
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A microscopic and spectroscopic study of rapid antimonite sequestration by a poorly crystalline phyllomanganate: differences from passivated arsenite oxidation†
Huawei Wangab,
Ya-nan Wangc,
Yingjie Sun *a,
Yiu Fai Tsangd,
Daoyong Zhangb and
Xiangliang Pan*b
*a,
Yiu Fai Tsangd,
Daoyong Zhangb and
Xiangliang Pan*b
aCollege of Environmental and Municipal Engineering, Qingdao University of Technology, Qingdao 266033, PR China. E-mail: yjsun1971@126.com; sunyingjie@qtech.edu.cn; Fax: +86-532-85071255; Tel: +86-532-85071255
bXinjiang Key Laboratory of Environmental Pollution and Bioremediation, Xinjiang Institute of Ecology and Geography, Chinese Academy of Sciences, Urumqi 830011, PR China. E-mail: xiangliangpan@163.com; panxl@ms.xjb.ac.cn
cCollege of Environmental Science and Engineering, Tongji University, Shanghai 200092, PR China
dDepartment of Science and Environmental Studies, The Education University of Hong Kong, Tai Po, New Territories, Hong Kong SAR, China
First published on 4th August 2017
Abstract
During the reaction of arsenite (As(III)) with δ-MnO2 (a typically poorly crystalline phyllomanganate), a significant decrease in the oxidation rate is frequently observed, which is mainly attributed to the surface passivation of δ-MnO2. However, whether surface passivation also occurs during the antimonite (Sb(III)) oxidation process is unclear. In this study, the behavior and mechanisms of Sb(III) oxidation were compared with those of As(III) during their reactions with δ-MnO2. The experimental kinetics results indicated that the oxidation rate of Sb(III) was 6.14–44.71 times faster than that of As(III) with initial concentrations ranging from 100 to 1000 μM. The macroscopic and spectroscopic results suggested that surface passivation during the adsorption of Mn(II) and the formation of Mn(III) were the predominant causes for the decrease in the As(III) oxidation rate, whereas surface passivation may not have been the limiting factor during Sb(III) oxidation. Compared to As(III) oxidation, the rapid oxidation of Sb(III) by δ-MnO2 led to significant changes in the structure and properties of δ-MnO2, and contributed to the precipitation of Mn(II) antimonate (MnSb2O6). The results of this study facilitate a better understanding of the environmental behavior of Sb and As on metal-oxide surfaces in aquatic environments.
1. Introduction
Arsenic (As) and antimony (Sb) are two metalloids that belong to group VA of the periodic table. Elevated levels of As and Sb in soils and groundwater from geological and anthropogenic sources have attracted worldwide attention due to their high toxicity to humans, plants and microorganisms.1–4 The predominant forms of As and Sb in contaminated environments are inorganic species. These species are most commonly present in the pentavalent state in the forms of antimonates (Sb(OH)6−, pKa = 2.72) and arsenates (H2AsO4−, HAsO42−, and AsO43−, pKa1–3 = 2.20, 6.97 and 11.53, respectively) in oxidizing and aerobic surroundings, while in anoxic environments, antimonites (Sb(OH)3, pKa = 11.9) and arsenites (H2AsO3−, HAsO32−, AsO33−, pKa1–3 = 9.22, 12.13 and 13.4) are present in the trivalent state.5 As(III) is considered more mobile in soils and sediments than As(V) because of its weak binding to metal mineral surfaces,6,7 and it is also more toxic because it binds to sulfhydryl (–SH) groups, affecting the function of many proteins.8 The toxicity of Sb is similar to that of As, i.e., Sb(V) < Sb(III),9–11 but its environmental behavior is not as well understood.The oxidation of As(III) to As(V) is an important reaction because this process can decrease not only the mobility of As(III) by increasing its tendency to be absorbed onto mineral surfaces but also its toxicity.10,11 Manganese (Mn) oxides are powerful oxidizing and detoxifying agents, and they can play an important role in many elemental geochemical processes.12–14 The reactivity of Mn oxides varies with their mineralogy, and previous studies have suggested that layered Mn oxides (i.e., phyllomanganates) are more reactive than Mn oxides with tunnel structures (e.g., pyrolusite and romanechite).15,16 Due to their high specific surface areas and structures with highly reactive vacancy sites, these phyllomanganates can absorb metals, such as Zn(II), Cu(II), Pb(II) and Ni(II).17–19 However, these materials can also readily oxidize many reduced toxic metals and metalloids, such as As(III), Cr(III), and Co(II),20–23 as well as organic contaminants.24,25
Many Mn oxides found in surface environments are poorly crystalline, of biogenic origin and highly reactive. Specifically, δ-MnO2, an amorphous crystalline form of hexagonal birnessite, is quite reactive with respect to As(III) oxidation. However, during reactions with As(III), the δ-MnO2 surface is usually passivated, i.e., the oxidation initially proceeds rapidly followed by a significant decrease in the oxidation rate.20–22 The passivation of δ-MnO2 is primarily caused by the retention of the reaction products, including the sorption of Mn(II) and As(V) and the formation of Mn(III).20,26 The sorption of As(V) and Mn(II) at the edge sites, which are the location at which As(III) oxidation occurs, can cover the reactive Mn(IV) sites, thus physically blocking them.21,27 In addition, the adsorption of Mn(II) onto the δ-MnO2 surface may undergo a cascade of reactions that gradually lead to the formation of less reactive Mn(III) sites as a result of Mn(II)/Mn(IV) comproportionation.26,28,29 Another possible passivation pathway is the formation of Mn(II) arsenate precipitates on the δ-MnO2 surface.27
The surface passivation of δ-MnO2 during the oxidation of Cr(III) and U(IV) has also been reported in the literature,30,31 but the related data for Sb(III) oxidation are rather limited. The decrease in δ-MnO2 reactivity when it reacts with Cr(III) can be attributed to the surface precipitation of Cr(OH)3, which coats the δ-MnO2 surface, whereas U(VI) adsorption on the mineral surface can decrease the reactivity of δ-MnO2 during U(IV) oxidation.30,31 Although surface passivation has been observed for many toxic metals, such as As(III) and Cr(III) (including metalloids), during the oxidation process, to the best of our knowledge, a study that focuses on the mechanisms that control Sb(III) oxidation by δ-MnO2 has not been published.
This study aims to investigate the oxidation behavior and mechanisms of Sb(III) during its reaction with δ-MnO2. The kinetics of the reaction of Sb(III) with δ-MnO2 were examined and compared with those of As(III) using batch kinetic experiments. A combined microscopic and spectroscopic approach, including X-ray photoelectron spectroscopy (XPS), scanning electron microscopy coupled with energy dispersive spectroscopy (SEM-EDS), Fourier transform infrared spectrometry (FTIR), Raman spectroscopy, and X-ray diffraction (XRD), was used to characterize the changes in the δ-MnO2 mineral composition and the properties involved in the As(III) and Sb(III) oxidation process and to identify the newly formed secondary precipitates. In addition, the oxidative mechanism and passivation behavior of As(III) and Sb(III) during their respective reactions with δ-MnO2 were discussed.
2. Materials and methods
2.1 Chemicals and materials
All chemicals used in this study were of analytical grade or guaranteed grade. Sodium arsenite (NaAsO2) (>98.0%) and sodium arsenate (Na3AsO4·12H2O) (>98.0%) were obtained from Xiya Reagent Co., Ltd, China. Potassium antimony(III)-tartrate hemihydrate (>99.0%), antimony pentoxide (>98.0%), and potassium pyroantimonate (K2H2Sb2O7·4H2O) (>98.0%) were purchased from Sinopharm Chemical Reagent Co., Ltd., China. The stock solutions of As(III) and Sb(III) (10 mM) and working solutions of various concentrations were prepared using deionized water.The δ-MnO2 used in this study was prepared following the methods described in a previous study.32 The properties of δ-MnO2 were analyzed using XRD, SEM-EDS, and a particle size analysis and the detailed results were published in our recent report.24
2.2 Batch kinetic experiments
Batch experiments to evaluate As(III) and Sb(III) oxidation by δ-MnO2 were conducted in 500 mL Erlenmeyer flasks. A series of As(III) or Sb(III) stock solutions was added to 300 mL of the solution containing δ-MnO2. The initial As(III) or Sb(III) concentrations ranged from 100 to 1000 μM (100, 200, 500 and 1000 μM), and the dosage of δ-MnO2 was 100 mg L−1. The background electrolyte was 0.01 M KCl, and the pH of all test solutions during the kinetic experiments was maintained at 5.0 using a 0.1 M acetic acid and sodium acetate buffer system. All reaction flasks for the kinetic experiments were sealed and then shaken using a reciprocating shaker at 200 rpm and 25 °C. At different time intervals (0, 5, 10, 15, 20, 25, 30, 45 and 60 min), 2 mL samples of the suspension were collected and filtered through 0.22 μm hydrophilic polyvinylidene fluoride membrane filters (Durapore PVDF, Millipore) to quench the oxidation reaction. All filtered samples were stored at 4 °C in the dark prior to instrumental analysis. The kinetic experiments were performed in triplicate. All glassware used in this study was soaked overnight in 10% HNO3 and washed thoroughly with deionized water before use.2.3 Solid phase analysis
After the kinetic experiments, the δ-MnO2 suspensions that were exposed to various As(III) and Sb(III) concentrations were centrifuged at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 10 min. The residual samples were washed three times with deionized water and freeze dried at −60 °C for 12 h in a vacuum freeze dryer (FD-1B-50, Shanghai BiLang Instruments Co., Ltd. China). The freeze-dried samples were further analyzed using XPS, SEM-EDS, XRD, FTIR, and Raman spectroscopy.
000 rpm for 10 min. The residual samples were washed three times with deionized water and freeze dried at −60 °C for 12 h in a vacuum freeze dryer (FD-1B-50, Shanghai BiLang Instruments Co., Ltd. China). The freeze-dried samples were further analyzed using XPS, SEM-EDS, XRD, FTIR, and Raman spectroscopy.
SEM images of the original δ-MnO2 and δ-MnO2 after reacting with As(III) or Sb(III) were recorded using a Zeiss Super 55 VP SEM coupled with a Bruker XFlash 5010 EDS system. The samples were fixed onto the SEM stage prior to SEM-EDS analysis using double-sided adhesive carbon tape. A thin gold coating (<5 nm) was sputtered onto the carbon to prevent surface charging during the SEM analyses. The SEM images were acquired at magnifications ranging from 1000× to 100000× with an accelerating voltage of 20 kV. The coated samples were also examined using the EDS system to obtain a rapid semi-quantitative elemental analysis. The EDS spectra were measured obtained at selected areas of the particles with a counting time of 60 s. The detection limit was approximately 0.1% (wt) for most elements and the relative percentage content of the elements was normalized. The amorphous state and any crystalline phases of δ-MnO2 before and after reacting with As(III) or Sb(III) were determined using an X-ray diffractometer (Bruker D8 advance, Germany). Approximately 100 mg of the sample was ground into a fine powder prior to the XRD measurements. The XRD spectra were recorded using a Cu Kα radiation source with an accelerating voltage of 40 kV and an electron-generating current of 30 mA. The diffraction angles were scanned from 10 to 80° (2θ) at 0.05° (2θ) per step. The possible crystalline phases and secondary products were identified according to the standard patterns published by the International Center for Diffraction Data.
To determine the various functional structures of δ-MnO2 involved in the As(III) or Sb(III) oxidation process, the samples were analyzed comparatively with the FTIR spectra. The spectra were recorded using a Thermo Nicolet Nexus 5700 spectrophotometer in the range of 4000–400 cm−1 with a resolution of 2 cm−1. Prior to the FTIR analysis, the freeze-dried samples were mixed and ground with spectrum-pure potassium bromide at a mass of 1% and then compressed into pellets. The spectrum of the pure potassium bromide was subtracted from each experimental spectrum to correct for the background. In addition, the pristine and reacted δ-MnO2 samples were further examined using a Renishaw microprobe RM2000 confocal Raman microscope equipped with 10× and 50× objectives. Raman spectra were collected at wavenumbers ranging from 50 to 4000 cm−1 at a nominal resolution of 1 cm−1 using a 532 nm laser for excitation. The samples were analyzed through a 50× microscope objective lens, and the acquisition time was set to 300 s with 2–4 accumulations. The sampling depths were 1.5–2.0 μm. A low laser power (1 mW) was selected to avoid mineral transformations and radiation damage during the Raman measurements. At least three independent measurements were carried out and the results did not exhibit significant differences.
XPS was used to quantify the surface element composition on the δ-MnO2 surface before and after reacting with Sb(III) and As(III). Prior to the XPS measurements, approximately 50 mg of each powder sample was adhered to the surface of double-sided adhesive tape and placed under vacuum. A Thermo Fisher Scientific ESCALAB 250Xi X-ray photoelectron spectrometer with a monochromatized Al Kα radiation source (1486.6 eV) at 200 W under a residual pressure of 1 × 10−9 mbar was used to record the data. A survey scan was collected using binding energies ranging from 0 to 1200 eV with a dwell time of 100 ms and a pass energy of 150 eV at a step of 1 eV per sweep. High-resolution spectra for As 3d, Sb 3d, Mn 3s, Mn 3p, Mn 2p and O 1s were obtained using a 20 eV pass energy at a step of 0.1 eV, and the dwell time was increased to 250 ms. To compensate for the charging effects, the spectra were calibrated C 1s as the conducting reference at a fixed value of 284.6 eV.
Curve fitting, data analysis and the quantification of the Mn 3p and O 1s high-resolution XPS spectra were performed using CasaXPS software (V2.3.17PR1.1). The spectra were well fitted using non-linear Shirley-type background subtraction with Flattened Lorentzian (FL) function to determine the optimal spectral baseline and peaks. The oxidation state of Mn was identified by analyses of the Mn 3s multiplet splitting and the position and shape of the Mn 3p regions. For determining the binding energy of the samples in the Mn 3p region, gold (Au) was vacuum-deposited on the Mn standards to charge reference the binding energy with respect to the Au 4f7/2 peak position (84.0 eV). After non-linear Shirley-type background subtraction, the data were normalized by comparison with Mn(II), Mn(III) and Mn(IV) reference standards. For each sample, a direction set method was conducted to adjust every fitting parameter (FWHM, the full width at half maximum; H, the peak height; E, the peak position center) and minimize the value of residual standard deviation (STD). The peak position and relevant parameters from curve fitting of Mn 3s, Mn 3p and O 1s high resolution spectra are listed in Tables S1–S3.†
The zeta potentials and particle sizes of the fresh δ-MnO2 were determined using a Malvern Zetasizer Nano analyzer (Malvern, UK). The BET surface of δ-MnO2 was examined using an ASAP 2010 specific surface area and porosity analyzer (Micromeritics, USA).
2.4 Determination of As, Sb and Mn(II) in solution
The concentrations of As(III/V) and Sb(III/V) in solution were determined using liquid chromatography combined with an AFS 830 hydride generation atomic florescence spectroscope (LC-HG-AFS) (Beijing Jitian, China). The inorganic As and Sb species were separated using a Hamilton PRP-X100 anion exchange column (250 mm × 4.1 mm, 10 μm) with a flow rate of 1 mL min−1 and a 100 μL sample injection volume. The mobile phases for the As(III)/As(V) and Sb(III)/Sb(V) separation were 15 mM (NH4)2HPO4 (pH adjusted to 6.0 using 10% formic acid) and 1 mM potassium hydrogen phthalate with 10 mM EDTA–2Na, respectively. The retention times of the As(III) and As(V) species were 2.54 min and 10.16 min, respectively, while those of Sb(III) and Sb(V) were 4.03 min and 2.63 min, respectively. The concentrations for the standard curves of As and Sb ranged from 10 to 100 μg L−1. The current-carrying and reducing agents used were 5% HNO3 (v/v) and 2% KBH4 (m/v) (prepared in 0.5% KOH), respectively. The recovery rates from the Sb(III), Sb(V), As(III) and As(V) analyses using the LC-HG-AFS system were 97.5%, 99.1%, 102.4% and 99.5%, respectively. The amounts of As or Sb in the solid during the oxidation process were determined by calculating the difference between the initial concentrations of As(III) or Sb(III) and the residual concentrations of As or Sb in solution with the reaction time. The Mn(II) concentration in solution was determined using manganese formaldehyde oxime spectrophotometry.333. Results and discussion
3.1 Kinetics of As(III) and Sb(III) oxidation by δ-MnO2
The kinetic experiments in this study were performed by reacting 100, 200, 500 and 1000 μM As(III) or Sb(III) with 100 mg L−1 δ-MnO2 at pH 5.0 (Fig. 1). The residual As(III) concentrations in solution decreased as a function of the reaction time, and the As(III) oxidation rate decreased with the increasing initial As(III) concentrations. The reaction process for Sb(III) oxidation showed a similar pattern. However, the oxidation rate of Sb(III) was more rapid than that of As(III).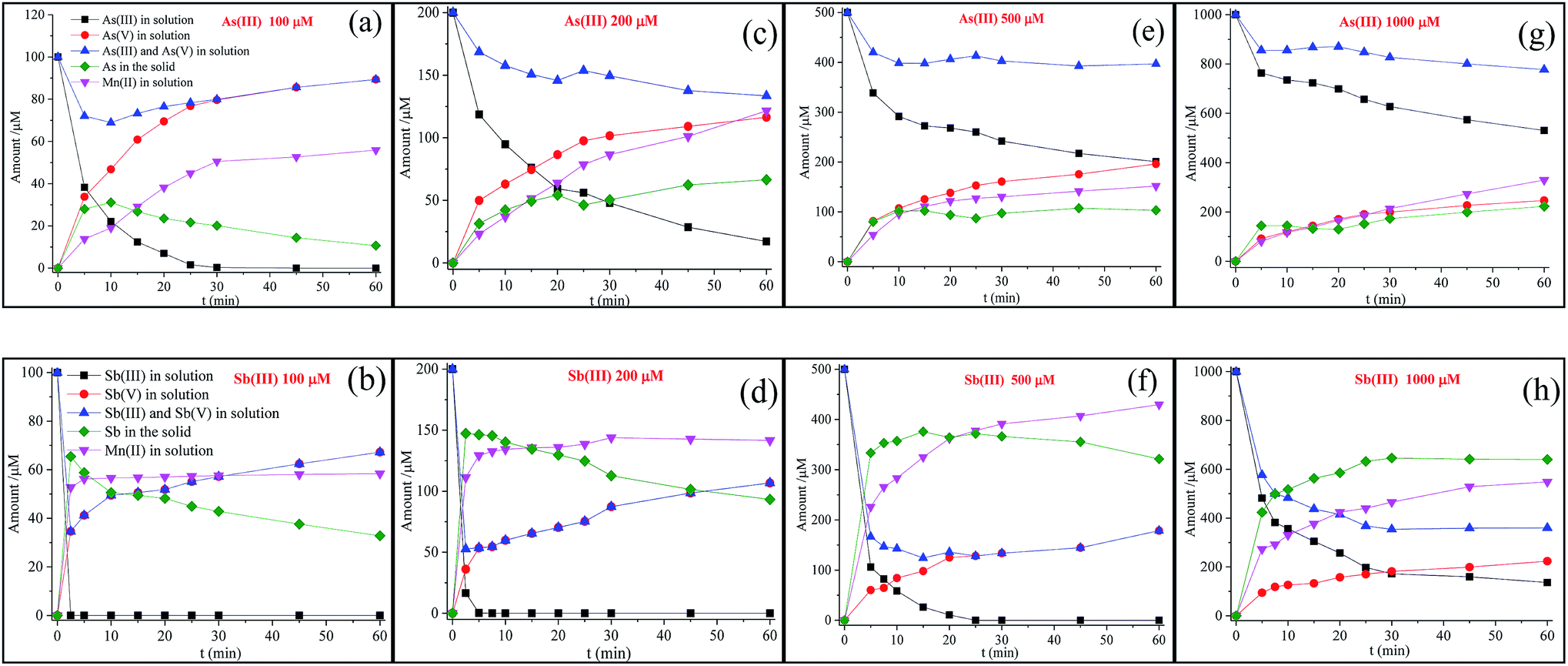 | ||
| Fig. 1 Changes in As and Sb species and the releasing of Mn(II) in the solution versus reaction time, where 100, 200, 500, and 1000 μM As(III) or Sb(III) reacted with 100 mg L−1 δ-MnO2 at pH 5.0. | ||
The first-order rate equation is frequently used to describe the depletion of As(III) by δ-MnO2 (eqn (1) and (2)).24,27
| ln(Ct/C0) = −kobst | (1) |
|
t1/2 = ln2/kobs
| (2) |
The kinetics data were generally well fitted for As(III) (R2 = 0.930–0.993) and Sb(III) (R2 = 0.870–0.996) after oxidation by δ-MnO2 (Table 1). The kobs values for the Sb(III) concentrations from 100 to 1000 μM were 6.14–44.71 times higher than those for As(III). In contrast, the half-life values for Sb(III) oxidation were only 2.20–15.97% of those of As(III). The disparities in the oxidation rates between Sb(III) and As(III) were possibly due to the significant surface passivation of δ-MnO2 during As(III) oxidation. During the oxidation process of As(III), As(V) and Mn(II) were produced according to the following reaction (eqn (3)).
| MnO2(s) + H3AsO3(aq) + H(aq)+ → Mn(aq)2+ + H2AsO4(aq)− + H2O(aq) | (3) |
| Items | Con. (μM) | kobs | t1/2 (min) | R2 |
|---|---|---|---|---|
| As(III) | 100 | 0.188 | 3.68 | 0.930 |
| 200 | 0.034 | 20.32 | 0.993 | |
| 500 | 0.009 | 80.58 | 0.932 | |
| 1000 | 0.007 | 100.43 | 0.987 | |
| Sb(III) | 100 | 2.923 | 0.23 | 0.996 |
| 200 | 1.520 | 0.45 | 0.870 | |
| 500 | 0.145 | 4.79 | 0.982 | |
| 1000 | 0.043 | 16.04 | 0.962 |
As shown in Fig. 1, at a low As(III) concentration (100 μM), the release of As(V) into the solution increased with the reaction time, and the concentrations of As(V) were significantly higher than those of Mn(II) in solution. This observation was attributed to the Mn(II) adsorption at the reactive sites, leading to the inhibition of As(V) adsorption.21 With the increasing initial As(III) concentration, the difference between the As(V) and Mn(II) concentrations diminished and eventually disappeared at a high As(III) concentration (1000 μM). These results suggest that once the δ-MnO2 reactive sites were completely occupied, freshly formed Mn(II) and As(V) tended to fully release into the solution at a mole ratio of 1:1. In addition, a slow oxidation rate was observed for δ-MnO2 with a high concentration of As(III). This result is consistent with the study reported by Lafferty et al. (2010a); in their study, during the As(III) reaction process, the oxidation rate significantly decreased but still continued in the Mn(II)-saturated δ-MnO2 system.26
As(III) must adsorb onto mineral surfaces prior to oxidation, but a number of studies have indicated that only As(V) can be retained at the reactive sites of δ-MnO2.20,21,26 In the high-resolution XPS spectra (Text S1–S9, Fig. 2 and S1 and Table S4†), the peaks correlated with the binding energies at 45.4 ± 0.1 eV and 540.4 ± 0.2 eV were assigned to As 3d and Sb 3d3/2, respectively, which indicated that only As(V) and Sb(V) were present on the surface of δ-MnO2.34,35
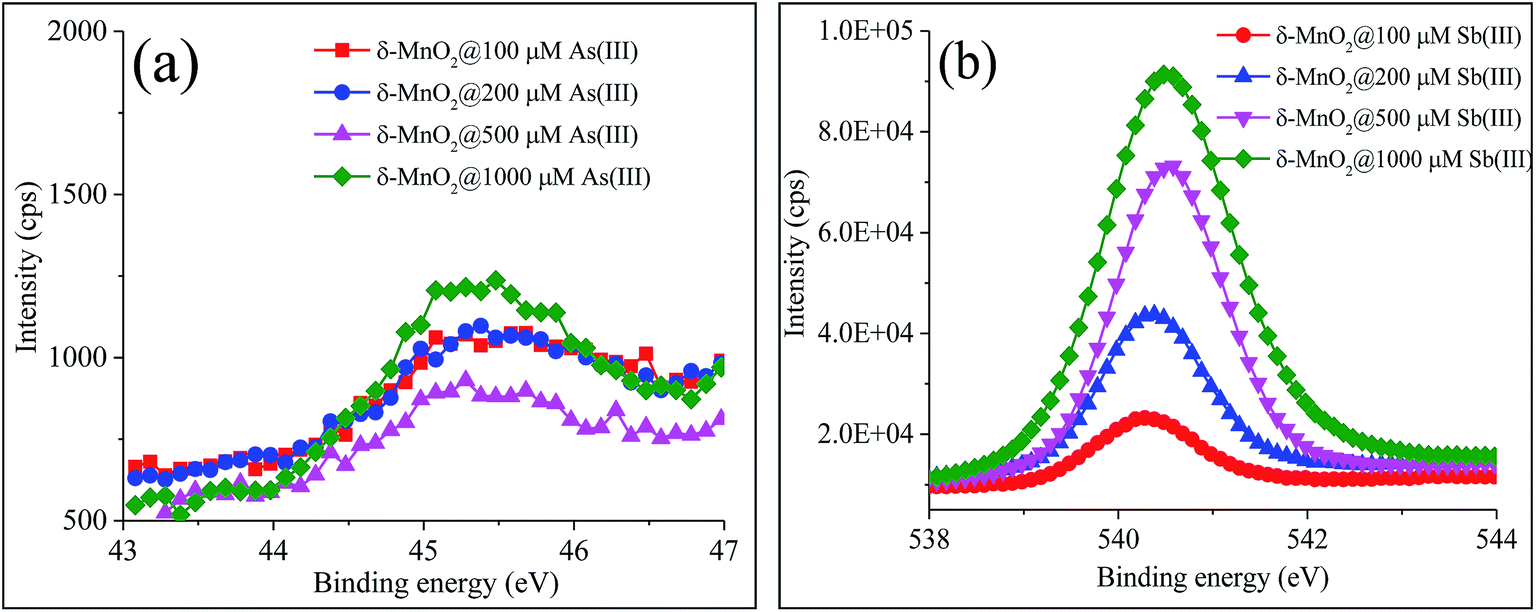 | ||
| Fig. 2 High-resolution XPS spectra of (a) As 3d and (b) Sb 3d3/2. | ||
The δ-MnO2 has two types of reactive sites: vacant sites within the Mn(IV) octahedral layers and edge sites at the Mn(IV) octahedral layer edges.36,37 As(III) oxidation and As(V) adsorption occur primarily at the edge sites, while Mn(II) tends to adsorb strongly at both the vacant and edge sites of δ-MnO2.27,38 The surface passivation of δ-MnO2 was primarily caused by the retention of secondary products such as Mn(II) on the δ-MnO2 reactive sites.26,39 The vacant sites were preferentially occupied by Mn(II) until all sites were occupied, and then, Mn(II) was expected to compete with As(V) for the edge sites. Previous studies have also indicated that Mn(II) adsorption on the δ-MnO2 surface is crucial for initiating passivation during the early stages of the reaction with As(III).21,26
For poorly crystallized δ-MnO2, the edge sites hold a relatively high proportion of the total particle charge, and thus, similar to As(III), the particle edge sites of δ-MnO2 may contribute significantly to Sb(III) adsorption prior to Sb(III) oxidation.20,27,40,41 After Sb(III) reacted with δ-MnO2 (Fig. 1), the release of Sb(V) and Mn(II) was quite different from that of the As(III) reaction. Although the oxidation rate of Sb(III) was faster than that of As(III), significantly less Sb(V) was released into the solution than As(V). The liberated Mn(II) rapidly achieved equilibrium (i.e., in less than 10 min) at low Sb(III) concentrations (100 μM and 200 μM), whereas the Mn(II) concentration increased continuously during the As(III) oxidation process. In addition, during the Sb(III) reaction with δ-MnO2, the Mn(II) concentrations in solution were clearly higher than those from As(III) oxidation. These results suggest that the mechanisms of the Sb(III) reaction with δ-MnO2 might be completely different from those of As(III) oxidation.
In addition, as shown in Fig. 1(a), at low As(III) concentrations, the adsorption of As(V) onto the solid quickly increased within 10 min, followed by a continuous decrease in As(V) adsorption until the end of the experiments. The decrease in the As(V) adsorption on the solid was attributed to the competition between As(V) and Mn(II) for the reactive sites. The changes in Sb(V) in the solid also exhibited a similar tendency with the Sb(III) concentrations of 100 and 200 μΜ (Fig. 1(b) and (d)). At high As(III) or Sb(III) concentrations (Fig. 1(e)–(h)), As(V) or Sb(V) in the solid slightly increased with the reaction time. The Sb(V) content in the solid increased more significantly than the As content (Fig. 2).
3.2 Mn oxidation state analysis
To better understand the mechanisms of the Sb(III) and As(III) reactions with δ-MnO2, the changes in the Mn oxidation states were investigated. The magnitude of the Mn 3s XPS multiplet splitting was used to determine changes in the δ-MnO2 oxidation state during the As(III) and Sb(III) reactions (Fig. S2 and S3 and Table S5†). The pristine δ-MnO2 sample showed multiplet splitting at 4.86 eV. According to the linear correlation between the Mn 3s multiplet splitting and the average oxidation state of Mn (Fig. S3†), the Mn in δ-MnO2 exhibited an average oxidation state at 3.56 eV, indicating the presence of Mn(III) and Mn(II) impurities.42,43 After reacting with As(III), the values for the Mn 3s multiplet splitting increased slightly with the increasing initial As(III) concentrations. The average Mn oxidation states ranged from 3.36 to 3.13, which coincided with the decrease in the Mn oxidation state from Mn(IV) to Mn(III/IV). The oxidation of 1000 μM Sb(III) by δ-MnO2 led to a more significant increase in the multiplet splitting from 4.86 to 5.82 than that observed in the As(III) reaction, indicating the rapid reduction of Mn(IV) to Mn(II).The changes in the relative content of Mn(IV, III and II) in the pristine and reacted δ-MnO2 were quantified by conducting a high-resolution XPS spectral analysis of the Mn 3p level (Fig. S4† and Table 2). The percent contents of Mn(IV), Mn(III) and Mn(II) in the pristine δ-MnO2 were 57.6%, 28.43% and 13.97%, respectively. After reacting with As(III) or Sb(III), the Mn 3p high-resolution spectra shifted toward a lower binding energy, indicating a decrease in the percentage of Mn(IV) on the δ-MnO2 surface. During the reaction of δ-MnO2 with various As(III) decrease in the percentage of, the relative content of Mn(III) and Mn(II) increased, while that of Mn(IV) decreased. These data were consistent with the average oxidation determined from fitting the Mn 3s multiplet splitting data. The increased concentration of Mn(II) on the δ-MnO2 surface was one of the major causes of early δ-MnO2 passivation. In addition, the formation of less reactive Mn(III) sites during the comproportionation reaction of the adsorbed Mn(II) with the original Mn(IV) was also a primary reason for the passivation of δ-MnO2. These results were consistent with those from previous reports, suggesting that the adsorption of Mn(II) and Mn(III) on the δ-MnO2 surface passivates δ-MnO2 and simultaneously decreases the As(III) oxidation rate.21,26
| Items | Con. (μM) | Mn(IV) (%) | Mn(III) (%) | Mn(II) (%) |
|---|---|---|---|---|
| δ-MnO2 | 0 | 57.6 | 28.43 | 13.97 |
| δ-MnO2@As(III) | 100 | 47.03 | 32.68 | 20.29 |
| 200 | 42.83 | 35.01 | 22.16 | |
| 500 | 34.29 | 37.54 | 28.17 | |
| 1000 | 32.03 | 39.35 | 28.64 | |
| δ-MnO2@Sb(III) | 100 | 42.15 | 34.65 | 23.20 |
| 200 | 41.76 | 33.25 | 24.99 | |
| 500 | 28.00 | 32.18 | 39.82 | |
| 1000 | 8.60 | 20.48 | 69.92 |
After reacting with Sb(III), a more significant decrease in the Mn(IV) percentage and an increase in the Mn(II) percentage was observed with the increasing initial Sb(III) concentration (Table 2). The percentage of Mn(IV) dropped to 8.60%, while that of Mn(II) reached 69.92% after reacting with 1000 μM Sb(III). More importantly, the Mn(III) and Mn(IV) content in δ-MnO2 proportionally decreased together with the increasing initial Sb(III) concentration. This trend was entirely different from that observed during the As(III) oxidation. The surface passivation of δ-MnO2 appeared to be the rate-limiting step for the As(III) oxidation, but it may not have been the limiting factor for controlling Sb(III) oxidation. Additional O 1s high-resolution spectra were collected to better understand the oxygen binding characteristics of δ-MnO2 during As(III) and Sb(III) oxidation.
The O 1s high-resolution spectra were divided into three peaks located at 530.0, 531.4 and 532.4 eV, which corresponded to lattice oxygen (Olatt), surface-absorbed oxygen (Oads) and chemisorbed oxygen species (OH2O), respectively44 (Fig. 3 and Table S6†). The predominant source of Olatt originated from the δ-MnO2 itself, and it changed during the oxidation process. Oads was primarily derived from As(III) and Sb(III) and thus existed as an experimental condition. After the reactions with Sb(III) and As(III), the percentage of Olatt significantly decreased along with the increasing percentage of Oads, indicating that the reactions of Sb(III) and As(III) with δ-MnO2 led to changes in the δ-MnO2 structure. The higher values of Oads/Olatt after Sb(III) oxidation than those obtained after As(III) oxidation were primarily attributed to the fact that more Sb(V) was retained on the δ-MnO2 surface. The percentage of OH2O in the δ-MnO2 changed slightly during the reactions with Sb(III) and As(III), indicating that the adsorbed H2O did not participate in the reaction between δ-MnO2 and Sb(III) or As(III).45
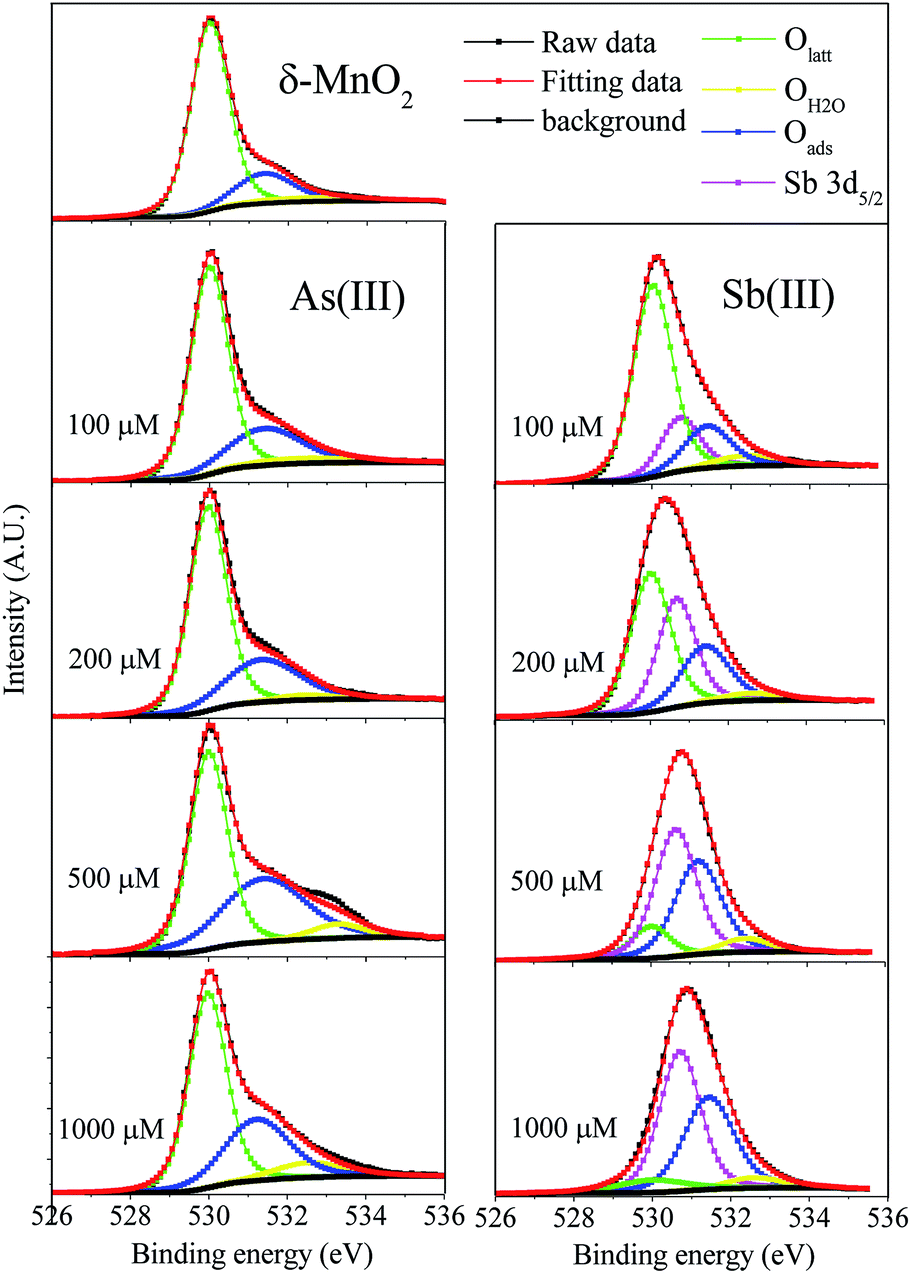 | ||
| Fig. 3 Fitting of O 1s XPS high resolution spectra for δ-MnO2 before and after reaction with As(III) and Sb(III). Olatt (surface lattice oxygen), OH2O (chemisorbed oxygen species) and Oads (surface absorb oxygen). | ||
3.3 δ-MnO2 property analysis
The changes in the properties of δ-MnO2 before and after the As(III) and Sb(III) oxidation were investigated using FTIR, Raman spectroscopy, SEM-EDS and XRD. The results are shown in Fig. 4 and 5.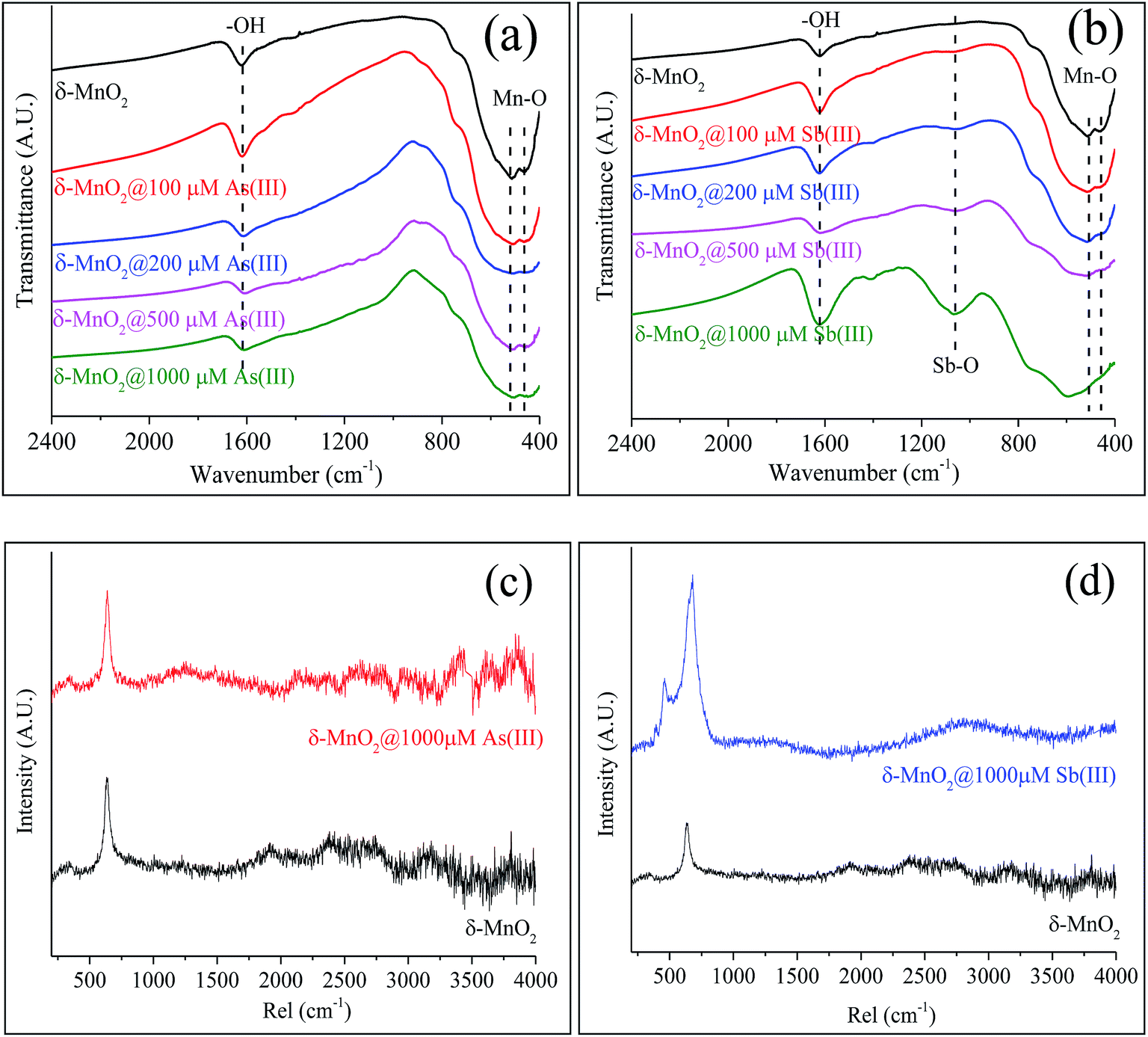 | ||
| Fig. 4 FTIR and Raman spectra of δ-MnO2 before and after reacting with As(III) and Sb(III). IR spectra of δ-MnO2 reacted with (a) As(III) and (b) Sb(III); Raman spectra of δ-MnO2 reacted with (c) As(III) and (d) Sb(III). | ||
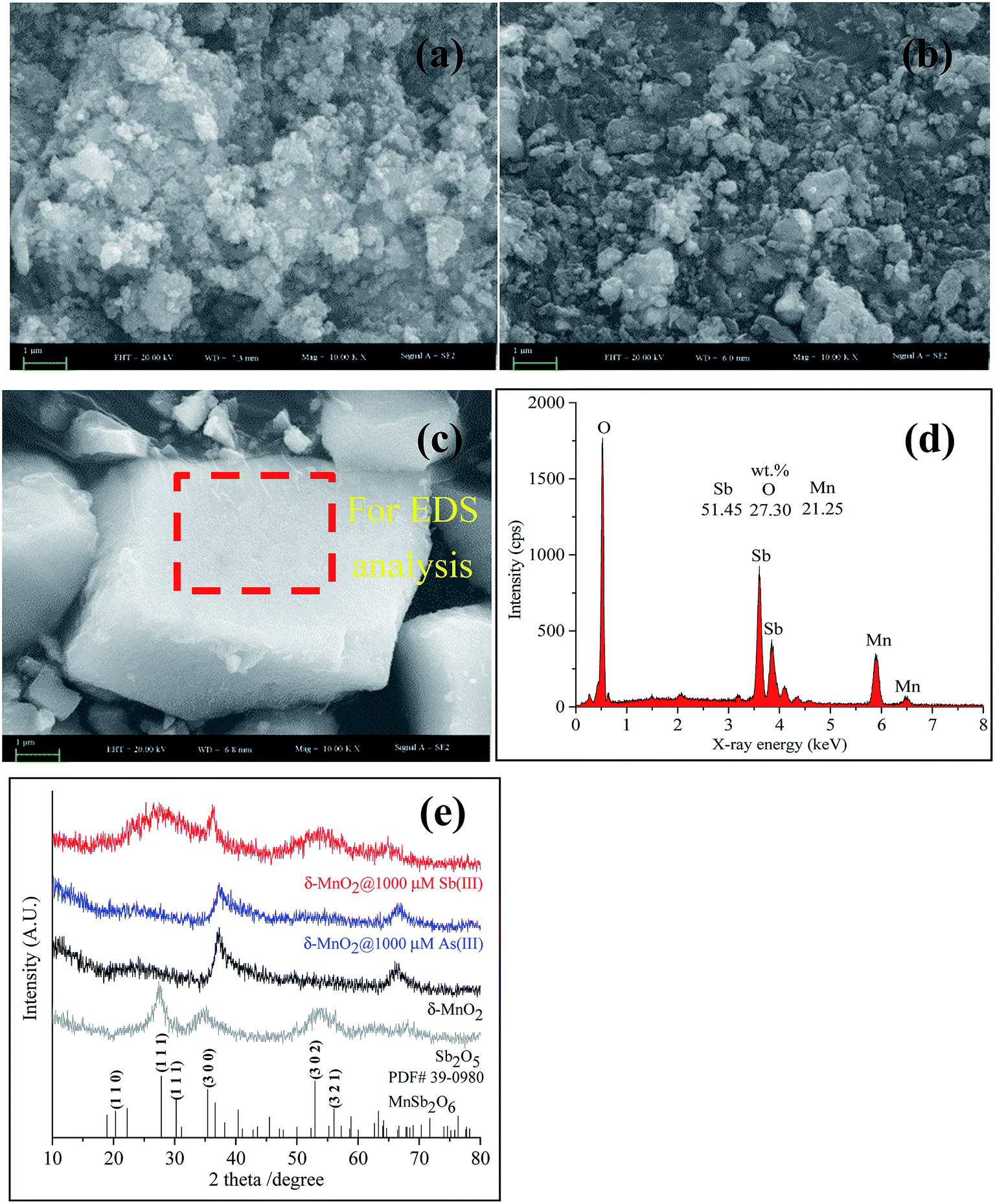 | ||
| Fig. 5 SEM-EDS and XRD of δ-MnO2 before and after reacting with As(III) and Sb(III). (a) SEM of pristine δ-MnO2; (b) SEM of δ-MnO2 reacted with As(III); (c) SEM and (d) EDS of δ-MnO2 reacted with Sb(III); (e) XRD patterns of δ-MnO2 before and after reacting with As(III) and Sb(III). | ||
FTIR and Raman spectra analyses were used to identify the functional groups of δ-MnO2 involved in the reactions with As(III) and Sb(III). As shown in Fig. 4a, several of the typical absorption bands for pristine δ-MnO2 that were observed in the absorption band at 1624 cm−1 were ascribed to the hydroxyl (–OH) groups of the physically adsorbed water molecules,46 and two broad features at 520 and 460 cm−1 were assigned to the Mn–O lattice vibration of the Mn oxides.47 The changes in the bond positions of δ-MnO2 in the FTIR spectra were insignificant during its reaction with various concentrations of As(III) (Fig. 4a). After the reactions between δ-MnO2 and Sb(III), significant changes in the FTIR spectra were observed (Fig. 4b). The Mn–O located in the “fingerprint zone” disappeared in the presence of high concentrations of Sb(III) (500 and 1000 μM). A new peak near 1060 cm−1 appeared, and the relative adsorption intensity increased with the Sb(III) concentration, which was ascribed to the formation of the Sb–O band (Fig. 4b and S5†). The spectral results indicate that remarkable changes to δ-MnO2 occurred during the reaction with Sb(III), whereas the δ-MnO2 structure was not significantly affected during the As(III) oxidation process.
Fig. 4c and d shows the Raman spectra of δ-MnO2 before and after reacting with As(III) and Sb(III). For the pristine δ-MnO2, the sharp peak near 640 cm−1 was assigned to the characteristic peak of the Mn oxides.48,49 Significant changes were not observed in δ-MnO2 during its reaction with 1000 μM As(III) (Fig. 4c), whereas the Raman spectra clearly changed after Sb(III) reacted with δ-MnO2 (Fig. 4d). The characteristic peak of the Mn oxides widened and became more intense, and a new peak located near 470 cm−1 appeared.
SEM coupled with EDS is a useful technique for identifying mineral structures, wherein major elements on the surface of the tested sample can be determined simultaneously. The high-magnification SEM image (Fig. 5a) reveals that the pristine δ-MnO2 particles were approximately 30–50 nm in diameter and poorly crystalline. The EDS spectrum showed that pristine δ-MnO2 was predominantly composed of O (48.72%, wt) and Mn (47.28%, wt) (data not shown). During the reaction of As(III) with δ-MnO2, the δ-MnO2 surface structure did not significantly change (Fig. 5b). A similar study also confirmed the particle morphology did not change after δ-MnO2 reacted with As(III), as shown in the transmission electron microscopy images.21 In addition, negligible amounts of As on the δ-MnO2 surface were detected using EDS (data not shown), similar to the results of the XPS survey analysis. After reacting with Sb(III), δ-MnO2 exhibited a well-crystallized cubic morphology with large particles (approximately 5.9 × 3.1 × 1.5 μm) (Fig. 5c) that consisted of elemental Sb (54.14%, wt), O (27.30%, wt) and Mn (21.25%, wt) (Fig. 5d). Distinct changes in the δ-MnO2 morphology and elemental composition were observed during the reaction process with Sb(III) that were not observed using As(III). These results also suggest that the surface morphology of δ-MnO2 did not significantly change after becoming passivated during the As(III) oxidation process.
The XRD patterns of the pristine δ-MnO2 exhibited only two main peaks at 37° and 66° (2θ) (Fig. 5e), which are characteristic of poorly crystalline phyllomanganates.50,51 The XRD patterns were slightly different for δ-MnO2 before and after reacting with As(III). However, changes in the XRD patterns were apparent after Sb(III) reacted with δ-MnO2. The two typical peaks for δ-MnO2 disappeared, and two broad peaks at approximately 27° (2θ) and 54° (2θ) and a sharp peak at 36.3° (2θ) appeared, indicating that the δ-MnO2 structures were altered after reacting with Sb(III). A further XRD analysis showed that the newly formed mineral was Mn(II) antimonate (MnSb2O6, PDF#39-0980). The results showed a similar crystal structure as Sb2O5, but low and broad peaks located at 27.5° and 54° (2θ) were observed. The crystal structure of Mn(II) antimonate could be related to the ratio of Mn/Sb. A similar study reported by Zhang et al. (2016) also confirmed that the formation of Mn(II) antimonate during reaction of Sb(III) with KMnO4.52 The high ratio of Mn/Sb contributes to a poorly crystallized structure with low and broad diffraction peaks. In addition, a cubic morphology Mn(II) antimonate with a particle size of 50–150 nm was observed by TEM analysis, which is in accordance with the SEM observations in Fig. 5c. These results suggesting that the δ-MnO2 properties changed more significantly during the reaction with Sb(III) than with As(III).
3.4 Mechanisms of the As(III) and Sb(III) reactions with δ-MnO2
The mechanisms of the As(III) oxidation by δ-MnO2 were well revealed. Namely, one mole of As(III) molecules reacted with one mole of Mn(IV) reactive sites to generate one mole of As(V) anions and one mole of Mn(II) ions (eqn (3)).20,21,27 In this study, the oxidation rate for Sb(III) by δ-MnO2 was faster than that for As(III). A decreasing As(III) oxidation rate was observed after the reaction of 100–1000 μM As(III); the decreased rate was mainly caused by the surface passivation of δ-MnO2 by secondary products. During the reactions with low concentrations of As(III) (100 and 200 μM), the releases ratio of As(V) and Mn(II) from the solution was not equal to the theoretical value of 1 (Fig. S6†). The ratio Mn(II)/As(V) was less than 1, which was primarily attributed to the adsorption of Mn(II) onto the δ-MnO2 surface. Mn(II) could compete with As(V) for the reactive sites, and when the reactive sites were occupied completely by Mn(II), freshly formed Mn(II) and As(V) tended to completely release into the solution at a mole ratio of 1:1. Increasing values of Mn(II)/As(V) approaching 1 were observed when using high concentrations of As(III) (500 and 1000 μM). Previous studies have indicated that the adsorption of Mn(II) onto the δ-MnO2 surface is one of the major causes of early δ-MnO2 passivation.21,26 Meanwhile, less reactive Mn(III) sites could be formed via comproportionation between the adsorbed Mn(II) and Mn(IV) at the δ-MnO2 surface, passivating the δ-MnO2.21,26 The results from the Mn 3s and 3p XPS analyses agreed with the increase in the Mn(II) and Mn(III) content on the δ-MnO2 surface. In addition, the macroscopic and spectroscopic analyses conducted using the SEM-EDS, XRD, FTIR and Raman spectra indicated that surface passivation did not significantly change the surface structure of δ-MnO2 during the reaction with As(III).
In contrast to the As(III) oxidation, surface passivation might be not have been a key factor in controlling the oxidation rate of Sb(III). The surface passivation of δ-MnO2 by the adsorption of Mn(II) did not contribute to the remarkable depletion of Mn(IV) or the change in the δ-MnO2 structure, but the Mn(IV) percentage decreased from 57.6% to 8.60% after reacting with 1000 μM Sb(III). Moreover, the increasing Mn(III) content on the δ-MnO2 surface could also induce surface passivation during the As(III) oxidation process, but the Mn(III) content decreased with the increasing initial Sb(III) concentration. This observation suggests that Mn(III) may have had the same ability as Mn(IV) to oxidize Sb(III). These results indicated that the surface passivation of δ-MnO2 might not have occurred during the oxidation of Sb(III).
According to the XRD results in this study and a previous study,53 the reaction of Sb(III) with δ-MnO2 can be expressed as shown in eqn (4).
| MnO2(s) + Sb(OH)3(aq) + H(aq)+ → 0.5Mn(aq)2+ + 0.5MnSb2O6(s) + 2H2O(aq) | (4) |
During the δ-MnO2 reaction with Sb(III), the transfer of two electrons was responsible for the Sb(III) oxidation, i.e., one mole of As(III) reacted with one mole of Mn(IV) to produce one mole of Sb(V) and one mole of Mn(II). As shown in Fig. S6,† the Mn(II)/As(V) ratios in solution during the Sb(III) oxidation were higher than those during the As(III) oxidation. According to the two-electron transfer reaction, the oxidation of one mole of Sb(III) produces one mole of Sb(V) and Mn(II), but the formation of one mole of precipitated Mn(II) antimonate requires one mole of Sb(V) and 0.5 mole of Mn(II) (eqn (4)). The unprecipitated Mn(II) could be absorbed onto the δ-MnO2 surface or released into the solution. At low Sb(III) concentrations, the low values of Mn(II)/Sb(V) were attributed to the adsorption of Mn(II) onto the δ-MnO2 surface. Due to the decrease in the number of Mn(IV) reactive sites, the adsorption of Mn(II) could be inhibited when reacting with high concentrations of Sb(III) (from 200 to 1000 μM). Interestingly, the maximum Mn(II)/Sb(V) ratios were observed in the presence of 500 μM Sb(III). This result was ascribed to the adsorption of Mn(II) onto the newly formed Mn(II) antimonate precipitates; this phenomenon reduced the concentration of Mn(II) in the solution. Previous studies have suggested that Mn(II) antimonate can effectively adsorb a series of divalent metals including Sr(II), Co(II), Ca(II) and Ba(II).52,54
In addition, Sb(V) was also released into the solution. This release was attributed to two causes. First, during the Sb(III) oxidation process, Sb(V) could adsorb onto the surface of δ-MnO2. A similar study reported by Wang et al. (2012) suggested that the oxidation of Sb(III) by manganite (γ-MnOOH) occurred within 10 min, and Sb(V) was absorbed by forming inner-sphere complexes at the manganite surface.55 Second, the dissolution of the Mn(II) antimonate precipitates might have contributed to the release of Sb(V) into the solution.
4. Conclusions
In geochemical processes, the migration and transformation of Sb and As are critically determined by minerals present in the environments. Poorly crystalline phyllomanganates such as δ-MnO2 can act as powerful oxidizers and scavengers for Sb(III) and As(III) in these environments. Therefore, understanding the retention mechanism between δ-MnO2 and Sb(III) or As(III) has important environmental implications. In this study, As(III) and Sb(III) were both oxidized by δ-MnO2, but the oxidation rate of Sb(III) was more rapid than that of As(III). The decreasing As(III) oxidation rate was primarily attributed to the surface passivation of δ-MnO2 by the adsorption of Mn(II) and the formation of Mn(III). In contrast, the surface of δ-MnO2 was not significantly passivated during the Sb(III) oxidation. The rapid oxidation of Sb(III) by δ-MnO2 favors the precipitation of Mn(II) antimonate rather than the complete release of Sb(V) into the solution. Thus, the difference between the Sb(III) and As(III) oxidation mechanisms in this study facilitated a better understanding of the environmental behavior of Sb and As on Mn oxide surfaces in aquatic environments.Acknowledgements
This work was financially supported by the Shandong Province Natural Science Foundation (Grant ZR2016DQ08), the CPSF-CAS Joint Foundation for Excellent Postdoctoral Fellows (Grant 2016LH0048), the China Postdoctoral Science Foundation funded project (Grant 2016M600829), and the National Natural Science Foundation of China (Grant 51678315).References
- D. Zhang, X. Pan, G. Mu and J. Wang, J. Appl. Phycol., 2010, 22, 479–488 CrossRef CAS.
- S. Wang, D. Zhang and X. Pan, Ecotoxicol. Environ. Saf., 2012, 84, 104–111 CrossRef CAS PubMed.
- L. Paoli, E. Fiorini, S. Munzi, S. Sorbo, A. Basile and S. Loppi, Chemosphere, 2013, 93, 2269–2275 CrossRef CAS PubMed.
- J. Warnken, R. Ohlsson, D. T. Welsh, P. R. Teasdale, A. Chelsky and W. W. Bennett, Chemosphere, 2017, 180, 388–395 CrossRef CAS PubMed.
- S. C. Wilson, P. V. Lockwood, P. M. Ashley and M. Tighe, Environ. Pollut., 2010, 158, 1169–1181 CrossRef CAS PubMed.
- S. K. Gupta and K. Y. Chen, J. - Water Pollut. Control Fed., 1978, 50, 493–506 CAS.
- S. Dixit and J. G. Hering, Environ. Sci. Technol., 2003, 37, 4182–4189 CrossRef CAS PubMed.
- J. Jiang, I. Bauer, A. Paul and A. Kappler, Environ. Sci. Technol., 2009, 43, 3639 CrossRef CAS PubMed.
- T. Gebel, Chem.-Biol. Interact., 1997, 107, 131 CrossRef CAS PubMed.
- B. A. Manning and S. Goldberg, Environ. Sci. Technol., 1997, 31, 2005–2011 CrossRef CAS.
- H. W. Nesbitt, G. W. Canning and G. M. Bancroft, Geochim. Cosmochim. Acta, 1998, 2097–2110 CrossRef CAS.
- D. W. Oscarson, P. M. Huang, C. Defosse and A. Herbillon, Nature, 1981, 291, 50–51 CrossRef CAS.
- P. Z. Ray and H. J. Shipley, RSC Adv., 2015, 5, 29885–29907 RSC.
- F. L. Fu, Z. H. Cheng and J. W. Lu, RSC Adv., 2015, 5, 85395–85409 RSC.
- D. W. Oscarson, P. M. Huang, W. K. Liaw and U. T. Hammer, Soil Sci. Soc. Am. J., 1983, 47, 644–648 CrossRef CAS.
- R. M. Weaver and F. M. Hochella, Am. Mineral., 2003, 88, 2016–2027 CrossRef CAS.
- K. D. Kwon, K. Refson and G. Sposito, Geochim. Cosmochim. Acta, 2010, 74, 6731–6740 CrossRef CAS.
- D. M. Sherman and C. L. Peacock, Geochim. Cosmochim. Acta, 2010, 74, 6721–6730 CrossRef CAS.
- J. P. Lefkowitz and E. J. Elzinga, Environ. Sci. Technol., 2015, 49, 4886–4893 CrossRef CAS PubMed.
- B. A. Manning, S. E. Fendorf, B. Bostick and D. L. Suarez, Environ. Sci. Technol., 2002, 36, 976 CrossRef CAS PubMed.
- B. J. Lafferty, M. Ginder-Vogel and D. L. Sparks, Environ. Sci. Technol., 2010, 44, 8460–8466 CrossRef CAS PubMed.
- G. Landrot, M. Ginder-Vogel, K. Livi, J. P. Fitts and D. L. Sparks, Environ. Sci. Technol., 2012, 46, 11594–11600 CrossRef CAS PubMed.
- J. F. Li, H. Gyoten, A. Sonoda, Q. Feng and M. Xue, RSC Adv., 2017, 7, 1490–1497 RSC.
- H. Wang, D. Zhang, S. Mou, W. Song, F. A. Al-Misned, M. M. Golam and X. Pan, Chemosphere, 2015, 136, 102–110 CrossRef CAS PubMed.
- L. Wang and H. Cheng, Environ. Sci. Technol., 2015, 49, 3473–3481 CrossRef CAS PubMed.
- B. J. Lafferty, M. Gindervogel, M. Zhu, K. J. T. Livi and D. L. Sparks, Environ. Sci. Technol., 2010, 44, 8467 CrossRef CAS PubMed.
- C. Tournassat, L. Charlet, D. Bosbach and A. Manceau, Environ. Sci. Technol., 2002, 36, 493 CrossRef CAS PubMed.
- M. Zhu, K. W. Paul, J. D. Kubicki and D. L. Sparks, Environ. Sci. Technol., 2009, 43, 6655 CrossRef CAS PubMed.
- B. J. Lafferty, M. Gindervogel and D. L. Sparks, Environ. Sci. Technol., 2011, 45, 9218–9223 CrossRef CAS PubMed.
- S. E. Fendorf and R. J. Zasoski, Environ. Sci. Technol., 1992, 26, 79–85 CrossRef CAS.
- Z. Wang, S. W. Lee, P. Kapoor, B. M. Tebo and D. E. Giammar, Geochim. Cosmochim. Acta, 2013, 100, 24–40 CrossRef CAS.
- M. Villalobos, B. Toner, J. Bargar and G. Sposito, Geochim. Cosmochim. Acta, 2003, 67, 2649–2662 CrossRef CAS.
- P. G. Brewer and D. W. Spencer, Limnol. Oceanogr., 1971, 16, 107–110 CrossRef CAS.
- D. Fullston, A. D. Fornasiero and J. Ralston, Langmuir, 1999, 15, 4530–4536 CrossRef CAS.
- J. P. Nair, R. Jayakrishnan, N. B. Chaure, A. Lobo, S. K. Kulkarni and R. K. Pandey, Thin Solid Films, 1999, 347, 39–45 CrossRef CAS.
- V. A. Drits, E. Silvester, A. I. Gorshkov and A. Manceau, Am. Mineral., 1996, 82, 946–961 Search PubMed.
- E. Silvester, A. Manceau and A. V. Drits, Am. Mineral., 1997, 82, 962–978 CAS.
- C. L. Peacock and D. M. Sherman, Chem. Geol., 2007, 238, 94–106 CrossRef CAS.
- Z. Wang and D. Giammar, ACS Symposium Series, Advances in the Environmental Biogeochemistry of Manganese Oxides, 2015, ch. 2, vol. 1197, pp. 29–50 Search PubMed.
- A. L. Foster Jr, G. E. Brown and G. A. Parks, Geochim. Cosmochim. Acta, 2003, 67, 1937–1953 CrossRef.
- A. Manceau, M. Lanson and N. Geoffroy, Geochim. Cosmochim. Acta, 2007, 71(1), 95–128 CrossRef CAS.
- P. S. Nico and R. J. Zasoski, Environ. Sci. Technol., 2001, 35, 3338–3343 CrossRef CAS PubMed.
- K. J. Livi, B. Lafferty, M. Zhu, S. Zhang, A. C. Gaillot and D. L. Sparks, Environ. Sci. Technol., 2012, 46, 970–976 CrossRef CAS PubMed.
- W. Si, Y. Wang, Y. Peng, X. Li, K. Li and J. Li, Chem. Commun., 2015, 51(81), 14977–14980 RSC.
- X. Ge, J. Liu, X. Song, G. Wang, H. Zhang, Y. Zhang and H. Zhao, Chem. Eng. J., 2016, 301, 139–148 CrossRef CAS.
- R. Liu, F. Liu, C. Hu, Z. He, H. Liu and J. Qu, J. Hazard. Mater., 2015, 300, 847–854 CrossRef CAS PubMed.
- R. M. Potter and G. R. Rossman, Am. Mineral., 1979, 64, 1199–1218 CAS.
- C. Julien, M. Massot, R. Baddour-Hadjean, S. Franger, S. Bach and J. P. Pereira-Ramos, Solid State Ionics, 2003, 159, 345–356 CrossRef CAS.
- N. Mironova-Ulmane, A. Kuzmin and M. Grube, J. Alloys Compd., 2009, 480, 97–99 CrossRef CAS.
- M. Villalobos, B. Lanson, A. Manceau, B. Toner and G. Sposito, Am. Mineral., 2006, 91, 489–502 CrossRef CAS.
- V. A. Drits, B. Lanson and A.-C. Gaillot, Am. Mineral., 2007, 92, 771–788 CrossRef CAS.
- L. Zhang, J. Wei, X. Zhao, F. Li, F. Jiang, M. Zhang and X. Cheng, Chem. Eng. J., 2016, 302, 733–743 CrossRef CAS.
- N. Belzile, Y. W. Chen and Z. Wang, Chem. Geol., 2001, 174, 379–387 CrossRef CAS.
- M. Abe, J. Inorg. Nucl. Chem., 1979, 41, 85–89 CrossRef CAS.
- X. Wang, M. He, C. Lin, Y. Gao and L. Zheng, Chem. Erde, 2012, 72, 41–47 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional information is noted in the main text of the current paper including Text S1–S9, Tables S1–S6, and Fig. S1–S6. See DOI: 10.1039/c7ra05939f |
| This journal is © The Royal Society of Chemistry 2017 |