frese-Schaper, J. A. Schardt, T. Sakai, G. L. Carboni, R. A. Schmid and S. Frese, Inhibition of tissue transglutaminase sensitizes TRAIL-resistant lung cancer cells through upregulation of death receptor 5, FEBS Lett., 2010, 584(13), 2867–2871 CrossRef CAS PubMed.
- W. Zhou and J. Yuan, Necroptosis in health and diseases, Semin. Cell Dev. Biol., 2014, 35, 14–23 CrossRef CAS PubMed.
- M. T. Lai, C. H. Hua, M. H. Tsai, L. Wan, Y. J. Lin and C. M. Chen, et al., Talin-1 overexpression defines high risk for aggressive oral squamous cell carcinoma and promotes cancer metastasis, J. Pathol., 2011, 224(3), 367–376 CrossRef CAS PubMed.
- K. P. Fang, W. Dai, Y. H. Ren, Y. C. Xu, S. M. Zhang and Y. B. Qian, Both Talin-1 and Talin-2 correlate with malignancy potential of the human hepatocellular carcinoma MHCC-97 L cell, BMC Cancer, 2016, 16, 45 CrossRef PubMed.
- A. Alsebaey and E. A. Ahmedy, Talin-1 and Non-invasive Fibrosis Models in the Assessment of Patients with Hepatocellular Carcinoma, Asian Pac. J. Cancer Prev., 2016, 17(8), 4077–4082 Search PubMed.
- I. M. Ghobrial, T. E. Witzig and A. A. Adjei, Targeting apoptosis pathways in cancer therapy, Ca-Cancer J. Clin., 2005, 55(3), 178–194 CrossRef PubMed.
- E. Szegezdi, S. E. Logue, A. M. Gorman and A. Samali, Mediators of endoplasmic reticulum stress-induced apoptosis, EMBO Rep., 2006, 7(9), 880–885 CrossRef CAS PubMed.
- M. Hassan, H. Watari, A. AbuAlmaaty, Y. Ohba and N. Sakuragi, Apoptosis and molecular targeting therapy in cancer, BioMed Res. Int., 2014, 2014, 150845 Search PubMed.
- M. Los, M. Mozoluk, D. Ferrari, A. Stepczynska, C. Stroh and A. Renz, et al., Activation and caspase-mediated inhibition of PARP: a molecular switch between fibroblast necrosis and apoptosis in death receptor signaling, Mol. Biol. Cell, 2002, 13(3), 978–988 CrossRef CAS PubMed.
- K. A. Sarosiek, X. Chi, J. A. Bachman, J. J. Sims, J. Montero and L. Patel, et al., BID preferentially activates BAK while BIM preferentially activates BAX, affecting chemotherapy response, Mol. Cell, 2013, 51(6), 751–765 CrossRef CAS PubMed.
- F. Van Herreweghe, N. Festjens, W. Declercq and P. Vandenabeele, Tumor necrosis factor-mediated cell death: to break or to burst, that's the question, Cell. Mol. Life Sci., 2010, 67(10), 1567–1579 CrossRef PubMed.
- N. Festjens, T. Vanden Berghe, S. Cornelis and P. Vandenabeele, RIP1, a kinase on the crossroads of a cell's decision to live or die, Cell Death Differ., 2007, 14(3), 400–410 CrossRef CAS PubMed.
- A. L. Mahul-Mellier, E. Pazarentzos, C. Datler, R. Iwasawa, G. AbuAli and B. Lin, et al., De-ubiquitinating protease USP2a targets RIP1 and TRAF2 to mediate cell death by TNF, Cell Death Differ., 2012, 19(5), 891–899 CrossRef CAS PubMed.
- I. Fabregat, C. Roncero and M. Fernandez, Survival and apoptosis: a dysregulated balance in liver cancer, Liver Int., 2007, 27(2), 155–162 CrossRef CAS PubMed.
- K. J. Schmitz, J. Wohlschlaeger, H. Lang, G. C. Sotiropoulos, M. Malago and K. Steveling, et al., Activation of the ERK and AKT signalling pathway predicts poor prognosis in hepatocellular carcinoma and ERK activation in cancer tissue is associated with hepatitis C virus infection, J. Hepatol., 2008, 48(1), 83–90 CrossRef CAS PubMed.
- A. R. Hussain, S. O. Ahmed, M. Ahmed, O. S. Khan, S. Al Abdulmohsen and L. C. Platanias, et al., Cross-talk between NFkB and the PI3-kinase/AKT pathway can be targeted in primary effusion lymphoma (PEL) cell lines for efficient apoptosis, PLoS One, 2012, 7(6), e39945 CAS.
- G. Sethi, K. S. Ahn and B. B. Aggarwal, Targeting nuclear factor-kappa B activation pathway by thymoquinone: role in suppression of antiapoptotic gene products and enhancement of apoptosis, Mol. Cancer Res., 2008, 6(6), 1059–1070 CrossRef CAS PubMed.
- J. A. Schmid and A. Birbach, IkappaB kinase beta (IKKbeta/IKK2/IKBKB)–a key molecule in signaling to the transcription factor NF-kappaB, Cytokine Growth Factor Rev., 2008, 19(2), 157–165 CrossRef CAS PubMed.
- S. Basak, H. Kim, J. D. Kearns, V. Tergaonkar, E. O'Dea and S. L. Werner, et al., A fourth IkappaB protein within the NF-kappaB signaling module, Cell, 2007, 128(2), 369–381 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |