
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Interactions between sirtuins and fluorogenic small-molecule substrates offer insights into inhibitor design†
Hua-Li Wang‡
,
Sha Liu‡,
Zhu-Jun Yu,
Chengyong Wu,
Linna Cheng,
Yuxi Wang,
Kai Chen,
Shu Zhou,
Qiang Chen,
Yamei Yu* and
Guo-Bo Li *
*
Key Laboratory of Drug Targeting and Drug Delivery System of Ministry of Education, West China School of Pharmacy, and State Key Laboratory of Biotherapy, Sichuan University, Chengdu, 610041, China. E-mail: liguobo@scu.edu.cn; yamei_yu@scu.edu.cn
First published on 20th July 2017
Abstract
Sirtuins are nicotinamide adenine dinucleotide (NAD+)-dependent lysine deacylases regulating metabolism and stress responses and are involved in human pathologies such as neurodegeneration. In this study, four fluorogenic small-molecule substrates, i.e., acetyl-(AcBKA), crotonyl-(CrBKA), succinyl-(SuBKA), and myristoyl-(MyBKA)-containing substrates, were synthesized and tested against three representative sirtuin isoforms (i.e., SIRT2, SIRT5, and SIRT6). Enzyme kinetic results indicate that the fluorogenic small-molecule substrates have similar sirtuin-isoform preference as compared to peptide substrates. ITC analyses reveal that AcBKA or MyBKA binding to SIRT2 is mainly driven by entropy, whereas SuBKA binding to SIRT5 is driven by enthalpy. The SIRT5:SuBKA complex crystal structure reveals a new substrate-binding mode that is different from peptide substrate binding modes, but involves Tyr102, Arg105, and other catalytically important residues on Loop S; this indicates that SuBKA is desuccinylated by SIRT5 probably through the catalytic mechanism proposed for peptide substrates. The biophysical and structural results presented herein will provide thermodynamic insights and key pharmacophore features for the development of selective sirtuin isoform-specific inhibitors.
Introduction
Sirtuins are nicotinamide adenine dinucleotide (NAD+)-dependent lysine deacylases (Fig. 1).1 Humans have seven sirtuin enzymes (SIRT1-7) that are divided into four classes: SIRT1-3 belong to class I, SIRT4 belongs to class II, SIRT5 belongs to class III, and SIRT6-7 belong to class IV.2 All sirtuins have a conserved catalytic core domain, but distinct N- or C-terminal extensions.3 Originally, sirtuins were known as deacetylases acting on histones, transcription factors, and metabolic enzymes.1,2 However, recently, sirtuins have been found to be able to catalyze the enzymatic removal of other acyl modifications besides acetyl moieties (Table S1†). For example, SIRT1-3 can remove propionyl and butyryl groups;4 more interestingly, they can also remove long-chain acyl groups and crotonyl moieties.5–7 Especially, SIRT5 can highly efficiently hydrolyze acidic acyl modifications such as succinylation, malonylation, and glutarylation,3,8 whereas SIRT6 prefers long-chain fatty acyl groups such as myristoyl-lysine residues.9,10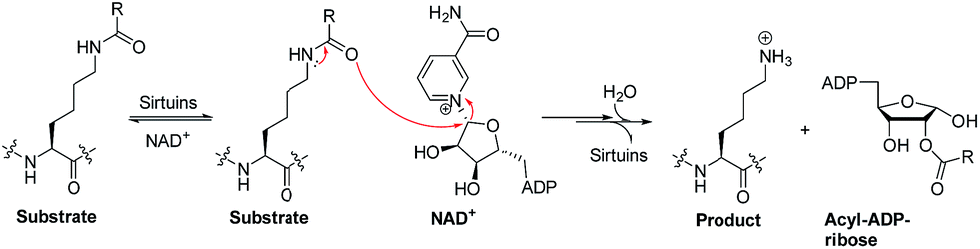 | ||
| Fig. 1 Outlined mechanisms for sirtuin-catalysed deacylations. | ||
Due to their multifaceted catalytic activities on various substrate proteins (Table S1†), sirtuins are pivotal regulators in various cellular processes, including transcription, genome stability, and energy metabolism,1,2 and are involved in human pathologies such as diabetes,11,12 cancer,13–16 and neurodegeneration.2,17 These features make sirtuins potentially attractive targets for drug discovery. Thus, sirtuin modulators are sought as chemical tools and potential therapeutics.14,18–20
Current activity assays for the identification of new sirtuin modulators are based on quantitative analysis of substrates/products via HPLC/CE,21,22 fluorescence resonance energy transfer (FRET)-based peptide substrates,23 or fluorogenic peptide substrates.18,24,25 The latter has been most widely used and requires a trypsin-coupled assay in which the release of a fluorophore, e.g. 7-amino-4-methylcoumarin (AMC), is continuously monitored (Fig. S1†). Compared with fluorogenic peptide substrates that are derived from sirtuin isoform-specific substrate proteins, small-molecule substrates for sirtuins have been rarely studied. In this study, we synthesized four types of fluorogenic small-molecule substrates, i.e. acetyl-(AcBKA), crotonyl-(CrBKA), succinyl-(SuBKA), and myristoyl-(MyBKA)-containing substrates (Scheme 1 and Fig. S2†), and tested them against three representative sirtuin proteins, i.e. SIRT2 (class I), SIRT5 (class III), and SIRT6 (class IV), with the aim of comparing their sensitivity and selectivity with those of peptide substrates. We then used isothermal titration calorimetry (ITC) analyses to investigate the inner drives of substrate binding and performed crystallographic analyses to investigate how SuBKA binds to its preferred sirtuin isoform SIRT5.
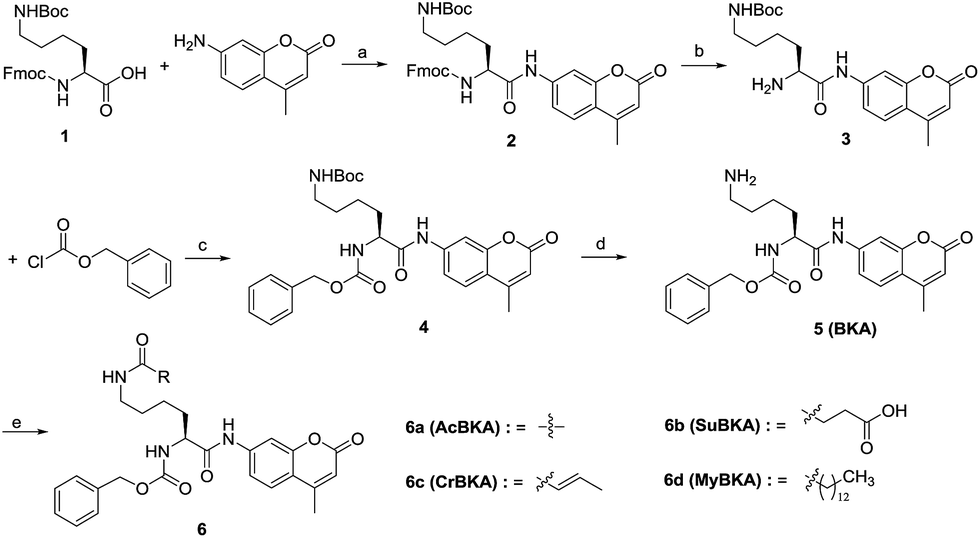 | ||
| Scheme 1 Reagents and conditions: (a) pyridine, POCl3, THF, 0 °C–RT, yield: 83%; (b) piperidine, MeCN, 4 °C, 16 h; DCM, RT, 8 h, yield: 80%; (c) Na2CO3, H2O/1,4-dioxane, 0 °C–RT, yield: 75%; (d) TFA, DCM, 0 °C–RT, yield: 71%; (e) for 6a: acetyl chloride, pyridine, THF, RT, yield: 43%; for 6b: DIPEA, succinic anhydride, THF, 0 °C–RT, yield: 38.5%; for 6c/6d: crotonic acid/myristic acid, EDCI, HOBT, NMM, DCM, yield: 48–54%. | ||
Results and discussion
The template molecule BKA (5) and the substrates AcBKA (6a), SuBKA (6b), CrBKA (6c), and MyBKA (6d) were synthesized via the synthetic route shown in Scheme 1. Briefly, all target compounds were prepared from the commercially available N2-(((9H-fluoren-9-yl)methoxy)carbonyl)-N6-(tert-butoxycarbonyl)-L-lysine (1), which reacted with 7-amino-4-methyl-2H-chromen-2-one to afford compound 2 in the presence of POCl3. Then, 2 was successively subjected to de-protection reaction and condensation reaction with benzyl carbonochloridate to obtain the intermediate 4 in good yield. Subsequently, 4 was treated with trifluoroacetic acid in DCM to obtain the key intermediate 5, which reacted with acetyl chloride, succinic anhydride, crotonic acid, and myristic acid, respectively, to produce the corresponding target compounds 6a, b, c, and d in moderate yields.We first tested whether BKA could be efficiently hydrolysed by trypsin. When treated with saturating trypsin to different concentrations of BKA for 24 h, we observed a clear linear relationship between BKA concentrations and measured fluorescence values (RFU) (Fig. S3†); this implied that BKA concentrations could be quantitatively monitored via fluorescence values (Fig. S3†). To examine hydrolysis efficiency, we obtained the initial rate velocities of trypsin (2 U μL−1) hydrolyzing BKA (30 μM to 0.05 μM) by acquiring the fluorescence values every 30 seconds. We observed that the reaction rate increases with the increasing concentrations of BKA (Fig. S4†); for 30 μM BKA, the rate of hydrolysis by trypsin is 0.038 ± 0.001 μM s−1 (Fig. S4†), indicating that trypsin can efficiently hydrolyze BKA.
We then examined the deacylation activities of SIRT2, SIRT5, and SIRT6 on the substrates AcBKA, CrBKA, SuBKA, and MyBKA (see the Experimental section). Preliminarily, these substrates were tested at the concentrations of 100 μM and 10 μM, reacted with sirtuins for 4 h at 37 °C, and then trypsin was added (2 U μL−1) to release the AMC fluorophore. The results revealed that SIRT2 could hydrolyse the acetyl and myristoyl moieties from AcBKA and MyBKA, respectively, at both 100 μM and 10 μM (Fig. 2a); obviously, SIRT2 had much stronger deacetylation activity than demyristoylation activity. No or very weak catalytic activity on CrBKA and SuBKA was observed for SIRT2 (Fig. 2a). In contrast, SIRT5 showed robust desuccinylation activity for SuBKA (Fig. 2b), but had no or very weak deacylation activities for AcBKA, CrBKA, and MyBKA (Fig. 2b); these findings reflected a highly selective desuccinylation activity of SIRT5. Note that for SIRT6, we only observed weak catalytic activities for AcBKA and MyBKA (Fig. 2c); this finding indicated that all these small-molecule substrates might not be suitable for high-throughput screening assays for SIRT6.
 | ||
| Fig. 2 Deacylase activities of SIRT2, SIRT5, and SIRT6 on small-molecule substrates. (a, b, and c) Fluorescence values obtained by treating SIRT2, SIRT5, and SIRT6 with 100 μM and 10 μM substrates, respectively. (d–f) Michaelis–Menten kinetic curves of SIRT2 on AcBKA (300 μM–0.58 μM), SIRT2 on MyBKA (75 μM–0.58 μM), and SIRT5 on SuBKA (500 μM–0.98 μM). | ||
Then, enzyme kinetic studies were carried out for SIRT2 and SIRT5 with their sensitive small-molecule substrates. The kinetic constants were determined by measuring reaction rate velocities at varying substrate concentrations, and the data was fitted to the Michaelis–Menten equation to obtain KM and Vmax values (Table 1); the kcat values were calculated from the relationship kcat = Vmax/[Enzyme]. For SIRT2, we observed that the KM values for AcBKA and MyBKA were ∼273.5 μM (unsaturation curve, Fig. 2d) and 1.7 μM (Fig. 2e and Table 1), respectively, which were similar to those for Ac-ETDKAc (KM = 750 μM)26 and Ac-ETDKMy (KM = 1.8 μM)26 peptide substrates, respectively. The catalytic efficiencies (kcat/KM) for AcBKA and MyBKA were 3.5 × 101 M−1 s−1 and 3.2 × 101 M−1 s−1 (Table 1), respectively. The observed SIRT2 deacetylation activity for AcBKA is similar to that for the peptide substrate Ac-ETDKAc (kcat/KM = 8.1 × 101 M−1 s−1),26 but the demyristoylation activity for MyBKA (kcat/KM = 8.5 × 103 M−1 s−1)26 is apparently lower than that for Ac-ETDKMy (Table 1) probably due to low water solubility of MyBKA. In contrast, SIRT5 was shown to have excellent catalytic efficiency for its sensitive substrate SuBKA (KM = 13.3 μM, kcat/KM = 1.6 × 102 M−1 s−1) (Fig. 2f and Table 1), which is similar to that for the peptide substrate Ac-LGKSu (KM = 33 μM, kcat/KM = 9.2 × 102 M−1 s−1).27,28 In addition, we observed that sirtuin-catalysed deacylations for AcBKA and MyBKA are highly associated with the concentrations of NAD+ (Fig. S5†); this further revealed the evidence for the NAD+-dependent catalysis nature of sirtuins.17,29 The enzyme kinetic results indicate that small-molecule substrates (along with precise acyl substituents) have similar sirtuin-isoform sensitivity as compared to peptide substrates.
| Entry | Enzyme | Substrate | [Enzyme] (μM) | KM (μM) | Vmax (nM min−1) | kcat (s−1) | kcat/KM (M−1 s−1) | Literature data for peptide substrates | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Peptide substrate | KM (μM) | kcat/KM (M−1 s−1) | Ref. | ||||||||
| 1 | SIRT2 | AcBKA | 0.5 | 273.5 | 29.3 | 9.8 × 10−4 | 3.5 × 101 | Ac-ETDKAc | 750 | 8.1 × 101 | 26 |
| 2 | MyBKA | 0.5 | 1.7 | 1.63 | 5.4 × 10−5 | 3.2 × 101 | Ac-ETDKMy | 1.8 | 8.5 × 103 | 26 | |
| 3 | SIRT5 | SuBKA | 0.2 | 13.3 | 21.7 | 2.6 × 10−3 | 1.6 × 102 | Ac-LGKSu | 33 | 9.2 × 102 | 27 |
We then used isothermal titration calorimetry (ITC) method to determine the binding affinity and thermodynamic parameters (e.g., enthalpy/entropy contributions) of AcBKA and MyBKA binding to SIRT2 as well as SuBKA binding to SIRT5 (for details, please check the Experimental section). The curves of molar ratio to thermal effects are shown in Fig. 3a–c. From the fitting data, we obtained the equilibrium dissociation constants (Ka) of AcBKA and MyBKA binding to SIRT2; the Ka values of AcBKA and MyBKA were 4.41 × 104 ± 2.84 × 104 M−1 (Fig. 3a) and 1.24 × 105 ± 5.26 × 104 M−1 (Fig. 3b), respectively, and the corresponding binding affinities (Kd = 1/Ka) were 22.67 μM and 8.06 μM, respectively, consistent with the KM values of AcBKA (273.5 μM, Table 1) and MyBKA (1.7 μM, Table 1), respectively. By contrast, the equilibrium dissociation constant Ka of SuBKA binding with SIRT5 is 1.11 × 105 ± 2.83 × 104 M−1 (Fig. 3c), and the binding affinity Kd is 9.01 μM, which is close to the KM value of SIRT5 hydrolyzing SuBKA (13.3 μM, Table 1).
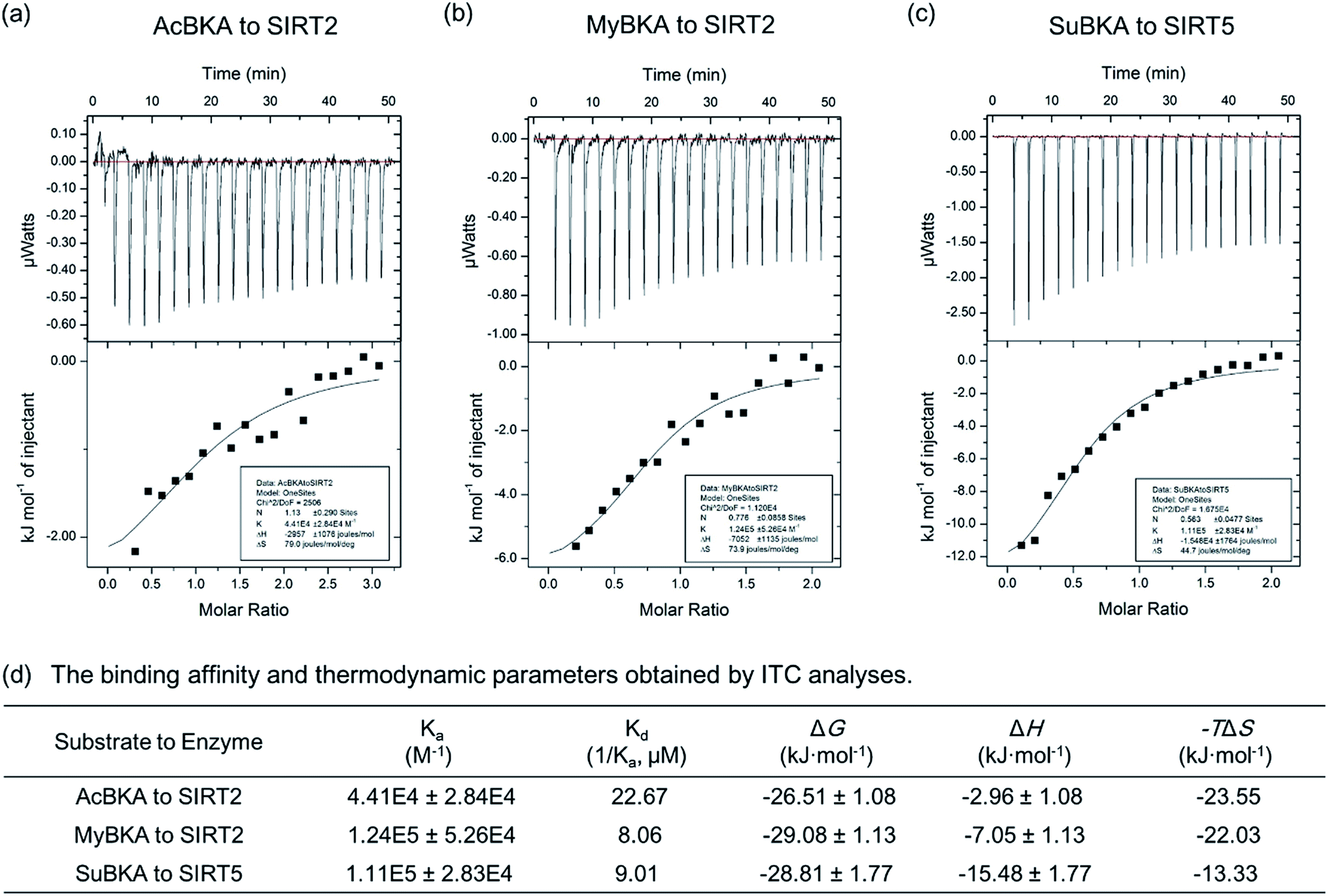 | ||
| Fig. 3 ITC analyses for small-molecule substrates to the preferred sirtuin isoforms. The curves of molar ratio to thermal effects of (a) AcBKA titrating to SIRT2, (b) MyBKA to SIRT2, (c) SuBKA to SIRT5, and (d) specific thermodynamic parameters, showing that the binding of AcBKA/MyBKA to SIRT2 is mainly driven by entropy, whereas SuBKA binding to SIRT5 is driven by enthalpy. | ||
The ITC analyses reveal that the enthalpy (ΔH) changes of AcBKA and MyBKA binding to SIRT2 are −2.96 ± 1.08 kJ mol−1 and −7.05 ± 1.13 kJ mol−1 (Fig. 3d), respectively; the entropy (−TΔS) changes are −23.55 kJ mol−1 and −22.03 kJ mol−1 (Fig. 3d); and the calculated binding free energy ΔG (ΔG = ΔH − TΔS) is −26.51 ± 1.08 kJ mol−1 and −29.08 ± 1.13 kJ mol−1 (Fig. 3d). The results indicate that both AcBKA and MyBKA binding to SIRT2 are mainly driven by entropy (|−TΔS| > |ΔH|), reflecting the exposure of buried hydrophobic surfaces and hydrophobic interactions of AcBKA/MyBKA with SIRT2. The enthalpy and entropy changes of SuBKA binding to SIRT5 are −15.48 kJ mol−1 and −13.33 kJ mol−1, respectively, and the calculated binding free energy is −28.81 kJ mol−1 (Fig. 3d). The results reveal that the binding of SuBKA to SIRT5 is driven by enthalpy (|ΔH| > |−TΔS|), uncovering the main contributions of hydrogen-bonding/electrostatic interactions and complementarity between SuBKA and SIRT5, consistent with that observed by subsequent crystallographic analyses (Fig. 4). In addition, these results can offer thermodynamic insights into the design of sirtuin isoform-specific inhibitors.
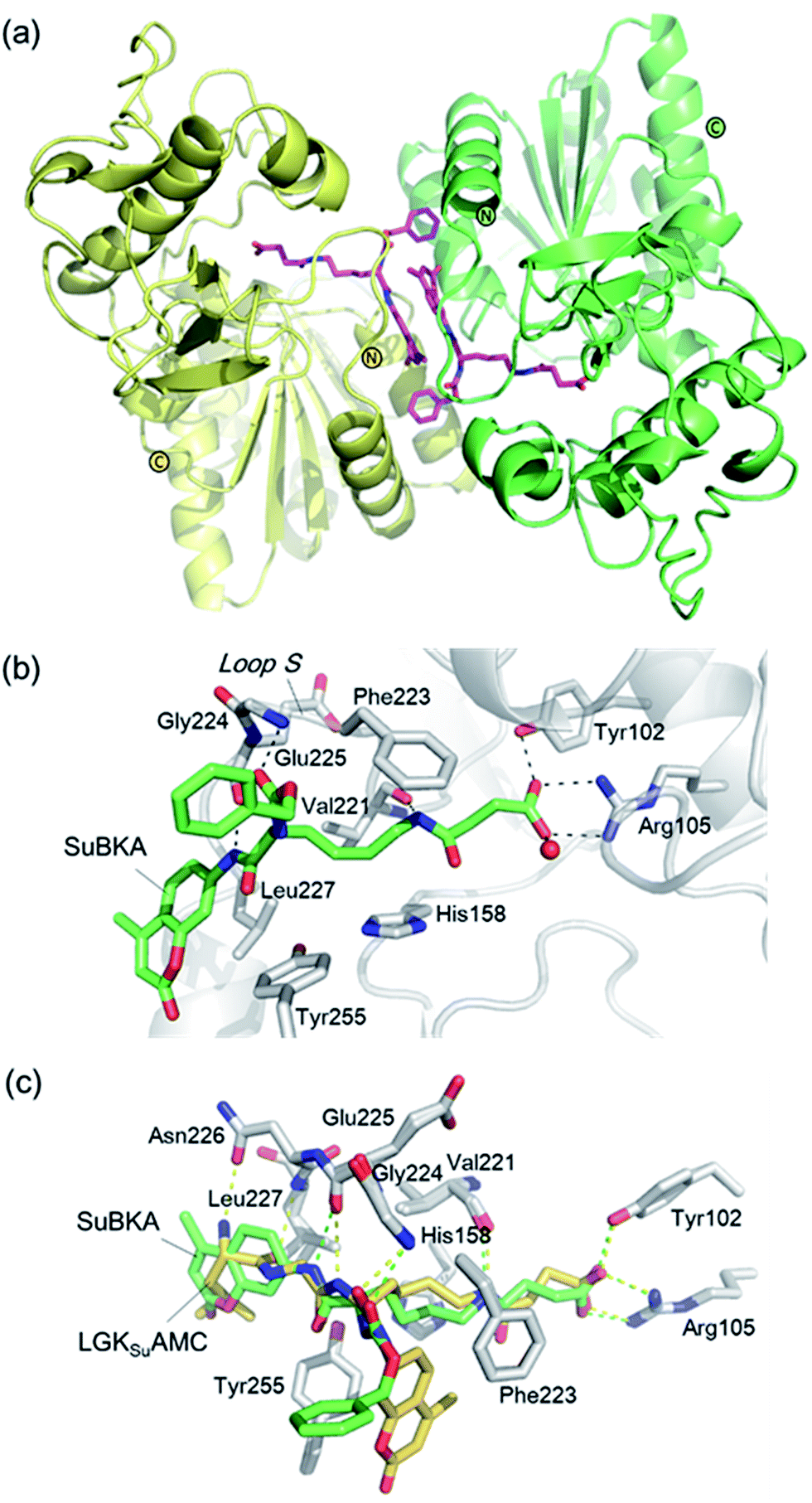 | ||
| Fig. 4 Crystallographic analyses reveals how SuBKA binds to SIRT5. (a) The AMC moieties of SuBKA in two neighboring SIRT5 molecules were observed to form π–π stacking interactions (PDB ID 5XHS). (b) View from the SIRT5:SuBKA complex structure, in which SuBKA is positioned to make hydrogen-bonding interactions with SIRT5-specific residues Tyr102 and Arg105, and the residues Val221, Gly224, and Glu225 on Loop S. (c) Comparison of SIRT5:SuBKA and SIRT5:LGKsuAMC structures (PDB ID 5BWL) reveals that although AMC moieties of SuBKA and LGKsuAMC bind in different manners, SuBKA and LGKsuAMC have similar hydrogen-bonding/electrostatic interactions with SIRT5-specific residues and other catalytically important residues on Loop S. SuBKA is shown by green sticks, and LGKsuAMC is shown by orange sticks. | ||
We then performed crystallographic and molecular docking analyses to investigate how the small-molecule substrates bind to their preferred sirtuin isoforms. We attempted co-crystallization and soaking, but failed to obtain a SIRT2 structure complexed with AcBKA or MyBKA probably due to their limited solubility under the used crystallizing conditions. The crystal structure of SIRT5:SuBKA complex was obtained via co-crystallization (see Experimental section) and determined to 2.19 Å resolution (Table S2†). The SIRT5:SuBKA structure crystallized with one molecule in the asymmetric unit (space group P21221) (PDB ID 5XHS, Table S2†). Interestingly, the AMC moieties of SuBKA in two neighboring SIRT5 molecules formed π–π stacking interactions, which might contribute to crystal packing (Fig. 4a).
In the SIRT5:SuBKA structure, we observed that SuBKA was positioned to make hydrogen-bonding interactions with the main chains of Gly224, Glu225, and Val221 on the Loop S and hydrophobic interactions with the side chains of Val221 and Phe223 (Fig. 4b). The carboxylate group of SuBKA was observed to form hydrogen bonding and electrostatic interactions with Tyr102 and Arg105 (Fig. 4b), both of which were unique residues of SIRT5. This, to a large extent, explained the specificity of SIRT5 for catalytically recognizing/hydrolyzing SuBKA.
On superimposing the SIRT5:SuBKA structure with the SIRT5:LGKsuAMC (a fluorogenic peptide substrate; PDB ID 5BWL) structure, we observed that SuBKA bound to SIRT5 via a new mode, which was different from that of LGKsuAMC (Fig. 4c). The lysine N-/C-terminus of SuBKA and LGKsuAMC bound in opposite directions (Fig. 4c); this suggested the possibility of incorporation of D-lysine as sirtuin substrates. Note that SuBKA and LGKsuAMC form hydrogen-bonding/electrostatic interactions with the residues (Val221, Gly224, and Glu225) on the Loop S and the SIRT5-specific residues (Tyr102 and Arg105) (Fig. 4c). This may reflect the most important binding features of the SIRT5 substrates and also provide possible common pharmacophore features for the design of new small-molecule inhibitors for SIRT5.
Comparison of SIRT5:SuBKA (PDB ID 5XHS) and SIRT5:Succ-IDH2:Carba-NAD (PDB ID 4G1C)30 structures reveals that like that of the peptide substrate Succ-IDH2, the amide bond of SuBKA approaches Carba-NAD (Fig. 5a). Through superimposition of the SIRT5:SuBKA structure with the SIRT5:bicyclic-intermediate (PDB ID 4F56)31 structure, we observed that SuBKA appears likely to form a bicyclic intermediate with the cofactor NAD+ (Fig. 5b), which is highly similar with the peptide substrate GTSKsuRAT. Together, the crystallographic analyses indicate that SuBKA is desuccinylated by SIRT5 probably through the catalytic mechanism proposed for peptide substrates.27,28
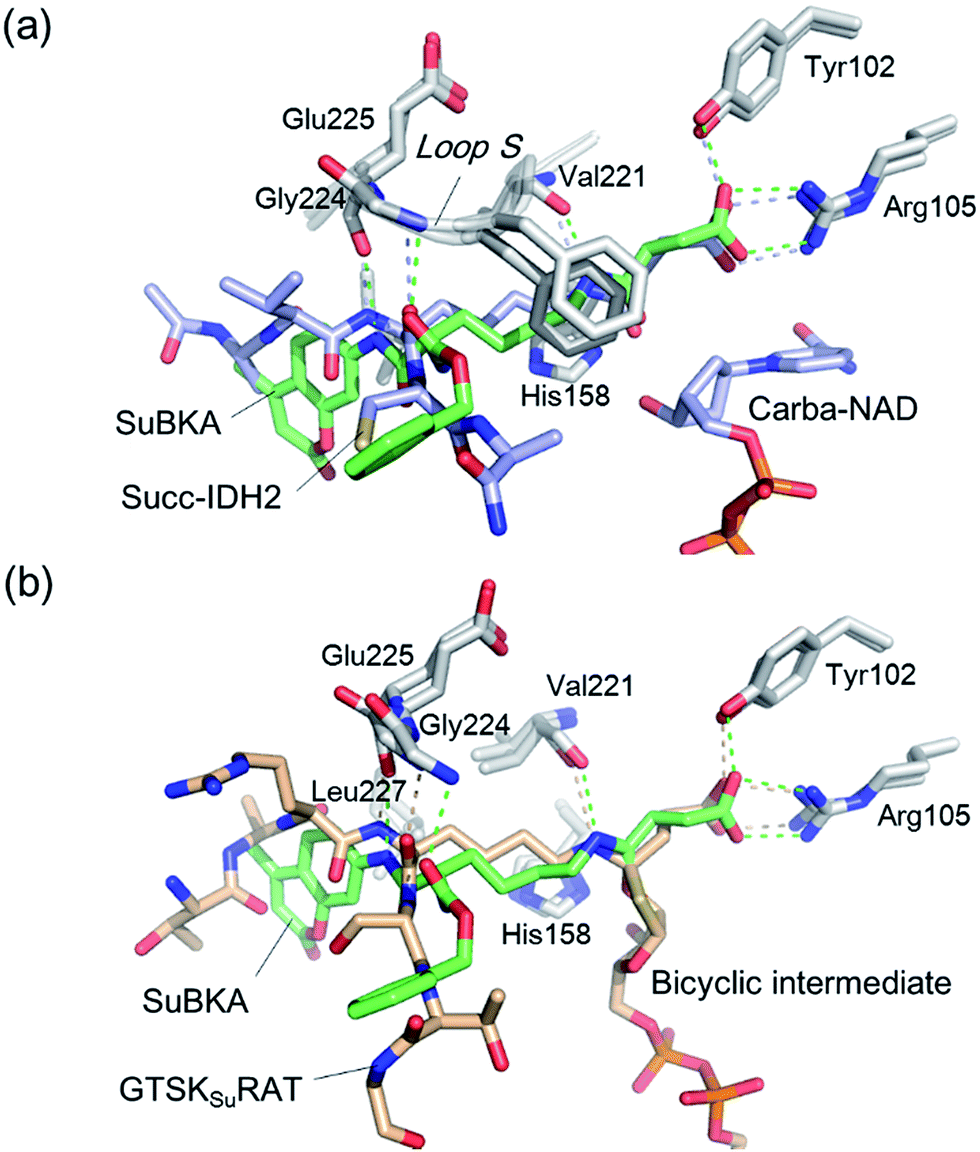 | ||
| Fig. 5 Comparisons of the SIRT5:SuBKA structure with those of (a) the SIRT5:Succ-IDH2:Carba-NAD (PDB ID 4G1C)30 and (b) SIRT5:bicyclic-intermediate (PDB ID 4F56)31 reveal that SuBKA appears likely to undergo the same catalytic mechanism as the peptide substrates. | ||
We used molecular docking to predict the binding modes of AcBKA and MyBKA with SIRT2. The results show that AcBKA and MyBKA are likely positioned to interact with the catalytically important residues of SIRT2 such as Phe235, Val233, Glu237, and Gln267 (Fig. 6a and b). Compared to AcBKA, MyBKA likely formed stronger interactions with the residues (e.g., Ile169, Phe119, Tyr139, and Phe96) at the SIRT2 hydrophobic site (Fig. 6b); this may explain why MyBKA binds to SIRT2 more tightly (Fig. 3d). Moreover, the molecular docking results revealed the similarity between the SIRT2-catalyzed mechanisms for AcBKA/MyBKA and the peptide substrates (e.g. H3K9myr, Fig. S6†).32
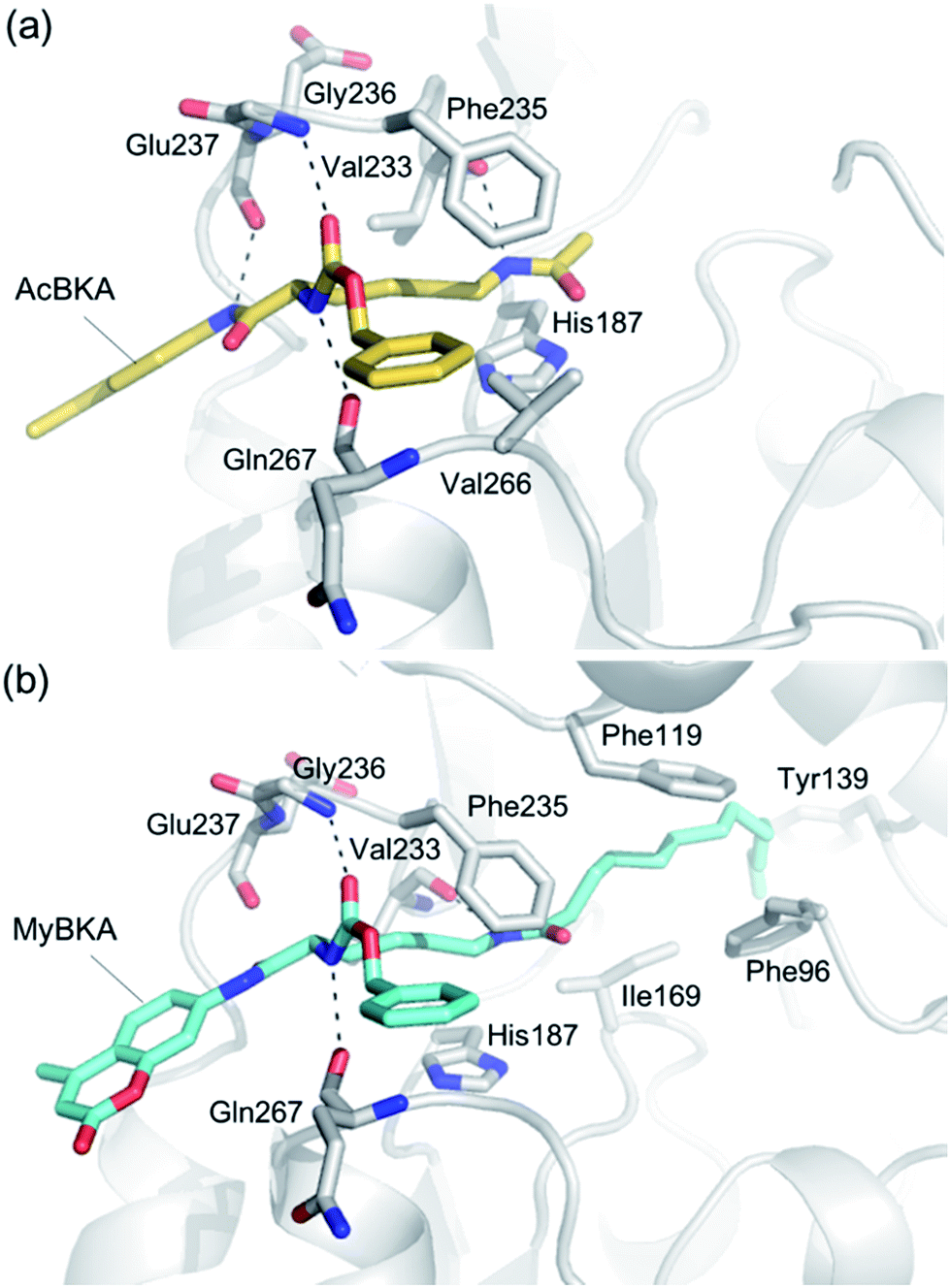 | ||
| Fig. 6 Predicted binding modes of (a) AcBKA and (b) MyBKA with SIRT2 using molecular docking simulations (for details, please check the Experimental section). | ||
Conclusions
In summary, enzyme kinetic analyses reveal that the four types of small-molecule substrates presented in this study have similar sirtuin-isoform sensitivity to peptide substrates. ITC analyses reveal that the binding of AcBKA or MyBKA to SIRT2 is mainly driven by entropy, whereas SuBKA binding to SIRT5 is driven by enthalpy, which also provide thermodynamic insights into inhibitor design. The SIRT5:SuBKA complex structure reveals that SuBKA binds to SIRT5 via a mode different from that of the peptide substrates, but it involves SIRT5-specific residues Tyr102 and Arg105 and other catalytically important residues on Loop S. The crystallographic analyses also indicate that SuBKA undergoes the same desucciylation mechanism as peptide substrates. Comparison analyses of the SIRT5:SuBKA structure and other SIRT5 complex structures revealed the common pharmacophore features of the SIRT5 substrates, which offer insights into the design of selective SIRT5 inhibitors. This study will aid further efforts not only for developing new small-molecule substrates but also for designing new selective sirtuin isoform-specific inhibitors as tools and therapeutics.Experimental
Chemical synthesis
All chemicals and solvents were commercially purchased and used as supplied without further purification. Room temperature refers to 20–25 °C. All reactions were monitored using thin layer chromatography (TLC) on silica gel F-254 TLC plates. Column chromatography was carried out using silica gel (300−400 mesh). 1H-NMR and 13C-NMR spectra were obtained using a Bruker AV-400 spectrometer or Bruker DRX 600 spectrometer. Chemical shifts (δ) were obtained in parts per million (ppm) relative to tetramethylsilane as the internal standard, and coupling constants (J) were obtained in hertz (Hz). Low-resolution mass spectral (MS) data were acquired using an Agilent 1100 series LC-MS instrument with UV detection at 254 nm in the electrospray ionization (ESI) mode. All the target compounds were purified to >95% purity, as determined by high-performance liquid chromatography (HPLC). HPLC analysis was performed via a Waters 2695 HPLC system equipped with a Kromasil C18 column (4.6 mm × 250 mm, 5 μm).![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), and the organic solvent was removed under reduced pressure and partitioned between water and ethyl acetate (3×). The organic layer was dried over anhydrous MgSO4 and concentrated in vacuo. The residue was purified by column chromatography (eluent gradient DCM/MeOH = 200/1) to obtain (9H-fluoren-9-yl)methyl tert-butyl (6-((4-methyl-2-oxo-2H-chromen-7-yl)amino)-6-oxohexane-1,5-diyl) (S)-dicarbamate (2, 1.102 g) in 83% yield.
:1), and the organic solvent was removed under reduced pressure and partitioned between water and ethyl acetate (3×). The organic layer was dried over anhydrous MgSO4 and concentrated in vacuo. The residue was purified by column chromatography (eluent gradient DCM/MeOH = 200/1) to obtain (9H-fluoren-9-yl)methyl tert-butyl (6-((4-methyl-2-oxo-2H-chromen-7-yl)amino)-6-oxohexane-1,5-diyl) (S)-dicarbamate (2, 1.102 g) in 83% yield.A mixture of the intermediate 2 (1.0 g) and catalytic amount of piperidine (0.184 mL) in 33 mL MeCN was stirred for 16 h at 4 °C. After the addition of dichloromethane (16 mL), the mixture was allowed to warm to room temperature and stirred for another 8 h. The resulting solution was concentrated to dryness and the residue was purified by silica gel column chromatography (eluent gradient DCM/MeOH/NH4OH = 100/1/0.25) to obtain the white compound 3 (526 mg) in 80% yield. 1H-NMR (400 MHz, DMSO-d6) δ 7.83 (s, 1H), 7.76 (d, J = 8.4 Hz, 1H), 7.57 (d, J = 8.4 Hz, 1H), 6.77 (br s, 1H), 6.29 (s, 1H), 3.85 (t, J = 6.0 Hz, 1H), 2.89 (s, 2H), 2.40 (s, 3H), 1.79–1.73 (m, 2H), 1.41–1.29 (m, 13H) ppm. LCMS m/z: 404.2 [M + H]+.
Protein cloning, expression, and purification
000 rpm for 30 min. The supernatant was applied onto a Ni-NTA column (Roche), and the resin was washed with 20 volumes of the buffer B (20 mM Tris–HCl, 250 mM NaCl, 10 mM imidazole, and pH 8.0) to remove nonspecifically binding proteins. Recombinant proteins were eluted with buffer C (20 mM Tris–HCl, 250 mM NaCl, 250 mM imidazole, and pH 8.0). The proteins of human SIRT2 (residues 56–356), human SIRT5 (residues 34–269), and human SIRT6 (residues 1–355) were pooled, concentrated, and desalted using a HiTrap Desalting column (GE Healthcare) into reaction buffer for enzyme kinetic analyses (SIRT2: 25 mM Tris–HCl, 150 mM NaCl, and pH 8.0; SIRT5: 20 mM Tris–HCl, 150 mM NaCl and 5% (w/v) glycerol, and pH 8.0; SIRT6: 20 mM Tris–HCl, 150 mM NaCl, 5% (w/v) glycerol and 0.5 mM TCEP, and pH 8.0). The target proteins were concentrated to 24 mg mL−1 using Amicon Ultra 10K (Millipore) and stored at −80 °C. The human SIRT5 (residues 34–269) for crystallization were digested with TEV protease (1:50) in the presence of 0.1% β-ME (β-mercaptoethanol) overnight (4 °C). The results of the TEV digestion were verified by 12% SDS-PAGE. His-Tags, TEV protease, and uncleaved protein removed by an Ni-NTA column (HiTrap™, chelating HP, GE Healthcare). The digests were concentrated using an Amicon Ultra 10K (Millipore), loaded onto a gel-filtration column (Superdex 75 10/300 GL, GE Healthcare), and eluted at 0.5 mL min−1 with buffer D (20 mM Tris–HCl 150 mM NaCl, and pH 8.0) using an ÄKTA explorer device (GE Healthcare). Fractions containing SIRT5 were pooled and concentrated to 13 mg mL−1 and stored at −80 °C. All purification steps were monitored via 12% SDS-PAGE, and the concentration was determined via a NanoDrop 2000 spectrophotometer (Thermo Scientific).Enzyme kinetic assays
In enzyme kinetic assays, all enzymes, cofactor NAD+, and substrates were dissolved in the assay buffer: 50 mM Tris, 137 mM NaCl, 2.7 mM KCl, and pH 8.0. The assays were performed in a 96-well black microplate (Corning No. 3603) with a reaction volume of 60 μL per well. Fluorescence intensity was measured using a microplate reader (BioTek Cytation 3, λex = 390 nm, and λem = 460 nm).Crystallographic analyses
The SIRT5 proteins (13 mg mL−1 in the buffer of 20 mM Tris/HCl pH 8.0, 150 mM NaCl) were co-crystallized with SuBKA (100 mM stocks dissolved in DMSO) at 16 °C. The protein and SuBKA (2% final DMSO concentration for crystallization) mixtures were prepared in a 1:5 molar ratio and incubated for 2 h at 4 °C. The 2 μL crystallization drops constituted of a 1:1 ratio of protein:reservoir solutions (reservoir: 23–25% PEG 3350, 0.1 M Bis-Tris, pH 5.5–6.5, and 0.2 M NaCl). Crystals appeared in 2–3 days. The crystals were transferred to a cryo-protectant drop composed of the reservoir, supplemented with 19% glycerol prior to flash-freezing using liquid nitrogen. Data were obtained at the Shanghai Synchrotron Radiation Facility and processed using HKL2000 (Otinowski and Minor, 1997).
Isothermal titration calorimetry (ITC) analysis
The ITC binding assay was carried out via a MicroCal ITC200 calorimeter (GE Healthcare) at 25 °C. AcBKA (500 μM) and MyBKA (750 μM) were titrated with SIRT2 (50 μM), separately, using a buffer of 20 mM HEPES, 100 mM NaCl, and pH 7.4; SuBKA (600 μM) was titrated with SIRT5 (60 μM) using the SIRT5 buffer (20 mM Tris, pH 8.0, 20 mM NaCl, and 5% glycerol). Both the cell and syringe of the calorimeter contained 0.6% DMSO. The system was equilibrated until the cell temperature reached 25 °C, and an additional delay of 60 s was applied. All titrations were conducted using a preliminary injection of 0.2 μL of 600 μM SuBKA and then a series of 19 individual injections of 2 μL at time intervals of 150 s. The titration cell was stirred continuously at 750 rpm. The obtained curve fitting to a single binding site model was performed by ITC data analysis module of Origin 7.5 (OriginLab).Molecular docking analyses
The AutoDock Vina program33 was used for molecular docking simulations. AcBKA and MyBKA were prepared as pdbqt file using AutoDockTools. The crystal structure of SIRT2 complexed with myristoylated peptide (H3K9myr) (PDB ID: 4Y6L)32 was selected as the protein model. Then, all the water molecules and solvent molecules were removed, Gasteiger–Marsili charges were added, and non-polar hydrogens were then merged onto their respective heavy atoms, as previously described.34,35 A total of 20 docking poses were generated for AcBKA and MyBKA, and the other parameters for Vina were set as default. The docking results were viewed using the PyMOL program.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors thank the staff of BL19U1 beamline of the National Center for Protein Science Shanghai at Shanghai Synchrotron Radiation Facility for assistance during data collection. This work was supported by the National Natural Science Foundation of China (Grant No.: 81502989 and 81501368), the Scientific Research Foundation of Sichuan University (Grant No.: 20822041A4193), and the China Postdoctoral Science Foundation Funded Project (Grant No.: 2015M570789).Notes and references
- B. Chen, W. Zang, J. Wang, Y. Huang, Y. He, L. Yan, J. Liu and W. Zheng, Chem. Soc. Rev., 2015, 44, 5246–5264 RSC.
- R. H. Houtkooper, E. Pirinen and J. Auwerx, Nat. Rev. Mol. Cell Biol., 2012, 13, 225–238 CrossRef CAS PubMed.
- L. Yang, X. Ma, Y. He, C. Yuan, Q. Chen, G. Li and X. Chen, Sci. China: Life Sci., 2017, 60, 249–256 CrossRef CAS PubMed.
- Y. Chen, R. Sprung, Y. Tang, H. Ball, B. Sangras, S. C. Kim, J. R. Falck, J. Peng, W. Gu and Y. Zhao, Mol. Cell. Proteomics, 2007, 6, 812–819 CAS.
- Y.-B. Teng, H. Jing, P. Aramsangtienchai, B. He, S. Khan, J. Hu, H. Lin and Q. Hao, Sci. Rep., 2015, 5, 8529 CrossRef CAS PubMed.
- X. Bao, Y. Wang, X. Li, X.-M. Li, Z. Liu, T. Yang, C. F. Wong, J. Zhang, Q. Hao and X. D. Li, eLife, 2014, 3, e02999 Search PubMed.
- J. L. Feldman, J. Baeza and J. M. Denu, J. Biol. Chem., 2013, 288, 31350–31356 CrossRef CAS PubMed.
- J. Du, Y. Zhou, X. Su, J. J. Yu, S. Khan, H. Jiang, J. Kim, J. Woo, J. H. Kim, B. H. Choi, B. He, W. Chen, S. Zhang, R. A. Cerione, J. Auwerx, Q. Hao and H. Lin, Science, 2011, 334, 806–809 CrossRef CAS PubMed.
- X. Zhang, S. Khan, H. Jiang, M. A. Antonyak, X. Chen, N. A. Spiegelman, J. H. Shrimp, R. A. Cerione and H. Lin, Nat. Chem. Biol., 2016, 12, 614–620 CrossRef CAS PubMed.
- H. Jiang, S. Khan, Y. Wang, G. Charron, B. He, C. Sebastian, J. Du, R. Kim, E. Ge, R. Mostoslavsky, H. C. Hang, Q. Hao and H. Lin, Nature, 2013, 496, 110–113 CrossRef CAS PubMed.
- J. Park, Y. Chen, D. X. Tishkoff, C. Peng, M. Tan, L. Dai, Z. Xie, Y. Zhang, B. M. Zwaans, M. E. Skinner, D. B. Lombard and Y. Zhao, Mol. Cell, 2013, 50, 919–930 CrossRef CAS PubMed.
- Y. Nishida, M. J. Rardin, C. Carrico, W. He, A. K. Sahu, P. Gut, R. Najjar, M. Fitch, M. Hellerstein, B. W. Gibson and E. Verdin, Mol. Cell, 2015, 59, 321–332 CrossRef CAS PubMed.
- A. Chalkiadaki and L. Guarente, Nat. Rev. Cancer, 2015, 15, 608–624 CrossRef CAS PubMed.
- L. Yang, X. Ma, C. Yuan, Y. He, L. Li, S. Fang, W. Xia, T. He, S. Qian, Z. Xu, G. Li and Z. Wang, Eur. J. Med. Chem., 2017, 134, 230–241 CrossRef CAS PubMed.
- H. Jing, J. Hu, B. He, Y. L. Negrón Abril, J. Stupinski, K. Weiser, M. Carbonaro, Y.-L. Chiang, T. Southard, P. Giannakakou, R. S. Weiss and H. Lin, Cancer Cell, 2016, 29, 297–310 CrossRef CAS PubMed.
- S. Xu, C.-X. Liu, W. Xu, L. Huang, J.-Y. Zhao and S.-M. Zhao, Signal Transduct. Target. Ther., 2017, 2, 16035 CrossRef.
- M. S. Bonkowski and D. A. Sinclair, Nat. Rev. Mol. Cell Biol., 2016, 17, 679–690 CrossRef CAS PubMed.
- M. Schiedel, T. Rumpf, B. Karaman, A. Lehotzky, S. Gerhardt, J. Ovádi, W. Sippl, O. Einsle and M. Jung, Angew. Chem., Int. Ed., 2016, 55, 2252–2256 CrossRef CAS PubMed.
- W. You, D. Rotili, T.-M. Li, C. Kambach, M. Meleshin, M. Schutkowski, K. F. Chua, A. Mai and C. Steegborn, Angew. Chem., Int. Ed., 2017, 56, 1007–1011 CrossRef CAS PubMed.
- H. Jing, J. Hu, B. He, Y. L. Negron Abril, J. Stupinski, K. Weiser, M. Carbonaro, Y. L. Chiang, T. Southard, P. Giannakakou, R. S. Weiss and H. Lin, Cancer Cell, 2016, 29, 297–310 CrossRef CAS PubMed.
- Y. Fan and G. K. E. Scriba, Electrophoresis, 2010, 31, 3874–3880 CrossRef CAS PubMed.
- C. Roessler, T. Nowak, M. Pannek, M. Gertz, G. T. Nguyen, M. Scharfe, I. Born, W. Sippl, C. Steegborn and M. Schutkowski, Angew. Chem., Int. Ed., 2014, 53, 10728–10732 CrossRef CAS PubMed.
- Y. Li, W. Huang, L. You, T. Xie and B. He, Bioorg. Med. Chem. Lett., 2015, 25, 1671–1674 CrossRef CAS PubMed.
- P. A. Marcotte, P. R. Richardson, J. Guo, L. W. Barrett, N. Xu, A. Gunasekera and K. B. Glaser, Anal. Biochem., 2004, 332, 90–99 CrossRef CAS PubMed.
- B. C. Smith, W. C. Hallows and J. M. Denu, Anal. Biochem., 2009, 394, 101–109 CrossRef CAS PubMed.
- I. Galleano, M. Schiedel, M. Jung, A. S. Madsen and C. A. Olsen, J. Med. Chem., 2016, 59, 1021–1031 CrossRef CAS PubMed.
- C. Roessler, C. Tuting, M. Meleshin, C. Steegborn and M. Schutkowski, J. Med. Chem., 2015, 58, 7217–7223 CrossRef CAS PubMed.
- A. S. Madsen and C. A. Olsen, J. Med. Chem., 2012, 55, 5582 CrossRef CAS PubMed.
- A. A. Sauve and D. Y. Youn, Curr. Opin. Chem. Biol., 2012, 16, 535–543 CrossRef CAS PubMed.
- B. G. Szczepankiewicz, H. Dai, K. J. Koppetsch, D. Qian, F. Jiang, C. Mao and R. B. Perni, J. Org. Chem., 2012, 77, 7319–7329 CrossRef CAS PubMed.
- Y. Zhou, H. Zhang, B. He, J. Du, H. Lin, R. A. Cerione and Q. Hao, J. Biol. Chem., 2012, 287, 28307–28314 CrossRef CAS PubMed.
- J. L. Feldman, K. E. Dittenhafer-Reed, N. Kudo, J. N. Thelen, A. Ito, M. Yoshida and J. M. Denu, Biochemistry, 2015, 54, 3037–3050 CrossRef CAS PubMed.
- O. Trott and A. J. Olson, J. Comput. Chem., 2010, 31, 455–461 CAS.
- G.-B. Li, L.-Y. Huang, H. Li, S. Ji, L.-L. Li and S.-Y. Yang, RSC Adv., 2016, 6, 61137–61140 RSC.
- G.-B. Li, M. I. Abboud, J. Brem, H. Someya, C. T. Lohans, S.-Y. Yang, J. Spencer, D. W. Wareham, M. A. McDonough and C. J. Schofield, Chem. Sci., 2017, 8, 928–937 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Crystallographic data collection and refinement statistics, additional figures and tables. See DOI: 10.1039/c7ra05824a |
| ‡ H.-L. Wang and S. Liu contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |